
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePenicimutamides D–E: two new prenylated indole alkaloids from a mutant of the marine-derived Penicillium purpurogenum G59†
Chang-Jing Wuab,
Chang-Wei Li *a,
Hao Gaoc,
Xiao-Jun Huangc and
Cheng-Bin Cuia
*a,
Hao Gaoc,
Xiao-Jun Huangc and
Cheng-Bin Cuia
aState Key Laboratory of Toxicology and Medical Countermeasures, Beijing Institute of Pharmacology and Toxicology, Beijing 100850, China. E-mail: sdrlcw@126.com; Fax: +86-10-68211656; Tel: +86-10-68211656
bCollege of Life Science and Agronomy, Zhoukou Normal University, Zhoukou 466001, China
cInstitute of Traditional Chinese Medicine & Natural Products, College of Pharmacy, Jinan University, Guangzhou 510632, China
First published on 8th May 2017
Abstract
Three prenylated indole alkaloids (1–3), including two new penicimutamides D–E (1–2), were isolated from a diethyl sulfate mutant of the marine-derived fungus Penicillium purpurogenum G59. The structures of 1–3, and their absolute configurations, were determined by spectroscopic methods, including X-ray crystallography and CD analyses. HPLC-UV and HPLC-MS analyses showing that 1–3 were only produced in the mutant evidenced that the silent biosynthetic pathways that produce 1–3 in the parental strain are activated by DES mutagenesis.
Prenylated indole alkaloids (PIAs) are a broad class of secondary fungal metabolites with a bicyclo[2.2.2]diazaoctane core ring system.1 Included in this group are brevianamides,2 notoamides,3 stephacidins,4 versicolamides,5 paraherquamides,6 malbrancheamides7 and marcfortines.8 Total syntheses,9 biomimetic syntheses10 and biosyntheses11 of PIAs, focusing on formation of the core ring system, have been extensively investigated because of the interesting structures of the PIAs.
In our previous work,12 three rare carbamate-containing PIAs, penicimutamides A–C, were isolated from a fungal mutant AD-2-1 of a marine-derived Penicillium purpurogenum G59 via diethyl sulfate (DES) mutagenesis. As a continuation of this, we herein report on three other PIAs, including two new penicimutamides D–E (1–2 in Fig. 1) and a known one (3). These compounds were produced in the same solid culture by the mutant AD-2-1 by activating silent pathways in parent G59 strain. The mutant AD-2-1 was selected by treating Penicillium purpurogenum G59 spores with 1% (v/v) DES in 50% (v/v) DMSO at 4 °C for 1 d. This produced a series of novel lipopeptides in liquid culture via the activation of pathways that were silent in the G59 strain.13
 | ||
| Fig. 1 Structures of compounds 1–3. | ||
To obtain new fungal secondary metabolites, the one-strain-many-compounds approach,14 chemical epigenetics15 and co-cultivation16 have been used to activate silent pathways by varying environmental factors for growth of the producing strains. In our previous work,17 a series of new methods17a–c based on ribosome engineering18 were developed for fungi, and several new secondary metabolites were isolated from the mutants.17c–e During this study, a practical mutagenesis strategy using DES was developed to activate silent fungal pathways.13,19 A diverse range of secondary metabolites were isolated from the DES mutant of Penicillium purpurogenum G59 by activating pathways that were silent in the parent strain via DES mutagenisis.19a
As reported in our previous work, the mutant AD-2-1 and parental G59 strains were fermented concurrently under the same conditions at 28 °C for 50 d using rice as a solid substrate fermentation medium to obtain methanol (MeOH) extracts of their cultures. These cultures inhibited K562 cells with inhibition rates of 62.5% and 6.1% at 100 μg mL−1, respectively. Production of new metabolites was tracked to guide separation of the mutant extract, and resulted in the isolation of 1–3 (Table 1).
| No. | 1 | 2 | 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δCb | δH (J in Hz) | δCb | δH (J in Hz) | δCb | ||||
| a Chemical shift (δH and δC) were recorded using the solvent signals of CD3OD (δH 3.31/δC 49.00) as references. Signals assignments were based on the results of DEPT, 1H–1H COSY, HMQC and HMBC experiments.b Multiplicities of the carbon signals were determined by DEPT experiments and are indicated by s (singlet), d (doublet), t (triplet) and q (quartet). | |||||||||
| 2 | — | 192.6 s | — | 142.7 s | — | 142.2 s | |||
| 3 | — | 41.6 s | — | 29.1 s | — | 35.4 s | |||
| 4 | 2.00 dd (9.0, 5.4) | 51.6 d | 2.19 dd (10.1, 3.7) | 47.6 d | 2.00 dd (10.8, 5.4) | 48.5 d | |||
| 5 | Hβ | 2.10 dd (12.6, 9.0) | 32.9 t | Hβ | 2.14 dd (13.4,10.1) | 31.9 t | Hα | 1.99 dd (13.2, 10.8) | 32.5 t |
| Hα | 1.97 dd (12.6, 5.4) | Hα | 1.94 dd (13.4,3.7) | Hβ | 1.94 dd (13.2, 5.4) | ||||
| 6 | — | 67.2 d | — | 66.5 d | — | 66.2 d | |||
| 7 | Hα | 2.57 dt (12.6, 6.6) | 28.1 t | Hα | 2.52 ddd (12.6, 9.0, 4.8) | 28.09 t | Hα | 2.51 ddd (13.2, 9.0, 6.0) | 28.2 t |
| Hβ | 1.47 td (12.6, 7.8) | Hβ | 1.47 ddd (12.6, 10.8, 7.2) | Hβ | 1.44 ddd (13.2, 10.8, 7.2) | ||||
| 8 | 1.95–1.89 2H, m | 23.4 t | 1.96–1.89 2H, m | 23.4 t | 1.89–1.82 2H, m | 23.6 t | |||
| 9 | Hα | 3.10 dt (9.0, 5.4) | 54.7 t | Hβ | 3.115 td (9.0, 4.2) | 54.3 t | Hβ | 3.04 ddd (9.0, 7.2, 3.6) | 55.4 t |
| Hβ | 2.31 q (9.0) | Hα | 2.39 q (9.0) | Hα | 2.15 br q (9.0) | ||||
| 11 | Hβ | 2.89 d (10.2) | 62.4 t | Hβ | 3.110 d (10.2) | 62.4 t | Hβ | 3.45 d (10.2) | 59.5 t |
| Hα | 2.49 d (10.2) | Hα | 2.67 d (10.2) | Hα | 2.24 dd (10.2, 1.8) | ||||
| 12 | — | 58.0 s | — | 56.8 s | — | 57.7 s | |||
| 13 | Hα | 2.68 d (14.7) | 43.1 t | Hα | 2.99 d (16.8) | 29.1 t | Hβ | 2.88 d (15.0) | 30.5 t |
| Hβ | 1.49 d (14.7) | Hβ | 2.85 d (16.8) | Hα | 2.85 d (15.0) | ||||
| 14 | — | 83.7 s | — | 103.7 s | — | 104.5 s | |||
| 15 | — | 143.0 s | — | 128.4 s | — | 128.2 s | |||
| 16 | 7.45 br d (7.8) | 123.3 d | 7.38 br d (7.7) | 118.5 d | 7.33 br d (8.0) | 118.3 d | |||
| 17 | 7.26 td (7.8, 0.8) | 127.6 d | 6.96 ddd (7.7, 7.1, 0.8) | 119.5 d | 6.93 td (8.0, 0.9) | 119.5 d | |||
| 18 | 7.37 td (7.8, 1.2) | 130.6 d | 7.04 ddd (8.1, 7.1, 1.2) | 122.0 d | 7.02 td (8.0, 1.1) | 122.0 d | |||
| 19 | 7.48 br d (7.8) | 121.2 d | 7.27 br d (8.1) | 111.7 d | 7.25 br d (8.0) | 111.6 d | |||
| 20 | — | 152.9 s | — | 138.4 s | — | 138.5 s | |||
| 21 | 1.30 3H, s | 26.9 q | 1.31 3H, s | 28.06 q | 1.42 3H, s | 24.4 q | |||
| 22 | 1.41 3H, s | 19.9 q | 1.20 3H, s | 24.6 q | 1.32 3H, s | 30.9 q | |||
| 23 | — | 175.2 s | — | 175.9 s | — | 176.8 s | |||
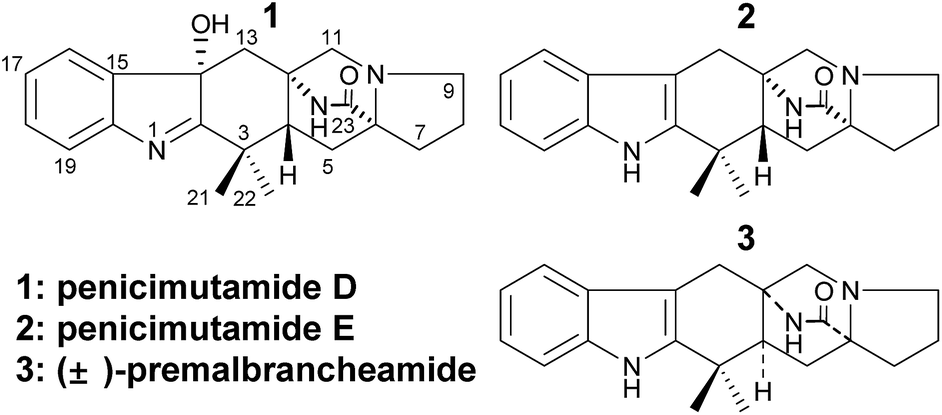
After analysis by HR-ESI-MS, penicimutamide D (1) was assigned the molecular formula C21H25N3O2 (m/z 352.2025 [M + H]+, calcd for C21H26N3O2 352.2025), which has one more oxygen atom than the known compound 3. UV absorptions at 220 and 264 nm showed it contained the indole structure. The 1H NMR data for H-16 (δ 7.53, br d, J = 7.8 Hz), H-19 (δ 7.52, br d, J = 7.8 Hz), H-18 (δ 7.46, td, J = 7.8, 1.2 Hz) and H-17 (δ 7.31, td, J = 7.8, 1.2 Hz) supported the presence of a disubstituted phenyl group in the indole structure. The 1H NMR, HMQC and 1H–1H COSY spectra showed five spin systems of H3-21–C-3–H3-22, –H2-13–, –H2-11–, –H-4–H2-5–, and –H2-7–H2-8–H2-9– (Fig. 2). All of the above spin systems were the same as those for the known compound 3, which suggested 1 had the same skeleton as 3. When the 13C NMR spectrum of 1 was compared with that of 3, the biggest differences were the absence of a sp2 carbon signal (C-14 of 3) and appearance of an oxygenated sp3 carbon signal (C-14 of 1). So, it could be deduced that 1 is an oxidative product of 3 with a hydroxyl group at C-14. In addition the double bond was transferred from C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C14 to C1C2, which resulted in a high-field shift of C-2 from δC 142.2 (in 3) to 192.6 (in 1). This gave a planar structure (Fig. 1), which was also supported by further NMR data (Fig. 2).
C14 to C1C2, which resulted in a high-field shift of C-2 from δC 142.2 (in 3) to 192.6 (in 1). This gave a planar structure (Fig. 1), which was also supported by further NMR data (Fig. 2).
 | ||
| Fig. 2 Structures and key NMR data for 1. | ||
The NOEs observed in the NOESY of 1 between H-4/Hβ-11, H-4/Hβ-13, H-4/Hβ-5, H-4/H3-21 and H3-21/Hα,β-5 indicated that ring C had a chair conformation and ring D had a boat conformation with CH3-22, HO-C14 groups and the lactam group located on the same face. The NOEs between Hβ-5/Hβ-7 and Hα-9/Hα-11 showed that the E ring had an envelope conformation with N-10 upwarped as shown in Fig. 2. Thus, the relative configuration was established.
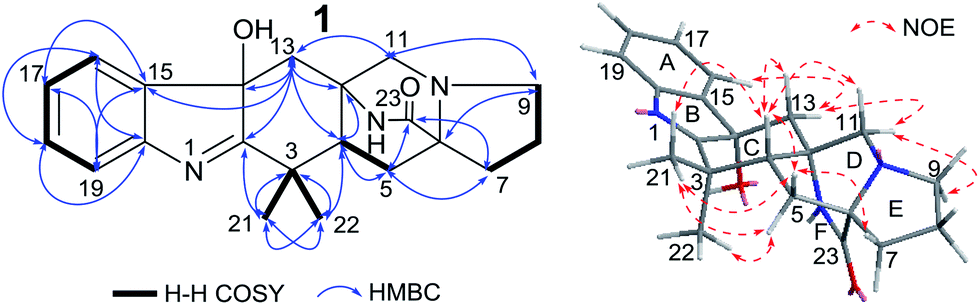
To determine the absolute configuration of 1, single-crystal X-ray diffraction using Cu Kα radiation for 1 was performed. An ORTEP drawing of the crystallographically determined structure of 1 is depicted in Fig. 3. The relative configuration of 1 determined was in accordance with the result based on NOEs. But the Flack parameter was −0.2 (2), which was not perfect enough to determine the absolute configuration.
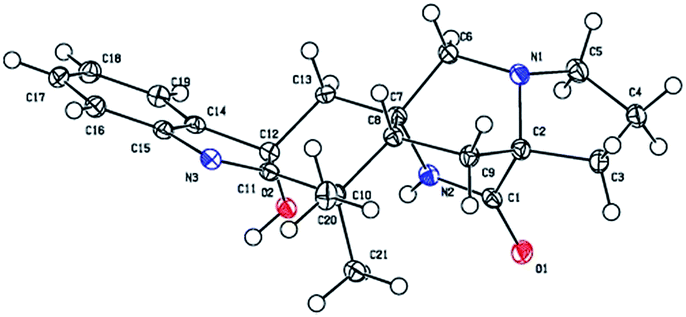 | ||
| Fig. 3 The crystal structure of 1. | ||
To confirm the absolute configuration of 1, TDDFT electronic CD (ECD) calculations20,21 for 1 and its enantiomer were performed. The calculated ECD of 1 agreed with the measured CD data (Fig. 4), and showed that the absolute configuration was 4R, 6S, 12S, 14S.
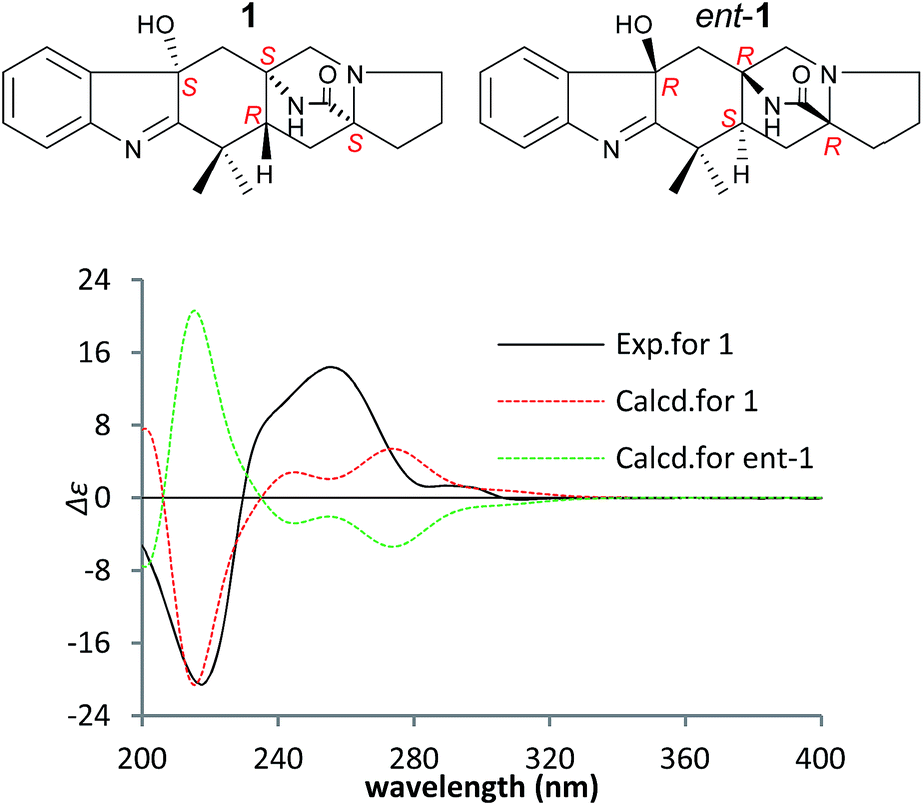 | ||
| Fig. 4 Measured and calculated ECD spectra of 1 and its enantiomer in MeOH. | ||
Penicimutamide E (2) was assigned the molecular formula C21H25N3O1 by HR-ESI-MS (m/z 336.2070 [M + H]+, calcd for C21H26N3O2 336.2072), which was the same as that of the known compound 3. The UV and IR spectra were also identical to those of 3. The similar 1H and almost identical 13C NMR data indicated that 2 had the same planar structure as 3, which was also supported by other NMR data.
The NOEs observed in the NOESY of 2 between H-4/Hβ-11, H-4/Hβ-13, H-4/Hβ-5, H-4/H3-21, H3-21/Hα,β-5, Hβ-5/Hβ-7 and Hα-9/Hα-11 established the relative configuration of 2 as shown in Fig. 5. To determine the absolute configuration of 2, TDDFT ECD calculations20,21 for 2 and its enantiomer were performed. The calculated ECD of 2 agreed with the measured CD data (Fig. 6), and the absolute configuration of 2 was determined as 4R, 6S, 12S.
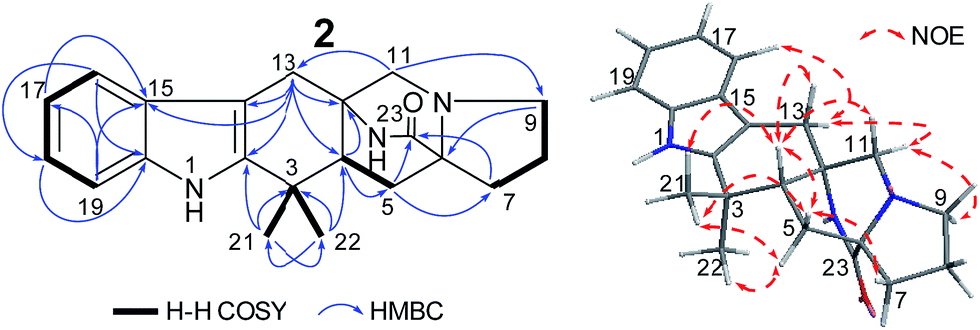 | ||
| Fig. 5 Structures and key NMR data for 2. | ||
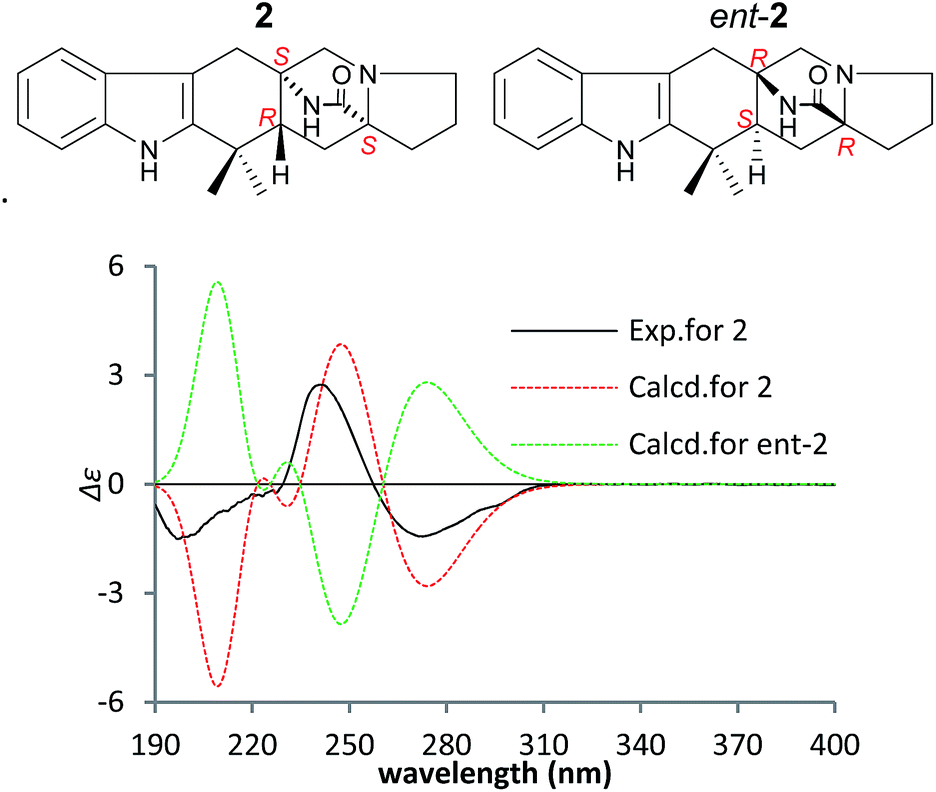 | ||
| Fig. 6 Measured and calculated ECD spectra of 2 and its enantiomer in MeOH. | ||
Compound 3 was obtained as a crystalline powder. The 1H and 13C NMR data were almost identical to the published data of (+)-premalbrancheamide,22 which indicated that 3 had the same planar structure as (+)-premalbrancheamide. The NOEs observed in the NOESY spectrum (Fig. 7) also indicated that 3 had the same relative stereochemistry as (+)-premalbrancheamide (Fig. 6). The [α]D of 3 (+3.2°) was smaller than that of (+)-premalbrancheamide (+15°),23 and there was no Cotton effect evident in the CD spectrum of 3. This indicates that 3 is a racemic mixture of (±)-premalbrancheamide. According to the HPLC analysis of compound 3 on a CHIRALPAK IE column with 65% MeOH, the (+)-premalbrancheamide content was about 56%, and the (−)-premalbrancheamide content was about 44% (Fig. 8). The racemic mixture was not separated because we only had a small quantity of 3.
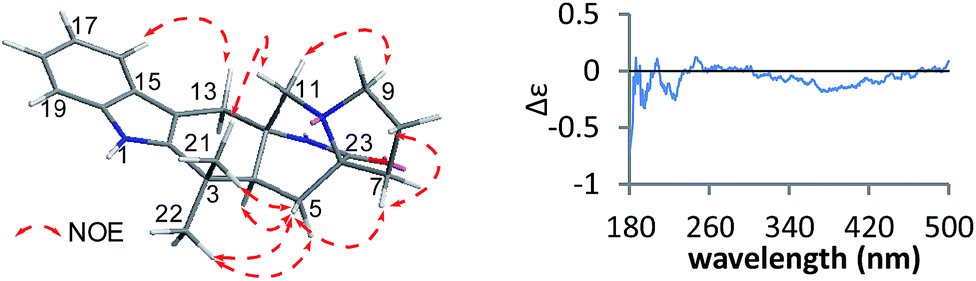 | ||
| Fig. 7 Key NOE data and the CD spectrum for 3. | ||
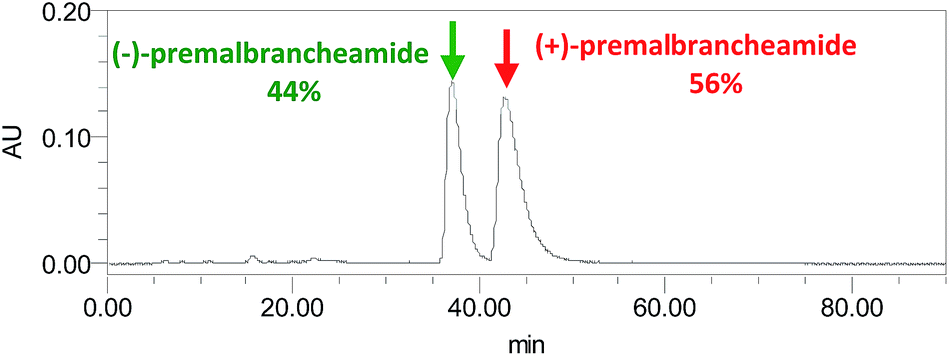 | ||
| Fig. 8 The HPLC chromatograph of 3 on a CHIRALPAK IE column with 65% MeOH. | ||
The MeOH extracts from the mutant AD-2-1 and parent G59 strains were analyzed by HPLC with a photodiode array detector and HPLC-ESI-MS using 1–3 as reference standards. The retention times and the UV and MS spectra (Fig. S2 and S3 in the ESI†) showed that 1–3 were only present in the mutant extract and not in the parent extract. This is evidence that 1–3 are produced in mutant AD-2-1 following activation of biosynthetic pathways that are silent in the parent G59 strain and subsequently activated by the DES mutagenesis process in the mutant.
As reported in our previous work,12 1 and 2 were both intermediates in plausible biosynthetic pathways for penicimutamides A–C. Plausible biosynthetic pathways for 3 have been reported in earlier studies.22,23 (+)-Premalbrancheamide was detected in one study,22 and was subsequently isolated from the fermentation product of Malbranchea aurantiaca.24 (−)-Premalbrancheamide was isolated for the first time in the present study.
To evaluate the inhibitory effect on human cancer cell lines, 1–3 and 5-fluorouracil were tested against human K562, HL-60, HeLa and BGC-823 cell lines at 100 μg mL−1 by the MTT assay. Compounds 1–3 only showed weak inhibition of the above four cell lines with inhibition rates ranging from 2% to 27%.
Conclusions
In our previous work, three rare carbamate-containing alkaloids, penicimutamides A–C, were isolated from the DES mutant AD-2-1 obtained from the marine-derived fungus Penicillium purpurogenum G59, and plausible biosynthetic pathways from the precursor deoxybrevianamide E were reported.12 In the present work, two new prenylated indole alkaloids, penicimutamides D–E, which might be intermediates in the biosynthesis of penicimutamides A–C, were isolated from the mutant AD-2-1. The discovery of penicimutamides A–E from the mutant AD-2-1 strain demonstrates the effectiveness of our previously reported DES mutagenesis strategy19 for obtaining new bioactive compounds from silenced fungal pathways. The present work confirms the proposed biosynthetic pathway of penicimutamides A–C.Conflicts of interest
The authors declare no conflict interest.Acknowledgements
This work was supported by the grants from the NSFC (81573300, 30973631), NHTRDP (2013AA092901, 2007AA09Z411), NSTMP (2009ZX09301-002, 2012ZX09301-003) and AMMS (2008), China.References
- S.-M. Li, Nat. Prod. Rep., 2010, 27, 57 RSC.
- (a) A. J. Birch and J. J. Wright, J. Chem. Soc., Chem. Commun., 1969, 644 RSC; (b) A. J. Birch and J. J. Wright, Tetrahedron, 1970, 26, 2329 CrossRef CAS PubMed; (c) A. J. Birch and R. A. Russell, Tetrahedron, 1972, 28, 2999 CrossRef CAS.
- (a) H. Kato, T. Yoshida, T. Tokue, Y. Nojiri, H. Hirota, T. Ohta, R. M. Williams and S. Tsukamoto, Angew. Chem., Int. Ed., 2007, 46, 2254 CrossRef CAS PubMed; (b) S. Tsukamoto, H. Umaoka, K. Yoshikawa, T. Ikeda and H. Hirota, J. Nat. Prod., 2010, 73, 1438 CrossRef CAS PubMed; (c) S. Tsukamoto, H. Kato, M. Samizo, Y. Nojiri, H. Onuki, H. Hirota and T. Ohta, J. Nat. Prod., 2008, 71, 2064 CrossRef CAS PubMed.
- J. Qian-Cutrone, S. Huang, Y.-Z. Shu, D. Vyas, C. Fairchild, A. Menendez, K. Krampitz, R. Dalterio, S. E. Klohr and Q. Gao, J. Am. Chem. Soc., 2002, 124, 14556 CrossRef CAS PubMed.
- (a) T. J. Greshock, A. W. Grubbs, P. Jiao, D. T. Wicklow, J. B. Gloer and R. M. Williams, Angew. Chem., Int. Ed., 2008, 47, 3573 CrossRef CAS PubMed; (b) S. Tsukamoto, T. Kawabata, H. Kato, T. J. Greshock, H. Hirota, T. Ohta and R. M. Williams, Org. Lett., 2009, 11, 1297 CrossRef CAS PubMed.
- (a) M. Yamazaki and E. Okuyama, Tetrahedron Lett., 1981, 22, 135 CrossRef CAS; (b) S. E. Blanchflower, R. M. Banks, J. R. Everett, B. R. Manger and C. J. Reading, Antibiotics, 1991, 44, 492 CrossRef CAS; (c) S. E. Blanchflower, R. M. Banks, J. R. Everett and C. J. Reading, Antibiotics, 1993, 46, 1355 CrossRef CAS.
- (a) S. Martínez-Luis, R. Rodríguez, L. Acevedo, M. C. González, A. Lira-Rocha and R. Mata, Tetrahedron, 2006, 62, 1817 CrossRef; (b) M. Figueroa, M. D. C. González and R. Mata, Nat. Prod. Res., 2008, 22, 709 CrossRef CAS PubMed; (c) K. R. Watts, S. T. Loveridge, K. Tenney, J. Media, F. A. Valeriote and P. Crews, J. Org. Chem., 2011, 76, 6201 CrossRef CAS PubMed; (d) Y. Ding, T. J. Greshock, K. A. Miller, D. H. Sherman and R. M. Williams, Org. Lett., 2008, 10, 4863 CrossRef CAS PubMed.
- (a) J. Polonsky, M.-A. Merrien, T. Prangé and C. Pascard, J. Chem. Soc., Chem. Commun., 1980, 13, 601 RSC; (b) T. Prangé, M.-A. Billion, M. Vuilhorgne, C. Pascard and J. Poponsky, Tetrahedron Lett., 1981, 22, 1977 CrossRef.
- (a) R. M. Williams, T. Glinka and E. Kwast, J. Am. Chem. Soc., 1988, 110, 5927 CrossRef CAS; (b) R. M. Williams, T. Glinka, E. Kwast, H. Coffman and J. K. Stille, J. Am. Chem. Soc., 1990, 112, 808 CrossRef CAS; (c) C. Escolano, Angew. Chem., Int. Ed., 2005, 44, 7670 CrossRef CAS PubMed; (d) F. C. Frebault and N. S. Simpkins, Tetrahedron, 2010, 66, 6585 CrossRef CAS.
- (a) T. J. Greshock and R. M. Williams, Org. Lett., 2007, 9, 4255 CrossRef CAS PubMed; (b) R. M. Williams, J. F. Sanz-Cervera, F. Sancenón, J. A. Marco and K. M. Halligan, Bioorg. Med. Chem., 1998, 6, 1233 CrossRef CAS PubMed; (c) T. J. Greshock, A. W. Grubbs and R. M. Williams, Tetrahedron, 2007, 63, 6124 CrossRef CAS PubMed.
- (a) R. M. Williams, E. Kwast, H. Coffman and T. Glinka, J. Am. Chem. Soc., 1989, 111, 3046 CrossRef; (b) L. R. Domingo, R. J. Zaragozá and R. M. Williams, J. Org. Chem., 2003, 68, 2895 CrossRef CAS PubMed; (c) J. D. Sunderhaus, D. H. Sherman and R. M. Williams, Isr. J. Chem., 2011, 51, 442 CrossRef CAS PubMed; (d) J. M. Finefield, H. Kato, T. J. Greshock, D. H. Sherman, S. Tsukamoto and R. M. Williams, Org. Lett., 2011, 13, 3082 CrossRef PubMed; (e) H. Kato, T. Hakahara, K. Sugimoto, K. Matsuo, I. Kagiyama, J. C. Frisvad, D. H. Sherman, R. M. Williams and S. Tsukamoto, Org. Lett., 2015, 17, 700 CrossRef CAS PubMed.
- C.-W. Li, C.-J. Wu, C.-B. Cui, L.-L. Xu, F. Cao and H.-J. Zhu, RSC Adv., 2016, 6, 73383 RSC.
- C.-J. Wu, C.-W. Li and C.-B. Cui, Mar. Drugs, 2014, 12, 1815 CrossRef PubMed.
- H. B. Bode, B. Bethe, R. Hofs and A. Zeeck, ChemBioChem, 2002, 3, 619 CrossRef CAS PubMed.
- R. B. Williams, J. C. Henrikson, A. R. Hoover, A. E. Lee and R. H. Cichewicz, Org. Biomol. Chem., 2008, 6, 1895 CAS.
- A. Marmann, A. H. Aly, W. Lin, B. Wang and P. Proksch, Mar. Drugs, 2014, 12, 1043 CrossRef PubMed.
- (a) Y.-J. Chai, C.-B. Cui, C.-W. Li, C.-J. Wu, C.-K. Tian and W. Hua, Mar. Drugs, 2012, 10, 559 CrossRef CAS PubMed; (b) C.-J. Wu, L. Yi, C.-B. Cui, C.-W. Li, N. Wang and X. Han, Mar. Drugs, 2015, 13, 2465 CrossRef CAS PubMed; (c) Y. Dong, C.-B. Cui, C.-W. Li, W. Hua, C.-J. Wu, T.-J. Zhu and Q.-Q. Gu, Mar. Drugs, 2014, 12, 4326 CrossRef CAS PubMed; (d) N. Wang, C.-B. Cui and C.-W. Li, Arch. Pharmacal Res., 2016, 39, 762 CrossRef CAS PubMed; (e) L. Yi, C.-B. Cui, C.-W. Li, J.-X. Peng and Q.-Q. Gu, RSC Adv., 2016, 6, 43975 RSC.
- (a) K. Ochi, S. Okamoto, Y. Tozawa, T. Inaoka, T. Hosaka, J. Xu and K. Kurosawa, Adv. Appl. Microbiol., 2004, 56, 155 CrossRef CAS PubMed; (b) K. Ochi, Biosci., Biotechnol., Biochem., 2007, 71, 1373 CrossRef CAS PubMed.
- (a) S.-M. Fang, C.-J. Wu, C.-W. Li and C.-B. Cui, Mar. Drugs, 2014, 12, 1788 CrossRef PubMed; (b) S.-M. Fang, C.-B. Cui, C.-W. Li, C.-J. Wu, Z.-J. Zhang, L. Li, X. J. Huang and W. C. Ye, Mar. Drugs, 2012, 10, 1266 CrossRef CAS PubMed; (c) M.-W. Xia, C.-B. Cui, C.-W. Li and C.-J. Wu, Mar. Drugs, 2014, 12, 1545 CrossRef PubMed; (d) M.-W. Xia, C.-B. Cui, C.-W. Li, C.-J. Wu, J.-X. Peng and D.-H. Li, Mar. Drugs, 2015, 13, 5219 CrossRef CAS PubMed.
- (a) M. J. Frisch, G. W. Trucks and H. B. Schlegel, et al., Gaussian, Inc., Wallingford CT, 2010; (b) T. Bruhn, A. Schaumlöffel, Y. Hemberger, and G. Bringmann, Version 1.61, University of Würzburg, Würzburg, Germany, 2013 Search PubMed; (c) T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243 CrossRef CAS PubMed.
- The TDDFT ECD calculations for 1 and 2 was performed using the Gaussian 09 software package, Gaussian 09, Revision A.02, Gaussian, Wallingford, CT, USA, 2010, see details in the ESI†.
- Y. Ding, T. J. Greshock, K. A. Miller, D. H. Sherman and R. M. Williams, Org. Lett., 2008, 10, 4863 CrossRef CAS PubMed.
- K. R. Watts, S. T. Loveridge, K. Tenney, J. Media, F. A. Valeriote and P. Crews, J. Org. Chem., 2011, 76, 6201 CrossRef CAS PubMed.
- M. Figueroa, M. González-Andrade, A. Sosa-Peinado, A. Madariaga-Mazón, F. Del Río-Portilla, M. Del Carmen González and R. Mata, J. Enzyme Inhib. Med. Chem., 2011, 26, 378 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR data for 1–3, DFT-optimized structures of the low-energy conformers for 1 and 2, figures for the HPLC-UV and HPLC-MS analyses, various spectra for 1–3, and X-ray data of 1 (CIF file). CCDC 1532020. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra02446k |
| This journal is © The Royal Society of Chemistry 2017 |