
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and surface characterization of well-defined amphiphilic block copolymers composed of polydimethylsiloxane and poly[oligo(ethylene glycol) methacrylate]†
Raita Goseki a,
Ling Honga,
Manabu Inutsukab,
Hideaki Yokoyama*b,
Kohzo Itob and
Takashi Ishizone*a
a,
Ling Honga,
Manabu Inutsukab,
Hideaki Yokoyama*b,
Kohzo Itob and
Takashi Ishizone*a
aDepartment of Chemical Science and Engineering, Tokyo Institute of Technology, 2-12-1-S1-13 O-okayama, Meguro-ku, Tokyo, 152-8552, Japan. E-mail: tishizon@polymer.titech.ac.jp
bDepartment of Advanced Materials Science, School of Frontier Sciences, The University of Tokyo, 5-1-5 Kashiwano-ha, Kashiwa, Chiba 277-8561, Japan
First published on 10th May 2017
Abstract
A series of amphiphilic polydimethylsiloxane-b-poly[tri(ethylene glycol) methyl ether methacrylate] (PDMS-b-PM3) diblock copolymers were prepared with varying PM3 compositions. The well-defined PDMS-b-PM3 was synthesized by using a coupling reaction between the living PM3 and ω-benzyl bromide-functionalized PDMS. The obtained polymers had narrow molecular weight distributions (Mw/Mn ≤ 1.08) and predictable molecular weights. The bulk-state self-assembly of the polymers was studied by transmission electron microscopy (TEM) and small-angle X-ray scattering (SAXS) measurements. The microphase separated morphologies in these amphiphilic block copolymers varied from the PDMS-cylinder to lamellar, even if the molecular fraction of the PM3 was progressively reduced from 70 wt% to 17 wt%. We also performed the detailed surface structural characterization of the amphiphilic PDMS-b-PM3 thin-films by X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), atomic force microscopy (AFM), and static water contact angle measurement. Based on these structural analyses, we proposed that the block copolymer self-assembled into a cylinder-like nanostructure at the top of thin film surface without any tedious annealing method in the case of the block copolymer possessing high PM3 content (≥70 wt%).
Introduction
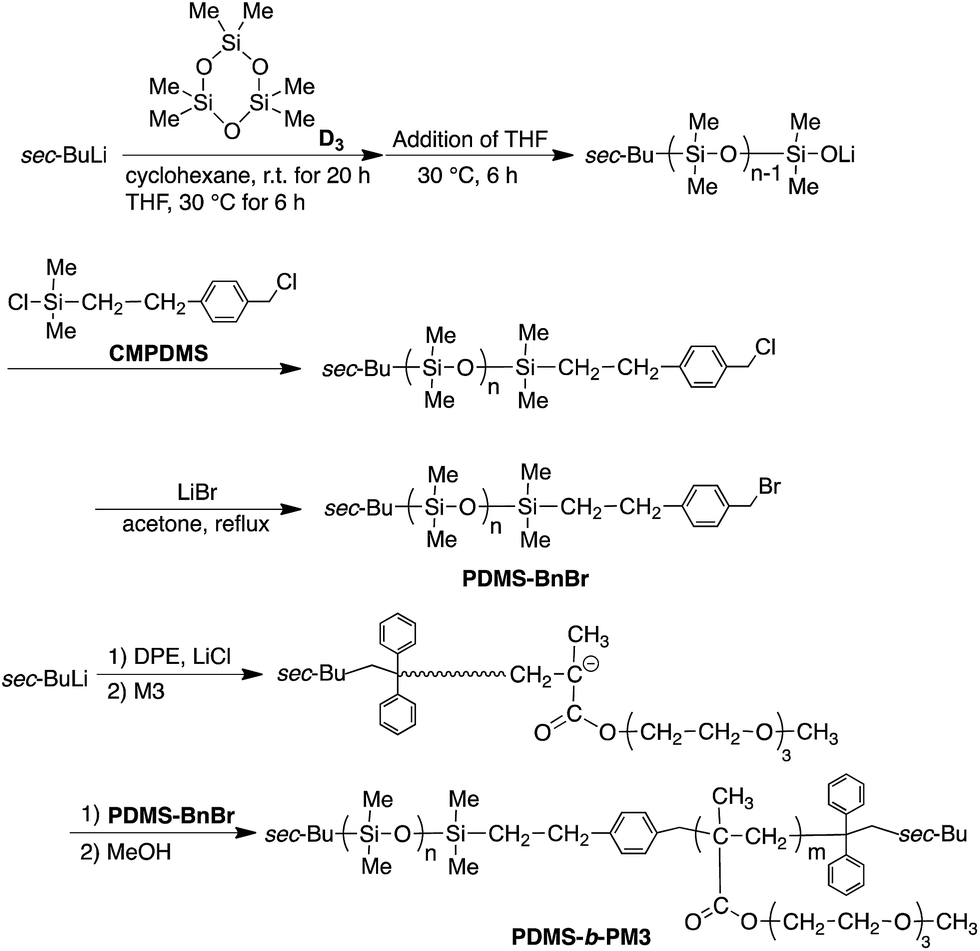
Amphiphilic block copolymers capable of self-assembling into periodic nanostructures in the bulk as well as in selective solvents have attracted much attention over the past decades due to their promising applications in biomedical materials, nano- and micro-templates, and other nanotechnologies.1–13 Thus, a vast majority of research work on self-assembly has been devoted to determine the properties of a wide variety of functionalized block copolymer segments.8–13 As a hydrophobic polymer, polydimethylsiloxane (PDMS) is well known to possess characteristic properties including a high stability, low glass transition temperature (Tg = −123 °C), low surface energy (19.8 mN m−1), high resistance to oxygen reactive ion etching (O2-RIE) and biocompatibility.14–19 Therefore, the incorporation PDMS segments into block copolymers are widely studied as biomaterials for a controlled drug delivery system18 and multifunctional materials such as nanolithographic applications.19Basically, the outermost surface of a block copolymer under dry conditions is usually covered by a low surface energy component. Therefore, PDMS-based diblock copolymers can form well-ordered microphase-separated nanostructures, but the surface segregation of PDMS easily occurs due to the low surface energy. This often induces difficulty in the formation of the desired structural morphology at the top of film surface. By the same principle, in amphiphilic block copolymers under dry conditions, hydrophobic segments spontaneously segregate to the surface and form a monolayer at the surface.20–27 However, a few exceptions have been reported.28–30 In a polymer having side chains of methoxy-terminated oligo (ethylene glycol) pendant groups, it has been revealed that the side chains were concentrated at the surface and reduced the contact angle of water. Previously, we reported the synthesis of a series of poly[oligo(ethylene glycol) methacrylate]s by the living anionic polymerization30–34 and the surface characterization of well-defined amphiphilic block copolymers composed of polystyrene (PS) and poly[tri(ethylene glycol) methyl ether methacrylate] (PM3) segments.29,30 In the study, we determined that a water-soluble hydrophilic PM3 segment selectively segregated on the top of most of the surface layer under dry conditions. In addition, it was suggested that the hydrophobic methyl terminus of the oligo (ethylene glycol) played an important role in decreasing the surface energy even if the chain length of the ethylene glycol is short. Although the PM3 has characteristic properties, such as water-solubility, thermosensitivity, and blood compatibility,35 there are few reports about the self-assembly of the PM3-containing block copolymer in the bulk as well as in a thin film. Needless to say, controlling the surface properties and composition of a polymer film is important for practical applications. A better understanding of the surface behavior of block copolymers with characteristic PM3 segments will lead to the better design of polymer materials. Nevertheless, the combination of PDMS and polymethacrylates (PMA) including PM3 are typically challenging to synthesize, therefore, there are only several reports about these well-defined block copolymers.36–43 Especially, despite these unique features as mentioned above, only a PM3-b-PDMS-b-PM3 triblock copolymer was synthesized by atom transfer radical polymerization using α,ω-dihydroxy-functionalized PDMS oligomer (Mn = 1000 g mol−1) to study the aggregation behaviors in the aqueous solution to date.41 A more versatile approach for the synthesis of the well-defined PDMS-b-PMA is still strongly desired.
In this study, we demonstrated a novel synthetic procedure for synthesizing a well-defined amphiphilic block copolymer composed of PDMS and PM3 using the coupling reaction based on the living anionic polymerization. The basic physical properties, self-assembled structure in the bulk, and surface structure of a thin film in the amphiphilic block copolymer with various compositions were also investigated.
Experimental section
Materials
sec-Butyllithium (sec-BuLi 1.4 M cyclohexane) was purchased from Kanto Chemical Co. 2-(Chloromethylphenyl)ethyl dimethylchlorosilane (CMPDMS) was purchased from Gelest Inc. The other reagents were purchased from Aldrich Japan Co. Tetrahydrofuran (THF) was refluxed over Na wire for 12 h and distilled over LiAlH4 under nitrogen. It was finally distilled from its sodium naphthalenide solution on a high vacuum line (10−6 Torr). 1,1-Diphenylethylene (DPE) was distilled over n-butyllithium, and hexamethylcyclotrisiloxane (D3) was distilled over CaH2. Lithium chloride (LiCl) and lithium bromide (LiBr) were heated under vacuum at 180 °C for 24 h. Acetone was distilled from molecular sieves 4A under nitrogen. Tri(ethylene glycol) methyl ether methacrylate (M3) was synthesized according to the previous report.32Measurements
1H and 13C NMR spectra were recorded on a BRUKER DPX-300 spectrometer at 300 MHz and 75 MHz, respectively. Molecular weights (Mn) and molecular weight distributions (Mw/Mn) were measured on an Asahi Techneion AT-2002 equipped with a Viscotek TDA model 302 triple detector array using THF as a carrier solvent at a flow rate of 1.0 mL min−1 at 40 °C. Three PS gel column (pore size (bead size)) were used: 650 Å (5 μm), 200 Å (5 μm), 75 Å (5 μm). The relative molecular weights were determined by SEC with RI detection using standard polystyrene calibration curve. Polymer mixtures after coupling reactions were separated by preparative SEC (KNAUER, Smart-line) equipped with four columns (TOSOH TSKgel G5000HHRx2, G4000HHRx2) and RI detector using THF as an eluent at a flow rate of 5.0 mL min−1 at room temperature. Glass transition temperature (Tg) and melting temperature (Tm) were measured with a Seiko instrument DSC6220 under nitrogen (heating rate 10 °C min−1, nitrogen flow rate 50 mL min−1). Turbidity measurements were performed on an ultraviolet-visible spectrometer (Jasco V-550) equipped with a Peltier temperature controller. Heating and cooling rates were 0.5 °C min−1 for all measurements. Dynamic light scattering (DLS) spectra were recorded on an Otsuka ELSZ spectrometer. Small angle X-ray scattering (SAXS) was carried out using a Bruker NanoSTAR (50 kV/100 mA) with a 2D-PSPC detector (camera length 1055 nm). XPS (JPS-90SX, JEOL) was carried out using AlKα radiation as the photosource and was used to investigate the surface composition of the sample. SEM image was carried out using a Hitachi SU9000 SEM with a field-emission source at 1.0 kV. For TEM measurements, bulk samples were embedded in epoxy resin and cured at 70 °C for 24 h. The embedded samples were then microtomed by a diamond knife at room temperature into a preset thickness of 70 nm using cryotome. The sections were picked up by the TEM grids and viewed directly with a JEOL JEM-2010F at 200 kV. Tapping mode atomic force microscopy (AFM-TM) measurements were performed on a Seiko model SPA-400.Bulk sample preparation
Samples of amphiphilic PDMS-b-PM3 diblock copolymer for investigating the bulk morphology were prepared by thermally annealed at 40 °C under vacuum for 24 h.Film sample preparation
Thin films were prepared by spin casting from a 20 μL polymer solution in acetone (3.0 wt%) onto silicon wafers (15 × 15 mm2) covered with native oxide at 2000 rpm for 20 s. After the complete removal of the solvent, subsequently the films were thermally annealed at 40 °C under vacuum for 24 h. The film thickness was measured by elipsometer.General procedure for synthesis of PDMS-b-PM3
All of polymerizations and coupling reactions were carried out under high vacuum condition (10−6 Torr) in sealed glass reactors equipped with break-seals. All reactors were prewashed with red-colored 1,1-diphenylhexyllithium in heptane after being sealed off from a vacuum line. The living PM3 was prepared by the polymerization of M3 with 1,1-diphenyl-3-methylpentyllithium in the presence of LiCl (3–5 equivalents to the initiator) in THF at −78 °C for 2.5 h. A small portion of the living PM3 was always taken to determine the Mn and Mw/Mn values prior to the next reaction.Synthesis of ω-chain-ended benzyl bromide (BnBr)-functionalized PDMS (PDMS-BnBr)
All reactors were prewashed with 1,1-diphenylhexyllithium (ca. 0.05 M) in heptane after being sealed off from a vacuum line. D3 (3.82 mmol) in cyclohexane (6.80 mL) was added at once with shaking to sec-BuLi (0.164 mmol) in heptane (2.90 mL) at 30 °C and the reaction was allow to stand at 30 °C for 20 h. Then, THF (7.0 mL) was added, and the reaction mixture was allowed to stand for additional 6 h. After that, CMPDMS (0.214 mmol) in heptane (6.80 mL) was added to quench at −78 °C and allowed to stand for 30 min. The reaction solution was poured into methanol to precipitate a polymer. The polymer was reprecipitated twice from THF to methanol, and freeze-dried from its dry benzene solution in vacuo for 24 h. The polymer (0.71 g) was obtained in 87% yield and characterized by SEC and 1H NMR spectroscopy. 1H NMR (300 MHz, CDCl3, δ, ppm): 7.27–7.21 (br, aromatic –CH2–Ar–CH2Cl), 4.57 (s, –Ar–CH2–Cl), 2.69–2.62 (m, Si–CH2–CH2), 0.96–0.87 (m, isobutyl –C(CH3)2), 0.11–0.01 (m, –Si(CH3)2, PDMS).The transformation reaction was carried out under nitrogen atmosphere. A solution of ω-chain-ended benzyl chloride-functionalized PDMS (0.71 g, 0.158 mmol) dissolved in a mixture of acetone (38 mL) and heptane (3.2 mL) in the presence of LiBr (1.89 g, 21.7 mmol) was refluxed for 6 h. The reaction mixture was poured into methanol. It was purified by reprecipitation twice from THF into methanol and freeze-drying from its absolute benzene solution (PDMS-BnBr: 0.55 g, yield: 77%). 1H NMR (300 MHz, CDCl3, δ, ppm): 7.27–7.23 (br, aromatic –CH2–Ar–CH2Br) 4.45–4.50 (br, –Ar–CH2–Br), 2.70–2.59 (m, Si–CH2–CH2), 0.97–0.85 (m, isobutyl –C(CH3)2), 0.14 to −0.03 (m, –Si(CH3)2, PDMS).
Synthesis of PDMS-b-PM3
A THF solution (11.1 mL) of ω-chain-ended BnBr-functionalized PDMS (0.275 g, 0.0511 mmol, Mn = 5400) pre-cooled at −78 °C, was added to a prior prepared living PM3 (0.0920 mmol, Mn = 8600) at −78 °C. The reaction mixture was allowed to stand at −40 °C for 24 h. The reaction was terminated by a small amount of degassed methanol, and the reaction mixture was poured into a large amount of methanol to precipitate the polymer. After checking the linking efficiency by the SEC to be quantitative, the objective block copolymer was isolated in 73% yield by preparative SEC (THF). 1H NMR (300 MHz, CDCl3, δ, ppm): 7.27–7.22 (br, aromatic –CH2–Ar–CH2–), 4.09 (s, –COO–CH2–), 3.65 (m, –COO–CH2–CH2–O–CH2–CH2–O–CH2–CH2–O–CH3), 3.56 (s, –CH2–O–CH3), 3.38 (s, –CH2–O–CH3), 2.02–1.67 (m, backbone), 1.09–0.79 (m, isobutyl –C(CH3)2), 0.14–0.03 (m, –Si(CH3)2, PDMS).Results and discussion
Synthesis of amphiphilic block copolymers by living anionic polymerization and coupling reaction
Living anionic polymerization technique is a promising way for synthesizing well-defined block copolymers by the sequential addition of monomers. However, there are pairs of block copolymers, such as PDMS and PMA, which cannot be prepared using sequential addition of monomers, because the nucleophilicity of the growing chain-end anions does not match the electron affinity of the other monomer. Recently, Hadjichristidis et al. developed a synthetic method for the well-defined PDMS-b-poly(tert-butyl methacrylate) (PtBMA) via a “one pot” process by the coupling reaction between ω-chain ended-benzyl chloride (BnCl) functionalized-PDMS and PtBMA anion in the presence of cesium iodide42 (Scheme S1†). Although this coupling method is very useful for preparing the synthetically difficult block copolymer, it required a long reaction time (3 day) and was unsuitable for synthesizing various block copolymers based on preparing the chain-ended functionalized pre-polymer due to the one-pot process. Therefore, there is still room for improvement of this valuable coupling reaction.To provide a more versatile approach, we propose a new synthetic route for PDMS-b-PM3 using ω-chain ended-benzyl bromide (BnBr)-functionalized PDMS based on the above mentioned coupling reaction because the BnBr group was capable of quantitative reacting with a variety of living polymer anions, such as polystyrene derivative, polydiene, and PMA anions, within several hours44–47 (Scheme 1). As can be seen, the synthesis is initiated from the ω-chain end-benzyl chloride (BnCl) functionalized-PDMS, which is obtained by the anionic ring-opening polymerization of hexamethylcyclotrisiloxane (D3) and the following termination reaction with 2-(chloromethylphenyl)ethyl dimethylchlorosilane (CMPDMS), by following the previous method.42 The resulting polymer was transformed into the corresponding benzyl bromide (BnBr) functionalized polymers by treating with LiBr in acetone/heptane (3/1, v/v). The 1H NMR spectrum showed the appearance of a new signal at 4.47 ppm, assigned to –Ar–CH2–Br with reasonable integral ratios (Fig. S1†). The SEC peaks of the polymers, reflecting their shapes and elution volume, were found to be almost similar both before and after the transformation reaction (Fig. 1a and b). The Mn and Mw/Mn values were 4500 g mol−1, 1.06 and 5400 g mol−1, 1.05, respectively. The MALDI-TOF-MS spectra of these polymers showed three series of peaks with an interval of 74 mass units corresponding to a 1/3 D3 repeating unit, and each peak exactly matched the total molar mass of the obtained PDMS possessing BnCl (PDMS-BnCl) and BnBr termini (PDMS-BnBr). This result strongly indicated that the termination reaction of the PDMS anion with CMPDMS quantitatively occurred. In addition, the mass difference of 44 (Da) in the PDMS-BnCl and PDMS-BnBr confirmed the quantitative conversion from BnCl to BnBr without any undesirable side reaction (Fig. S2†).
 | ||
| Scheme 1 Synthetic scheme of amphiphilic PDMS-b-PM3. | ||
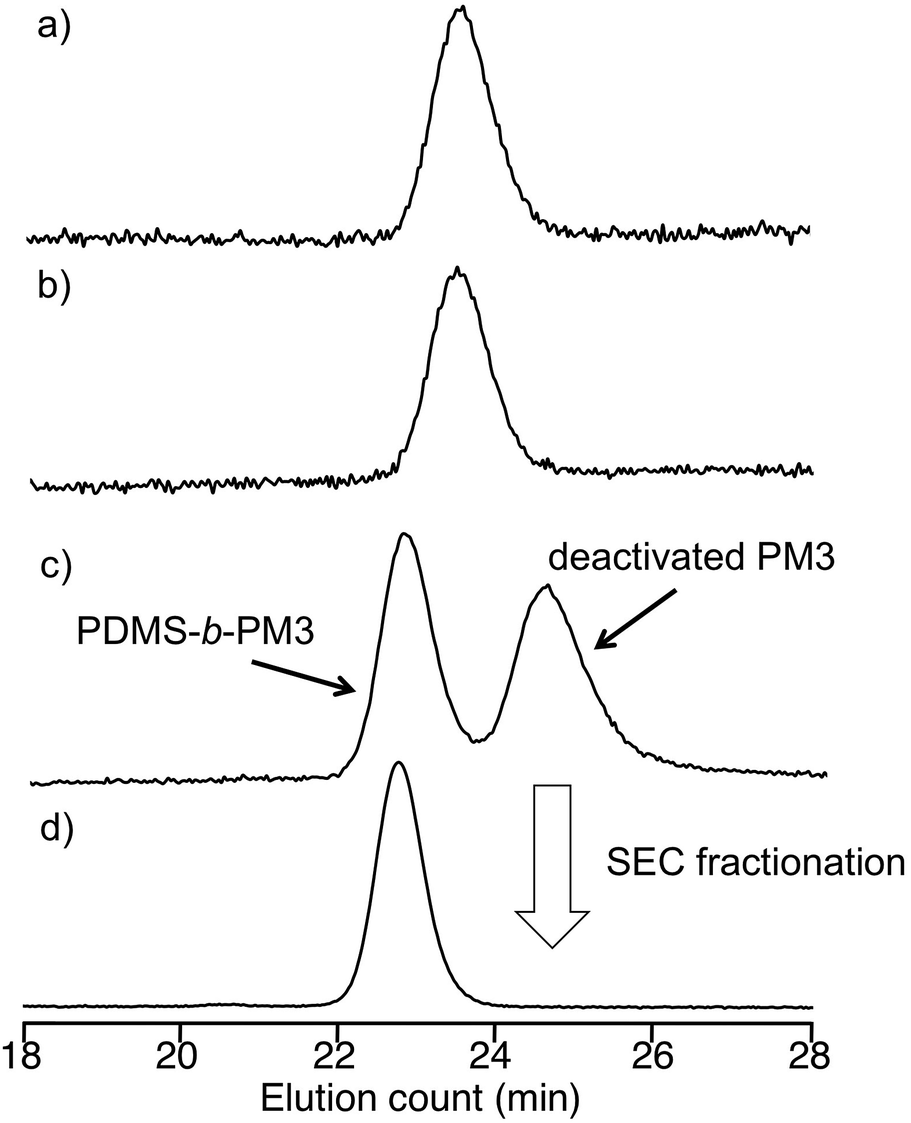 | ||
| Fig. 1 SEC curves of (a) PDMS-BnCl, (b) PDMS-BnBr, (c) after the coupling reaction of PDMS-BnBr and living PM3 anion, and (d) PDMS-b-PM3. | ||
In the final step, these PDMS-BnBr polymers were reacted with a 1.2–2.0 fold excess of living PM3 anion31 at −40 °C for 24 h. The results of coupling reaction are listed in the Table 1. The chemical structure of the PDMS-b-PM3 was characterized by 1H NMR, and the molecular weight was measured by SEC. The SEC chromatograms after coupling reaction displayed two distinct sharp peaks corresponding to the desired coupling product and the residual PM3 used in this reaction (Fig. 1c and d). No signal in the high molecular weight region was observed, indicating that the undesired side reaction did not occur under this reaction condition. The quantitative reaction was confirmed by disappearance of the peak for the precursory PDMS-BnBr. The desired coupling product was isolated by SEC fractionation. The SEC curve of the isolated polymer clearly shifted to the higher molecular weight region, and the molecular weight distribution was narrow (Mw/Mn < 1.06). The 1H NMR spectrum of PDMS-b-PM3 clearly displayed the successful incorporation of each block, indicated by the appearance of methyl (−OSi(CH3)2) protons corresponding to PDMS at 0.05 ppm, and methoxy (−OCH2CH2OCH3) protons corresponding to PM3 at 3.38 ppm (Fig. S1†). Hence, the composition of PDMS-b-PM3 was ascertained by using the integration ratios from 1H NMR spectrum of the methyl protons for PDMS and the methoxy protons for PM3 (Table 1). The resulting molecular weights and compositions fairly agreed with the calculated values. Thus, the linking reaction satisfactorily proceeded at −40 °C within 24 h, and the well-defined and expected structures of the resulting amphiphilic PDMS-b-PM3 were confirmed.
| Polymer | PM3a Mn (kg mol−1) | PDMS-BnBr Mna (kg mol−1) | Mn (kg mol−1) | Mw/Mn | PM3 (wt%) | |||
|---|---|---|---|---|---|---|---|---|
| Calcdb | SECa | obsdc | Calcd | Obsdc | ||||
| a Estimated by SEC with standard polystyrene samples.b Mncalcd. = (Mw of M3) × DP + (Mn of PDMS-BnBr).c Determined by comparison of integrations of methyl protons corresponding to PDMS at 0.05 ppm versus methoxy protons corresponding to PM3 at 3.38 ppm in the 1H NMR spectrum and molecular weights. | ||||||||
| PDMS-b-PM3 (70 wt%) | 8.6 | 5.4 | 14.0 | 15.1 | 18.1 | 1.02 | 61 | 70 |
| PDMS-b-PM3 (61 wt%) | 8.0 | 6.0 | 15.0 | 20.0 | 15.0 | 1.03 | 61 | 61 |
| PDMS-b-PM3 (48 wt%) | 5.1 | 6.0 | 12.2 | 16.2 | 13.0 | 1.03 | 50 | 48 |
| PDMS-b-PM3 (42 wt%) | 7.7 | 13.0 | 23.1 | 24.0 | 22.3 | 1.04 | 43 | 42 |
| PDMS-b-PM3 (30 wt%) | 6.0 | 13.0 | 20.4 | 24.1 | 18.0 | 1.04 | 32 | 30 |
| PDMS-b-PM3 (17 wt%) | 3.7 | 19.4 | 23.0 | 23.2 | 23.1 | 1.08 | 19 | 17 |
Therefore, it was found that the proposed synthetic procedure for the amphiphilic PDMS-b-PM3 block copolymer effectively and satisfactorily worked as shown in Scheme 1. In other words, the PDMS-BnBr carrying the electrophilic BnBr moiety is proved to be a usuful synthetic precursor for the well-defined block copolymers containing PMA segments.
Solubility and thermal property of block copolymer
The solubilities of the amphiphilic PDMS-b-PM3 (17, 30, 42, 48, 61, and 70 wt%: the numbers indicate the weight content of PM3) are summarized in Table S1† along with those of the corresponding homopolymer as references. The PDMS-b-PM3 was soluble in the common solvents, such as benzene, toluene, CHCl3, and THF. On the other hand, the solubility in hexane and methanol apparently reflected the content of the hydrophobic PDMS and hydrophilic PM3. Interestingly, the block copolymers with over 42 wt% PM3 were barely soluble in water, though the PS-b-PM3 (PM3: 55 wt%) was not soluble in water.30The obtained amphiphilic PDMS-b-PM3s were viscous colorless liquids. Thermal property of the obtained PDMS-b-PM3s was examined by DSC (Table 2). To eliminate the effect of the thermal history of the sample transitions, the sample was heated to 40 °C and held for 10 min at this temperature before cooling to −170 °C at the rate of 10 °C min−1. The reported DSC data were obtained from the second run. For the PDMS-b-PM3 (61 wt%), two distinct transitions were observed at −138 °C and −50 °C in the heating cycle, which correspond to the Tg of PDMS and PM3, respectively (Fig. S3†). On the other hand, by decreasing the PM3 fraction to below 48 wt%, the additional endothermic and exothermic peaks were newly observed in the heating cycle, which corresponded to the crystallization temperature of PDMS (Tc) and Tm of PDMS, respectively48 (Fig. S3b†).
| Polymers | Mn (kg mol−1)a | Tg (°C)b | Tm (°C)b |
|---|---|---|---|
| a Total molecular weight of PDMS-b-PM3.b Determined by DSC at the heating rate 10 °C min−1 on a second scan under N2. | |||
| PM3 | 12 | −50 | — |
| PDMS-b-PM3 (70 wt%) | 18 | −151, −48 | — |
| PDMS-b-PM3 (61 wt%) | 15 | −138, −56 | — |
| PDMS-b-PM3 (48 wt%) | 13 | −132, n.d | −56 |
| PDMS-b-PM3 (42 wt%) | 22 | −130, n.d | −56 |
| PDMS-b-PM3 (30 wt%) | 19 | −131, n.d | −55 |
| PDMS-b-PM3 (17 wt%) | 23 | −130, n.d | −51 |
| PDMS | 5.4 | −131 | −57 |
Thermosensitivity of block copolymer
Next, we investigated the thermosensitivity of the water-soluble PDMS-b-PM3 (42–70 wt%). Since the PM3 homopolymer shows a lower critical solution temperature (LCST) behavior (at 52 °C) in water, it is expected that these water-soluble block copolymers show a thermosensitivity. The LCST value was determined by turbidity measurements and dynamic light scattering (DLS). The LCST was defined as the onset of the slope of the turbidity curves, and the intersection of the tangent to the DLS curve at the second inflection with the horizontal tangent.The aqueous solution of the block copolymer (0.2 mg mL−1) was almost transparent at room temperature and the transmittance sharply decreased when the temperature reached a critical temperature (33–38 °C). The phase transition behavior was sensitive (ΔT = 3–4 °C) and reversible, which indicated that the PDMS-b-PM3s had a thermosensitivity as expected (Fig. S4†). It is noteworthy that the hysteresis of the transition was observed between the heating and cooling cycles due to morphological change depending on the time. The composition of PM3 and the molecular weight of PDMS did not affect the LCST value. In contrast, the LCST value of the block copolymer was significantly different from that of the PM3 homopolymer (52 °C). Considering these results, the formation of the copolymer micelles is facilitated by the intermolecular interaction of the polymer chains due to the effect of the hydrophobic PDMS. Indeed, it was observed that the initial transmittance gradually decreased from 100% with the increasing composition of the PDMS segments (Table 3).
| Polymers | Mna (kg mol−1) | Mn PDMSb (kg mol−1) | Cloud point (°C) | T0e (%) | Diameterc (nm) | ||
|---|---|---|---|---|---|---|---|
| DLSc | UV/visd | T < LCST | T > LCST | ||||
| a Total molecular weight of each polymer sample.b Estimated by SEC with standard polystyrene samples.c Determined by DLS measurement (0.2 mg mL−1) at the heating rate of 0.25 °C min−1.d Determined by UV/vis spectroscopy at 500 nm at the heating rate of 0.5 °C min−1 in water (0.2 mg mL−1).e Initial value of transmittance at 20 °C. | |||||||
| PM3 | 12 | 0 | 51 | 53 | 100 | 5 | 1091 |
| PDMS-b-PM3 (70 wt%) | 18 | 5.4 | 36 | 38 | 99 | 36 | 280 |
| PDMS-b-PM3 (61 wt%) | 15 | 6.0 | 32 | 35 | 97 | 43 | 2000 |
| PDMS-b-PM3 (48 wt%) | 13 | 6.0 | 32 | 35 | 89 | 49 | 760 |
| PDMS-b-PM3 (42 wt%) | 22 | 13.0 | 36 | 33 | 59 | 64 | 320 |
Next, the micelle behavior of PDMS-b-PM3 in water was investigated by DLS measurement. The micelle size of PDMS-b-PM3 in water was estimated to be 36–64 nm below LCST (Table 3). On the other hand, the micelle rapidly assembled into large aggregates above a certain temperature (32–36 °C), and the estimated size of aggregates was 280–760 nm (Fig. 2). The estimated LCST values of the resulting DLS measurement were in good agreement with the results of the turbidity measurements. Meanwhile, PM3 showed very small size (∼5 nm) below 50 °C, indicating that, under LCST, the block copolymer formed a micelle in water, and the PM3 homopolymer existed as unimers without aggregation.
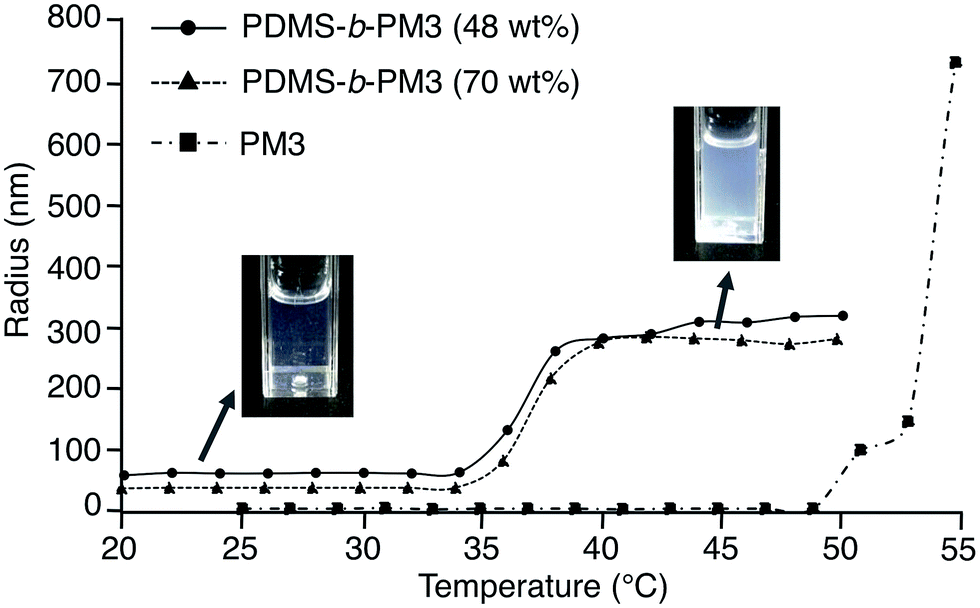 | ||
| Fig. 2 Variation in particle radius versus temperature at 2.0 °C min−1 in 0.2 mg mL−1 PDMS-b-PM3 aqueous solution. | ||
Bulk morphological characterization
The morphology of the PDMS-b-PM3 in the bulk state was characterized by small-angle X-ray scattering (SAXS) and transmission electron microscopy (TEM). The bulk samples were prepared by slow evaporation from chloroform at room temperature, then subsequently thermally annealed at 40 °C for 24 h in the vacuum oven, and quenched to room temperature. For the TEM measurement, all samples were imaged without staining, as the high electron density associated with the main chain Si atoms provided sufficient contrast for the TEM micrograph. As a result, the PDMS areas are dark, whereas the PM3 areas are bright.Fig. 3 shows the SAXS results of the PDMS-b-PM3 (70–48 wt%). It was observed for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :31/2:71/2 (or 1:31/2:41/2) set of peaks for the SAXS pattern of PDMS-b-PM3 (70, 61, and 48 wt%), which was consistent with spherical or cylindrical morphology. From the first order peak position, the long period spacing of the ordered structure is found to be equal to 20.5, 26.3 and 22.3 nm, respectively. Fig. 3a shows a TEM image of the PDMS-b-PM3 (70 wt%), spherical nanostructure was observed but not well-ordered. By directly measuring the width between the center to center PDMS domain (dark region) on the micrograph, the measured width is approximately consistent with the long period spacing measured from that corresponding to SAXS result. With a further decreasing of the content of PM3 from 42 wt% to 17 wt%, the SAXS diffraction shows reflections with the relative peak positions in the ratio 1:2: 3(:4:5), likely indicating a microphase separated lamellar nanostructure. In the TEM image of PDMS-b-PM3 (42 wt%), the PDMS domain exhibits as a dark lamellar layer in the image (Fig. 3b). The width between the PDMS domain centers on the images by directly measuring, the measured average length (28.2 nm) was very close to the SAXS results (29.4 nm). While, the TEM images of PDMS-b-PM3 (17 wt%) exhibited not lamellar nanostructure, but irregular island-sea like texture with 100 nm sized spherical aggregates (Fig. 3c). It was considered that the disordered structure formation was caused by a structural reconstruction when the thin film sample was prepared for measuring the TEM because both block segments have a sufficient molecular mobility to adjust the surface energy (TgPDMS = −138 °C, TgPM3 = −50 °C). It was suggested that the structure of the cryomicrotomed film sample was different from that of the original bulk structure.
:31/2:71/2 (or 1:31/2:41/2) set of peaks for the SAXS pattern of PDMS-b-PM3 (70, 61, and 48 wt%), which was consistent with spherical or cylindrical morphology. From the first order peak position, the long period spacing of the ordered structure is found to be equal to 20.5, 26.3 and 22.3 nm, respectively. Fig. 3a shows a TEM image of the PDMS-b-PM3 (70 wt%), spherical nanostructure was observed but not well-ordered. By directly measuring the width between the center to center PDMS domain (dark region) on the micrograph, the measured width is approximately consistent with the long period spacing measured from that corresponding to SAXS result. With a further decreasing of the content of PM3 from 42 wt% to 17 wt%, the SAXS diffraction shows reflections with the relative peak positions in the ratio 1:2: 3(:4:5), likely indicating a microphase separated lamellar nanostructure. In the TEM image of PDMS-b-PM3 (42 wt%), the PDMS domain exhibits as a dark lamellar layer in the image (Fig. 3b). The width between the PDMS domain centers on the images by directly measuring, the measured average length (28.2 nm) was very close to the SAXS results (29.4 nm). While, the TEM images of PDMS-b-PM3 (17 wt%) exhibited not lamellar nanostructure, but irregular island-sea like texture with 100 nm sized spherical aggregates (Fig. 3c). It was considered that the disordered structure formation was caused by a structural reconstruction when the thin film sample was prepared for measuring the TEM because both block segments have a sufficient molecular mobility to adjust the surface energy (TgPDMS = −138 °C, TgPM3 = −50 °C). It was suggested that the structure of the cryomicrotomed film sample was different from that of the original bulk structure.
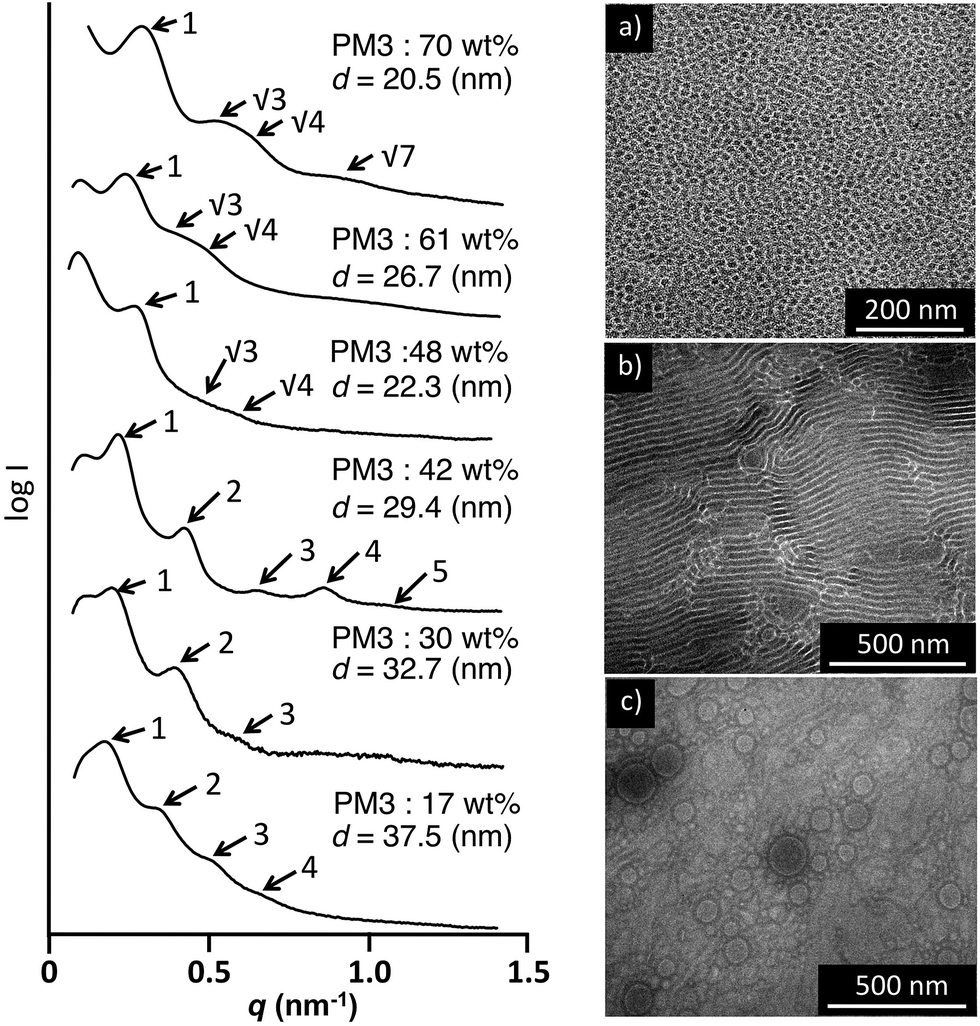 | ||
| Fig. 3 SAXS profiles and TEM images of PDMS-b-PM3 block copolymers (a) PDMS-b-PM3 (70 wt%), (b) PDMS-b-PM3 (42 wt%), and (c) PDMS-b-PM3 (17 wt%). | ||
Surface structure investigation of block copolymer
The surface compositions of the block copolymers were not always the same as the bulk composition, but can be practically dominated by the component with the lower surface energy. In the PDMS-containing block copolymer system, such as PS-b-PDMS, the PDMS segment usually exclusively covers the surface and reduces the surface energy of the air/polymer interface.49 However, since the surface energy of PM3 might be higher than that of PS, it is very interesting to evaluate the surface structure of the PDMS-b-PM3. Therefore, the surface composition of the block copolymer films with different PM3 compositions were investigated using X-ray photoelectron spectroscopy (XPS) at various take-off angles (TOA), scanning electron microscopy (SEM), and atomic force microscopy (AFM). The amphiphilic block copolymer thin films were prepared by spin-coating of the solutions of PDMS-b-PM3 in acetone (3.0 wt%) onto silicon wafers, subjected to thermal annealing at 40 °C for 24 h, in order to reach thermodynamic equilibrium. The thickness of the films was estimated by elipsometry, and found to be from 40 to 60 nm. The PDMS-b-PM3 copolymers (70, 42, and 17 wt%) were used to compare the results of the bulk morphology.Fig. 4a presents the typical XPS survey spectrum of PDMS-b-PM3 at TOA 90°. Three elements were detectable, O1S, C1S, Si2S, and Si2P atomic signals assigned at 533, 285, 153, and 102 eV, respectively. Since the signal depending on the PM3 composition was observed in the range based on the carbon peak of 280–295 eV, we estimated the top of the surface structure of block copolymer thin film by comparing the area ratio of C–C, C–O, and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peaks. In Fig. 4c, a fairly sharp C1s peak is displayed. While, the C–O and O–CO peaks are barely visible with the increasing composition of PM3 segment (Fig. 4c–e, red line), suggesting that the PM3 existed near the surface despite the fact that the PDMS has low surface energy. Therefore, in order to further investigate the surface composition, XPS depth-profile measurement was carried out.
O peaks. In Fig. 4c, a fairly sharp C1s peak is displayed. While, the C–O and O–CO peaks are barely visible with the increasing composition of PM3 segment (Fig. 4c–e, red line), suggesting that the PM3 existed near the surface despite the fact that the PDMS has low surface energy. Therefore, in order to further investigate the surface composition, XPS depth-profile measurement was carried out.
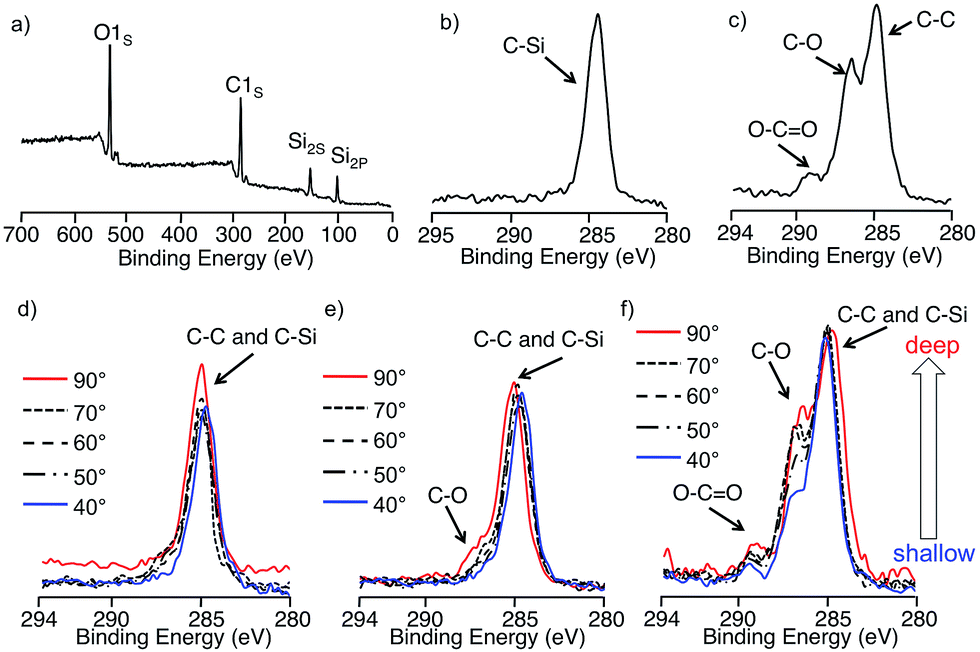 | ||
| Fig. 4 XPS spectra of (a) typical PDMS-b-PM3 sample in the range of 700–0 eV (TOA = 90°), (b) PDMS homopolymer in the range of 295–280 eV (TOA = 90°), (c) PM3 homopolymer in the range of 294–280 eV (TOA = 90°), and C 1s photoelectron intensities with various TOAs ranging from 40° to 90° of (d) PDMS-b-PM3 (17 wt%), (e) PDMS-b-PM3 (42 wt%), and (f) PDMS-b-PM3 (70 wt%). PDMS functionalized polymer anions. | ||
Higher TOA meant that X-ray could penetrate more deeply into the surface of the sample. In Fig. 4c, the hydrocarbon peaks at 286 eV dominated the spectra at all the TOAs, and these spectra are almost similar that of the PDMS homopolymer (Fig. 4b), indicating that the surface of PDMS-b-PM3 (17 wt%) is exclusively covered with PDMS. In the case of the increasing PM3 content, which is 42 wt%, the C–O and O–CO signals slightly appeared at TOA 90°, but the signals gradually decreased and were not observed at TOA 40° (Fig. 4d). In contrast, these signals for PDMS-b-PM3 (70wt%) were still definitely confirmed, although the signal intensity decreased at TOA 40° (Fig. 4e). Hence, to quantitatively evaluate the surface composition, the content of the C, O, and Si elements was estimated by comparing the peak ratio of C1S, O1S, and Si2P (Fig. S5†). As the result, the estimated value was the almost same as that of the PDMS homopolymer regardless of the different composition ratio of the block copolymer. This means that the top surface was covered with the hydrophobic PDMS segment. Interestingly, the Si content value slightly decreased when the content of PM3 was higher than 60% (PDMS; 34%, PDMS-b-PM3 (70 wt%); obsd. 29%). These results indicated that the top of the surface structure of the PDMS-b-PM3 (70 wt%) thin film consisted of both PDMS and PM3 segments.
Next, to further investigate the surface morphology of the PDMS-b-PM3 thin films, SEM and AFM observations were carried out. The thin film of PDMS-b-PM3 (17 wt%) with the thickness of 44 nm was smooth and almost featureless, which was strongly supported by the XPS results (Fig. 5a, d, and e), indicating that the top surface was fully consistent with a continuous PDMS layer. With the PM3 content increasing, the phase-separated structure was clearly observed (Fig. 5b and c). In particular, the PDMS-b-PM3 (70 wt%) film with a thickness of 48 nm showed the coexistence of line and dot disordered cylinder-like structures (Fig. 5c, f, and g). The line structures are derived from the cylindrical PDMS microdomains oriented parallel to the substrate, while the dot structures are most likely the PDMS cylinder oriented vertical to the substrate. The domain spacing was 26 nm, which is close to the SAXS results (21 nm, Fig. 3). To confirm the surface component, oxygen reactive ion etching (O2-RIE) treatment was carried out for 30 s on the same block copolymer thin film. The AFM image of the exposed film clearly showed the remaining microphase-separated nanostructure with protruded island oxidized PDMS patterns due to the high etch resistance (Fig. S6†). Conclusively, the brighter (matrix) and darker (dot) regions in the AFM images could be regarded as PM3 and PDMS components, respectively. Considering the composition ratio, this observation image was a reasonable structure. Thus, in the case of PDMS-b-PM3 (70 wt%), the existence of the PM3 segment on the top surface is a thermodynamically-stable structure, which was not seen in the thermally-annealed PDMS-b-PMMA and PDMS-b-PS thin film having almost the same PDMS weight fraction (29 and 34 wt%).43,49 From direct observation of the top surface by SEM, it was considered that the surface structure of the PDMS-containing block copolymer could be tuned by controlling the composition of the PM3 segment.
 | ||
| Fig. 5 SEM images of PDMS-b-PM3 thin films (a) PDMS-b-PM3 (17 wt%), (b) PDMS-b-PM3 (42 wt%), (c) PDMS-b-PM3 (70 wt%). AFM height and phase images of thin films (d), (e) PDMS-b-PM3 (17 wt%), and, (f), (g) PDMS-b-PM3 (70 wt%). | ||
Finally, to evaluate the macroscopic surface property, the static water contact angle measurement was performed. Since the surface structure of the block copolymer affected its chemical composition, the surface free energy of the block copolymer would be different. In addition, the rapid reconstruction of the surface response to an environmental change is expected because both segments have very low Tgs. When a water droplet was placed on the PDMS-b-PM3 (17 wt%) film, the contact angle showed a large contact angle (88°) nearly equal to that of PDMS homopolymer (100°), because the outermost surface is covered with the PDMS segment. Interestingly, the shape of the water droplet changed within 10 s, and the contact angle obviously changed after 10 s from 88° to 66°. In other words, the wetting ability of the top surface changed to more hydrophilic than that of the initial state by responding to the environment condition (Fig. 6). It was considered that this phenomenon may be attributed to the rapid reconstruction from the PDMS layer to PM3 domain, when the water absorbs into the near surface region. Indeed, the initial hydrophobicity recovered after a thermal treatment at 40 °C for 60 min in vacuo. This behavior was also repeatedly observed through the following process: (1) soaking in water for 60 s, and (2) thermal annealing at 40 °C for 60 min in a vacuum oven (Fig. S7†). In contrast, the wetting ability was dramatically changed to hydrophilic with the increasing composition of PM3. The PDMS-b-PM3 (70 wt% and 42 wt%) showed the low contact angles, 24 and 25°, respectively. This indicated that the surface composition of the PDMS-b-PM3 (70 wt%) and (42 wt%) is significantly different from PDMS-b-PM3 (17 wt%). Thus, it was confirmed that the surface morphology with a different composition of the block copolymer strongly affected the surface property, and that the surface reconstruction quickly occurred in response to the environmental change.
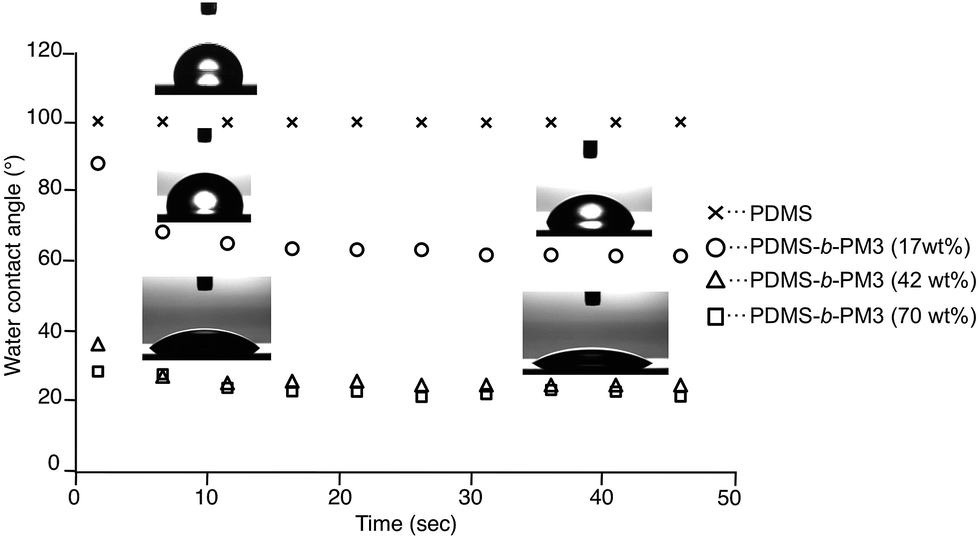 | ||
| Fig. 6 Water contact angle of the surfaces of PDMS homopolymer and PDMS-b-PM3 block copolymer thin films. | ||
Conclusions
We have demonstrated the successful synthesis of a series of novel amphiphilic block copolymers composed of PDMS and water-soluble poly [tri(ethylene glycol) methyl ether methacrylate] (PM3). Well-defined PDMS-b-PM3 block copolymers were synthesized by the living anionic polymerization and coupling reaction between the ω-chain ended-benzyl bromide-functionalized PDMS and living PM3 anion. Under the conditions employed in this study, all the coupling reactions were observed to almost quantitatively proceed within 24 h. The resulting block copolymers all had narrow molecular weight distributions (Mw/Mn ≤ 1.08) and controlled molecular weights. The solubility of the amphiphilic PDMS-b-PM3s was strongly affected by its composition, and the polymer was soluble in water when the content of PM3 was over 42 wt%. The water-soluble PDMS-b-PM3 showed an LCST behavior at around 30 °C, and the value was significantly lower than that of the PM3 homopolymer (52 °C). We also confirmed the self-assembling characteristics in the bulk by SAXS and TEM, and the surface structure of the thin film was characterized by XPS, SEM and AFM. In thin films, we revealed that an amphiphilic PDMS-b-PM3 (70 wt%) could form a self-assembled nanostructure on the top of the surface, and that the wetting ability of water drastically changed with the different PM3 compositions. Therefore, our findings not only provide a versatile synthetic route for the PDMS-containing block copolymer, but also develop an efficient scaffold for nanopatterning materials. Moreover, the results of this study can help in the application of amphiphilic block copolymers in various areas by demonstrating control of the surface structure.Acknowledgements
This work was partly supported by a Grant-in-Aid for Scientific Research from the Japan Society of the Promotion of Science (No. 18550105 for T. I.) and Mizuho Foundation for the Promotion of Sciences (2016–2018 for R. G.). We also thank Rohei Kikuchi of the National University Corporation, Tokyo Institute of Technology Center for Ascended Materials Analysis, for the measurement of TEM. We would like to thank Prof. Teruaki Hayakawa (Tokyo Institute of Technology, Japan) for many helpful discussions and support concerning about oxygen plasma ashing.References
- Y. Zhang and H. Zhao, Langmuir, 2016, 32, 3567–3579 CrossRef CAS PubMed.
- A. B. Kutikov and J. Song, ACS Biomater. Sci. Eng., 2015, 1, 463–480 CrossRef CAS PubMed.
- O. Onaca, D. W. Hughes, V. Balasubramanian, M. Grzelakowski, W. Meier and C. G. Palivan, SOD Antioxidant Nanorears, Macromol. Biosci., 2010, 10, 531–538 CAS.
- D. Li, C. Li, G. Wan and W. Hou, Colloids Surf., A, 2010, 372, 1–8 CrossRef CAS.
- L. Chuenchom, R. Kraehnert and B. M. Smarsly, Soft Matter, 2012, 8, 10801–10812 RSC.
- Y. Bakkour, V. Darcos, F. Coumes, S. Li and J. Coudane, Polymer, 2013, 54, 1746–1754 CrossRef CAS.
- S. H. Kim, M. J. Minster, M. Kimura, T. Xu and T. P. Russell, Adv. Mater., 2004, 16, 226 CrossRef CAS.
- Q. Zhou, L. Xu, F. Liu and W. Zhang, Polymer, 2016, 97, 323–334 CrossRef CAS.
- B. H. Tan, H. Hussain and C. B. He, Macromolecules, 2011, 44, 622–631 CrossRef CAS.
- R. Goseki, T. Hirai, Y. Ishida, M. Kakimoto and T. Hayakawa, Polym. J., 2012, 44, 658–664 CrossRef CAS.
- C. Zuo, X. Dai, S. Zhao, X. Liu, S. Ding, L. Ma, M. Liu and H. Wei, ACS Macro Lett., 2016, 5, 873–878 CrossRef CAS.
- S. Junnila, N. Houbenov, A. Karatzas, N. Hadjichristidis, A. Hirao, H. Iatrou and O. Ikkala, Macromolecules, 2012, 45, 2850–2856 CrossRef CAS.
- C. Wei, Y. Zhang, H. Xu, Y. Xu, Y. Xu and M. Lang, J. Mater. Chem. B, 2016, 4, 5059–5067 RSC.
- G. Kickelbick, J. Bauer, N. Hüsing, M. Andersson and A. Palmqvist, Langmuir, 2003, 19, 3198–3201 CrossRef CAS.
- G. Kickelbick, J. Bauer, N. Hüsing, M. Andersson and A. Palmqvist, Langmuir, 2003, 19, 10073–10076 CrossRef CAS.
- P. G. Fragouli, H. Iatrou, N. Hadjichristidis, T. Sakurai and A. Hirao, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 614–619 CrossRef CAS.
- W. Zhao, P. Fonsny, P. FltzGerald, G. G. Warr and S. Perrier, Polym. Chem., 2013, 4, 2140–2150 RSC.
- A. Car, P. Baumann, J. T. Duskey, M. Chami, N. Bruns and W. Meier, Biomacromolecules, 2014, 15, 3235–3245 CrossRef CAS PubMed.
- Y. S. Jung and C. A. Ross, Nano Lett., 2007, 7, 2046–2050 CrossRef CAS PubMed.
- T. Ishizone, K. Sugiyama, Y. Sakano, H. Mori, A. Hirao and S. Nakahama, Polym. J., 1999, 31, 983–988 CrossRef CAS.
- T. Teraya, A. Takahara and T. Kajiyama, Polymer, 1990, 31, 1149–1153 CrossRef CAS.
- K. R. Shull, K. I. Winey, E. L. Thomas and E. J. Kramer, Macromolecules, 1991, 24, 2748–2751 CrossRef CAS.
- C. Creton, E. J. Kramer, C.-Y. Hui and H. R. Brown, Macromolecules, 1992, 25, 3075–3088 CrossRef CAS.
- K. Senshu, S. Yamashita, M. Ito, A. Hirao and S. Nakahama, Langmuir, 1995, 11, 2293–2300 CrossRef CAS.
- K. Senshu, S. Yamashita, H. Mori, M. Ito, A. Hirao and S. Nakahama, Langmuir, 1999, 15, 1754–1762 CrossRef CAS.
- K. Sensyu, M. Kobayashi, N. Ikawa, S. Yamashita, A. Hirao and S. Nakahama, Langmuir, 1999, 15, 1763–1769 CrossRef.
- H. Mori, A. Hirao, S. Nakahama and K. Senshu, Macromolecules, 1994, 27, 4093–4100 CrossRef CAS.
- A. Takahara, T. Teraya and T. Kajiyama, Polym. Bull., 1990, 24, 333 CrossRef.
- H. Yokoyama, T. Miyamae, S. Han, T. Ishizone, K. Tanaka, A. Takahara and N. Torikai, Macromolecules, 2005, 38, 5180–5189 CrossRef CAS.
- T. Ishizone, S. Han, M. Hagiwara and H. Yokoyama, Macromolecules, 2006, 39, 962–970 CrossRef CAS.
- T. Ishizone, S. Han, S. Okuyama and S. Nakahama, Macromolecules, 2003, 36, 42–49 CrossRef CAS.
- S. Han, M. Hagiwara and T. Ishizone, Macromolecules, 2003, 36, 8312–8319 CrossRef CAS.
- J. Yamanaka, T. Kayasuga, M. Ito, H. Yokoyama and T. Ishizone, Polym. Chem., 2011, 2, 1837–1848 RSC.
- T. Ishizone, A. Seki, M. Hagiwara, S. Han, H. Yokoyama, A. Oyane, A. Deffieux and S. Carlotti, Macromolecules, 2008, 41, 2963–2967 CrossRef CAS.
- A. Oyane, T. Ishizone, M. Uchida, K. Furukawa, T. Ushida and H. Yokoyama, Adv. Mater., 2005, 17, 2329–2332 CrossRef CAS.
- Y. Nakagawa, P. J. Miller and K. Matyjaszewski, Polymer, 1998, 39, 5163–5170 CrossRef CAS.
- K. Huan, L. Bes, D. M. Haddleton and E. J. Khoshdel, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1833–1842 CrossRef CAS.
- L. Bes, K. Huan, E. Khoshdel, M. J. Lowe, C. F. McConville and D. M. Haddleton, Eur. Polym. J., 2003, 39, 5–13 CrossRef CAS.
- D. Pavlovic, J. G. Linhardt, J. F. Kunzler and D. A. Shipp, Macromol. Chem. Phys., 2010, 211, 1482–1487 CrossRef CAS.
- P. J. Miller and K. Matyjaszewski, Macromolecules, 1999, 32, 8760–8767 CrossRef CAS.
- G. Wang, M. Chen, S. Guo and A. Hu, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2684–2691 CrossRef CAS.
- V. Bellas, H. Iatrou, E. N. Pitsinos and N. Hadjichristidis, Macromolecules, 2001, 34, 5376–5378 CrossRef CAS.
- Y. Luo, D. Montarnal, S. Kim, W. Shi, K. P. Barteau, C. W. Pester, P. D. Husta, M. D. Christianson, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Macromolecules, 2015, 48, 3422–3430 CrossRef CAS.
- A. Hirao, K. Murano, R. Kurokawa, T. Watanabe and K. Sugiyama, Macromolecules, 2009, 42, 7820–7827 CrossRef CAS.
- H.-S. Yoo, T. Watanabe and A. Hirao, Macromolecules, 2009, 42, 4558–4570 CrossRef CAS.
- R. Goseki, A. Hirao, M. Kakimoto and T. Hayakawa, ACS Macro Lett., 2013, 2, 625–629 CrossRef CAS.
- T. Higashihara, T. Sakurai and A. Hirao, Macromolecules, 2009, 42, 6006–6016 CrossRef CAS.
- E. Vargün and A. Usanmaz, Polym. Int., 2010, 59, 1586–1597 CrossRef.
- D. Borah, S. Rasappa, R. Senthamaraikannan, J. D. Holmes and M. A. Morris, Langmuir, 2013, 29, 8959–8968 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, the solubility of PDMS-b-PM3 block copolymers, 1H NMR spectra, MALDI-TOF-MASS spectra, DSC charts, turbidity measurement, XPS measurement, AFM images of before and after O2-RIE treatment, and repeated measurement of water contact angle (Fig. S1–5). See DOI: 10.1039/c7ra02569f |
| This journal is © The Royal Society of Chemistry 2017 |