
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComplete genome sequencing of Arachidicoccus ginsenosidimutans sp. nov., and its application for production of minor ginsenosides by finding a novel ginsenoside-transforming β-glucosidase†
Muhammad Zubair Siddiqi*ab,
Siddiqi Muhammad Shafic and
Wan-Taek Im *ab
*ab
aDepartment of Biotechnology, Hankyong National University, 327 Chungang-no, Anseong-si, Kyonggi-do 17579, Republic of Korea. E-mail: mzsiddiqi1988@yahoo.com; wandra@hknu.ac.kr; Fax: +82-31-670-5339; Tel: +82-31-670-5335
bCenter for Genetic Information, Graduate School of Bio and Information Technology, Hankyong National University, 327 Chungang-no, Anseong-si, Kyonggi-do 17579, Republic of Korea
cGreen Planet Co. Ltd, Tsushima nishizaka 2 chome 5-41-203, Okayama, Okayama ken, Japan
First published on 3rd October 2017
Abstract
A novel bacterial strain (BS20T), which has ginsenoside-transforming ability, was whole genome sequenced for the identification of a target gene. After complete genome sequencing, phylogenetic, phenotypic and chemotaxonomic analyses, the strain BS20T (Arachidicoccus ginsenosidimutans sp. nov.) was placed within the genus Arachidicoccus of family Chitinophagaceae. The complete genome of strain BS20T comprised a circular chromosome of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 138017 bp. To find the target functional gene, 17 sets of four different glycoside hydrolases were cloned in E. coli BL21 (DE3) using the pGEX4T-1 vector and were characterized. Among these 17 sets of clones, only one, BglAg-762, exhibited ginsenoside-conversion ability. The BglAg-762 comprised 762 amino acid residues and belonged to the glycoside hydrolase family 3. The recombinant enzyme (GST-BglAg-762) was able to convert major ginsenosides Rb1 to F2 via gypenoside-XVII (Gyp-XVII), Rb2 to C-O, and Rb3, Rc, Rd, and Gyp-XVII to C-Mx1, C-Mc1, and F2, respectively. Finally, ginsenoside F2 was transformed into compound K (C-K). Besides, these pilot data demonstrate the identification of 17 sets of target/functional genes of 4 different glycoside hydrolases from a novel bacterial species via whole genome sequencing. Our results have shown that the recombinant BglAg-762 very quickly converts the major ginsenosides into minor ginsenosides, which can be used for the enhanced production of target minor ginsenosides. Furthermore, the web service of NCBI is suitable for any targeted gene identification, but based on our experimental analysis we concluded that the hypothetical protein present in NCBI should be considered as a putative or uncharacterized protein.
138017 bp. To find the target functional gene, 17 sets of four different glycoside hydrolases were cloned in E. coli BL21 (DE3) using the pGEX4T-1 vector and were characterized. Among these 17 sets of clones, only one, BglAg-762, exhibited ginsenoside-conversion ability. The BglAg-762 comprised 762 amino acid residues and belonged to the glycoside hydrolase family 3. The recombinant enzyme (GST-BglAg-762) was able to convert major ginsenosides Rb1 to F2 via gypenoside-XVII (Gyp-XVII), Rb2 to C-O, and Rb3, Rc, Rd, and Gyp-XVII to C-Mx1, C-Mc1, and F2, respectively. Finally, ginsenoside F2 was transformed into compound K (C-K). Besides, these pilot data demonstrate the identification of 17 sets of target/functional genes of 4 different glycoside hydrolases from a novel bacterial species via whole genome sequencing. Our results have shown that the recombinant BglAg-762 very quickly converts the major ginsenosides into minor ginsenosides, which can be used for the enhanced production of target minor ginsenosides. Furthermore, the web service of NCBI is suitable for any targeted gene identification, but based on our experimental analysis we concluded that the hypothetical protein present in NCBI should be considered as a putative or uncharacterized protein.
1. Introduction
Ginseng, the root of Panax ginseng C. A. Meyer, which has been used as herbal medicine in Asian countries (China, Korea, and Japan) for centuries, has gained fame in western countries in recent decades.1–3 The pharmacological active ingredients in ginseng have been reported to be ginsenosides, a class of distinctive triterpene glucosides.4–6 Based on differences in chemical constitution and dammarane skeleton, ginsenosides are categorized as protopanaxadiol (PPD), protopanaxatriol (PPT), and oleanane-type saponins.6 Major ginsenosides are natural high molecular-weight glycosides of triterpene; due to their size, low solubility, and poor permeability through the cell membrane, they are not easily absorbed by the human gastrointestinal system.7,8 It was reported previously that major ginsenosides (Rb1, Rb2, Rc, and Rd), when taken orally, are converted into minor ginsenosides (Rg3, Rh2, F2, and compound K [CK]) by intestinal bacteria.9 Many studies have been performed to investigate the conversion of major ginsenosides into minor ginsenosides, which possess desirable pharmaceutical activities and can be readily absorbed by the human body. So far, researchers have used heat treatment, chemical treatment (acid or base), microorganisms and highly-expressed recombinant glycoside hydrolase enzymes for converting major ginsenosides into minor ginsenosides for exploiting their pharmacological and cosmetic applications.10–14 Therefore, studies have been mainly focused on finding suitable microbes or enzymes to transform major ginsenosides into minor ginsenosides. Minor ginsenosides are derivatives of major ginsenosides, exist in smaller amounts in the total ginseng ginsenosides, and show desirable pharmacological effects such as anti-cancer, anti-oxidative, anti-diabetic, anti-aging, and anti-osteoporosis.15–22 Therefore, the initial biotransformation of PPD- or PPT-type ginsenosides is very important. Thus, for primary bioconversion of ginsenosides, 11 novel species were screened, as shown in Fig. S1.† After screening, based on the efficiency of bioconversion, strain BS20T was selected for complete genome sequencing to find the target functional gene. The novel strain BS20T was taxonomically characterized, and was found to belong to the genus Arachidicoccus.The genus Arachidicoccus was recently described by Madhaiyan et al. (2015) based on the description of a single species. Phylogenetically, the genus belongs to the phylum Bacteroidetes and is closely related to members of the family Chitinophagaceae. At the time of writing, the genus Arachidicoccus comprises only two recognized species, A. rhizosphaerae KCTC 22378T and A. ginsenosidivorans KCTC 22820T (http://www.bacterio.net). Both members of this genus are Gram-negative, non-motile, non-flagellated, and non-spore forming.22,23 Affiliates of this genus are negative for flexirubin-type reactions. The major quinone found in members of this genus was menaquinone MK-7 and the main polar lipid, phosphatidylethanolamine (PE). The described species of the genus contained iso-C15:0, iso-C15:1 G, iso-C17:0 3-OH, and summed feature 3 (comprising C16:1 ω7c and/or C16:1 ω6c) as the major cellular fatty acids.22,23 As the strain BS20T showed ginsenoside-conversion ability, it whole genome was sequenced to find the target gene responsible for this conversion.
Today, techniques for whole genome sequencing of bacterial strains are rapidly developing. In bacterial taxonomy, the complete genome sequence is considered the most important source of information for describing a species. In 1995, the first two complete bacterial genomes were sequenced and published.24,25 Thereafter, the study of bacteria has dramatically changed. Using third-generation DNA sequencing, it is possible to identify any gene of interest from the complete genome of a bacterial strain within a few hours along with the complete/whole genome as well.
In this study, we described the phenotypic and genomic properties of a novel Arachidicoccus strain, A. ginsenosidimutans BS20T. As the strain BS20T exhibited efficient conversion of major ginsenosides into minor ginsenosides. Therefore, after the complete genome analysis, 17 sets of 4 different glycoside hydrolases (β-glucosidase; α,β-xylosidase; L-rhamnosidase; and α,β-arabinofuranosidase) were isolated and cloned in E. coli BL21 (DE3). In addition, both the novel species, which was designated as strain BS20T and the novel recombinant β-glycosidase (BglAg-762), which belonged to the glycoside hydrolase family 3, were characterized.
2. Materials and methods
2.1. Chemicals and ginsenoside standards
The ginsenoside standards, Rb1, Rc, Rb2, Rb3, Rd, Re, Rg1, and Gyp-XVII, were purchased from Nanjing Zelang Medical Technology Co. Ltd. (China). PPD-type ginsenoside mixture extracted from the roots of Panax quinquefolius (American root saponins, mainly comprising Rb1, Rc, and Rd) was acquired from Hongjiou Biotech Co. Ltd. (China), and was used as the initial substrate in this study. The p-nitrophenyl-β-D-glucopyranoside (pNPGlc) and other substrates used in the study were purchased from Sigma.2.2. Strain isolation for ginsenoside biotransformation
During the course of a study to determine the types of aerobic bacteria in compost (made from foodstuff waste), the samples were collected from different places in Daejeon city, South Korea. Then, the samples were thoroughly suspended with 0.85% saline (sterilized), following serial dilution, and then were spread onto an R2A agar medium (BD). The plates were incubated at 30 °C for 7 days. Single colony was obtained by subculture. The strain BS20T was seek out, and then it was routinely cultured on R2A agar at 30 °C and preserved as glycerol suspensions (25%, v/v) at −80 °C.For screening β-glucosidase-positive bacteria, the colonies were isolated from different sources, including compost and ginseng soil. To check the β-glucosidase activities of the novel strains, the isolates were transferred to esculin-R2A agar plates (R2A agar containing 1.0 g L−1 esculin and 0.5 g L−1 ferric citrate), and incubated at 30 °C for 1–3 days. The β-glucosidase-positive strains, which released esculetin (reddish-brown zone) from esculin, were selected and used for the bioconversion of ginsenosides. Briefly, the liquid R2A broth cultures of these β-glucosidase-positive bacterial strains in their maximum growth phase were mixed with 200 μL of sterilized R2A broth containing 1 mg mL−1 of PPD-mix [(Rb1, Rb2, and Rc)] or PPT-mix [Re and Rg1] type ginsenosides in a 1.5 mL Eppendorf Tube and incubated at 30 °C for 24 hours. After 24 hours, 50 μL aliquots were taken, extracted with an equal volume of 80% water-saturated n-butanol, and then analyzed by thin layer chromatography [TLC] (Fig. S1†). The novel strain BS20T, which exhibited ginsenoside-hydrolyzing activity, was selected for complete genome sequencing and further experiments.
2.3. Taxonomic characterization of strain BS20T sp. nov.
2.4. Complete genome sequencing
2.5. Phylogenetic analysis of BglAg-762
Database homology search was performed with BLAST program provided by NCBI. Sequences of the characterized glycosyl hydrolases were obtained from the CAZY database [Carbohydrate-Active enZymes database (http://www.cazy.org)], and multiple alignments were performed using the CLUSTAL_X program.28 Gaps were edited in the BioEdit program,29 and evolutionary distances were calculated using a Poisson model. A phylogenetic tree was constructed using the neighbor-joining algorithm31 in the MEGA6 program,33 with bootstrap values based on 1000 replicates.352.6. Molecular cloning, expression, and purification of the novel recombinant BglAg-762 enzyme
A β-glucosidase belonging to the glycoside hydrolase family 3 can hydrolyze the glucose molecules attached to the C3, C6, or C20 position of the major ginsenosides. The gene encoding β-glucosidase was identified by analyzing the complete genome sequence, and the oligonucleotide primers used for cloning were designed based on the DNA sequence of BglAg-762 (GenBank accession no. CP015971). These primers were designed as forward (5′-GGTTCCGCGTGGATCCAAAAAATCAATCGTAAAAATTT-3′) and reverse (5′-GATGCGGCCGCTCGAGTTATTTGCTCCAATCAATAGCT-3′) by Macrogen Co. Ltd. Korea were used to introduce the BamHI and XhoI restriction sites, respectively. After amplification with PCR, the amplified DNA fragments were purified and inserted into the pGEX4T-1 GST fusion vector using an EzCloning Kit (Enzynomics Co. Ltd., Korea). The resultant recombinant pGEX-BglAg-762 was used to transform E. coli BL21 (DE3). The recombinant E. coli BL21 (DE3) containing a plasmid with the target gene (BglAg-762) was grown in an LB–ampicillin medium at 37 °C until the culture achieved an OD600 value of 0.4–0.6. At this point, the protein expression was induced by adding 0.1 mM isopropyl-β-D-thiogalactopyranoside (IPTG), and the cells were incubated for 24 hours at 28 °C. After this incubation, the cells were harvested through centrifugation and were then washed twice with 1% Triton solution (100 mM sodium phosphate and 1% Triton X-100 at pH 7.0) and the pallets (1 g/10 mL) were resuspended in 100 mM sodium phosphate (pH 7.0). Cell sonication was carried out using the Branson Digital Sonifier (400 W, 70% power, USA) using the following settings: low power (36%) output, 5 s ON/5 s OFF pulses, ±5 °C water bath but no floating ice. The total sonication time was 10 min. The crude cell extract was retained and the unwanted cell debris was removed by centrifugation (5000 rpm, for 10 min at 4 °C). The GST tag was purified using the GST-bind agarose resin (Elpisbiotech Co. Ltd, Korea) and the homogeneity of the protein was assessed using 10% SDS-PAGE and an EZStain Aqua solution (ATTO Corporation, Japan). The SDS PAGE analysis showed a difference between the molecular weights of the well-localized proteins in the gel (around 90 kDa) and the full-length sequence (102 kDa). After the purification of BglAg-762, the molecular weight of the recombinant enzyme was confirmed to be around 90 kDa.44–46Most enzymes are very sensitive to their surrounding pH and temperature, and very high or low pH values generally result in complete loss of activity. Therefore, the effect of pH on the activity of BglAg-762 was determined using 1.0 mM pNPG as a substrate in the following buffers, each at 50 mM: KCl–HCl (pH 2.0), glycine–HCl or citrate (pH 3.0), sodium acetate (pH 4.0 and 5.0), sodium phosphate (pH 6.0, 7.0, and 7.5), Tris–HCl (pH 8.0, and 9.0), and glycine–sodium hydroxide or carbonate (pH 10). pH stability was measured by incubating the mixture of the sample and the buffer (containing BglAg-762 enzyme and 2.0 mM pNPG in 50 mM potassium buffer) for 12 h at 4 °C. After 12 h, the percentage of activity gained at the optimum pH was measured. To investigate the effect of temperature on the activity of BglAg-762, and to determine the optimum temperature for its activity, the samples were incubated at the optimum pH for 5 min in 50 mM potassium phosphate buffer with 2.0 mM pNPG under various temperatures (4, 10, 20, 25, 30, 37, 40, 45, 50, 55, and 60 °C). As BglAg-762 was very sensitive to temperature, the thermo-stability of the enzyme was observed by incubating the samples in 50 mM potassium phosphate buffer for different periods of time (5 min, 10 min, 30 min, 1 h, 2 h, 4 h, and 16 h) at the different temperatures (4–60 °C). After the preset time intervals, the samples were collected and the enzymatic activity was determined using pNPG as the substrate.
Metals and heavy metals are naturally occurring elements with high atomic weight and density. Therefore, the enzyme activity is affected by heavy metal availability and concentration as well. High concentrations of heavy metals are toxic to the active site of protein and could inhibit enzyme activity. Thus, the effects of metals and other chemicals on BglAg-762 activity were also determined. BglAg-762 activity was assessed in the presence of 1 and 10 mM (final concentration) of NaCl, KCl, CaCl2, MgCl2, CoCl2, MnSO4, MgSO4, β-mercaptoethanol, and EDTA for 10 min at 50 °C. The retained activity was determined using pNPG as a substrate and expressed as a percentage of the activity in the absence of the compound.
It is commonly known that enzymes show different levels of activity for different substrates. Therefore, in this study, substrate preference of BglAg-762 was determined using 2.0 mM chromogenic o-nitrophenyl (oNP) and p-nitrophenyl (pNP) as substrates. The samples were incubated at 37 °C for 5 min, and the enzyme activity was observed. One unit of enzyme activity (U) was defined as the amount of enzyme required to release 1 μmol of oNP or pNP in 1 min. The following substrates were tested: pNP-β-D-galactopyranoside, pNP-β-D-glucopyranoside, pNP-β-D-fucopyranoside, pNP-β-L-arabinopyranoside, pNP-N-acetyl-β-D-glucosaminide, pNP-β-D-mannopyranoside, pNP-β-D-xylopyranoside, pNP-α-D-glucopyranoside, pNP-α-L-arabinofuranoside, pNP-α-L-arabinopyranoside, pNP-α-L-rhamnopyranoside, pNP-α-D-mannopyranoside, pNP-α-D-xylopyranoside, oNP-β-D-glucopyranoside, oNP-β-D-galactopyranoside, oNP-β-D-fucopyranoside, and oNP-α-D-galactopyranoside (all from Sigma).
:35:10, lower phase) as a solvent in 60 F254 silica gel plates (Merck, Germany). The spots on the TLC plates were visualized by spraying 10% (v/v) H2SO4 and heating at 110 °C for 5–10 min, and then compared with ginsenoside standards.3. Results and discussion
3.1. Characterization of Arachidicoccus ginsenosidimutans sp. nov.
Arachidicoccus ginsenosidimutans (gin.se.no.si.di.mu'tans. N.L.n. ginsenosidum ginsenoside; L. part. adj. mutans transforming, converting; N.L. part. adj. ginsenosidimutans ginsenoside-converting).After screening 15 novel bacterial species isolated from various sources, including ginseng soil and compost, the ability of these isolates to convert ginsenosides was tentatively determined by TLC analysis (data not shown). One of 15 novel species, designated as BS20T, was characterized using a polyphasic approach to clarify its taxonomic position. After complete genomic analysis of this strain, a novel ginsenoside-hydrolyzing β-glucosidase was identified and characterized.
Cells of the BS20T strain were Gram-negative, aerobic, non-motile, and short rod-shaped, approximately 0.5–2.0 μm in length and 0.4–0.6 μm in diameter (Fig. S2†). After 48 h of incubation on R2A agar medium, the colonies were 2–4 mm in diameter, flat, circular, smooth, entire, and had a milky color. Optimum growth occurred at 30 °C (15–37 °C) and the optimum pH was 6–7 (pH 6.0–9.0). The BS20T strain grew well on the R2A medium, weakly on nutrient agar, Luria Bertani, and DNase agar, and did not grow at all on trypticase soy agar and MacConkey agar. Growth occurred at NaCl concentrations ranging from 0 to 1%. BS20T cells were positive for catalase, oxidase, and esculin hydrolysis activities, but negative for hydrolysis of cellulose, Tween-80, starch, Tween-20, chitin, tyrosine and casein. When tested in API (20NE, 32GN, and API ZYM) systems, the cells were positive for alkaline phosphatase, esterase, leucine arylamidase, valine arylamidase, cystine arylamidase, acid phosphatase, naphtol-AS-BI-phosphohydrolase, α-galactosidase, β-galactosidase, α-glucosidase, β-glucosidase, N-acetyl-β-glucosaminidase, α-fucosidase, L-rhamnose, N-acetyl-glucose, D-saccharose, and D-maltose. The cells were negative for indole production, urea, gelatin, lipase, trypsin, potassium gluconate, capric acid, adipic acid, malic acid, phenylacetic acid, potassium 2-ketogluconate, 3-hydroxybutyric acid, 4-hydroxybenzoic acid, propionic acid, 3-hydroxybenzoic acid, valeric acid, trisodium citrate, L-fucose, D-sorbitol, L-serine, itaconic acid, suberic acid, sodium malonate, sodium acetate, lactic acid, L-alanine, potassium-5-ketogluconate, and inositol in these systems. The phenotypic differences between the BS20T and other closely related species in the same genus, based on phylogenetic analysis, are summarized in Table 1. The predominant fatty acids in these cells were iso-C15:0, iso-C15:1 G, iso-C17:0 3-OH, and summed feature 3 (comprising C16:1 ω7c and/or C16:1 ω6c). The cellular fatty acid profiles of BS20T and two reference strains grown under identical conditions are summarized in Table 2. These results suggested that this newly isolated bacterium belonged to the genus Arachidicoccus. The major polar lipids in these cells were PE and an unknown polar lipid (L2), while six unidentified minor polar lipids (L1, L3, L4, L5, L6, and L7), two unknown aminophospholipids (APLl and APL2), and two unknown phospholipids (PL1 and PL2) were found (Fig. S3†). The only predominant menaquinone in this strain was MK-7. The G + C content in the DNA was 39.4 mol%. Tables 1 and 2 show the characteristics used to distinguish BS20T from the other related species in the genus Arachidicoccus.
| Characteristic | 1 | 2 | 3 |
|---|---|---|---|
| a Data taken from: A. ginsenosidivorans KCTC 22820T (Siddiqi et al. 2017).b Data taken from: A. rhizosphaerae KCTC 22378T (Madhaiyan et al. 2015). | |||
| Isolation source | Compost | Ginseng soila | Rhizosphere soilb |
| Cell morphology | Rod | Roda | Coccoid rod b |
| Colony colour | Milkey | Light yellowa | Milkeyb |
| Salinity range for growth (% NaCl w/v) | 0.5–1 | 0–4a | 0–2b |
| Temperature range | 15–37 | 10–37a | 20–37b |
| pH range | 6–9 | 5–7a | 6–8b |
|
|||
| Enzymes activity | |||
| Esterase lipase | + | − | + |
| Arginine dihydrolase | − | − | + |
| α-Mannosidase | + | − | − |
| β-Glucuronidase | + | − | − |
| α-Chymotrypsin | − | + | − |
|
|||
| Utilization of | |||
| D-Ribose | + | − | − |
| Glycogen | + | − | + |
| D-Mannitol | + | − | − |
| D-Glucose | + | − | + |
| Salicin | + | − | − |
| D-Melibiose | + | − | + |
| L-Arabinose | + | − | + |
| L-Histidine | + | − | + |
| L-Proline | + | − | − |
| D-Mannose | + | − | − |
| D-Mannitol | − | + | − |
| N-Acetyl-glucosamine | + | − | + |
| D-Maltose | + | − | + |
| DNA G + C content (mol%) | 39.4 | 39.5a | 43b |
| Fatty acids | 1 | 2 | 3 |
|---|---|---|---|
| a Summed features represent groups of two or three fatty acids that could not be separated by gas chromatography (GLC) with the MIDI system. Summed features consist of: 3, C16:1 ω7c and/or C16:1 ω6c. | |||
| Saturated | |||
| C14:0 | 0.5 | 0.3 | 0.8 |
| C16:0 | 6.0 | 3.8 | 6.8 |
| C18:0 | — | 0.3 | 1.7 |
| C20:0 | — | — | 0.8 |
|
|||
| Branched | |||
| Iso-C15:0 | 39.9 | 42.7 | 39.5 |
| Iso-C15:0 3-OH | 0.7 | 2.7 | 2.2 |
| Iso-C15:1 G | 10.9 | 14.9 | 19.0 |
| Anteiso-C15:0 | 0.8 | 0.9 | 1.2 |
| Iso-C16:0 3-OH | 0.6 | — | 0.5 |
| Iso-C17:0 3-OH | 17.0 | 20.7 | 14.0 |
|
|||
| Hydroxy | |||
| C16:0 2-OH | 6.2 | 0.3 | 0.2 |
| C16:0 3-OH | 5.4 | 3.8 | 3.0 |
| C17:0 2-OH | 0.5 | — | 0.2 |
|
|||
| Summed featurea | |||
| 3; C16:1 ω7c and/or C16:1 ω6c | 10.8 | 9.3 | 9.8 |
The nearly complete 16S rRNA gene sequence (1450 nt) of the BS20T strain was determined and subjected to comparative analysis. Phylogenetic analysis using the maximum likelihood method based on 16S rRNA gene sequences indicated that BS20T was clustered within the genus Arachidicoccus and occupied an intra-genus clade with A. ginsenosidivorans KCTC 14980T and A. rhizosphaerae KCTC 22378T (Fig. 1). This relationship was also evident from the phylogenetic trees constructed with the neighbor-joining and maximum-parsimony methods. Further, the 16S rRNA gene of the strain BS20T exhibited the highest sequence similarity to A. ginsenosidivorans KCTC 22820T (95.0%) and A. rhizosphaerae KCTC 22378T (94.0%).
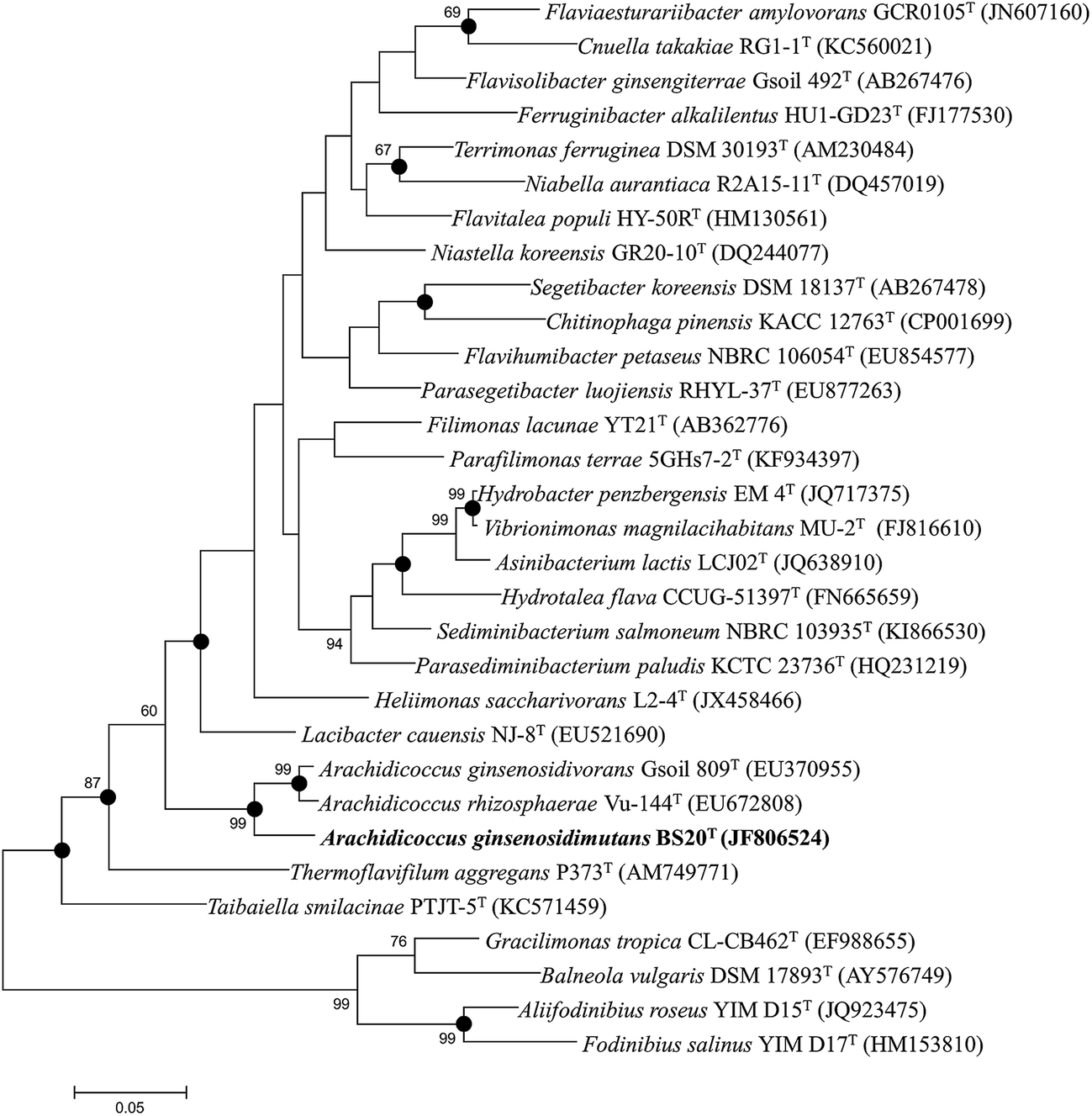 | ||
| Fig. 1 Phylogenetic relationship of strain BS20T, recognized as Arachidicoccus species, and other related genera in the family Chitinophagaceae. The tree was constructed using the maximum-likelihood method based on 16S rRNA gene sequences. Bootstrap values (expressed as percentages of 1000 replications) greater than 60% are shown at branch points. Filled circles indicate that the corresponding nodes were also recovered in the tree generated with maximum-parsimony and neighbor-joining algorithms. Bar represents 0.05 substitutions per nucleotide position. | ||
Based on the 16S rRNA gene sequence and the phylogenetic analysis, A. ginsenosidivorans KCTC 22820T and A. rhizosphaerae KCTC 22378T were selected as the closest recognized neighbors of the BS20T strain. These strains were then obtained from culture collections, grown under the same conditions, and used as reference strains in the subsequent phenotypic tests.
On the basis of low 16S rRNA-sequence similarity (<95%), phylogenetic position, and phenotypic and chemotaxonomic characteristics, a new taxon in the family Chitinophagaceae of phylum Bacteroidetes, named Arachidicoccus ginsenosidimutans sp. nov., was proposed, and the type strain for this species was proposed to be strain BS20T (=KACC 18624T = LMG 29195T). Phylogenetically, the novel isolate was related to the genus Arachidicoccus and shared several common features with members of this genus (MK-7 as the major respiratory quinone, PE as the major polar lipid, branched fatty acids with odd number of carbons, and the presence of a distinctive methoxy-fatty acid [iso-C17:0 3-OH]). The GenBank accession number for the 16S rRNA gene sequence of this strain is JF806524.
3.2. Genome properties of strain BS20T
The BS20T strain had a circular chromosome with 4138017 bp and a G + C content of 39.4 mol%. The genome sequence has been deposited in GenBank nucleotide database under the accession number CP015971. The genome statistics are shown in Table 3 and Fig. 2. Of the 3043 genes predicted, 2998 were protein-coding genes, and 45 were RNAs; 27 pseudogenes were also identified. The majority of the protein-coding genes (98.2%) were assigned a putative function, while the remaining was annotated as hypothetical or conserved hypothetical proteins (Table 3).
| Features | Chromosome |
|---|---|
| Length (bp) | 4138017 |
| DNA coding region (bp) | 3724510 |
| G + C content (%) | 39.4 |
| Genes | 3043 |
| Pseudo genes | 27 |
| CDS | 2998 |
| rRNA gene | 6 |
| tRNA genes | 39 |
 | ||
| Fig. 2 Graphical map of the chromosome of strain BS20T. From outside to the center: genes on the forward strand (colored by COG categories), genes on the reverse strand (colored by COG categories), RNA genes (tRNAs, green; rRNAs, red; other RNAs, black), and GC content and GC skew. | ||
3.3. Phylogenetic analysis of sequence of BglAg-762
The β-glycosidase gene (BglAg) consists of 2286 bp encoding 762 amino acids with a molecular mass of 102.0 kDa and a theoretical pI value of 6.15 (http://web.expasy.org/compute_pi/). Amino acid sequence analysis of the of BglAg-762 indicated that it has bellow than 69.0% similarity to the glycoside hydrolase of Niabella ginsenosidivorans (GenBank number WP 067760954) and other uncharacterized glycoside hydrolases, which belong to glycoside hydrolase family 3 (GH3). This means that BglAg-762 has homology to the protein domain of GH3. The Carbohydrate-Active enZymes database (http://www.cazy.org) describes more than 5000 uncharacterized and 222 characterized GH3 members that are widespread across numerous organisms. The glycoside hydrolase family 3 (GH3) is subdivided into six subfamilies.48 In order to determine the evolutionary position of BglAg-762 within the characterized enzymes in glycoside hydrolases family 3, a phylogenetic analysis was conducted using the neighbor-joining method in the MEGA6 program with bootstrap values based on 1000 replications (Fig. 3). The phylogenetic tree analysis indicate that BglAg-762 clustered within subfamily 6 and formed a separate and well-supported group (bootstrap of 100) with β-glucosidase derived from Escherichia coli [(AAA60495) uncharacterized] and Chrysosporium lucknowense,47 which was characterized.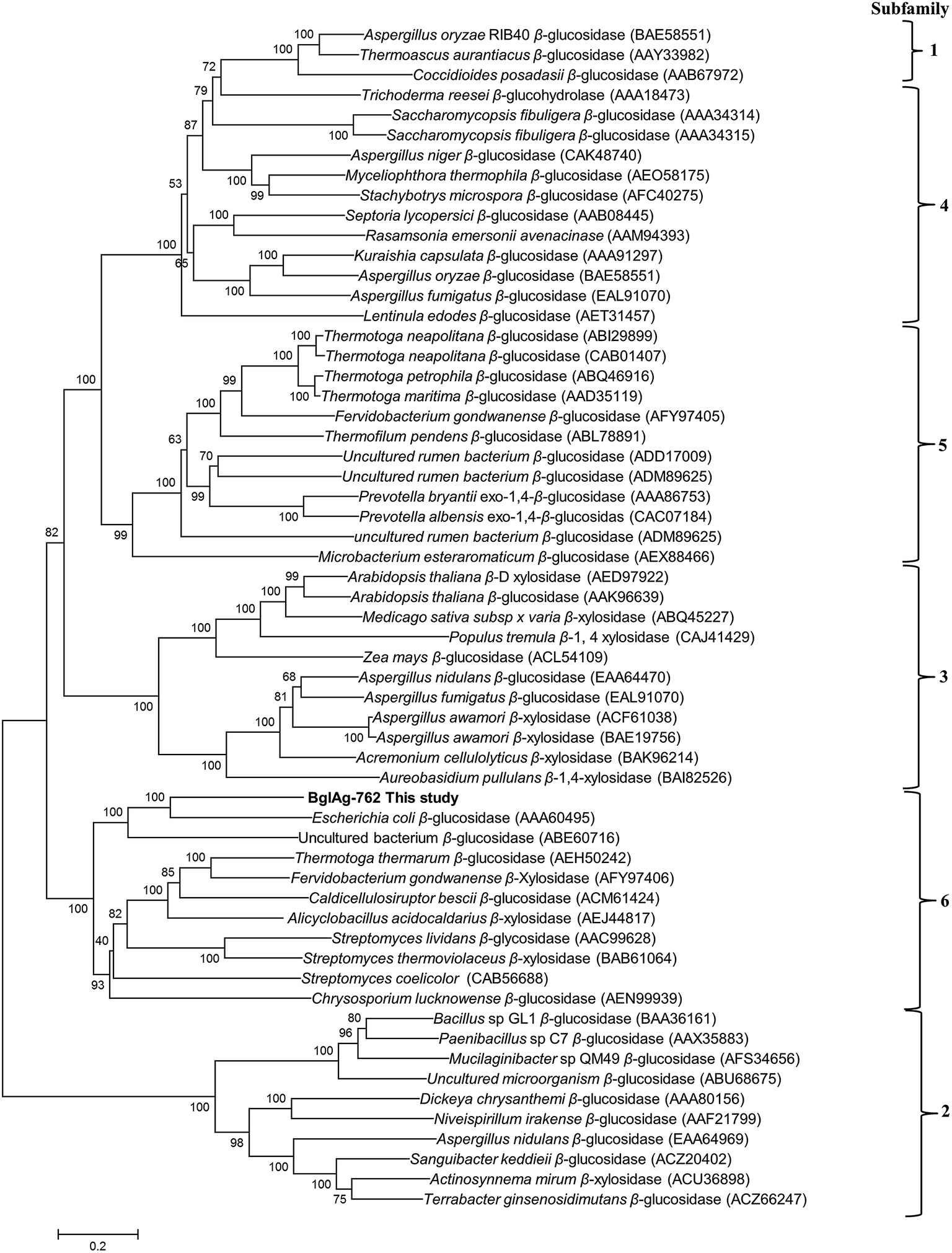 | ||
| Fig. 3 Phylogenetic analysis of characterized and/or uncharacterized glycoside hydrolases family 3 (GH3). Amino acid sequences were obtained from NCBI/EMBL database and CAZy database (accession numbers are indicated on the tree). This tree was made using the neighbor-joining method with a Poisson model and pairwise deletion. Bootstrap values expressed as percentages of 1000 replications. The bar represents 20 amino acid residues substitutions per 100 amino acid residues. | ||
3.4. Molecular cloning of β-glucosidase gene (BglAg-762) from strain BS20T
Genomic DNA from strain BS20T was isolated and subjected to complete genome sequencing by Macrogen, Korea. From this analysis, 17 sets of 4 different glycoside hydrolase genes, including β-glucosidase (8 sets), α,β-xylosidase (4 sets), L-rhamnosidase (2 sets), and α,β-arabinofuranosidase (3 sets) were selected and used for gene cloning. The sequences of the oligonucleotide primers used for the gene cloning were based on the DNA sequences of β-glucosidase, α,β-xylosides, L-rhamnosidase, and α,β-arabinofuranosidase (Table S1†). These primers were designed with BamHI and XhoI restriction sites, respectively, and were synthesized by Macrogen Co. Ltd. (Korea).After cloning, some sets of clones from the BS20T strain showed positive activities for pNP-β-D-glucopyranoside, pNP-β-D-galactopyranoside, pNP-β-D-fucopyranoside, pNP-N-acetyl-β-D-glucosaminide, pNP-β-L-arabinopyranoside, pNP-β-D-mannopyranoside, pNP-β-D-xylopyranoside, pNP-α-D-glucopyranoside, pNP-α-L-arabinofuranoside, pNP-α-D-fucopyranoside, pNP-α-L-rhamnopyranoside, pNP-α-D-mannopyranoside, oNP-β-D-glucopyranoside, oNP-β-D-fucopyranoside, and oNP-α-D-galactopyranoside (Table 4). Among the 17 sets of clones, only one (BglAg-762) was found to be positive for the ability to convert ginsenosides (Table 4), and was selected for use in further experiments. Based on amino acid-sequence analysis, BglAg-762 (GenBank accession number: CP015771) was predicted to have a molecular weight of 102 kDa. The amino acid-sequence analysis also indicated that it was 69% identical to the glycoside hydrolase of Niabella ginsenosidivorans (GenBank accession number: CP015772), which also belongs to glycoside hydrolase family 3. The enzymatic activity of the glycoside hydrolase from Niabella ginsenosidivorans has not yet been characterized. The glycoside hydrolase are classified according to amino acid-sequence similarity, as it has been shown to reflect the structural features of the enzymes, in addition to their substrate specificity (http://www.cazy.org/fam/acc_GH.html).49 Kinetic studies were performed with freshly purified enzymes using pNPG at 1–20 mM. Specific activity was expressed as units per milligram of protein. Protein concentrations were determined using the bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL), with bovine serum albumin (Sigma Aldrich, USA) as the standard. All assays were performed in triplicate.
| Substratesa | Enzyme activities of 17 different clones of Arachidicoccus ginsenosidivorans strain BS20 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2c | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | |
| a Final concentration, 2.0 mM.b Activity toward pNP-β-D-glucopyranoside was set as 100%.c One unit of activity was defined as the amount of protein required to generate 1 μmol of p-nitrophenol per min. Enzymes were purified ones. | |||||||||||||||||
| pNP-β-D-glucopyranosideb | w | ++ | w | w | + | vw | w | w | w | — | — | — | w | — | — | — | — |
| pNP-β-D-galactopyranoside | — | — | w | — | — | — | — | w | w | — | w | — | — | — | — | — | — |
| pNP-β-D-fucopyranoside | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
| pNP-β-D-glucosaminide | — | — | w | — | — | vw | — | — | — | vw | vw | — | vw | — | — | w | — |
| pNP-β-L-arabinofuranoside | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
| pNP-β-D-mannopyranoside | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
| pNP-β-D-xylopyranoside | — | — | — | — | vw | — | — | — | — | — | — | — | — | — | — | — | — |
| pNP-α-D-glucopyranoside | vw | — | vw | — | w | vw | — | — | vw | — | w | — | vw | w | — | vw | — |
| pNP-α-L-arabinofuranoside | — | — | — | — | — | — | — | — | vw | — | — | — | — | — | — | — | — |
| pNP-α-D-fucopyranoside | — | — | — | — | — | — | — | — | — | vw | — | — | — | — | — | — | — |
| pNP-α-L-rhamnnopyranoside | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
| pNP-α-D-mannopyranoside | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
| oNP-β-D-glucopyranoside | w | w | — | — | w | vw | — | — | vw | — | — | — | — | — | — | — | — |
| oNP-β-D-fucopyranoside | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
| oNP-β-D-galactopyranoside | — | — | — | — | — | — | — | — | vw | — | — | — | — | — | — | — | — |
| Ginsenosides transformation | — | ++ | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
3.5. Recombinant protein expression and ginsenoside transformation by BglAg-762
The recombinant pGEX-BglAg-762 was used to transform E. coli BL21 (DE3). The protein expression was then induced by the addition of 0.1 mM IPTG. In order to maximize the yield of the fusion protein, samples of the recombinant E. coli BL21 (DE3) grown in LB + amp (1 g mL−1) broth were kept in different temperatures (18, 22, 25, and 28 °C) with different induction conditions (0.05, 0.1, and 0.5 mM IPTG [data not shown]). Incubation with 0.5 mM IPTG at 28 °C for 18 h produced the maximum level of soluble active fused enzyme, as shown in Fig. 4 and S4.† The ability of BglAg-762 to convert PPD-type (Rb1, Rb2, Rb3, Rc, Rd, and GypXVII) and PPT-type (Re and Rg1) major ginsenosides was assessed by performing TLC analysis of the enzyme hydrolysates at regular intervals. Based on the Rf values, it was clear that BglAg-762 could transform only PPD-type major ginsenosides, as it showed no activity in converting PPT-type ginsenosides, Re and Rg1 (Fig. 5). BglAg-762 could efficiently hydrolyze the glucose attached at the C3 and C20 positions of all six PPD-type ginsenosides. The proposed pathways for the biotransformation of 1 mg mL−1 of PPD-type ginsenosides by BglAg-762 are as follows: Rb1 → Gyp-XVII → F2 → C-k; Rb2 → C-O; Rb3 → C-Mx1; Rc → C-Mc1 → C-Mc; Rd → F2; and Gyp-XVII → F2. The biotransformation occurs through the stepwise hydrolysis of the outer and inner glucose moieties at C3 and C20 positions of aglycon (Fig. 6). However, BglAg-762 could not further hydrolyze C-O, C-Mx1, and C-Mc1 even after incubating longer at 37 °C. The ginsenoside Rb1 was initially transformed into Gyp-XVII and then converted into F2 within 10 min and finally convert into C-k after 4 h incubation at 37 °C. Further incubation of C-k together with recombinant enzyme (BglAg-762) show no more hydrolysis of C-k, indicating that the recombinant BglAg-762 preferred the outer glucose at the C3 or C20 position. Moreover, the rate of conversion was much faster than previously published crude enzyme extracts (β-glucosidase) obtained from Caulobacter leidyia GP45 (ref. 50) and Bifidobacterium sp. SH5,51 which converted 1 mg mL−1 of Rb1 to Rd in 24 hours.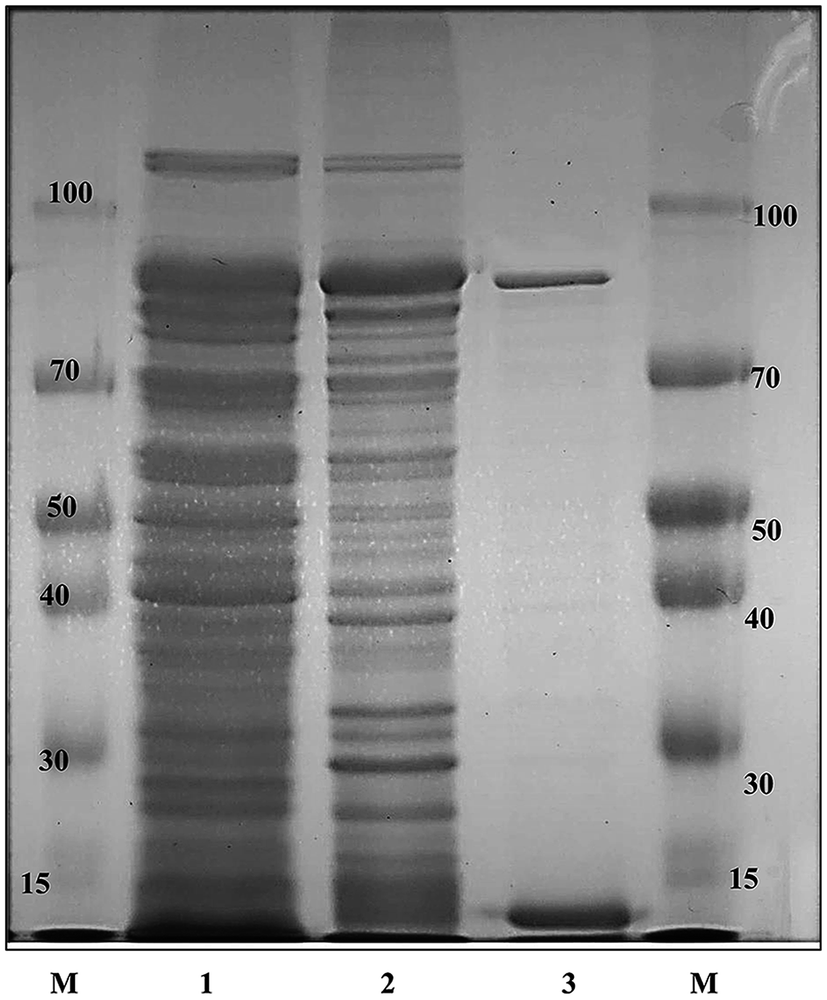 | ||
| Fig. 4 SDS-PAGE analysis of recombinant BglAg-762. Lanes: (M) molecular weight standard; (1) crude extract of BL21 (DE3) carrying pGEX-BglAg-762 before induction; (2) soluble fraction of crude extract of BL21 (DE3) carrying pGEX-BglAg-762 after induction; (3) GST-BglAg-762 enzyme fraction after purification with the GST-bind agarose resin. | ||
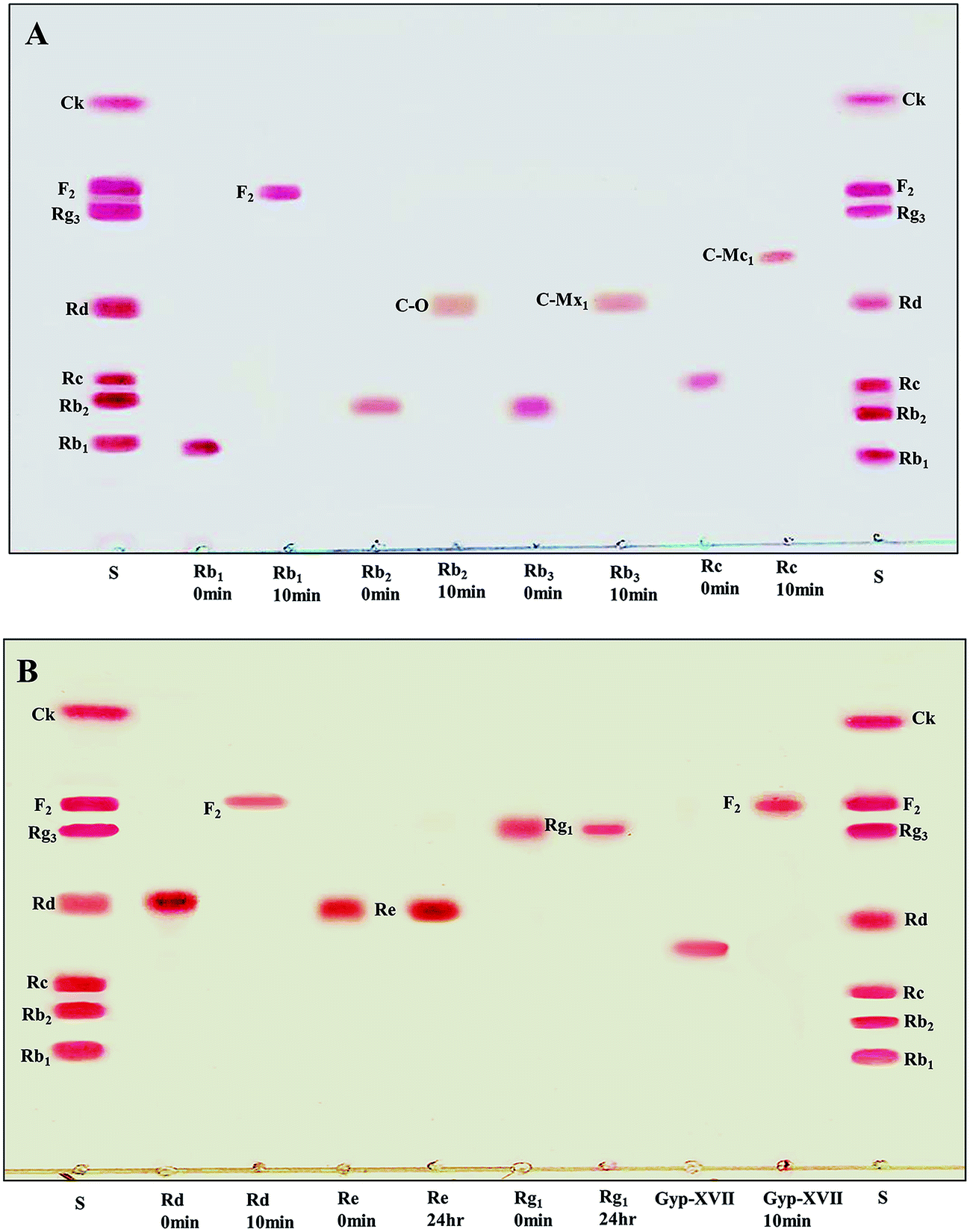 | ||
| Fig. 5 (A and B) TLC analyses of time course of ginsenoside bioconversion by BglAg-762 at an enzyme concentration of 1 mg mL−1. (A) Enzymatic transformation of ginsenosides Rb1, Rb2, and Rc. (B) Enzymatic transformation of ginsenosides Rd and Gyp-XVII. PPT-type ginsenosides Re and Rg1 did not show any change. Developing solvent: CHCl3–CH3OH–H2O (65:35:10, v/v, lower phase). Lane S, standards (Rb1, Rb2, Rc, Rd, Rg3, F2, and C-K). | ||
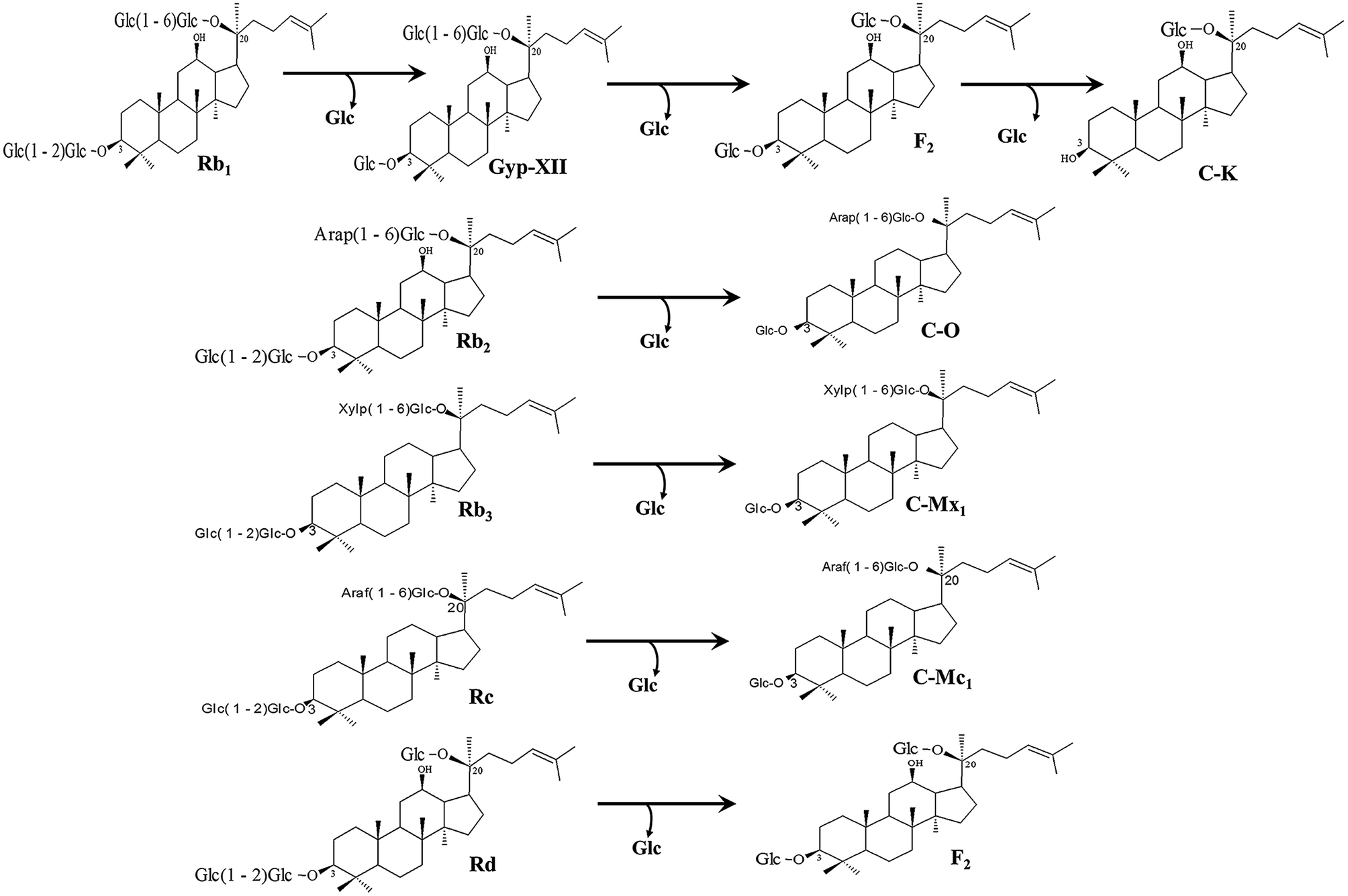 | ||
| Fig. 6 Pathways for enzymatic biotransformation of ginsenosides Rb1, Rb2, Rb3, Rc, Rd, and Gyp-XVII by recombinant BglAg-762, respectively. | ||
3.6. Purification and characterization of recombinant BglAg-762
GST-bind agarose resin was used to purify the recombinant enzyme, GST-BglAg-762. The supernatant from cell lysate as well as the purified protein was subjected to 10% SDS-PAGE (Fig. 4 and S4†). From the SDS PAGE analysis, we found a difference between the molecular weights of the well-localized proteins in the gel (around 90 kDa) and the full-length sequence (102 kDa). After purifying BglAg-762, the molecular weight of the recombinant enzyme was confirmed to be around 90 kDa, as shown in Fig. 4 and S4.† The difference between the molecular weight of the recombinant protein BglAg-762 calculated from amino acid-sequence analysis and that calculated after protein purification might be due to self-proteolysis of the bacterial strain protein.44–46The optimum pH and temperature for biotransformation were determined using crude BglAg-762. The BglAg-762 had optimal activity at pH 7.5 in a sodium phosphate buffer, and it was stable at pH 5.0 to 10.0 (Fig. 7A). When the pH was less than 5.0 or more than 10.0, the enzyme stability decreased very quickly, and at pH 5.0 and 10.0, the enzyme activity decreased to 38.0% and 14.0% of the optimum activity (Fig. 7A). The optimal temperature was found to be 50 °C; at 45 °C, the enzyme had 95.0% relative activity, while thermo-stability decreased significantly below 45 °C and no activity was detected when the enzyme was incubated above 50 °C for 2 h (Fig. 7B). BglAg-762 was stable below 45 °C, and about 52% of the activity was lost after incubation at 50 °C for 2 h. Thus, the optimal temperature for the activity of BglAg-762 was similar to that of the β-glucosidase isolated from Terrabacter ginsenosidimutans (45 °C) and lower than that of the β-glucosidase from Pyrococcus furiosus (55 °C). Thus, perhaps BglAg-762 is mesophilic and stable at a neutral pH range. In addition, the near-neutral optimal pH and the mild optimal temperature of BglAg-762 are similar to those of ginsenoside-hydrolyzing GH3 from other bacteria.18,52–55 Although the optimum temperature of BglAg-762 against pNPGlc was 50 °C, during extended and stable transformation, the ginsenoside-conversion reaction occurred at 37 °C with an optimum pH of 7.0–7.5.
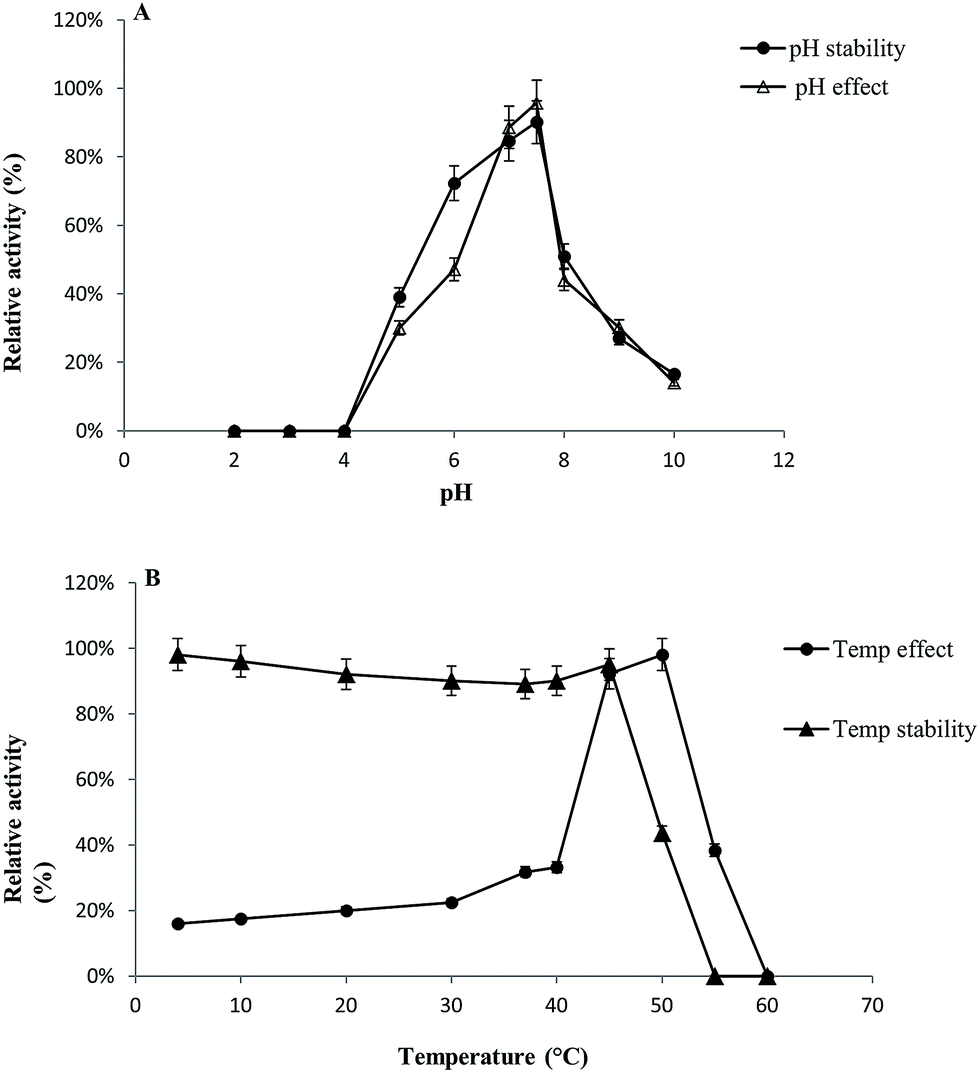 | ||
| Fig. 7 (A and B) Effect of pH on enzyme activity. The enzyme activities were measured under standard assay conditions. Enzyme solutions containing 2.0 mM pNPGlc were incubated with various buffers at pH 2.0 to 10.0 for 12 h at 4 °C. Stability of BglAg-762, the enzymes were incubated for 30 min at 50 °C in various buffers at pH 2.0 to 10.0 and the residual activities were measured (A). The effect of temperature on the stability and activity of recombinant BglAg-762 was measured under standard assay conditions. The thermo-dependence of BglAg-762 was assayed in 50 mM potassium phosphate buffer (pH 7.5) at varying temperatures ranging from 4 to 60 °C. Thermostability was tested by incubating aliquots of the enzyme in 50 mM potassium phosphate buffer (pH 7.5) for different periods of time at various temperatures. After cooling the sample, residual activity was determined (B). | ||
As mentioned in previous sections, heavy metals are atoms with high molecular weights and their concentration may affect enzyme activities. Therefore, the effects of metal ions, EDTA, and β-mercaptoethanol on the activity of GST-BglAg-762 were investigated. The results were expressed as a percentage of the activity in the absence of the test compound (Table S2†). The activity of BglAg-762 was not affected by 1 mM of metal ions Na+, K+, Mg2+, Mn+, and Co2+, or EDTA and β-mercaptoethanol; however, its activity was enhanced by 1 mM Ca2+, Co2+, and Mn2+. In addition, BglAg-762 activity was decreased by 10 mM of all the tested metal ions (except Co2+ and Mn2+), EDTA, and β-mercaptoethanol. The chelating agent, EDTA (10 mM), inhibited BglAg-762 activity, which indicated that divalent cations might be required for its activity. However, no dramatic positive effect on the activity of the BglAg-762 was found for any of the tested metal ions.
To investigate the substrate specificity of BglAg-762, it was treated with α- and β-configurations of 2.0 mM pNP- and oNP-glycosides. The results, which are summarized in Table 4, showed that BglAg-762 was most active against pNPGlc, followed by oNPGlc, but had no effect on any of the other pNP- and oNP-glycosides.
In this study, we encountered a problem with the sequence similarity through the NCBI protein blast (blastp). Using the data of complete genome sequence of the BS20T strain, we succeeded in cloning of 17 sets of four (4) different glycoside hydrolases in E. coli. However, when we analyzed the sequence homology, analogy, and paralogy for our functional gene using blastp, the results did not reflect the interesting aspects of the conservation of our target gene. The results of the NCBI blast showed that our gene had a sequence similarity of more than 60 and 70% to the different glycoside hydrolases (hypothetical proteins), as shown in Table S3.† However, after enzyme expression, most of the enzymes did not show specific activity against a substrate (Table 4), indicating that the corresponding results of blastp for our protein were not accurate and that the hypothetical protein will be considered as a putative, uncharacterized protein.
3.7. Ginsenoside-transformation characteristics of BglAg-762
For verifying the bioconversion pathways of the six PPD-type ginsenosides (Rb1, Rb2, Rb3, Rc, Rd, and Gyp-XVII) by BglAg-762, TLC analyses were performed at regular intervals. GST-BglAg-762 was found to transform six ginsenosides, as shown by the Rf values from the TLC analysis (Fig. 5A and B). During the biotransformation of Rb1, two types of metabolites (Gyp-XVII and F2) were detected (Fig. S5†). Thus, BglAg-762 successively hydrolyzed the outer and inner glucose moieties at position C20 and the inner glucose at position C3. It also transformed all PPD-type major ginsenosides into minor ginsenosides within 10 min. During the conversion of all the PPD-type ginsenosides, it was confirmed through transformation pathways that BglAg-762 preferred to hydrolyze glucose moieties at C3, rather than those at C20. From TLC analysis, it was also observed that BglAg-762 could not hydrolyze the PPT-type ginsenosides, Re and Rg1, indicating that BglAg-762 could not hydrolyze single glucose molecules attached to C6 and C20 positions.The proposed pathways for the biotransformation of ginsenosides by BglAg-762 are as follows: Rb1 → Gyp-XVII → F2 → C-k; Rb2 → C-O; Rb3 → C-Mx1; Rc → C-Mc1; Rd → F2; and Gyp-XVII → F2. The biotransformation occurs through the stepwise hydrolysis of glucose moieties at the C3 and C20 positions of aglycon (Fig. 6).
4. Conclusion
Based on the phylogenetic position and phenotypic and chemotaxonomic characteristics, a novel species in the genus Arachidicoccus, isolated from compost, was identified. We proposed BS20T (=KACC 18624T = LMG 29195T) as the type strain of the novels species, Arachidicoccus ginsenosidimutans sp. nov. Complete genome sequencing (GenBank accession number: CP015971) and optical mapping of the strain BS20T was performed to identify the gene encoding glycoside hydrolase, responsible for ginsenoside transformation. Through complete genome analysis, a novel ginsenoside-transforming β-glucosidase, termed as BglAg-762, was identified. It belonged to the glycoside hydrolase superfamily 3. The optimum reaction condition for BglAg-762 was 50 °C at pH 7.5. BglAg-762 could convert various major ginsenosides (Rb1, Rb2, Rb3, Rc, Rd, and Gyp-XVII) by selective hydrolysis of glucose moieties. During the conversion of the different PPD-type ginsenosides, BglAg-762 efficiently hydrolyzed the inner glucose moieties at the C3 position. This is the first study to describe this novel species (Arachidicoccus ginsenosidimutans sp. nov.), to sequence its whole genome, and to characterize a novel ginsenoside-transforming β-glucosidase (BglAg-762) isolated from it. Furthermore, due to its quick and efficient reaction with the major ginsenoside, Rb1, BglAg-762 might be useful for the mass production of the minor ginsenoside, F2.Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
This research was supported by the project on survey and excavation of Korean indigenous species of the National Institute of Biological Resources (NIBR) under the Ministry of Environment and by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2015R1D1A1A01060255), Republic of Korea. There is no person employed by the funders but support the authors and this research work.References
- I. H. Cho, Effects of Panax ginseng in neurodegenerative diseases, J. Ginseng Res., 2012, 36(4), 342–353 CrossRef CAS PubMed.
- H. J. Park, D. H. Kim, S. J. Park, J. M. Kim and J. H. Ryu, Ginseng in traditional herbal prescriptions, J. Ginseng Res., 2012, 36, 225–241 CrossRef PubMed.
- S. Sengupta, S. A. Toh, L. A. Sellers, J. N. Skepper, P. Koolwijk, H. W. Leung, H. W. Yeung, R. N. Wong, R. Sasisekharan and T. P. Fan, Modulating angiogenesis: The yin and the yang in ginseng, Circulation, 2004, 110, 1219–1225 CrossRef CAS PubMed.
- A. S. Attele, J. A. Wu and C. S. Yuan, Ginseng pharmacology: Multiple constituents and multiple actions, Biol. Pharm. Bull., 1999, 58, 1685–1693 CAS.
- C. S. Yuan, J. A. Wu and J. Osinski, Ginsenoside variability in American ginseng samples, Am. J. Clin. Nutr., 2002, 75, 600–601 CAS.
- L. P. Christensen, Ginsenosides chemistry, biosynthesis, analysis, and potential health effects, Adv. Food Nutr. Res., 2009, 55, 1–99 CAS.
- M. A. Tawab, U. Bahr, M. Karas, M. Wurglics and M. Schubert-Zsilavecz, Degradation of ginsenosides in humans after oral administration, Drug Metab. Dispos., 2003, 31(8), 1065–1071 CrossRef PubMed.
- Q. F. Xu, X. L. Fang and D. F. Chen, Pharmacokinetics and bioavailability of ginsenoside Rb1 and Rg1 from Panax notoginseng in rats, J. Ethnopharmacol., 2003, 84, 187–192 CrossRef CAS PubMed.
- T. Akao, M. Kanaoka and K. Kobashi, Appearance of compound K, a major metabolite of ginsenoside Rb1 by intestinal bacterial in rat plasma after oral administration-measurement of compound K by enzyme immunoassay, Biol. Pharm. Bull., 1998, 21, 245e9 Search PubMed.
- M. Z. Siddiqi, M. S. Shafi, K. D. Choi and W. T. Im, Panacibacter ginsenosidivorans gen. nov., sp. nov., with ginsenoside converting activity isolated from soil of a ginseng field, Int. J. Syst. Evol. Microbiol., 2016a, 66, 4039–4045 Search PubMed.
- M. Z. Siddiqi, M. S. Shafi, K. D. Choi and W. T. Im, Sphingobacterium jejuense sp. nov., with ginsenosides converting activity, isolated from compost, Int. J. Syst. Evol. Microbiol., 2016b, 66, 4433–4439 Search PubMed.
- S. R. Ko, K. J. Choi, K. Suzuki and Y. Suzuki, Enzymatic preparation of ginsenosides Rg2, Rh1, and F1, Chem. Pharm. Bull., 2003, 51(4), 404–408 CrossRef CAS PubMed.
- C. H. Cui, J. K. Kim, S. C. Kim and W. T. Im, Characterization of a ginsenoside-transforming beta-glucosidase from Paenibacillus mucilaginosus and its application for enhanced production of minor ginsenoside F(2), PLoS One, 2014, 9, e85727 Search PubMed.
- C. H. Cui, S. C. Kim and W. T. Im, Characterization of the ginsenoside-transforming recombinant beta-glucosidase from Actinosynnema mirum and bioconversion of major ginsenosides into minor ginsenosides, Appl. Microbiol. Biotechnol., 2013a, 97, 649–659 Search PubMed.
- J. H. Lee, J. Y. Ahn, T. J. Shin, S. H. Choi, B. H. Lee, S. H. Hwang, J. Kang, H. J. Kim, C. W. Park and S. Y. Nah, Effects of minor ginsenosides, ginsenoside metabolites, and ginsenoside epimers on the growth of Caenorhabditis elegans, J. Ginseng Res., 2011, 35, 375–383 CrossRef CAS PubMed.
- K. W. Leung and A. S. Wong, Pharmacology of ginsenosides: a literature review, Chin. Med., 2010, 5, 20–22 CrossRef PubMed.
- S. Choi, T. W. Kim and S. V. Singh, Ginsenoside Rh2-mediated G1 phase cell cycle arrest in human breast cancer cells is caused by p15 Ink4B and p27 Kip1-dependent inhibition of cyclin-dependent kinases, Pharm. Res., 2009, 26, 2280–2288 CrossRef CAS PubMed.
- S. H. Choi, T. J. Shin, S. H. Hwang, B. H. Lee, J. Kang, J. W. Oh, C. S. Bae, S. H. Lee and S. Y. Nah, Differential effects of ginsenoside metabolites on HERG k channel currents, J. Ginseng Res., 2011, 35, 191–199 CrossRef CAS PubMed.
- Y. J. Kim, N. Yamabe, P. Choi, J. W. Lee, J. Ham and K. S. Kang, Efficient thermal deglycosylation of ginsenoside Rd and its contribution to the improved anticancer activity of ginseng, J. Agric. Food Chem., 2013, 61(38), 9185–9191 CrossRef CAS PubMed.
- M. H. Siddiqi, M. Z. Siddiqi, S. Ahn, S. Kang, Y. J. Kim, K. Veerappan, D. U. Yang and D. C. Yang, Stimulative Effect of Ginsenosides Rg5:Rk1 on Murine Osteoblastic MC3T3-E1 Cells, Phytother. Res., 2014, 28, 1447–1455 CrossRef CAS PubMed.
- M. Z. Siddiqi, M. H. Siddiqi, Y. J. Kim, Y. Jin, M. A. Huq and D. C. Yang, Effect of Fermented Red Ginseng Extract Enriched in Ginsenoside Rg3 on the Differentiation and Mineralization of Preosteoblastic MC3T3-E1 Cells, J. Med. Food, 2015, 18, 1–7 CrossRef PubMed.
- M. Madhaiyan, S. Poonguzhali, M. Senthilkumar, D. Pragatheswari, J. S. Lee and K. C. Lee, Arachidicoccus rhizosphaerae gen. nov., sp. nov., a plant-growth-promoting bacterium in the family Chitinophagaceae isolated from rhizosphere soil, Int. J. Syst. Evol. Microbiol., 2015, 65, 578–586 CrossRef CAS PubMed.
- M. Z. Siddiqi, Z. Aslam and W. T. Im, Arachidicoccus ginsenosidivorans sp. nov., with ginsenoside converting activity isolated from ginseng cultivating soil, Int. J. Syst. Evol. Microbiol., 2017, 67, 1005–1010 CrossRef PubMed.
- R. D. Fleischmann, M. D. Adams, O. White, R. A. Clayton, E. F. Kirkness, A. R. Kerlavage, C. J. Bult, J. F. Tomb, B. A. Dougherty and J. M. Merrick, Whole-genome random sequencing and assembly of Haemophilus influenzae Rd, Science, 1995, 269, 496–512 CAS.
- C. M. Fraser, J. D. Gocayne, O. White, M. D. Adams, R. A. Clayton, R. D. Fleischmann, C. J. Bult, A. R. Kerlavage, G. Sutton and J. M. Kelley, The minimal gene complement of Mycoplasma genitalium, Science, 1995, 270, 397–404 CAS.
- W. T. Im, Q. M. Liu, J. E. Yang, M. S. Kim, S. Y. Kim, S. T. Lee and T. H. Yi, Panacagrimonas perspica gen. nov., sp. nov., a novel member of Gammaproteobacteria isolated from soil of a ginseng field, J. Microbiol., 2010, 48, 262–266 CrossRef PubMed.
- O. S. Kim, Y. J. Cho, K. Lee, S. H. Yoon, M. Kim and H. Na, et al., Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species, Int. J. Syst. Evol. Microbiol., 2012, 62, 716–721 CrossRef CAS PubMed.
- J. D. Thompson, T. J. Gibson, F. Plewniak, F. Jeanmougin and D. G. Higgins, The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools, Nucleic Acids Res., 1997, 25, 4876–4882 CrossRef CAS PubMed.
- T. A. Hall, BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT, Nucleic Acids Symp. Ser., 1999, 41, 95–98 CAS.
- M. Kimura, The neutral theory of molecular evolution, Cambridge University Press, 1983 Search PubMed.
- N. Saitou and M. Nei, The neighbor-joining method: a new method for reconstructing phylogenetic trees, Mol. Biol. Evol., 1987, 4, 406–425 CAS.
- W. M. Fitch, Toward defining the course of evolution: minimum change for a specific tree topology, Syst. Zool., 1971, 20, 406–416 CrossRef.
- K. Tamura, G. Stecher, D. Peterson, A. Filipski and S. Kumar, MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0, Mol. Biol. Evol., 2013, 30, 2725–2729 CrossRef CAS PubMed.
- J. Felsenstein, Confidence limits on phylogenies: an approach using the bootstrap, Evolution, 1985, 39, 783–791 CrossRef PubMed.
- P. Gerhardt, R. G. E. Murray, R. N. Costilow, E. W. Nester, W. A. Woods and N. R. Krieg, et al., Manual of Methods for General Bacteriology, American Society for Microbiology, Washington DC, 1981 Search PubMed.
- J. F. Bernardet, Y. Nakagawa and B. Holmes, Proposed minimal standards for describing new taxa of the family Flavobacteriaceae and emended description of the family, Int. J. Syst. Evol. Microbiol., 2002, 52, 1049–1070 CAS.
- A. Hiraishi, Y. Ueda, J. Ishihara and T. Mori, Comparative lipoquinone analysis of influent sewage and activated sludge by high-performance liquid chromatography and photodiode array detection, J. Gen. Appl. Microbiol., 1996, 42, 457–469 CrossRef CAS.
- M. Sasser, Identification of bacteria by gas chromatography of cellular fatty acids, USFCC News Lett., 1990, 20, 16 Search PubMed.
- D. E Minnikin, A. G. O'Donnell, M. Goodfellow, G. Alderson and M. Athalye, et al., An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids, J. Microbiol. Methods., 1984, 29, 233–241 CrossRef.
- A. L. Delcher, D. Harmon, S. Kasif, O. White and S. L. Salzberg, Improved microbial gene identification with GLIMMER, Nucleic Acids Res., 1999, 27(23), 4636–4641 CrossRef CAS PubMed.
- R. Overbeek, R. Olson, G. D. Pusch, G. J. Olsen, J. J. Davis and T. Disz, et al., The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST), Nucleic Acids Res., 2014, 42(Database issue), D206–D214 CrossRef CAS PubMed.
- V. M. Markowitz, K. Mavromatis, N. N. Ivanova, I. M. Chen, K. Chu and N. C. Kyrpides, IMG ER: a system for microbial genome annotation expert review and curation, Bioinformatics, 2009, 25(17), 2271–2278 CrossRef CAS PubMed.
- I. Pagani, K. Liolios, J. Jansson, I. M. a. Chen, T. Smirnova, B. Nosrat, V. M. Markowitz and N. C. Kyrpides, The Genomes OnLine Database (GOLD) v. 4: status of genomic and metagenomic projects and their associated metadata, Nucleic Acids Res., 2012, 40(Database issue), D571–D579 CrossRef CAS PubMed.
- J. Miyake, S. Ochiai-Yanagi, T. Kasumi and T. Takagi, Isolation of a Membrane Protein from R. rubrum Chromatophores and Its Abnormal Behavior in SDS-Polyacrylamide Gel Electrophoresis Due to a High Binding Capacity for SDS, J. Biochem., 1978, 83, 1679–1686 CrossRef CAS PubMed.
- Q. R. Ahmad, D. H. Nguyen, M. A. Wingerd, G. M. Church and M. A. Steffen, Molecular weight assessment of proteins in total proteome profiles using 1D-PAGE and LC/MS/MS, Proteome Sci., 2005, 3, 6, DOI:10.1186/1477-5956-3-6.
- A. Rath, M. Glibowicka, V. G. Nadeau, G. Chen and C. M. Deber, Detergent binding explains anomalous SDS-PAGE migration of membrane proteins, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1760–1765 CrossRef CAS PubMed.
- G. S. Dotsenko, O. A. Sinitsyna, S. W. Hinz, J. Wery and A. P. Sinitsyn, Characterization of a GH family 3 beta-glycoside hydrolase from Chrysosporium lucknowense and its application to the hydrolysis of beta-glucan and xylan, Bioresour. Technol., 2012, 112, 345–349 CrossRef CAS PubMed.
- A. J. Harvey, M. Hrmova, G. De, J. N. Varghese and G. B. Fincher, Comparative modeling of the three-dimensional structures of family 3 glycoside hydrolases, Proteins, 2000, 41, 257–269 CrossRef CAS.
- B. Henrissat and G. Davies, Structural and sequence-based classification of glycoside hydrolases, Curr. Opin. Struct. Biol., 1997, 7, 637–644 CrossRef CAS PubMed.
- L. Q. Cheng, M. K. Kim, J. W. Lee, Y. J. Lee and D. C. Yang, Conversion of major ginsenoside Rb1 to ginsenoside F2 by Caulobacter leidyia, Biotechnol. Lett., 2006, 28(14), 1121–1127 CrossRef CAS PubMed.
- H. Chi and G. E. Ji, Transformation of ginsenosides Rb1 and Re from Panax ginseng by food microorganism, Biotechnol. Lett., 2005, 27, 765–771 CrossRef CAS PubMed.
- D. S. An, C. H. Cui, H. G. Lee, L. Wang, S. C. Kim, S. T. Lee and F. Jin, et al., Identification and characterization of a novel Terrabacter ginsenosidimutans sp. nov. β-glucosidase that transforms ginsenoside Rb1 into the rare gypenosides-XVII and LXXV, Appl. Environ. Microbiol., 2010, 76, 5827–5836 CrossRef CAS PubMed.
- C. H. Cui, Q. M. Liu, J. K. Kim, B. H. Sung, S. G. Kim, S. C. Kim and W. T. Im, Identification and characterization of a Mucilaginibacter sp. strain QM49 beta-glucosidase and its use in the production of the pharmaceutically active minor ginsenosides (S)-Rh1 and (S)-Rg2, Appl. Environ. Microbiol., 2013b, 79, 5788–5798 Search PubMed.
- L. H. Quan, J. W. Min, Y. Jin, C. Wang, Y. J. Kim and D. C. Yang, Enzymatic biotransformation of ginsenoside Rb1 to compound K by recombinant beta-glucosidase from Microbacterium esteraromaticum, J. Agric. Food Chem., 2012, 60, 3776–3781 CrossRef CAS PubMed.
- C. C. Ruan, H. Zhang, L. X. Zhang, Z. Liu, G. Z. Sun and J. Lei, et al., Biotransformation of ginsenoside Rf to Rh1 by recombinant beta-glucosidase, Molecules, 2009, 14, 2043–2048 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02612a |
| This journal is © The Royal Society of Chemistry 2017 |