DOI:
10.1039/C7RA02738A
(Paper)
RSC Adv., 2017,
7, 42597-42605
Fangchinoline accumulates autophagosomes by inhibiting autophagic degradation and promoting TFEB nuclear translocation
Received
6th March 2017
, Accepted 29th August 2017
First published on 4th September 2017
Abstract
Autophagy, an evolutionarily conserved cellular self-digestive process, is associated with different diseases and can be inhibited or induced by a series of agents. In this study, we reported that fangchinoline (FCL), an alkaloid from Stephania tetrandra S. Moore, increased the expression of LC3-II and the formation of GFP-LC3 puncta in non-small cell lung cancer (NSCLC) cells. Numerous yellow puncta were observed in mRFP–EGFP-LC3 stably expressed NSCLC cells under FCL treatment and the FCL-increased expression of LC3-II was not further increased after co-treatment with bafilomycin A1, an autophagy inhibitor, suggesting that FCL inhibits autophagic flux. Results of co-localization of GFP-LC3 with LysoTracker Red or lysosomal-associated membrane protein 1 indicated that FCL inhibits autophagosomes–lysosomes fusion. Furthermore, FCL decreased the activities of cathepsin B and cathepsin D and affected the cellular acidification. Interestingly, FCL also increased the nuclear translocation of transcription factor EB (TFEB), a master regulator of autophagic and lysosomal genes, and the mRNA expressions of TFEB-targeted genes, such as SQSTM1, MAP1LC3B, and UVRAG. Knockdown of TFEB by using small inference RNA decreased the FCL-induced expression of LC3-II and the formation of GFP-LC3 puncta. Overall, we reported that FCL increases the expressions of autophagy markers via both inhibition (inhibition of autophagosomes–lysosomes fusion and dysfunction of lysosome) and induction (promotion of TFEB nuclear translocation) of autophagy, providing insights into the better understanding of the complexity of agent-mediated autophagy.
Introduction
Autophagy is a dynamic self-digestion process that degrades damaged or unnecessary organelles and proteins in a lysosome-dependent manner.1 The program of autophagy mainly consists of initiation, elongation, fusion, and degradation processes, which are accurately directed by numerous autophagy-related (ATG) proteins.2 Under stimulation, the isolated membranes generate in the cytoplasm,2,3 elongate to form autophagosomes and encircle the degradative cargos through two ubiquitination-like systems, namely, the phosphatidylethanolamine-modified microtubule-associated protein light-chain 3 (LC3-II) system and the ATG12–ATG5–ATG16 system.2,3 The autophagosomes then fuse with lysosomes to form autolysosomes and subsequently degrade their cargos, which are dependent on the lysosomal functions.4 During the program of autophagy, the LC3-II protein is located on the surface of autophagosomes and are degraded after autophagosome–lysosome fusion.5 In higher eukaryotes, the expression of LC3-II can be used as a protein marker for assessing the occurrence of autophagy.5,6
An increasing numbers of studies suggested that autophagy plays an important role in different diseases, including cancer, neurodegeneration, as well as inflammation etc.1 Thus far, numerous autophagy regulators have been identified by using different methods,7,8 such as liensinine,9 oblongifolin C,10 and andrographolide11 have been considered as autophagy inhibitors, whereas isorhynchophylline12 and corynoxine13 are autophagy inducers. In previous studies, we have also successfully identified some autophagy inducers, like platycodin D,14,15 glycerrhetinic acid,16,17 licochalcone A,18 chelerythrine,19 and baicalein20 etc.
Fangchinoline (FCL), a bis-benzylisoquinoline alkaloid that isolated from the dried root of Stephania tetrandra S. Moore, presents a wide spectrum of biological activities, such as anti-cancer,21 neuroprotection,22 anti-inflammatory,23 as well as anti-hyperglycaemic activities24 etc. In this study, FCL was identified as an autophagy regulator in non-small cell lung cancer (NSCLC) cells. Interesting, the mechanism of FCL-regulated autophagy was through both inhibition and induction of autophagic flux.
Materials and methods
Reagents
FCL was obtained from the National Institutes for Food and Drug Control (Beijing, China). The compound was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 40 mM and stored at −20 °C. Bafilomycin A1 (BAF), puromycin, paraformaldehyde (PFA), bovine serum albumin (BSA), acridine orange (AO), rapamycin (RAP), triton™ X-100, and DMSO were purchased from Sigma (St. Louis, MO, USA). LysoTracker Red was purchased from Beyotime Biotechnology Corporation-Shanghai (Shanghai, China). Alexa Fluor® 647 lysosomal-associated membrane protein 1 (LAMP1) antibody was purchased from BioLegend (San Diego, CA, USA). Roswell Park Memorial Institute (RPMI) 1640 medium, Dulbecco's Modified Eagle's Medium (DMEM) medium, penicillin, streptomycin, fetal bovine serum (FBS), and phosphate-buffered saline (PBS) were obtained from Gibco Life Technologies (Grand Island, NY, USA). The primary antibodies, i.e., LC3, cathepsin B (CTSB), cathepsin D (CTSD), transcriptional factor EB (TFEB), histone H3, tubulin, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and the responsive secondary antibodies were obtained from Cell Signaling Technology Inc. (Beverly, MA, USA).
Cell line and cell culture
A549 and NCI-H1299 cells were obtained from the American Type Culture Collection (Rockville, MD, USA) and cultured in a RPMI 1640 medium supplemented with 10% (v/v) FBS and antibiotics (100 units per mL penicillin and 100 μg mL−1 streptomycin). Normal human embryonic lung fibroblast (HELF) cells were obtained from Nanjing KeyGen Biotech Co. Ltd. (Nanjing, Jiangsu, China) and cultured in a DMEM medium supplemented with 10% (v/v) FBS and 1% (v/v) antibiotics (100 units per mL penicillin and 100 μg mL−1 streptomycin). A549 cells with mRFP–EGFP-LC3 stable expression were cultured in a DMEM medium supplemented with 10% (v/v) FBS, antibiotics (100 units per mL penicillin and 100 μg mL−1 streptomycin), and 1.5 μg mL−1 puromycin. All cells were grown in a 5% CO2 incubator at 37 °C.
Western blot assay
After treatment with the indicated methods, cells were washed with PBS, collected, lysed in a radioimmunoprecipitation lysis buffer containing 1% protease inhibitor cocktail and 1% phenylmethanesulfonyl fluoride for 20 min at 4 °C, and then centrifuged (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g) for 20 min at 4 °C. The supernatant fraction was collected as protein and the concentrations of proteins were determined with BCA™ Protein Assay Kit (Pierce, Rockford, IL, USA). Equal amounts of proteins were separated by using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. The membranes were blocked in 5% non-fat dried milk in phosphate buffer saline-tween at room temperature for 1 h and then incubated with specific primary antibodies overnight at 4 °C. The membranes were incubated with the corresponding secondary antibodies at room temperature for 1 h. The specific protein bands were visualized with an electrochemiluminescence-advanced western blot detection kit (BD Biosciences, Bedford, MA, USA). The expression levels of proteins were obtained as the following steps: (1) the grey level of each indicated protein was obtained by using ChemiDocTM MP imaging system. (2) The ratio of indicated protein/GAPDH, tubulin, or histone H3 was calculated. (3) The fold of control was obtained by calculating “treatment group value”/“control group value”. (4) Triple independent experiments were studied and the mean ± standard error (SE) was calculated.
000g) for 20 min at 4 °C. The supernatant fraction was collected as protein and the concentrations of proteins were determined with BCA™ Protein Assay Kit (Pierce, Rockford, IL, USA). Equal amounts of proteins were separated by using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. The membranes were blocked in 5% non-fat dried milk in phosphate buffer saline-tween at room temperature for 1 h and then incubated with specific primary antibodies overnight at 4 °C. The membranes were incubated with the corresponding secondary antibodies at room temperature for 1 h. The specific protein bands were visualized with an electrochemiluminescence-advanced western blot detection kit (BD Biosciences, Bedford, MA, USA). The expression levels of proteins were obtained as the following steps: (1) the grey level of each indicated protein was obtained by using ChemiDocTM MP imaging system. (2) The ratio of indicated protein/GAPDH, tubulin, or histone H3 was calculated. (3) The fold of control was obtained by calculating “treatment group value”/“control group value”. (4) Triple independent experiments were studied and the mean ± standard error (SE) was calculated.
Immunofluorescent staining assay
A549 cells were transiently transfected with GFP-LC3 plasmid (supplied by Toren Finkel, addgene plasmid #24920 (ref. 25)) for 24 h with or without subsequent transfection with small inference RNA (siRNA) of TFEB for 24 h by using Lipofectamine™ 2000 transfection reagent (Invitrogen Corp., Carlsbad, CA, USA) according to the manufacturer's instructions. The cells were then treated with FCL. To detect the formation of GFP-LC3 puncta, the cells were fixed with 4% PFA for 30 min at room temperature and washed with PBS. To detect the co-localization of GFP-LC3 puncta with LysoTracker Red, the cells were incubated with LysoTracker Red (50 nM) in the dark for 30 min at 37 °C. To detect the co-localization of GFP-LC3 puncta with LAMP1 protein, the cells were fixed with 4% PFA in PBS for 30 min at room temperature, washed with PBS, permeabilized with 0.5% Triton™ X-100 in PBS for 20 min at room temperature, blocked in a blocking solution consisting of 5% BSA and 0.2% Triton™ X-100 for 60 min at room temperature, and labeled with Alexa Fluor® 647 LAMP1 antibody overnight at 4 °C. Immunofluorescent images were obtained with a confocal laser scanning microscope (Leica TCS SP8, Solms, Germany).
Cathepsin activity assay
CTSB and CTSD activities were determined by using commercial assay kits (K140-100 and K143-100, BioVision, Mountain View, CA, USA) in accordance with the manufacturer's instructions. After treatment with 5 or 10 μM FCL for 24 h, cells were collected and lysed in chilled cell lysis buffer for 10 min at 4 °C. Cell lysate was then transferred into 96-well plates and mixed with reaction buffer and substrate at 37 °C for 1.5 h. Samples were read at 400 nm excitation and 505 nm emission for CTSB activity and at 328 nm excitation and 460 nm emission for CTSD activity. The activities of CTSB and CTSD were normalized with the protein concentration.
AO staining assay
After treatment with the 5 or 10 μM FCL for 24 h, the cells were incubated with AO dye (1.5 μg mL−1) for 30 min at 37 °C. The fluorescent cell images were obtained with an IN Cell Analyzer 2000 (GE Healthcare, Little Chalfont, UK). Statistical results were obtained with a flow cytometer (FACS-Canto, BD Bioscience, USA). AO produces red fluorescence (emission peak at 650 nm) in acidic or lysosomal compartments and green fluorescence (emission peak between 530 and 550 nm) in cytosolic and nuclear compartments.
LysoTracker Red staining assay
After treatment with the 5 or 10 μM FCL for 24 h, the cells were stained with 50 nM LysoTracker Red for 90 min at 37 °C. After washing with PBS, a total of 10000 stained cells were collected and analyzed by using a flow cytometer (FACS-Canto, BD Bioscience, USA).
Isolation of cytoplasmic and nuclear fractions assay
The cytosolic and nuclear fractions were isolated by using a commercial assay kit (Beyotime, Shanghai, China) in accordance with the manufacturer's instructions. Following 10 μM FCL treatment, the cells were collected, washed, and re-suspended in 200 μL of cytoplasmic extraction buffer A on ice for 15 min. A total of 10 μL cytoplasmic extraction buffer B was added in cell lysates with vortex for 1 min followed by subsequent centrifugation at 14000g for 5 min. The supernatant was the cytoplasmic extract. Before the cell lysates were centrifuged at 14000g for 10 min, the pellet (containing nuclei) was re-suspended in 50 μL of nuclear extraction buffer with vortex at 4 °C for 30 min. The supernatant liquid was the nuclear extract. The concentrations of proteins were determined with BCA™ Protein Assay Kit (Pierce, Rockford, IL, USA). Western blot assay was performed to determine the expression levels of the mentioned proteins.
Quantitative real-time polymerase chain reaction (qPCR) assay
The mRNA expressions of SQSTM1, MAP1LC3B, and UVARG were detected by using qPCR assay. After the cells were treated with 10 μM FCL for 3 h, the total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA). The RNA was then reverse-transcribed into the single-stranded cDNA with a SuperScript™ III first strand cDNA synthesis kit (Toyobo, Japan). qPCR was performed through SYBR Green PCR Master Mix (Life Technologies) on a Stratagene Mx3005P multiplex quantitative PCR system (Agilent Technologies). The primers that used in this study were obtained from Invitrogen Life Technologies (Shanghai, China) and listed in Table 1.26–28
Table 1 The related PCR primers
| Gene |
Forward |
Reverse |
| SQSTM1 |
5′-AGAGACAGCTCAGCAGCTCCT-3′ |
5′-GCCTTGTCAGCCTCCATCAG-3 |
| MAP1LC3B |
5′-ACCATGCCGTCGGAGAAG-3′ |
5′-ATCGTTCTATTATCACCGGGATTTT-3′ |
| UVARG |
5′-TGACAATTCGTTGCAGGCAGTTA-3′ |
5′-AGGCAACTTGACACCGCATACA-3′ |
| GAPDH |
5′-GCGACACCCACTCCTCCACCTTT-3′ |
5′-TGCTGTAGCCAAATTCGTTGTCATA-3′ |
siRNA transfection assay
The specific target sequences of TFEB (sense: 5′-GGAGACGAAGGUUCAACAUTT-3′, antisense: 5′-AUGUUGAACCUUCGUCUCCTT-3′) and scrambled siRNA (sense: 5′-UUCUCCGAACGUGUCACGUTT-3′, antisense: 5′-ACGUGACACGUUCGGAGAATT-3′) were synthesized by GenePharma (Shanghai, China). A549 cells were transfected with specific siRNA of TFEB or scrambled by using Lipofectamine™ 2000 transfection reagent (Invitrogen Corp., Carlsbad, CA, USA) in strict accordance with the manufacturer's instructions. The cells were then incubated with 10 μM FCL for 3 h. The expression levels of the mentioned proteins were determined via Western blot assay, and the formation of GFP-LC3 puncta were detected via immunofluorescent staining assay.
Statistical analysis
The mean ± SE was determined for each group. Statistical analysis was performed with one-way analysis of variance of Tukey's test and unpaired t-test. Differences were considered statistically significant for (*) P < 0.05 and (**) P < 0.01. The “ns” means “no statistical difference”.
Results
FCL triggers autophagy in NSCLC cells
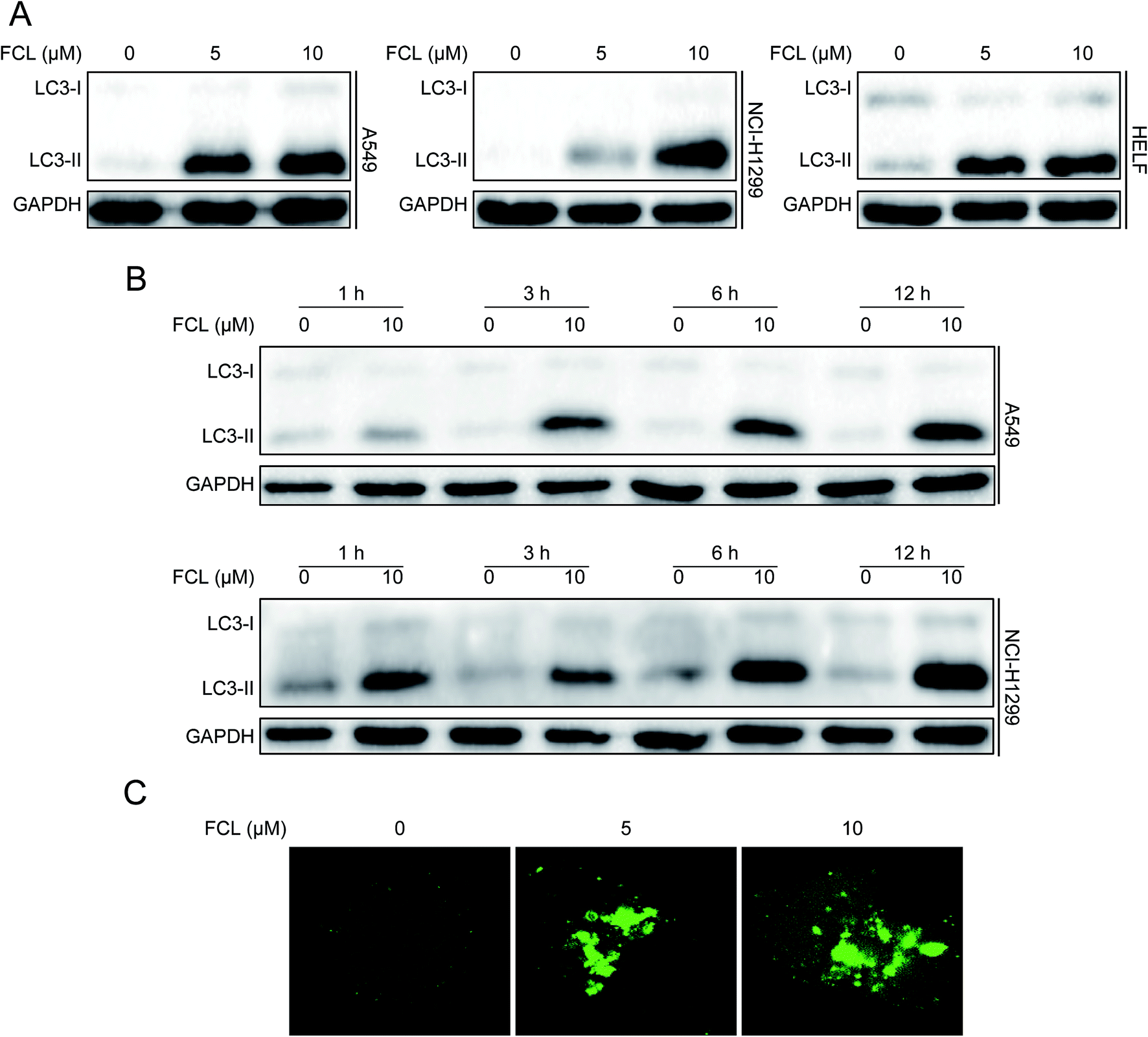
Increasing the protein expression of LC3-II is essential for autophagosome formation and usually used as a protein marker for autophagy happen.5,6 Therefore, we initially assessed the expression of LC3-II protein under FCL treatment by using Western blot assay. As shown in Fig. 1A and B, FCL increased the protein level of LC3-II in concentration- and time-dependent manners in NSCLC A549 and NCI-H1299 cells. FCL also increased the accumulation of LC3-II in HELF cells (Fig. 1A). Another reliable marker of autophagy is the formation of GFP-LC3 puncta.29 The cells were transiently transfected with GFP-LC3 plasmid before FCL treatment. As shown in Fig. 1C, FCL increased the accumulation of GFP-LC3 puncta in A549 cells in a concentration-dependent manner. These results indicated that FCL triggers autophagy in NSCLC cells.
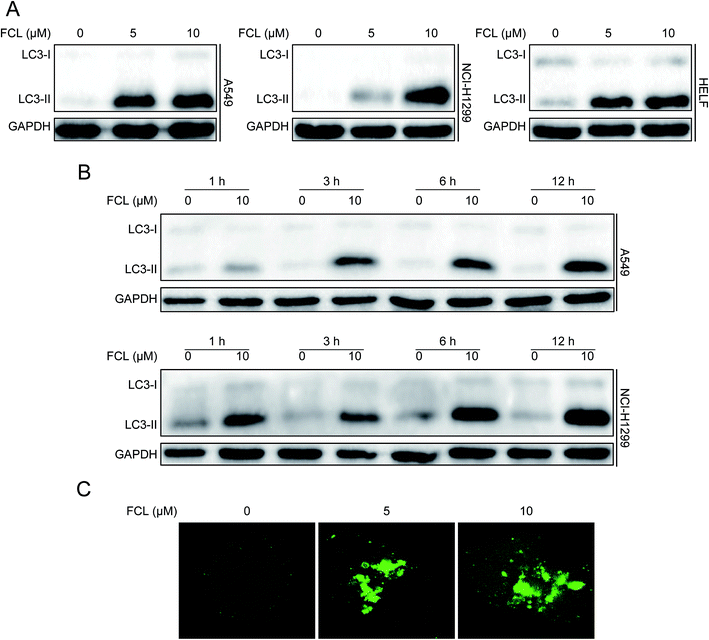 |
| | Fig. 1 FCL triggers autophagy in NSCLC. (A) Cells were treated with different concentrations of FCL for 24 h. Cellular proteins were extracted and analyzed to determine the expressions of LC3 and GAPDH by using Western blot assay. (B) A549 and NCI-H1299 cells were incubated with 10 μM FCL at the indicated times, and cell extracts were analyzed to determine the expressions of LC3 and GAPDH through Western blot assay. (C) A549 cells were transiently transfected with GFP-LC3 plasmid for 24 h. 10 μM FCL was added into the cells and cultured for another 24 h. GFP-LC3 puncta were observed with a confocal microscope, and typical images were presented. Bar: 10 μm. | |
FCL inhibited autophagic flux in NSCLC cells
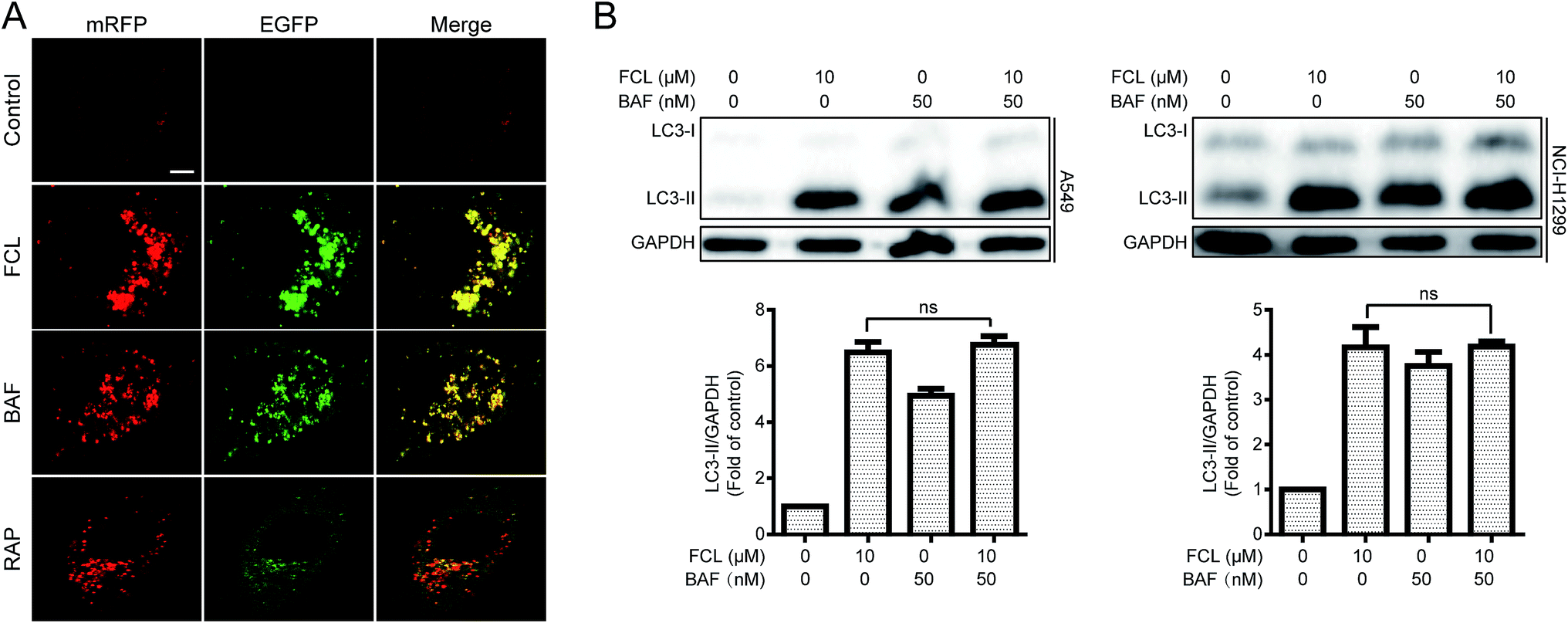
To determine the FCL-increased expression of LC3-II protein and the formation of GFP-LC3 puncta were due to induction or inhibition of autophagy process, the mRFP–EGFP-LC3 stably expressed A549 cells were used. When an agent induces autophagic flux (for example, RAP29), more red puncta are observed because of the easy-to-quench GFP and the relatively stable mRFP in acidic environment, such as autolysosomes and lysosomes. Meanwhile, inhibition of autophagosomes–lysosomes fusion and/or lysosomal function (for example, BAF30) results in the presentation of both green and red fluorescence of most puncta; these puncta show yellow fluorescence in the merged images.29 Similar to the result of BAF, FCL stimulated a large amount of yellow puncta, while RAP induced numerous red puncta in the mRFP–EGFP-LC3 stably expressed A549 cells (Fig. 2A). Moreover, the autophagy inducer-stimulated expression of LC3-II protein is further enhanced in combinative treatment with BAF, whereas the autophagy inhibitor-stimulated expression of LC3-II was not further increased by co-treatment with BAF.5 As shown in Fig. 2B, combinative treatment of FCL with BAF failed to further increase the FCL-induced expression of LC3-II protein in A549 and NCI-H1299 cells, suggesting that FCL inhibits autophagic flux in NSCLC cells.
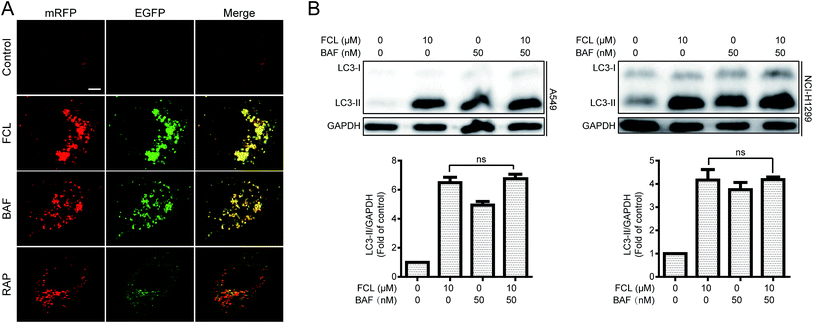 |
| | Fig. 2 FCL inhibited autophagic flux in NSCLC cells. (A) The mRFP–EGFP-LC3 stably expressed A549 cells were exposed to 10 μM FCL, 50 nM BAF, and 1 μM RAP for 24 h. The co-localizations of mRFP and EGFP-LC3 puncta were examined with a confocal laser scanning microscope. The typical images were shown. Bar: 10 μm. (B) A549 and NCI-H1299 cells were treated with 10 μM FCL for 24 h with or without BAF pretreatment (50 nM, 1 h). Cell extracts were analyzed for protein expression through Western blot assay. The “ns” means “no statistical difference”. | |
FCL inhibited autophagosome–lysosome fusion in NSCLC cells
At the late stage of autophagic flux, autophagosomes fuse with lysosomes to generate autolysosomes and then degrade cargos.4 To determine whether FCL inhibits autophagic flux through blockage of autophagosomes–lysosomes fusion, confocal microscopy assay was used to detect the co-localization of GFP-LC3 and LysoTracker Red, a specific dye for live cell lysosome labeling.31 As shown in Fig. 3A, similar to the result of BAF-treated cells, a remarkable separation of GFP-LC3 and LysoTracker Red was observed in FCL-exposed A549 cells. Meanwhile, GFP-LC3 was co-localized with LysoTracker Red in RAP-treated cells (Fig. 3A). The intensity of LysoTracker Red dye can be affected by changing the pH. Thus, the co-localizations of GFP-LC3 and LAMP1, a protein marker of lysosome,32 were determined through immunofluorescent assay to further confirm that FCL blocks autophagosomes–lysosomes fusion. As shown in Fig. 3B, as observed in BAF-exposed cells, FCL-treated cells also presented the separation of GFP-LC3 and LAMP1. Meanwhile, RAP induced the significant co-localizations of GFP-LC3 and LAMP1 (Fig. 3B). These findings suggested that FCL blocks autophagosomes–lysosomes fusion.
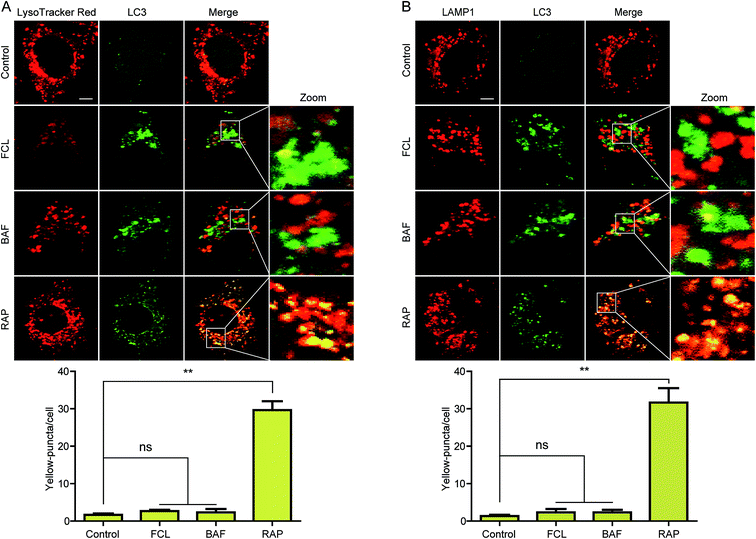 |
| | Fig. 3 FCL inhibited autophagosomes–lysosomes fusion in NSCLC cells. (A) A549 cells were transiently transfected with GFP-LC3 plasmid and treated with vehicle, FCL (10 μM), BAF (50 nM), and RAP (1 μM) for 24 h. The fluorescent signals were detected by confocal microscopy after staining with LysoTracker Red. Bar: 10 μm. (B) A549 cells were transiently transfected with GFP-LC3 plasmid and incubated with vehicle, FCL (10 μM), BAF (50 nM), and RAP (1 μM) for 24 h. Then, the cells were processed for LAMP1 immunostaining and observed with a confocal microscope. Typical images were presented. Bar: 10 μm. For quantification, at least 10 cells (per experiment) were randomly selected for counting the number of yellow puncta in each group. *P < 0.05, **P < 0.01. The “ns” means “no statistical difference”. | |
FCL inhibited the activities of cathepsins and affected lysosomal pH in NSCLC cells
Dysfunction of lysosome also results in the inhibition of autophagic flux. Cathepsins, such as CTSB and CTSD, are the major lysosomal proteases for degrading the lysosomal cargos and for maintaining lysosomal functions.33 In this study, we investigated whether FCL affects the expressions of mature CTSB and CTSD through Western blot assay, and results indicate that FCL decreases the expressions of mature CTSB and CTSD in A549 cells in a concentration-dependent manner (Fig. 4A). Moreover, the fluorescent substrate assay was applied to measure the enzymatic activities of CTSB and CTSD in FCL-treated A549 cells. As shown in Fig. 4B, FCL significantly decreased the enzymatic activities of CTSB and CTSD in a concentration-dependent manner, suggesting that the activities of CTSB and CTSD decreased in FCL-exposed A549 cells. The lysosomal pH is also required for lysosomal function.34 Therefore, an AO dye, which accumulates in the acidic or lysosomal vesicles and presents red fluorescence when excited by blue light,35 was applied to determine the effect of FCL on lysosomal pH. As shown in Fig. 4C, the red fluorescence was reduced in FCL-treated A549 cells. To eliminate the possibility of lowered intensity of red fluorescence caused by decreased AO loading, the ratio of red fluorescent and green fluorescent intensities was studied. Results indicate that FCL remarkably decreased the ratio of red fluorescent and green fluorescent intensities (Fig. 4D). In addition, the LysoTracker Red DND-99, another dye that can be used to evaluate the lysosomal pH,36,37 was further used. As shown in Fig. 4E and F, the fluorescence of LysoTracker Red was decreased after FCL treatment, suggesting that FCL affects the lysosomal pH in A549 cells.
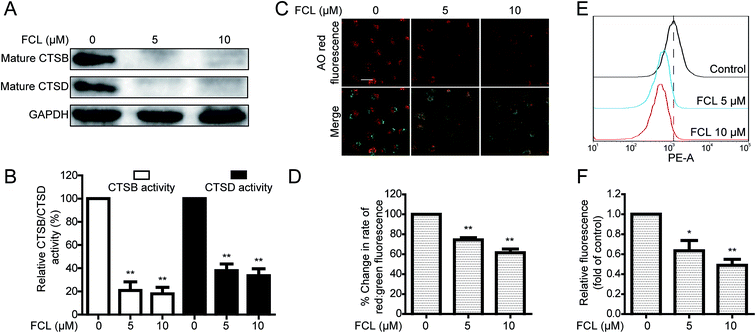 |
| | Fig. 4 FCL inhibited cathepsin activities and affected lysosomal pH in NSCLC cells. (A) A549 cells were treated with the indicated concentrations of FCL for 24 h, and the expression levels of the indicated proteins were evaluated through Western blot assay. (B) After treatment of A549 cells with different concentrations of FCL for 24 h, the activities of CTSB and CTSD were determined with a commercial analysis kit in accordance with the manufacturer's instructions. *P < 0.05 and **P < 0.01. (C and D) A549 cells were treated with 0, 5, and 10 μM FCL for 24 h and then stained with AO dye. The fluorescent cell images were obtained with an IN Cell Analyzer 2000, and statistical results were obtained with a flow cytometer. Bar: 50 μm. (E and F) A549 cells were treated with FCL for 24 h, and subsequently stained with LysoTracker Red. The fluorescent intense of cells were detected through flow cytometer. | |
The FCL-promoted nuclear translocation of TFEB partially contributed to FCL-increased autophagosomes formation in NSCLC cells
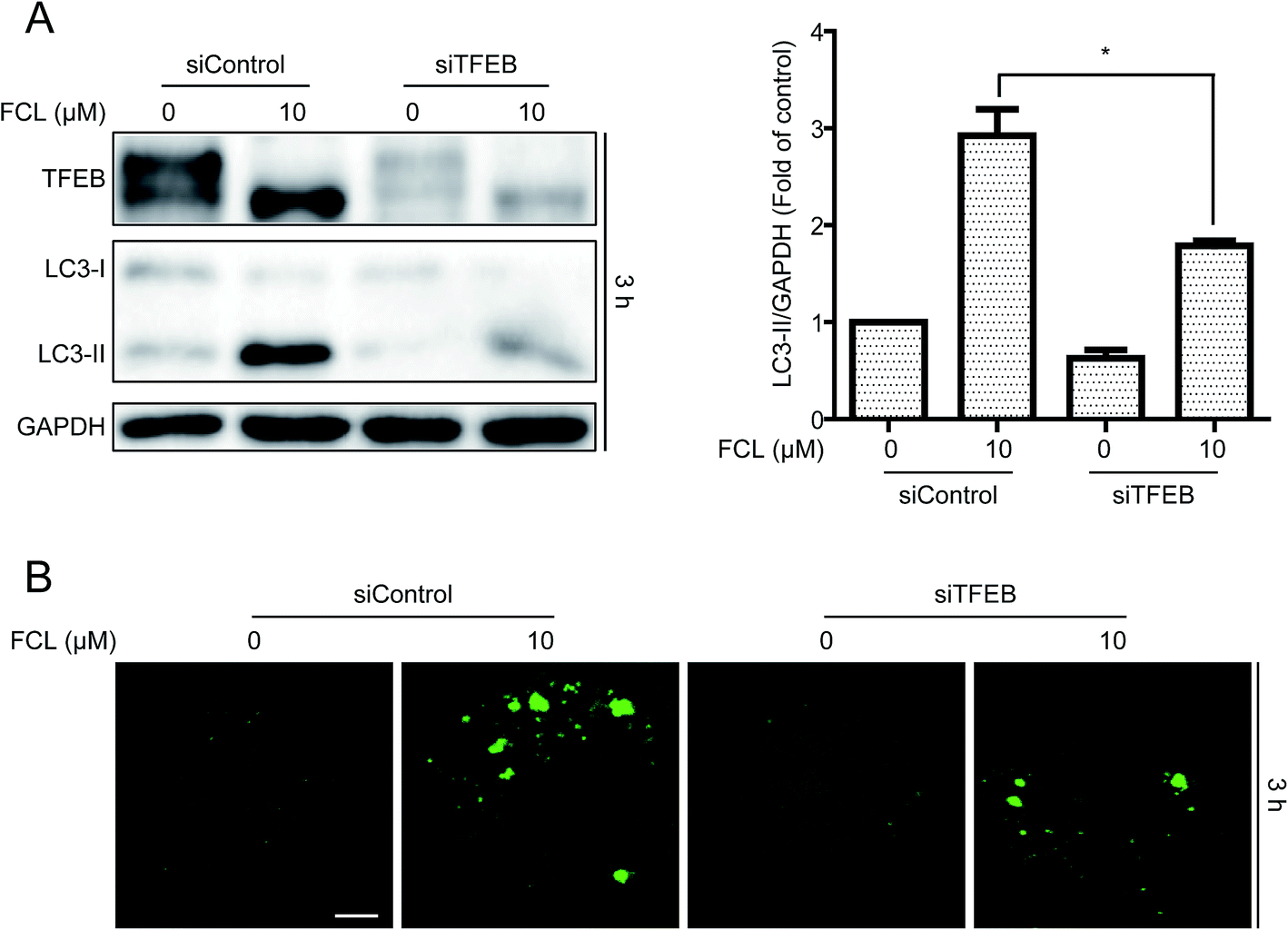
In addition to inhibition of autophagic degradation, we speculated whether the FCL-increased formation of autophagosomes is also due to the enhancement of autophagic flux. The TFEB, a master gene for autophagy and lysosomal biogenesis,38 was detected after treatment with FCL for various time points. As shown in Fig. 5A, FCL induced the downshift in TFEB molecular weight, especially in 3 h FCL treatment, and this phenomenon is associated with TFEB nuclear translocation and then expression of autophagic genes.38,39 However, this downshift in TFEB molecular weight was gradually attenuated after prolonging the FCL treatment. To assess whether FCL promotes the nuclear translocation of TFEB, the cytoplasmic and nuclear fractions were isolated after 3 h FCL treatment. As shown in Fig. 5B, Western blot assay indicated that FCL remarkably decreased the cytoplasmic TFEB protein level and increased the nuclear TFEB protein level in A549 and NCI-H1299 cells. Moreover, the mRNA levels of the TFEB-target genes, such as SQSTM1, MAP1LC3B, and UVARG,35 increased under FCL treatment for 3 h (Fig. 5C). To assess whether the FCL-promoted nuclear translocation of TFEB also contributes to FCL-accumulated autophagosomes, siRNA of TFEB assay was studied. As shown in Fig. 6A and B, knockdown of TFEB partly reversed the FCL-induced expression of LC3-II protein and formation of GFP-LC3 puncta under 3 h of FCL treatment. Results demonstrated that promotion of TFEB nuclear translocation also contributes to FCL-increased autophagosome accumulation.
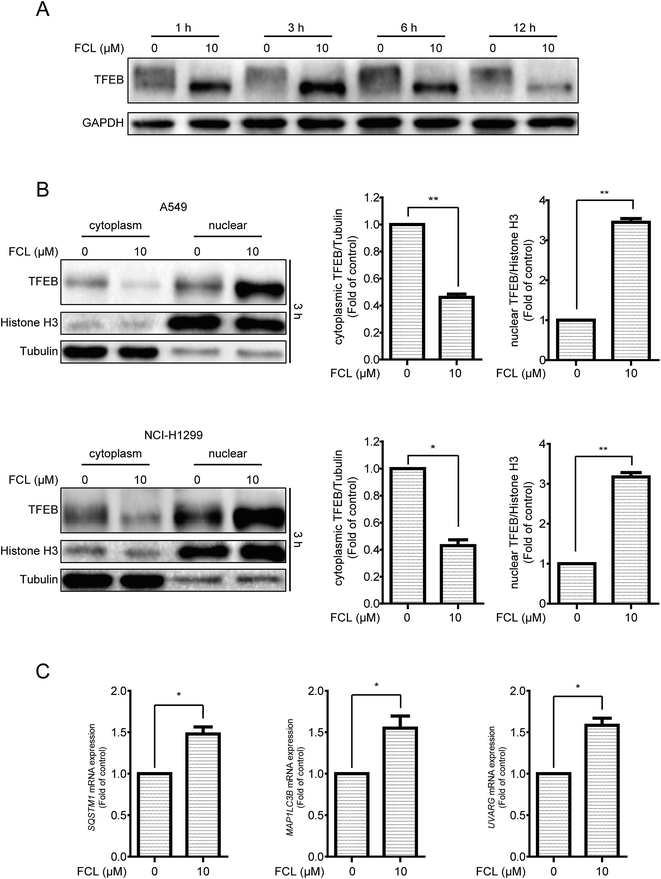 |
| | Fig. 5 FCL increased the nuclear translocation of TFEB and the expressions of TFEB-targeted genes in NSCLC cells. (A) Cells were treated with FCL (10 μM) at different times. The expression levels of TFEB and GAPDH proteins were evaluated through Western blot assay. (B) A549 and NCI-H1299 cells were treated with 10 μM FCL for 3 h; the cytosol and nuclear fractions were isolated with commercial assay kits in accordance with the manufacturer's instructions. The expression levels of the indicated proteins were evaluated by Western blot assay. *P < 0.05 and **P < 0.01. (C) A549 cells were treated with 10 μM FCL for 3 h. The mRNA levels of the indicated genes were determined by qPCR. *P < 0.05 and **P < 0.01. | |
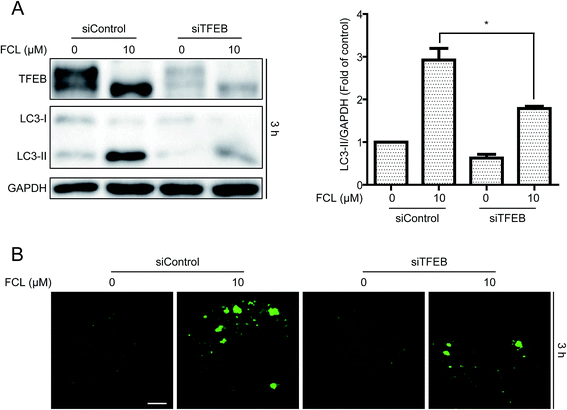 |
| | Fig. 6 Knockdown of TFEB reversed the FCL-induced expression of LC3-II protein and formation of GFP-LC3 puncta in NSCLC cells. (A) After transient transfection with scramble or TFEB siRNA for 24 h, A549 cells were treated with FCL (10 μM) for 3 h. The indicated protein expression was evaluated by Western blot assay. *P < 0.05 and **P < 0.01. (B) A549 cells were transiently transfected with GFP-LC3 for 24 h and TFEB siRNA for 24 h. Cells were then treated with FCL (10 μM) for 24 h. GFP-LC3 puncta were examined with a confocal microscope. Typical images were presented. Bar: 10 μm. | |
Discussion
Previous study indicated that FCL induces autophagy via the p53/sestrin2/AMPK pathway in hepatocellular carcinoma HepG2 and PLC/PRF/5 cells.40 We reported that FCL-regulated autophagy (inhibition or induction of autophagy) in NSCLC cells depends on the treatment time of FCL. TFEB coordinates the program of autophagy by increasing the expression of autophagic and lysosomal genes,38 such as SQSTM1, which is essential for binding the degradative cargos with autophagosomes,41 MAP1LC3B, which promotes the maturation of autophagosome,42 and UVARG, which interacts with Beclin 1 and initiates autophagy.43 Some autophagy stimulators, such as starvation, fisetin, and curcumin, induce autophagy by promoting the nuclear translocation of TFEB.38,44,45 In the early period of FCL treatment of NSCLC cells, the FCL-increased accumulation of autophagosomes was mainly due to the promotion of the nuclear translocation of TFEB and caused the expression of the ATG genes, including SQSTM1, MAP1LC3B, and UVARG. At the late stage of autophagy, the autophagosomes fuse with lysosomes to form autolysosomes and then degradation of cargos in autolysosomes in a lysosome-dependent manner.1 In the late period of FCL treatment, FCL increased the accumulation of autophagosomes mainly through the inhibition of autophagosomes–lysosomes fusion and dysfunction of lysosomes. Therefore, whether a compound is an autophagy inducer or inhibitor is a complex issue and depends on the treatment time. Thus far, numerous compounds have been identified as autophagy inducers or inhibitors at a specific time point. Whether these autophagy inducers or inhibitors have opposite effects on the regulation of autophagy at other time points remain unclear. Therefore, screening of autophagy inducers or inhibitors by using a real-time monitored method is necessary for further study.
Autophagy is a dynamic program consisting mainly of generation and degradation.2 Generation can be mediated through TFEB, PI3K, and ATG1 etc., whereas degradation mainly depends on autophagosomes–lysosomes fusion and lysosomal function.4 Inhibition of autophagy generation or degradation can remarkably inhibit autophagy; however, these two kinds of autophagy inhibition are quite different.29 When an autophagy inhibitor obstructs generation, the autophagosomes will not be generated and the cargos, for example, the damaged mitochondria, will not be surrounded by autophagosomes and remain exposed in the cytoplasm.29,46 By contrast, when an autophagy inhibitor blocks the degradation, the cargos can still be surrounded by autophagosomes.29 In this study, FCL promoted autophagosomes formation not only by blocking the degradative process of autophagy through inhibition of autophagosomes–lysosomes fusion and lysosomal function, but also by increasing the generative process of autophagy through promotion of TFEB nuclear translocation. Previous studies indicated that the accumulation of autophagosomes could increase the therapeutic effects of anti-cancer drugs.47–49 For example, accumulation of autophagosomes via treatment with the autophagy inhibitor chloroquine remarkably increased the anti-cancer effect of vorinostat, a histone deacetylase inhibitor, in the advanced solid tumors.49 However, the FCL-increased expression of LC3-II was also observed in the normal human embryonic lung fibroblast cells, suggesting that the phenomenon of FCL-regulated autophagy effect is not specific to cancer cells. Therefore, the side effects of combined treatment of FCL with the anti-cancer drugs should be considered.
In conclusion, we reported that FCL increased the expression of LC3-II protein and the formation of GFP-LC3 puncta by both stimulating the generation of autophagosomes through promotion of TFEB nuclear translocation and blocking the degradation of autophagosomes through inhibition of autophagosomes–lysosomes fusion and lysosomal function. Findings of this study may provide better understanding of agent-mediated autophagy.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by Science and Technology Development Fund, Macao S.A.R (FDCT) (038/2014/A1), the Research Fund of University of Macau (MYRG2015-00091-ICMS-QRCM and MYRG2015-00101-ICMS-QRCM).
References
- N. Mizushima, B. Levine, A. M. Cuervo and D. J. Klionsky, Nature, 2008, 451, 1069–1075 CrossRef CAS PubMed.
- J. H. Hurley and B. A. Schulman, Cell, 2014, 157, 300–311 CrossRef CAS PubMed.
- R. A. Nixon, Nat. Med., 2013, 19, 983–997 CrossRef CAS PubMed.
- D. J. Klionsky, K. Abdelmohsen, A. Abe, M. J. Abedin, H. Abeliovich and A. Acevedo Arozena, et al., Autophagy, 2016, 12, 1–222 CrossRef PubMed.
- N. Mizushima and T. Yoshimori, Autophagy, 2007, 3, 542–545 CrossRef CAS PubMed.
- D. C. Rubinsztein, A. M. Cuervo, B. Ravikumar, S. Sarkar, V. I. Korolchuk and S. Kaushik, et al., Autophagy, 2009, 5, 585–589 CrossRef CAS PubMed.
- H. Vakifahmetoglu-Norberg, H.-g. Xia and J. Yuan, J. Clin. Invest., 2015, 125, 5–13 CrossRef PubMed.
- Q. Ding, J. Bao, W. Zhao, Y. Hu, J. Lu and X. Chen, Phytochem. Rev., 2015, 14, 137–154 CrossRef CAS.
- J. Zhou, G. Li, Y. Zheng, H.-M. Shen, X. Hu and Q.-L. Ming, et al., Autophagy, 2015, 11, 1259–1279 CrossRef CAS PubMed.
- Y. Lao, G. Wan, Z. Liu, X. Wang, P. Ruan and W. Xu, et al., Autophagy, 2014, 10, 736–749 CrossRef CAS PubMed.
- J. Zhou, S.-E. Hu, S.-H. Tan, R. Cao, Y. Chen and D. Xia, et al., Autophagy, 2012, 8, 338–349 CrossRef CAS PubMed.
- J. H. Lu, J. Q. Tan, S. S. K. Durairajan, L. F. Liu, Z. H. Zhang and L. Ma, et al., Autophagy, 2012, 8, 98–108 CrossRef CAS PubMed.
- L. L. Chen, J. X. Song, J. H. Lu, Z. W. Yuan, L. F. Liu and S. S. K. Durairajan, et al., J. Neuroimmune Pharmacol., 2014, 9, 380–387 CrossRef PubMed.
- T. Li, X. h. Xu, Z. h. Tang, Y. f. Wang, C. h. Leung and D. l. Ma, et al., Acta Pharmacol. Sin., 2015, 36, 1503–1513 CrossRef CAS PubMed.
- T. Li, Z. H. Tang, W. S. Xu, G. S. Wu, Y. F. Wang and L. L. Chang, et al., Eur. J. Pharmacol., 2015, 749, 81–88 CrossRef CAS PubMed.
- Z. H. Tang, L. L. Zhang, T. Li, J. H. Lu, D. L. Ma and C. H. Leung, et al., Oncotarget, 2015, 6, 43911–43926 Search PubMed.
- Z. H. Tang, T. Li, L. L. Chang, H. Zhu, Y. G. Tong and X. P. Chen, et al., J. Agric. Food Chem., 2014, 62, 11910–11916 CrossRef CAS PubMed.
- Z. H. Tang, X. Chen, Z. Y. Wang, K. Chai, Y. F. Wang and X. H. Xu, et al., Sci. Rep., 2016, 6, srep26241 CrossRef PubMed.
- Z.-H. Tang, W.-X. Cao, Z.-Y. Wang, J.-H. Lu, B. Liu and X. Chen, et al., Redox Biol., 2017, 12, 367–376 CrossRef CAS PubMed.
- Y. F. Wang, T. Li, Z. H. Tang, L. L. Chang, H. Zhu and X. P. Chen, et al., Phytother. Res., 2015, 29, 674–679 CrossRef CAS PubMed.
- B. Guo, P. Xie, J. Su, T. Zhang, X. Li and G. Liang, Tumor Biol., 2016, 37, 2709–2719 CrossRef CAS PubMed.
- T. Y. Lin, C. W. Lu, L. T. Tien, S. H. Chuang, Y. R. Wang and W. H. Chang, et al., Neurochem. Int., 2009, 54, 506–512 CrossRef CAS PubMed.
- H. S. Choi, H. S. Kim, K. R. Min, Y. Kim, H. K. Lim and Y. K. Chang, et al., J. Ethnopharmacol., 2000, 69, 173–179 CrossRef CAS PubMed.
- T. Tsutsumi, S. Kobayashi, Y. Y. Liu and H. Kontani, Biol. Pharm. Bull., 2003, 26, 313–317 CAS.
- I. H. Lee, L. Cao, R. Mostoslavsky, D. B. Lombard, J. Liu and N. E. Bruns, et al., Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3374–3379 CrossRef CAS PubMed.
- Y. Xu, L. Jiang, J. Fang, R. Fang, H. C. Morse III and G. Ouyang, et al., J. Canc., 2015, 6, 953–961 CrossRef PubMed.
- L. Z. Liang, B. Ma, Y. J. Liang, H. C. Liu, G. S. Zheng and T. H. Zhang, et al., J. Oral Pathol. Med., 2012, 41, 621–629 CrossRef CAS PubMed.
- J. Kovsan, M. Blüher, T. Tarnovscki, N. Klöting, B. Kirshtein and L. Madar, et al., J. Clin. Endocrinol. Metab., 2010, 96, E268–E277 CrossRef PubMed.
- N. Mizushima, T. Yoshimori and B. Levine, Cell, 2010, 140, 313–326 CrossRef CAS PubMed.
- D. J. Klionsky, Z. Elazar, P. O. Seglen and D. C. Rubinsztein, Autophagy, 2008, 4, 849–850 CrossRef CAS PubMed.
- B. Lemieux, M. D. Percival and J. P. Falgueyret, Anal. Biochem., 2004, 327, 247–251 CrossRef CAS PubMed.
- E. L. Eskelinen, Mol. Aspects Med., 2006, 27, 495–502 CrossRef CAS PubMed.
- B. Turk, D. Turk and V. Turk, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2000, 1477, 98–111 CrossRef CAS.
- J. P. Luzio, P. R. Pryor and N. A. Bright, Nat. Rev. Mol. Cell Biol., 2007, 8, 622–632 CrossRef CAS PubMed.
- F. Traganos and Z. Darzynkiewicz, Methods Cell Biol., 1994, 41, 185–194 CrossRef CAS PubMed.
- K. Zheng, Y. Li, S. Wang, X. Wang, C. Liao and X. Hu, et al., Autophagy, 2016, 12, 1593–1613 CrossRef CAS PubMed.
- X. Zhao, Y. Fang, Y. Yang, Y. Qin, P. Wu and T. Wang, et al., Autophagy, 2015, 11, 1849–1863 CrossRef CAS PubMed.
- C. Settembre, C. Di Malta, V. A. Polito, M. G. Arencibia, F. Vetrini and S. Erdin, et al., Science, 2011, 332, 1429–1433 CrossRef CAS PubMed.
- D. L. Medina, S. Di Paola, I. Peluso, A. Armani, D. De Stefani and R. Venditti, et al., Nat. Cell Biol., 2015, 17, 288–299 CrossRef PubMed.
- N. Wang, W. Pan, M. Zhu, M. Zhang, X. Hao and G. Liang, et al., Br. J. Pharmacol., 2011, 164, 731–742 CrossRef CAS PubMed.
- S. Pankiv, T. H. Clausen, T. Lamark, A. Brech, J.-A. Bruun and H. Outzen, et al., J. Biol. Chem., 2007, 282, 24131–24145 CrossRef CAS PubMed.
- Y. Kabeya, N. Mizushima, T. Ueno, A. Yamamoto, T. Kirisako and T. Noda, et al., EMBO J., 2000, 19, 5720–5728 CrossRef CAS PubMed.
- C. Liang, P. Feng, B. Ku, I. Dotan, D. Canaani and B. H. Oh, et al., Nat. Cell Biol., 2006, 8, 688–698 CrossRef CAS PubMed.
- J. Zhang, J. Wang, J. Xu, Y. Lu, J. Jiang and L. Wang, et al., Oncotarget, 2016, 7, 75659–75671 Search PubMed.
- S. Kim, K. J. Choi, S. J. Cho, S. M. Yun, J. P. Jeon and Y. H. Koh, et al., Sci. Rep., 2016, 6, srep24933 CrossRef PubMed.
- K. Okamoto and N. Kondo-Okamoto, Biochim. Biophys. Acta, 2012, 5, 595–600 CrossRef PubMed.
- A. Thorburn, D. H. Thamm and D. L. Gustafson, Mol. Pharmacol., 2014, 85, 830–838 CrossRef PubMed.
- R. Rangwala, Y. C. Chang, J. Hu, K. M. Algazy, T. L. Evans and L. A. Fecher, et al., Autophagy, 2014, 10, 1391–1402 CrossRef CAS PubMed.
- D. Mahalingam, M. Mita, J. Sarantopoulos, L. Wood, R. K. Amaravadi and L. E. Davis, et al., Autophagy, 2014, 10, 1403–1414 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*