
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photodegradable hydrogels for external manipulation of cellular microenvironments with real-time monitoring†
Hanxu Ji a,
Kai Xib,
Qiuhong Zhangb and
Xudong Jia*a
a,
Kai Xib,
Qiuhong Zhangb and
Xudong Jia*a
aState Key Laboratory of Coordination Chemistry, Department of Polymer Science & Engineering, Nanjing National Laboratory of Microstructures, Nanjing University, Nanjing 210093, P. R. China. E-mail: jiaxd@nju.edu.cn
bDepartment of Polymer Science & Engineering, Nanjing University, Nanjing 210093, P. R. China
First published on 4th May 2017
Abstract
With the rapid progress of tissue engineering and regenerative medicine, the cell-friendly construction of a 3D extracellular matrix in a precisely controlled manner is needed. This biomimicry of the native extracellular matrix replicates major aspects of the native cellular microenvironment. Herein, we design a polyethylene glycol (PEG) based hydrogel combined with caging chemistry, which could achieve light-triggered local control of the hydrogel. The crosslinking density of the hydrogel could be tuned with UV exposure by decomposing the structure of the hydrogel. Meanwhile, by introduction of the releasable Rhodamine 110 molecule, we could conveniently monitor the change of the crosslinking density of the hydrogels via fluorescence. By this approach, adjustable user-defined stiffness patterns with different fluorescence intensity in a range of soft tissue microenvironments have been obtained. This novel hydrogel design could be useful for the manipulation of cell fate in various other contexts.
Introduction
In the last two decades, hydrogels, which usually possess inherent and excellent biocompatibility and biodegradability, have emerged as versatile biomaterials in various fields such as tissue engineering and cell biology.1–7 Such cell-compatible hydrogels have been prepared using a variety of polymeric materials, which can broadly be divided into two categories according to their origins: natural materials such as hyaluronic acid8–12 and chitosan, 13–16or synthetic materials such as poly(ethylene glycol) (PEG) and poly(vinyl alcohol) (PVA).17–24 The naturally derived hydrogels show excellent biocompatibility and biodegradability. However, limited tunability of degradation kinetics, relatively poor mechanical properties, batch-to-batch variations from manufacturers, and potential immunogenic reactions restrict the application of natural polymer based hydrogels.2,7,25 On the other side, synthetic polymers with commercial availability afford tunable mechanical properties, great flexibility in the working range of pH, ionic strength, and chemical conditions, including the introduction of degradable or biochemical moieties, which makes them excellent candidates for hydrogel preparation. Among all the synthetic polymers for hydrogels, PEG is the most popular one. The PEG macromolecule can easily be functionalized via its hydroxyl end groups to yield numerous homofunctional or heterofunctional terminal groups, including thiols,26 vinylsulfones,27 maleimides,28 acrylates,29 allyls,30 and norbornenes.31 Since PEG lacks any protein binding site due to its hydrophilic and uncharged structure, it forms highly hydrated layers that restrict protein adsorption.32 And the excellent biocompatibility and low toxicity of PEG-based hydrogels make them ideal candidates for various biomedical applications, including tissue engineering, cell encapsulation, controlled stem cell differentiation, and bioactive molecule delivery.33–38The high water content and tunable physicochemical properties of hydrogel have provided a powerful tool for researchers to mimic the physical properties of specific extracellular matrix (ECM) of native tissues and study cell behavior in vitro. However, the fundamental cell-ECM interactions are highly dynamic in nature, as cells interact with and respond to ECM signals and subsequently remodel their surroundings.39–42 It is urgent to control the dynamic properties of hydrogel at a desired time. To apply this concept, a major focus has been on light-responsive synthetic hydrogel modulated by a light beam.43–48 Through the activation of light-triggered compound, the stiffness of such polymer networks could be controlled in the micrometer scale designedly. Therefore, light-directed activation of caged compound could be considered as a powerful technique in order to spatiotemporally modify hydrogel properties. Irradiation of caged compounds, which releases the protected specific functional groups, has been employed to locally control biochemical properties of hydrogels.49–52 Hydrogels will be degraded when irradiated with cytocompatible doses of long wavelength UV, visible, or two-photon IR light (365, 405, and 740 nm, respectively), enabling precise control over hydrogel degradation profiles in situ.53 This photo-degradation scheme was successfully applied to control the spreading of human mesenchymal stem cell (hMSC) within light-eroded channels. The photo-degradation and the corresponding change in crosslink density of hydrogel lead to an increase in the mesh size and a decrease in the polymer density surrounding the cells, promoting encapsulated hMSC spreading as compared to non-irradiated control hydrogels.54 Although these approaches could control the stiffness of hydrogel, they were lack of monitoring the change of the stiffness in situ. The measurement of hydrogel stiffness always needs the help of instruments such as Atomic Force Microscope (AFM) or rheology, which was considered to be time-consuming and irreversible. Therefore, there is an urgent demand to develop a method, which could easily monitor the change of the hydrogel.
To implement this concept, herein we introduce a novel photo-cleavable compound as a linker to form the hydrogel.55–57 This compound caged the fluorescence Rhodamine 110 molecule with two oxirane groups has been synthesized. Because of the presence of nitrobenzene function group, the fluorescence of this caged compound is quenched. This caged compound is then introduced to form the hydrogel with 4-arm-PEG thiol by the traditional thiol–epoxy coupling reaction. Before the UV exposure, the hydrogel is well cross-linked and its fluorescence is greatly quenched. In the presence of UV light, the fluorescence of hydrogel is increased due to the releasing of fluorescence molecule Rhodamine 110 which is separated from the fluorescence quenching group of nitrobenzene. Meanwhile, the stiffness of hydrogel also decreases due to the decreasing of the crosslinking density, which is resulted from the decomposition of caged molecules. In this condition, the change of the hydrogel stiffness could be conveniently monitored by fluorescence on real time. With this strategy, we could easily monitor the change of the hydrogel by fluorescence, which provides a powerful tool in the investigation on the behavior of cells.
Experimental
Materials
3,4-Dimethoxy-6-nitrobenzaldehyde, vinyl magnesium bromide, m-chloroperbenzoic acid (MCPBA), carbonyldiimidazole (CDI), Rhodamine 110, tetrabutylammonium fluoride (TBAF) were purchased from Aldrich. 4arm-PEG thiol (10 kDa) was purchased from Laysan Bio, Inc. Poly-D-lysine (molecular weight: 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–300000) was purchased from Beyotime. Annexin V-FITC/PI Cell Apoptosis Kit was obtained from KeyGen Biotech. Co. Ltd. (Nanjing, China). All other reagents were analytical grade and used without further purification. The scanning electron microscope (SEM) was measured on S-3400N II Hitachi, Japan. The NMR spectra were measured on a 400 MHz Bruker instrument. Absorption spectra were recorded on a Hewlett-Packard 82152 diode array spectrophotometer. Fluorescence spectroscopy was performed on an SLM-AB2 fluorometer (Sopra, Buttlelborn, Germany). The rheological measurements were performed on a strain-controlled rotational rheometer (ARES of TA Instruments Corp.). Hela cells and A549 cells were obtained from the Institute of Biochemistry and Cell Biology, Shanghai Institute for Biological. Cytomics FC500 Flow Cytometry (Beckman Coulter Ltd.) was used to detect at least 10000 cells, and the data were analyzed with flowjo 10.2. The UV exposure was using a 40 W UV lamp (365 nm, PHILIPS). All syntheses were conducted in a darkened room or protected from room light with aluminum foil.
000–300000) was purchased from Beyotime. Annexin V-FITC/PI Cell Apoptosis Kit was obtained from KeyGen Biotech. Co. Ltd. (Nanjing, China). All other reagents were analytical grade and used without further purification. The scanning electron microscope (SEM) was measured on S-3400N II Hitachi, Japan. The NMR spectra were measured on a 400 MHz Bruker instrument. Absorption spectra were recorded on a Hewlett-Packard 82152 diode array spectrophotometer. Fluorescence spectroscopy was performed on an SLM-AB2 fluorometer (Sopra, Buttlelborn, Germany). The rheological measurements were performed on a strain-controlled rotational rheometer (ARES of TA Instruments Corp.). Hela cells and A549 cells were obtained from the Institute of Biochemistry and Cell Biology, Shanghai Institute for Biological. Cytomics FC500 Flow Cytometry (Beckman Coulter Ltd.) was used to detect at least 10000 cells, and the data were analyzed with flowjo 10.2. The UV exposure was using a 40 W UV lamp (365 nm, PHILIPS). All syntheses were conducted in a darkened room or protected from room light with aluminum foil.
Preparation of 1-hydroxy-1-(3,4-dimethoxy-6-nitrophenyl)-2-propene (1)
3, 4-Dimethoxy-6-nitrobenzaldehyde (1.88 g, 8.95 mmol) which was freshly recrystallized out of toluene, was dissolved in 50 mL of dry THF under an argon atmosphere. The yellow solution was cooled to −70 °C followed by adding dropwise 10.7 mL of a 1 M solution of vinyl magnesium bromide in THF. The deep red solution was stirred for 4 h and slowly warmed to 20 °C then added 40 mL of a saturated NH4Cl solution dropwisely. The red solution was extracted three times with ethyl acetate, and the organic phase was washed five times with saturated NaCl and dried over MgSO4. After filtration and concentration, the red oily residue was taken up in a little ethyl acetate and eluted through a silica gel column developed in 3:1 hexane/ethyl acetate. The solvent was removed from the product fractions to produce an orange-red powder. Yield: 1.61 g, 75%. 1HNMR [400 MHz, CDCl3, δ (ppm)]: 2.63 (s, 1H, ROH), 3.94 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 5.24 (dd, 1H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.42 (dd, 1H, CH2), 5.92 (d, 1H, CHCH2), 6.07 (ddd, 1H, CH), 7.20 (s, 1H, ArH), 7.57 (s, 1H, ArH) (Fig. S3†).
CH2), 5.42 (dd, 1H, CH2), 5.92 (d, 1H, CHCH2), 6.07 (ddd, 1H, CH), 7.20 (s, 1H, ArH), 7.57 (s, 1H, ArH) (Fig. S3†).
Photolytic isomerization of compound
A 100 μM solution of demand compound dissolved in ethanol was irradiated in a quartz cuvette with ultraviolet light. The absorption, fluorescence and infrared spectra of the sample were recorded with UV exposure of different time.Preparation of 1-(3,4-dimethoxy-6-nitrophenyl)-2,3-epoxypropyl hydroxide (2)
m-Chloroperoxybenzoic acid (MCPBA, technical grade, about 80% pure) was washed three times with phosphate buffer at pH 7.5 and dried. The above purified MCPBA (1.311 g, 7.60 mmol) was dissolved in 20 mL of CH2Cl2 and slowly dropped into a precooled solution of 1.80 g (7.52 mmol) of 1 in 15 mL of CH2Cl2. The bright yellow solution was stirred for 48 h at room temperature. The white precipitate was filtered and washed with CH2Cl2 followed by adding 70 mL of a saturated solution of NaHCO3 with stirring for another 30 min. The organic phase was washed seven times with a saturated solution of NaHCO3, then three times with a saturated solution of NaCl, and finally dried over MgSO4. After filtration and concentration, the yellow oily residue was recrystallized out of ethyl acetate/hexane. Yield: 1.50 g, 80%. 1HNMR [400 MHz, CDCl3, δ (ppm)]: 2.58–2.60 (s, 1H, OH), 2.62–2.71 (dd, 1H, O–CH2), 2.90–3.00 (dd, 1H, O–CH2), 3.3–3.6 (ddd, 1H, O–CH), 3.96–3.98 (s, 3H, OCH3), 3.98–4.02 (s, 3H, OCH3), 5.52–5.56 (dd, 1H, CH), 7.22–7.28 (s, 1H, ArH), 7.64–7.68 (s, 1H. ArH) (Fig. S4†).Preparation of 4-[1-(3,4-dimethoxy-6-nitrophenyl)-2,3-epoxypropyl-1-oxycarbonyl imidazole (3)
Under a dry argon atmosphere, a solution of carbonyldiimidazole (0.535 g, 3.3 mmol) dissolved in dichloromethane (4.0 mL) was added dropwise over 5 minutes to a solution of 1 (0.765 g, 3.0 mmol) dissolved in dichloromethane (3.0 mL). After 15 minutes, the reaction mixture was concentrated by rotary evaporation, and purified by flash chromatography (eluted with hexane/EtOAc of 1:2). Evaporation of solvent gave 3 (0.848 g, 81%) as a viscous yellow oil which was solidified on storage at −20 °C.1HNMR [400 MHz, CDCl3, δ (ppm)]: 2.30–2.40 (dd, 1H, O–CH2), 2.70–2.75 (dd, 1H, O–CH2), 3.40–3.50 (ddd, 1H, O–CH), 3.95–3.96 (s, 3H, OCH3), 3.96–3.97 (s, 3H, OCH3), 6.85–6.86 (t, 1H, CH), 6.88–6.89 (d, 1H, N–CHCH), 7.15–7.10 (s, 1H, N–CHCH), 7.44–7.46 (d, 1H, ArH), 7.60–7.65 (s, 1H, ArH), 8.12 (s, 1H, NCH–N) (Fig. S5†).
Preparation of bis[1-(3,4-dimethoxy-6-nitrophenyl)-2,3-epoxypropyl]-N,N′-rhodamine carbonate (4)
Compound 3 (349 mg, 1 mmol) dissolved in 2 mL of water free N,N-dimethylformamide (DMF) was treated with 250 μL of N-ethyldiisopropylamine (2.1 mmol) and 180 mg of Rhodamine 110 hydrochloride (0.49 mmol). The reaction mixture was stirred overnight at room temperature. Then the product was concentrated by rotary evaporation, and purified by flash chromatography (eluted with hexane/EtOAc of 1:2) yield: 290 mg (54.7%). 1HNMR [400 MHz, CD3OD, δ (ppm)]: 2.35 (dd, 1H, O–CH2), 3.06 (dd, 1H, O–CH2), 3.53 (ddd, 1H, OCH), 3.90 (s, 3H, OCH3), 3.95 (s, 3H, OCH3), 6.62 (d, 1H, CH), 7.01 (s, 1H, ArH), 6.68 (d, 1H, CH), 7.10 (s, 1H, ArH), 7.15 (d, 1H, ArH), 7.17 (d, 1H, ArH), 7.19 (dd, 1H, ArH), 7.30 (dd, 1H, ArH), 7.60 (s, 1H, ArH), 8.10 (d, 1H, NH–CO) (Fig. S6†).
Form of hydrogel
8.3 mg of compound 4 and 4 μl of TBAF (75% in H2O) were dissolved in 0.25 ml of DMF, subjected to a ultrasonic process to obtain a clear solution, and then added to an already prepared solution of 4arm-PEG thiol (10 kDa) (100 mg) in 4 ml of H2O. This formulation was subjected to an ultrasonic process to obtain a clear solution. This solution was heated to 70 °C for 120 minutes. After cooling to the room temperature, the crosslinked material was washed thoroughly with ethanol and PBS solution for several times to remove the excess organic solvent and then used for further studies.Cell study
All cells were seeded in Dulbecco's modified Eagle medium/high-glucose medium supplemented with 10% fetal bovine serum and 1% antibiotics (penicillin/streptomycin) at 37 °C under a humidified atmosphere containing 5% CO2. Subconfluent cell cultures were passaged by trypsin digestion. Before cell adhesion, the hydrogel was functioned with 0.1 mg mL−1 poly-D-lysine for 2 hours, and then washed with PBS for three times to remove the excess poly-D-lysine.Rheometry
The rheological measurements were performed on a strain-controlled rotational rheometer (ARES of TA Instruments Corp.). Cone-and-plate geometry of 50 mm diameter and 0.04 rad cone angle was chosen to ensure uniform shear throughout the samples. As measurement criterion, the strain amplitude was kept within the linear viscoelasticity region while being large enough for obtaining reasonable signal intensities in the frequency range (0.01–100 rad s−1). The dynamic storage modulus (G′) was measured at frequencies (ω) of 1 rad s−1.Results and discussion
The synthesis of caged compound
The Grignard reaction of vinyl-magnesium bromide on 3,4-dimethoxy-6-nitrobenzaldehyde created a hydroxyl and vinyl group off the benzylic carbon, which provided the starting point for the synthesis of the two different reactive groups of the cross-linking compound (Scheme 1). The first group was used to react with the fluorescence compound Rhodamine 110 with the help of CDI. Ottl et al. once used 4,6-diphenylthieno[3,4-d][1,3]dioxol-2-on-5,5-dioxide (TDO) to synthesize this caged compound.57 In our experiment, both TDO and CDI were employed. Compared with using only TDO, the synthesis by use of CDI showed better repeatability, easier purification and higher yield. While the Rhodamine 110 was attached to the nitrobenzene group, the fluorescence was quenched. The second group, epoxy, was used to alkylate thiol groups on PEG and served to crosslink the caged Rhodamine 110 compound into the hydrogel. This synthetic approach also allowed the physical and chemical separation of the two nucleophilic reactive groups from the photo-isomerization functionality (Scheme 1). This design also provided the compound with two oxirane groups, which could be a linker compound in the formation of hydrogel. With this design, the enhancement of fluorescence of hydrogel displays the change of cross-link intensity of hydrogel, which means that this caged fluorescence molecule Rhodamine 110 could act as a signal beacon to monitor the change of hydrogel.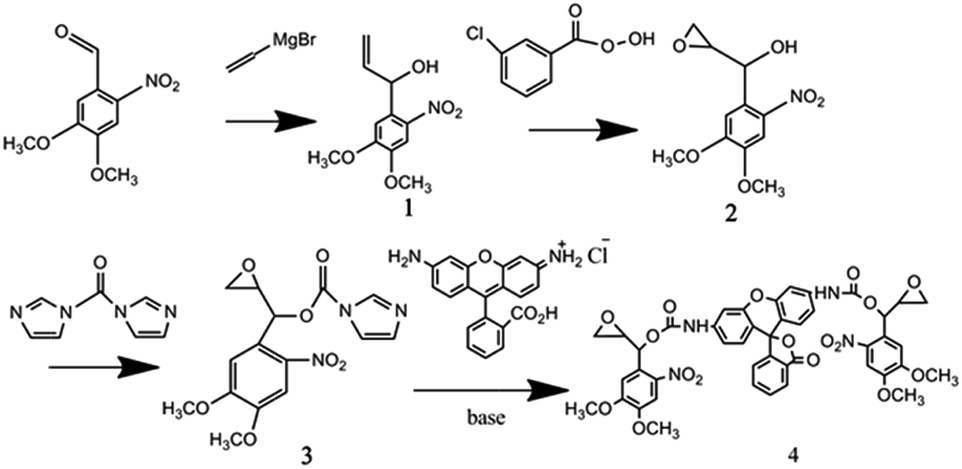 | ||
| Scheme 1 Synthetic procedures of caged Rhodamine 110 molecule. | ||
Spectral properties of compound 1
The absorption spectra of a defined concentration of an ethanolic solution of compound 1 (Fig. 1) showed that the chromophore had a broad n–π* transition centered at 350 nm. Excitation of this ethanolic solution with near-ultraviolet light (365 nm) led to a dose-dependent loss of the 350 nm band, while a new absorption transition were appeared at near 400 nm (Fig. 1a). These absorption bands probably belonged to the 3,4-dimethoxy-6-nitrosoacetophenone photoproduct (Fig. S1†). Additionally, after the exposure of UV light, the color of the solution of compound 1 turned from yellow to dark brown, which could also easily prove the structure change with the exposure of UV light. The evidence that this reaction occurred via a photo-isomerization of the nitrophenyl group was also provided by an analysis of the infrared spectra of a film of compound 1 recorded before and after UV irradiation (Fig. 1b), which showed a loss of the hydroxyl absorption band at 3600 cm−1 of compound 1 and a gain of ketone absorption band at 1720 cm−1.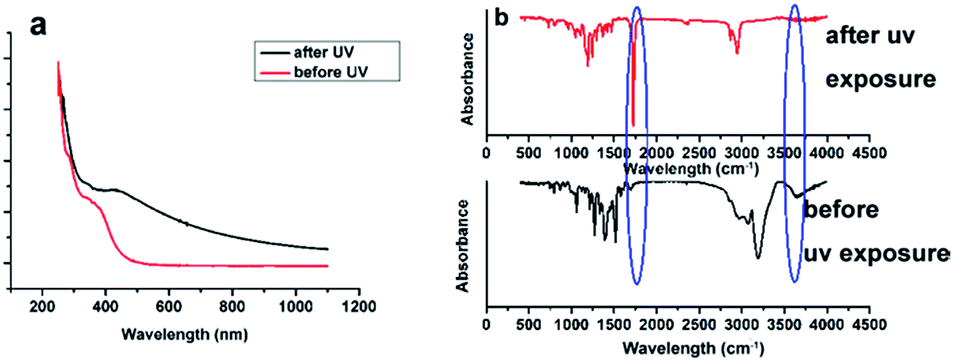 | ||
| Fig. 1 The absorption (a) and infrared (b) spectra of compound 1 before and after UV exposure. | ||
Photocleavable, cross-linking compounds and the preparation of a thiol-reactive caged Rhodamine110
With the help of CDI, compound 2 was shown to react with Rhodamine 110 to produce a non-fluorescent bis-substituted carbonate through a two-step reaction (compound 4, Scheme 1). Irradiation of the nonfluorescent (caged) Rhodamine 110 with near-ultraviolet light cleaved the two carbonate bonds and released free Rhodamine 110. Followed by the separation of Rhodamine 110 and nitrophenyl group, the fluorescence of the compounds was recovered. This statement was proved by ultraviolet absorption spectrum and fluorescence spectrum analysis (Fig. 2). As shown in Fig. 2a, with the increasing time of UV exposure, there was a concomitant recovery of the visible absorption band at nearly 500 nm, which was attributed to the release of Rhodamine 110. Similarly, in the fluorescence spectra, with the increasing time of UV exposure, fluorescence emission centered at 543 nm was increased, which was the emission peak of Rhodamine 110. These results showed that we had successfully made a thiol reactive caged Rhodamine 110 compound with excellent UV response.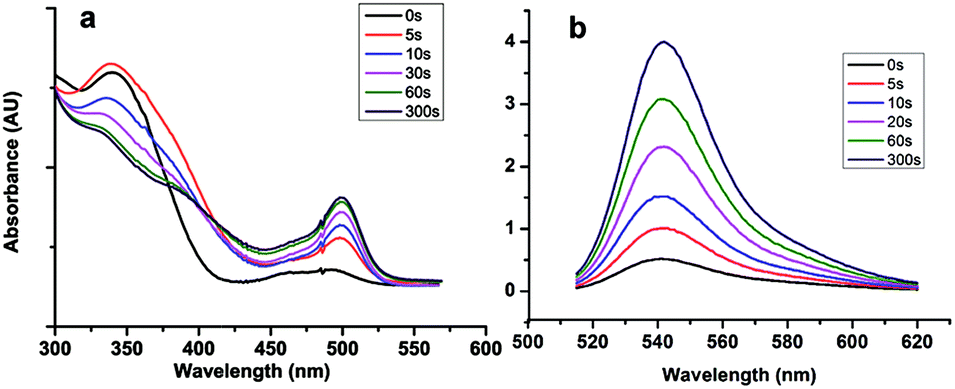 | ||
| Fig. 2 (a) Absorption spectra of a 50 μM alcohol solution of caged Rhodamine 110 before irradiation and after near-ultraviolet irradiation for different time (b) fluorescence emission spectra of a 50 μM alcohol solution of caged Rhodamine 110 with excitation at 488 nm of for different time. | ||
User-defined control of hydrogel stiffness
To form the hydrogel, the 4arm-PEG thiol was firstly reacted with compound 4 through the reaction of thiol and oxirane, and a nearly non-fluorescent hydrogel was obtained after 2 h incubation, which was characterized by SEM. As seen in Fig. S7,† the SEM picture revealed the well cross-linked network structure of such hydrogel. After UV exposure for a few minutes, the color of the hydrogel turned to be green, which was due to the release of fluorescent molecule Rhodamine 110.To a deep investigation of this hydrogel, we used confocal lasers to change the crosslink intensity of the hydrogel. When the irradiation of laser spotted on the hydrogel, a green fluorescence spot emerged, it was due to the release of fluorescent molecule Rhodamine 110. As the increasing time of UV exposure, the fluorescence intensity was also increased and reached a maximum at the exposure time of 600 s (Fig. 3c). By this approach, any desired patterns could simply be drawn by the using of confocal lasers as a pen, which could be further applied in the field of biography according to the needs of the users.
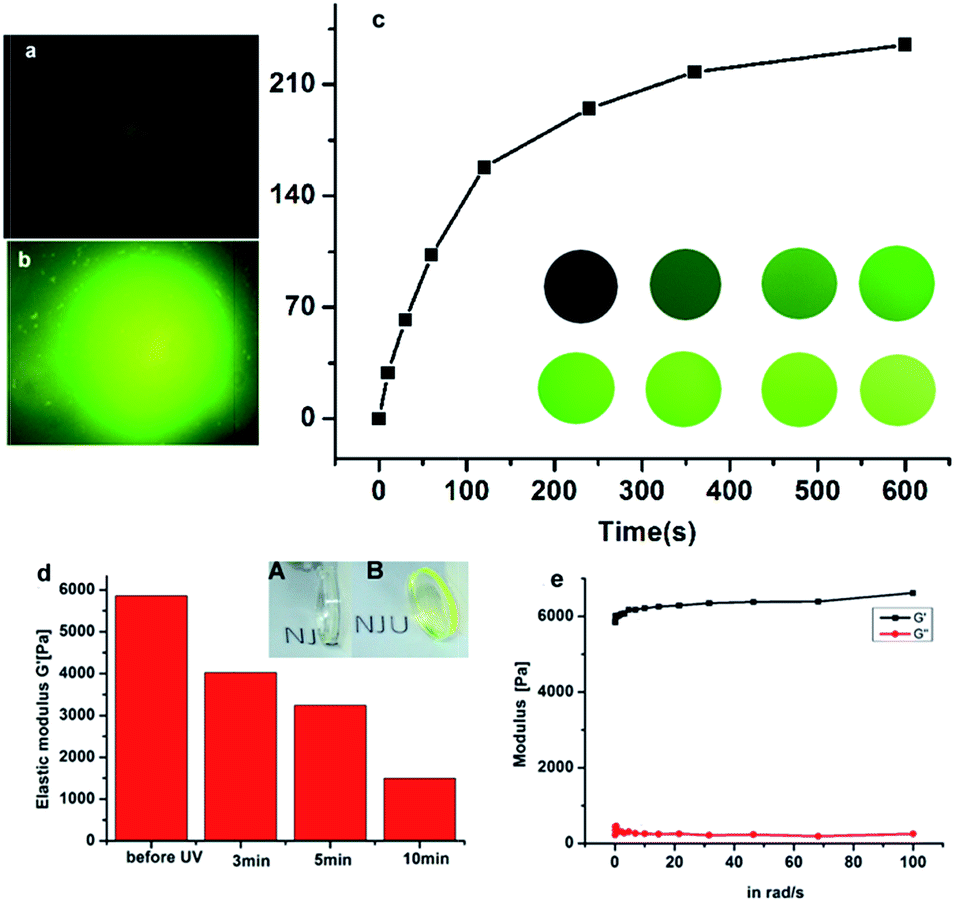 | ||
| Fig. 3 The images of hydrogel after being spotted by UV laser (a)10 s, (b): 600 s; (c) the change of fluorescence intensity of hydrogel after being spotted by UV laser for different time (inset: the spot images).(d): the change of elastic modulus of hydrogels with UV exposure for different time. Inset: (A) image of hydrogel before UV exposure (B): image of hydrogel after 10 min UV exposure (e): the rheological measurement of hydrogels before UV exposure. | ||
The change of fluorescence intensity was the result of the uncaging of the compound 4. Theoretically, as the linker of the hydrogel, the destruction of the compound 4 would also cause the degradation of hydrogel (Scheme 2). To prove this hypothesis, the colors of hydrogels before and after UV exposure were compared. It was shown that the color of hydrogels turned from air color to green, which was due to the releasing of Rhodamine 110 (Fig. 3d inset). Then, the stiffness of the hydrogel before and after UV exposure was measured. Before UV exposure, the storage modulus of well-formed hydrogel was nearly 6000 Pa. The difference of G′ (storage modulus) and G′′ (loss modulus) means that this well cross-linked hydrogel presents the characteristic of solid (Fig. 3e). After different times of exposure to the UV lamp, the G′ of the hydrogel showed a continuous decrease, which indicated the gradual degradation of the gel matrix upon light irradiation, and a series of hydrogels with different stiffness were obtained thereafter (Fig. 3d). Above all, upon UV exposure, the destruction of the compound 4 could both induce the increasing of fluorescence and the decreasing of stiffness. Moreover, as shown in Fig. S8,† there was a fine linear relationship between the increasing of fluorescence and the decreasing of stiffness. Through this method, we could observe the hydrogel degradation and softening in situ.
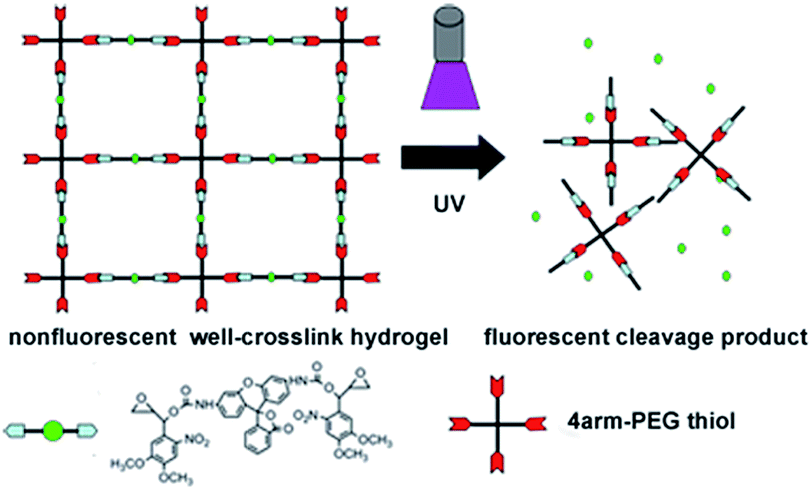 | ||
| Scheme 2 The diagram of the decomposition of hydrogel with UV light exposure. | ||
Influence of dynamic hydrogels change on cellular behavior
At first, we tested the biocompatibility of our pre-synthesis hydrogels. Since PEG lacks any protein binding sites, the hydrogels were firstly modified with 0.1 mg mL−1 poly-D-lysine to increase the adhesion ability of cells. After washed thoroughly with PBS buffer to remove the excess poly-D-lysine, the cells were incubated on the hydrogel for cell adhesion in cell media for 12 h at 37 °C with 5% CO2. The post incubated hydrogels were washed thoroughly with PBS buffer to remove non-adhered cells, and then imaged using a fluorescence microscope. Following that, flow cytometer was utilized to observe the biocompatibility of cells incubated on hydrogels. As shown in Fig. 4, with Annexin V-FITC/PI method, both of Hela cells and A549 cells had nearly 90% living cells, which proved the well cell-compatibility of the hydrogels. And the fluorescence spectroscopy pictures of the Hela cells and A549 cells also showed the well condition of cells incubated on hydrogels. Furthermore, the influence of the stiffness of hydrogels on cell adhesion was investigated. Before cell incubation, we first modified the hydrogels by spotting with UV laser (Fig. S2a†), and then compared the cell adhesion on different areas of hydrogels. We used the UV laser to modify the central area of the hydrogel. When exposed to UV laser, the central area of hydrogels became brighter due to the releasing of Rhodamine 110 which was separated from the fluorescence quenching group of nitrobenzene. Meanwhile, the stiffness of such area also decreased due to the decreasing of the crosslinking density, which was resulted from the decomposition of caged molecules. As shown in Fig. S2b,† the number of the cells incubated on bright area was obviously fewer than that on dark area. And we calculated from Fig. S2c,† it could be found that the A549 cells preferred to attach the stiff base, which meant that the matrix stiffness was a prominent effect for the promotion of A549 cell growth,58 while the stiffness of the environment had a contrary influence on the adhesion of Hela cells. The Hela cells preferred to adhere on the softer area, which was consistent with the reported.59 Briefly, during cell adhesion process, different cells had their own optimal supporting stiffness, which meant that the responses to substrate stiffness were cell specific.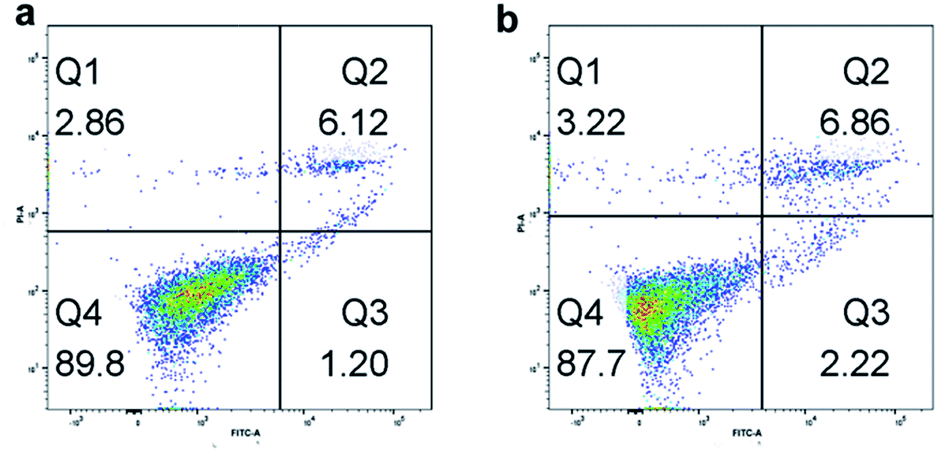 | ||
| Fig. 4 The flow cytometer images of (a) Hela cells and (b) A549 cells grown on hydrogels. | ||
Furthermore, we use Hela cells as a model to compare the morphology of cells grown on hydrogel with and without UV exposure. Fig. 5a represented the image of cells grown on hydrogel without UV exposure under visible light, Fig. 5b represented the image of cells grown on hydrogel with UV exposure under blue light, Fig. 5c represented the image of cells grown on hydrogel without UV exposure under visible light, and Fig. 5d represented the image of cells grown on hydrogel with UV exposure under blue light. By the comparison of Fig. 5a and c, we could find the cells incubated on the softer hydrogels with UV exposure showed a more circular morphology, which was consistent with the earlier report.7 By comparison of Fig. 5b and d, we could find the hydrogels without UV exposure (Fig. 5b) nearly appeared as a dark image with no fluorescence. It was due to the reason that the caged fluorescence molecules Rhodamine 110 in the hydrogels were quenched by the nitro-phenyl groups. With the help of caged fluorescence molecules Rhodamine 110, we could not only monitor the change of dynamic properties of the hydrogels by fluorescence, but also monitor the response of cells towards the changes of ECM. Additionally, from Fig. 5d we could find an interesting phenomenon that the cells were particularly bright when they were grown on the hydrogels with UV exposure. This was due to the reason that during the time when the hydrogels were incubated with the cell, the free fluorescence Rhodamine 110 molecules tended to enrich in the cells, which promoted the fluorescence of cells particularly high. Above all, with the as-prepared hydrogel, we could conveniently monitor the changes of cell morphology in growth, which had a great dependence upon the stiffness of basement. This unique convenient fluorescence monitor system could easily be applied in the cell study, and have great potential in other biology fields.
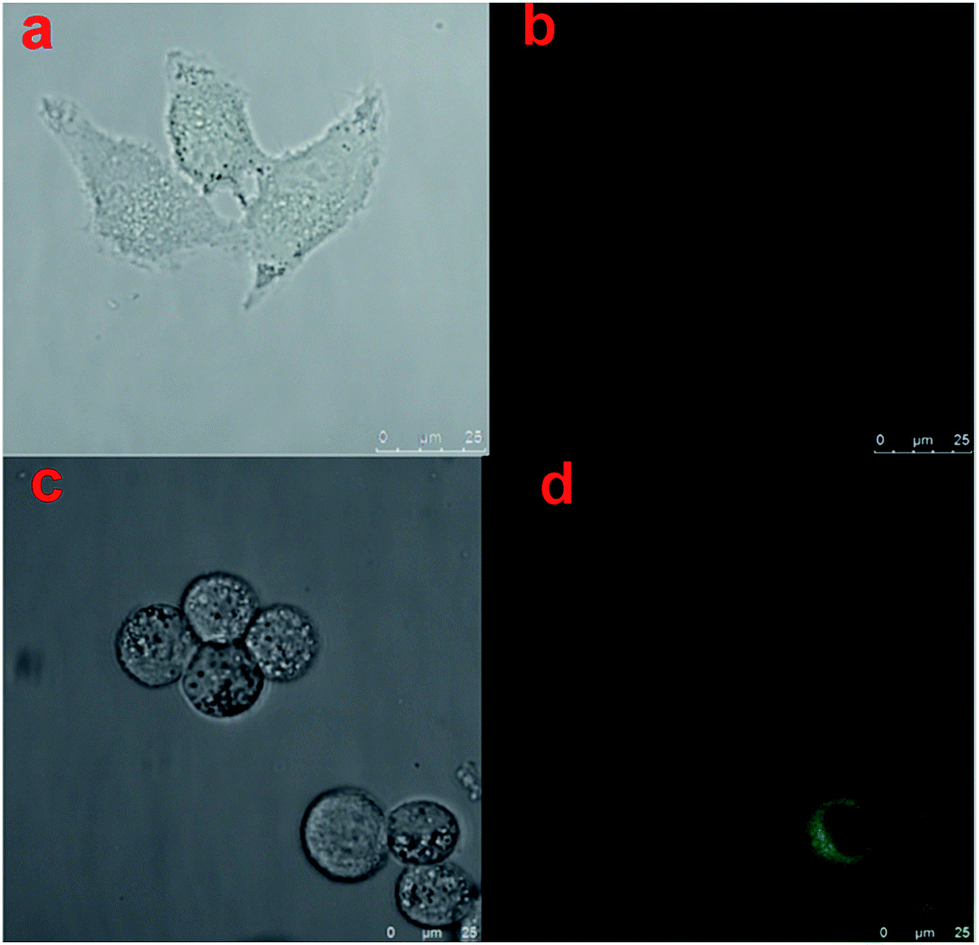 | ||
| Fig. 5 (a) Cell image on hydrogel without UV exposure under visible light, (b) cell image on hydrogel without UV exposure under blue light, (c) cell image on hydrogel with UV exposure under visible light, (d) cell image on hydrogel with UV exposure under blue light. | ||
Conclusions
In summary, we have developed a new type of hydrogel whose stiffness could not only be controlled with light but also be monitored in situ by the fluorescence conveniently. The practical use of this new type of photodegradable hydrogel was demonstrated by the photo-modulation of the microenvironment of cells. The unique convenient fluorescence monitor system made this hydrogel even more attractive as smart biomaterials for spatially defined modulation on cellular microenvironments. This hydrogel had great potential in investigating into precise control of the photo-patterned channels and different biological behaviors of the live cells on real time.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21104032), the Natural Science Foundation of Jiangsu Province (No. BK2011413) and the Fundamental Research Funds for the Central Universities (No. 1104020505). The authors would like to thank the Program for Chang jiang Scholars and Innovative Research Team in University for financially supporting this research. The authors thank Dr Dechen Jiang and Dr Gerard Marriott for the help in experiment.References
- M. P. Lutolf, P. M. Gilbert and H. M. Blau, Nature, 2009, 462, 433–441 CrossRef CAS PubMed.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487–492 CrossRef CAS PubMed.
- J. A. Burdick and G. Vunjak-Novakovic, Tissue Eng., Part A, 2009, 15, 205–219 CrossRef CAS PubMed.
- S. Gobaa, S. Hoehnel, M. Roccio, A. Negro, S. Kobel and M. P. Lutolf, Nat. Methods, 2011, 8, 949–955 CrossRef CAS PubMed.
- M. Guvendiren and J. A. Burdick, Nat. Commun., 2012, 3, 792 CrossRef PubMed.
- R. G. Wylie, S. Ahsan, Y. Aizawa, K. L. Maxwell, C. M. Morshead and M. S. Shoichet, Nat. Mater., 2011, 10, 799–806 CrossRef CAS PubMed.
- P. M. Kharkar, K. L. Kiick and A. M. Kloxin, Chem. Soc. Rev., 2013, 42, 7335–7372 RSC.
- F. Rehfeldt, A. E. X. Brown, M. Raab, S. Cai, A. L. Zajac, A. Zemel and D. E. Discher, Integr. Biol., 2012, 4, 422–430 RSC.
- X. Xu, A. K. Jha, D. A. Harrington, M. C. Farach-Carson and X. Jia, Soft Matter, 2012, 8, 3280–3294 RSC.
- S. Ekici, P. Ilgin, S. Butun and N. Sahiner, Carbohydr. Polym., 2011, 84, 1306–1313 CrossRef CAS.
- N. Izawa, M. Serata, T. Sone, T. Omasa and H. Ohtake, J. Biosci. Bioeng., 2011, 111, 665–670 CrossRef CAS PubMed.
- Y. Tao, L. Ai, H. Bai and X. Liu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3507–3516 CrossRef CAS.
- N. Bhattarai, J. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 83–99 CrossRef CAS PubMed.
- K. Murakami, H. Aoki, S. Nakamura, S. I. Nakamura, M. Takikawa, M. Hanzawa, S. Kishimoto, H. Hattori, Y. Tanaka, T. Kiyosawa, Y. Sato and M. Ishihara, Biomaterials, 2010, 31, 83–90 CrossRef CAS PubMed.
- J. K. F. Suh and H. W. T. Matthew, Biomaterials, 2000, 21, 2589–2598 CrossRef CAS PubMed.
- Y. Che, D. Li, Y. Liu, Q. Ma, Y. Tan, Q. Yue and F. Meng, RSC Adv., 2016, 6, 106035–106045 RSC.
- A. Alexander, Ajazuddin, J. Khan, S. Saraf and S. Saraf, J. Controlled Release, 2013, 172, 715–729 CrossRef CAS PubMed.
- A. D. Baldwin, K. G. Robinson, J. L. Militar, C. D. Derby, K. L. Kiick and R. E. Akins Jr, J. Biomed. Mater. Res., Part A, 2012, 100, 2106–2118 CrossRef PubMed.
- S. Seetharaman, S. Natesan, R. S. Stowers, C. Mullens, D. G. Baer, L. J. Suggs and R. J. Christy, Acta Biomater., 2011, 7, 2787–2796 CrossRef CAS PubMed.
- L. M. Weber, C. G. Lopez and K. S. Anseth, J. Biomed. Mater. Res., Part A, 2009, 90, 720–729 CrossRef PubMed.
- R. Webster, E. Didier, P. Harris, N. Siegel, J. Stadler, L. Tilbury and D. Smith, Drug Metab. Dispos., 2007, 35, 9–16 CrossRef CAS PubMed.
- S. J. Buwalda, A. Amgoune and D. Bourissou, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1222–1227 CrossRef CAS.
- C. Hou, K. Ma, T. Jiao, R. Xing, K. Li, J. Zhou and L. Zhang, RSC Adv., 2016, 6, 110799–110807 RSC.
- M. Sakthivel, D. S. Franklin, S. Sudarsan, G. Chitra and S. Guhanathan, RSC Adv., 2016, 6, 106821–106831 RSC.
- Z. Shi, X. Gao, M. W. Ullah, S. Li, Q. Wang and G. Yang, Biomaterials, 2016, 111, 40–54 CrossRef CAS PubMed.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 CrossRef CAS PubMed.
- M. P. Lutolf and J. A. Hubbell, Biomacromolecules, 2003, 4, 713–722 CrossRef CAS PubMed.
- K. M. Schultz, A. D. Baldwin, K. L. Kiick and E. M. Furst, Macromolecules, 2009, 42, 5310–5316 CrossRef CAS PubMed.
- A. Metters and J. Hubbell, Biomacromolecules, 2005, 6, 290–301 CrossRef CAS PubMed.
- A. E. Rydholm, S. K. Reddy, K. S. Anseth and C. N. Bowman, Polymer, 2007, 48, 4589–4600 CrossRef CAS PubMed.
- S. B. Anderson, C.-C. Lin, D. V. Kuntzler and K. S. Anseth, Biomaterials, 2011, 32, 3564–3574 CrossRef CAS PubMed.
- S. I. Jeon, J. H. Lee, J. D. Andrade and P. G. Degennes, J. Colloid Interface Sci., 1991, 142, 149–158 CrossRef CAS.
- F. P. Brandl, A. K. Seitz, J. K. V. Tessmar, T. Blunk and A. M. Goepferich, Biomaterials, 2010, 31, 3957–3966 CrossRef CAS PubMed.
- V. Chan, P. Zorlutuna, J. H. Jeong, H. Kong and R. Bashir, Lab Chip, 2010, 10, 2062–2070 RSC.
- D. S. W. Benoit, M. P. Schwartz, A. R. Durney and K. S. Anseth, Nat. Mater., 2008, 7, 816–823 CrossRef CAS PubMed.
- H. Park, X. Guo, J. S. Temenoff, Y. Tabata, A. I. Caplan, F. K. Kasper and A. G. Mikos, Biomacromolecules, 2009, 10, 541–546 CrossRef CAS PubMed.
- A. A. Aimetti, A. J. Machen and K. S. Anseth, Biomaterials, 2009, 30, 6048–6054 CrossRef CAS PubMed.
- C. Shu, E. M. B. Sabi-mouka, W. Yang, Z. Li and L. Ding, RSC Adv., 2016, 6, 100765–100771 RSC.
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Science, 2009, 324, 1673–1677 CrossRef CAS PubMed.
- J. B. Gurdon and P. Y. Bourillot, Nature, 2001, 413, 797–803 CrossRef CAS PubMed.
- F. Chowdhury, S. Na, D. Li, Y.-C. Poh, T. S. Tanaka, F. Wang and N. Wang, Nat. Mater., 2010, 9, 82–88 CrossRef CAS PubMed.
- T. Ingverud, E. Larsson, G. Hemmer, R. Rojas, M. Malkoch and A. Carlmark, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3415–3424 CrossRef CAS.
- S. Nemir, H. N. Hayenga and J. L. West, Biotechnol. Bioeng., 2010, 105, 636–644 CrossRef CAS PubMed.
- M. T. Thompson, M. C. Berg, I. S. Tobias, J. A. Lichter, M. F. Rubner and K. J. Van Vliet, Biomacromolecules, 2006, 7, 1990–1995 CrossRef CAS PubMed.
- K. T. Nguyen and J. L. West, Biomaterials, 2002, 23, 4307–4314 CrossRef CAS PubMed.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS PubMed.
- R. D. Cheng, J. G. Chen, Z. T. Liu, Z. W. Liu and J. Q. Jiang, Macromol. Rapid Commun., 2016, 37, 514–520 CrossRef CAS PubMed.
- S. Tamesue, S. Abe, T. Mitsumata, N. Tsubokawa and T. Yamauchi, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1317–1322 CrossRef CAS.
- E. E. Nekongo and V. V. Popik, J. Org. Chem., 2014, 79, 7665–7671 CrossRef CAS PubMed.
- S. S. Agasti, A. M. Laughney, R. H. Kohler and R. Weissleder, Chem. Commun., 2013, 49, 11050–11052 RSC.
- K. B. Joshi, A. Vlachos, V. Mikat, T. Deller and A. Heckel, Chem. Commun., 2012, 48, 2746–2748 RSC.
- G. C. R. Ellis-Davies, Nat. Methods, 2007, 4, 619–628 CrossRef CAS PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- A. M. Kloxin, M. W. Tibbitt, A. M. Kasko, J. A. Fairbairn and K. S. Anseth, Adv. Mater., 2010, 22, 61–66 CrossRef CAS PubMed.
- G. Marriott, Biochemistry, 1994, 33, 9092–9097 CrossRef CAS PubMed.
- G. Marriott and M. Heidecker, Biochemistry, 1996, 35, 3170–3174 CrossRef CAS PubMed.
- J. Ottl, D. Gabriel and G. Marriott, Bioconjugate Chem., 1998, 9, 143–151 CrossRef CAS PubMed.
- J. D. Mih, A. S. Sharif, F. Liu, A. Marinkovic, M. M. Symer and D. J. Tschumperlin, Plos One, 2011, 6, e19929 CAS.
- A. M. Jonker, S. A. Bode, A. H. Kusters, J. C. M. van Hest and D. Lowik, Macromol. Biosci., 2015, 15, 1338–1347 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02629c |
| This journal is © The Royal Society of Chemistry 2017 |