
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functional responsive superparamagnetic core/shell nanoparticles and their drug release properties†
Zied Ferjaoui ab,
Raphaël Schneiderc,
Abdelaziz Meftahb,
Eric Gaffeta and
Halima Alem*a
ab,
Raphaël Schneiderc,
Abdelaziz Meftahb,
Eric Gaffeta and
Halima Alem*a
aInstitut Jean Lamour (IJL), Université de Lorraine, Department N2EV, UMR CNRS 7198, Parc de Saurupt CS50840, 54011 Nancy, France. E-mail: halima.alem@univ-lorraine.fr
bUnite Nanamatériaux et Photonique, Faculty of Sciences of Tunis, Tunis El-Manar University, 2092 Tunis, Tunisia
cLaboratoire Réactions et Génie des Procédés (LRGP), Université de Lorraine, UMR CNRS 7274, 1 rue Grandville, 54001 Nancy, France
First published on 17th May 2017
Abstract
The preparation of responsive superparamagnetic iron oxide (SPIO) nanoparticles (NPs), able to carry the anticancer drug doxorubicin (DOX) and to release it in physiological media at the physiological temperature, is one of the major challenges in nanomedicine. In this work, two families of NPs were synthesized. The first one consists of superparamagnetic Fe3O4 NPs functionalized via covalent grafting by a biocompatible responsive copolymer based on 2-(2-methoxy)ethyl methacrylate (MEO2MA) and oligo(ethylene glycol) methacrylate (OEGMA). The second one consists of the same core/shell NPs but folic acid, a biological cancer targeting molecule, was grafted at the polymer chain end. The core/shell NPs were fully characterized by the combination of spectroscopic and microscopic techniques. The influence of the polymer chain structure in water and in physiological media was studied. We demonstrate that the magnetic properties of the NPs were only influenced by the amount of the grafted polymer and no influence of NP aggregation was evidenced. Indeed, the derived nanostructured materials displayed a combination of the physical properties of the core and the macromolecular behavior of the shell. The drug release experiments confirmed that DOX was largely released above the co-polymer LCST due to the presence of DOX. The nanomaterials developed in this work have high potential as multi-modal cancer therapy tools.
1. Introduction
One of the major challenges in nanomedicine is to develop materials able to serve as efficient diagnostic and/or therapeutic tools against severe diseases, such as infectious or neurodegenerative disorders.1 The most powerful diagnosis tool in medical science is MRI (magnetic resonance imaging).2 To enhance MRI images interpretation, contrast agents were developed to increase the signal/noise ratio. Among them, superparamagnetic iron oxide (SPIO) nanoparticles (NPs) have received a great attention since their development as a liver contrast agent twenty years ago.3,4 Their properties, originating from their nanosized dimensions, allow different biodistributions and opportunities compared to the conventional molecular imaging agents.3,5–8 SPIONPs are already approved by the US Food and Drug Administration (FDA) and have been greatly investigated due to their good biocompatibility, low toxicity and ease of synthesis.3,9 Below 27 nm, SPIONPs are superparamagnetic which make them better candidates as diagnostic and/or therapeutic tools compared to the ferri or ferromagnetic NPs.7 Indeed, SPIONPs do not retain their magnetization once the magnetic field is removed. The opportunity to coat those NPs with a polymer shell that can ensure a better stability of the materials in the body, enhance their biodistribution and give them new functionalities appears as very challenging for medicinal applications. Synthetic processes leading to SPIONPs displaying diameters smaller than 20 nm have emerged in the last decade.7 Owing to their physical properties, SPIONPs received a great attention in many biomedical applications such as drug delivery platforms,3,4,7,8 contrast agents,3 and detoxiquifying agents for biological fluids.5,10 Monitoring and controlling the drug loading and its release can be ensured by the development of core/shell NPs. The shell can either improve the drug/cancer cells interactions that can in turn reduce the amount of drugs that have to be administrated during the treatment.Smart polymers are a subclass of polymers capable to respond to environmental changes such as temperature, pH, ionic strength, electric field, or light, and are a very attractive coating for biomedical applications especially for the controlled release of drugs.11–13 Thermo-responsive polymers are known to exhibit either an Upper Critical Solution Temperature (UCST) or a Lower Critical Solution Temperature (LCST).14 When the UCST is reached upon heating, the polymers become hydrophilic while the LCST is the temperature at which the polymer chains shrunk leading to their volume decrease due to the release of the water molecule from its backbone and finally to their hydrophobicity. As a consequence, the use of thermoresponsive polymers displaying a LCST in a temperature range of 40–42 °C is of high interest for a further use in core/shell NPs design. At physiological temperature, once the responsive macromolecules grafted at the surface of SPIONPs, they could trap a cancer drug by supramolecular interactions while their hydrophilicity could allow their good dispersion and circulation in the blood. Upon heating locally at 40–42 °C, the drug release could be triggered in a temporal controlled way.14 Nowadays, many temperature responsive polymers were reported in the literature, but copolymers containing poly(ethylene glycol) moieties have been found to appear as ideal candidates as they are biocompatible and proteins repellent.15 Copolymers based on the 2-(2-methoxy)ethyl methacrylate (MOE2MA) and oligo(ethylene glycol) methacrylate (OEGMA) can exhibit variable LCST depending on the amount of each monomer. As a consequence, the target temperature range mentioned above (40–42 °C) can be reached by choosing the proper MOE2MA/OEGMA molar ratio.16,17 To prepare stable core/shell NPs, the covalent grafting of the copolymer is preferred to a weak electrostatic association that can be debilitated in physiological media. Responsive polymers or copolymers are generally grafted at the surface of SPIONPs by the “grafting-to” process, i.e. the copolymer is first synthesized and then grafted at the surface of the NPs. Hence this process could easily lead to very stable and functional nanomaterials, but as a drug will be trapped within the polymer chain, its quantity is directly related to the amount of polymer chains grafted at the surface, and thus to the grafting density.18,19 To increase the grafting density of the copolymer chains, the grafting-from process is a more suitable process as it allows the growth of the copolymer from the NPs surface.20 To initiate the polymerization from the surface, controlled polymerization processes (CRP) have become powerful methods to prepare core/shell NPs with well-defined structure.5,18,19 The recently developed Activator ReGenerated by Electron Transfer-Atom Transfer Radical Polymerization (ARGET-ATRP) is one of the most promising CRP as it can ensure the growth of the copolymer from NPs surface with less sensitivity to oxygen and requires only a ppm amount of the copper catalyst compared to the ATRP process.21–23
In this work, thermoresponsive core/shell NPs for drug delivery purposes were first prepared by growing a responsive statistical copolymer by surface initiated ARGET-ATRP from SPIONPs. Two family of NPs were synthesized. The first one consists in Fe3O4 NPs functionalized by a biocompatible responsive copolymer based on MEO2MA and OEGMA, the copolymers were marked as P(MEO2MAx-co-OEGMA100−x), where x and (100 − x) represent the molar fractions of MEO2MA and OEGMA, respectively. In a second step, to enhance the selective binding and the delivery of the drug to the targeted cells via the receptor mediated endocytosis, the introduction of specific antibodies or ligands appeared as essential. Among the various ligands available, folic acid (FA) is one of the best candidate owing to its stability, low cost, absence of immunogenicity and especially due its high affinity for folates receptors that are over expressed in various human carcinomas like breast, kidney, lung or ovary.24,25
To avoid the FA screening by the polymer chains, grafting FA at the chain end appeared as a good strategy.26 In the given work, to increase the FA accessibility with the cancer cell wall, a process has been developed that consists in first the growing of a responsive copolymer based on MEO2MA, OEGMA and the FA was subsequently grafted at the chain end (Fig. 1). The efficiency of the FA grafting and the final nanomaterials structure were characterized by the combination of spectroscopic and microscopic methods. The drug release capacities of the NPs were investigated by using the Doxorubicin drug.
 | ||
| Fig. 1 Schematic representation of the surface-initiated ARGET-ATRP polymerization of P(MOE2MAx-OEGMA100−x) from Fe3O4@silane. | ||
2. Materials and methods
2.1 Chemicals
All reagents were purchased from Sigma-Aldrich, excepted ((chloromethyl)phenylethyl)trimethoxysilane (CMPETMS) (Gelest, >95%). All the reagents were used as received.2.2 Synthesis of Fe3O4 NPs
Superparamagnetic Fe3O4 nanocrystals were synthesized by coprecipitation. A mixture of FeCl3·6H2O (6 mmol; 1.622 g) and FeSO4·7H2O (5 mmol; 1.39 g) was dissolved in 40 mL water in a three necked round bottom flask. 5 mL of a 28% (v/v) aqueous ammonia solution were subsequently added to the mixture and the final solution was heated at 90 °C under argon atmosphere with stirring. Then, 4.4 g (14.9 mmol) of sodium citrate in 15 mL water were added dropwise until getting a black solution. The reaction mixture was further stirred for 30 min. Fe3O4 SPIONPs were finally recovered by magnetic separation, washed several times with ethanol and redispersed in 100 mL water.2.3 Synthesis of CMPETMS-coated Fe3O4 NPs (Fe3O4@silane)
SPIONs were dispersed in 10 mL of toluene under argon. CMPETMS (0.2 mmol, V = 49.1 μL) was injected and the mixture stirred for 2 min. Then, 1 mL of an ethanolic solution of tetramethylammonium hydroxide pentahydrate (TMAH) (36.25 mg) was added and the mixture further stirred under argon for 15 min at 50 °C. The mixture was then cooled and SPIONs were separated by centrifugation and washed two times with toluene. The silanized SPIONs were dispersed in 10 mL of toluene and 2 mL of an ethanolic solution of TMAH (36.25 mg) were injected. The reaction was conducted under argon for 30 min at 50 °C under magnetic stirring. The mixture was cooled in a water bath and SPIONPs were separated by centrifugation and washed two times with toluene.2.4 Synthesis of Fe3O4@P(MEO2MAx-OEGMAy) and Fe3O4@P(MEO2MAx-co-OEGMAy)–FA NPs
The surface initiated polymerization of P(MEO2MAx-OEGMAy) from iron oxide NPs surface was conducted using an adapted protocol described by Alem et al.23 Briefly, in a 100 mL Schlenk flask, 50 mg Fe3O4@silane NPs were dispersed in 20 mL of a DMF/DMSO mixture (10/90, v/v). 1.6 mL of MEO2MA and 1.14 mL of OEGMA were added for the synthesis of Fe3O4@P(MEO2MA60-OEGMA40) 1.26 mL of MEO2MA, 1 mL of OEGMA for Fe3O4@P(MEO2MA65-OEGMA35). Once the NPs were completely dispersed, 200 μL of a CuBr2/TPMA (0.884 μmol CuBr2, 4.3 μmol TPMA) stock solution in DMSO were added to the mixture. The reaction mixture was stirred and heated to 65 °C. Next, 250 μL of a stock solution of hydrazine in DMSO (7.1 mg mL−1) were added and the mixture was stirred for 2 h at 65 °C with argon. At the end of the polymerization, the mixture was poured drop by drop into hot Milli-Q water to precipitate the insoluble components. The materials were purified by redispersion of the core/shell NPs in cold water followed by centrifugation of the solution after heating.Fe3O4@P(MEO2MAx-OEGMAy) NPs functionalized by FA (Fe3O4@P(MEO2MAx-OEGMAy)–FA) were prepared according to a one-pot synthesis protocol similar to that described with Fe3O4@P(MEO2MAx-OEGMAy). Briefly, 8.33 mg of Fe3O4@P(MEO2MAx-OEGMAy) NPs and 400 mg of modified FA (see the synthesis protocol in the ESI†) were dispersed in 20 mL ethanol and 200 μL of the CuBr2/TPMA (0.884 μmol CuBr2, 4.3 μmol TPMA). The mixture was stirred under argon for 30 min. Next, 250 μL of hydrazine (7.1 mg mL−1 in DMSO) were added and the mixture stirred under argon for 24 h. At the end of the polymerization, the mixture was poured into hot Milli-Q water to precipitate the insoluble components. The materials were purified by washing with hot Milli-Q water followed by centrifugation.
2.5 Association with DOX
Conjugation of DOX with the copolymer was achieved through formation of imine bond, also called Schiff base bond, between the primary amine group of DOX and the aldehyde group of the P(MEO2MAx-co-OEGMAy) polymer. Fe3O4@P(MEO2MAx-OEGMAy)–FA NPs (2.5 mg) were suspended in 5 mL PBS (pH 7.4). Next, 5 mg of DOX were added (the DOX concentration was 1 mg mL−1). The mixture was stirred in the dark for 12 h. Subsequently, the DOX-loaded Fe3O4@P(MEO2MAx-OEGMAy)–FA NPs were washed with PBS by magnetic decantation ten times until the supernatant solution was clear and that no signal of the DOX could be detected by UV-visible spectroscopy. Then, the DOX-loaded Fe3O4@P(MEO2MAx-OEGMAy)–FA NPs were dispersed in 5 mL of PBS. The concentration of released DOX vs. temperature (between 25 and 50 °C) is calculated by measuring the absorbance of the supernatant at 480 nm of the free DOX remaining in solution.2.6 Characterization methods
Dynamic light scattering (DLS) was performed at room temperature using a Malvern zetasizer HsA instrument with an He–Ne laser (4 × 10−3 W) at a wavelength of 633 nm. The NPs aqueous solutions were filtered through Millipore membranes (0.2 μm pore size). The data were analysed by the CONTIN method to obtain the hydrodynamic diameter and size distribution in each aqueous dispersion of NPs. Transmission electron microscopy characterization: for each sample, one drop of a dispersed solution (for each step of the synthesis) was deposited on holey carbon grids and imaged by transmission electron microscopy (TEM). The Microscope used is an ARM 200F. The crystallographic structure of VN thin films was identified by X-Ray Diffraction (XRD) using Philips PW3710 diffractometer with Cu Kα radiation. Fourier transform infrared (FT-IR) measurements were conducted by using a Nicolet 6700 FT-IR spectrometer equipped with a deuterated triglycine sulphate (DTGS) detector in the 400–7000 cm−1 wavelength range (i.e., 1.24–25 μm) in a reflexion mode. A SETSYS Supersonic thermobalance (SETARAM) was used for the thermogravimetric measurements. The furnace is made up of a graphite element operating from room temperature up to 1600 °C. The apparatus is controlled by software appointed Calisto. Dry samples of 30 mg were put in an alumina crucible with a volume of 30 μL. The samples were heated from room temperature to 600 °C at a heating rate of 5 °C min−1 under argon atmosphere. The magnetic properties of the core/shell NPs were studied by superconducting quantum interference device SQUID-VSM combined to Vibrating Sample Magnetometer (Fig. 5 and 6).3. Results and discussion
3.1 Physicochemical properties of the superparamagnetic NPs
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) 0 and C–O bonds stretching, respectively. The anchorage of FA was confirmed by the presence of a typical large NH stretching peak at 3400 cm−1 originating from the N–H bond involved in hydrogen bond. The typical aromatic CC stretchings between 1600 and 1450 cm−1 can also be seen in the spectrum of Fe3O4@P(MEO2MAx-OEGMAy)–FA NPs. The sharp aromatic C–N peak at 1350 cm−1 and the N–H out of plane vibration at 700 cm−1 further confirmed the anchorage of FA at the periphery of Fe3O4@P(MEO2MAx-OEGMAy) NPs.
0 and C–O bonds stretching, respectively. The anchorage of FA was confirmed by the presence of a typical large NH stretching peak at 3400 cm−1 originating from the N–H bond involved in hydrogen bond. The typical aromatic CC stretchings between 1600 and 1450 cm−1 can also be seen in the spectrum of Fe3O4@P(MEO2MAx-OEGMAy)–FA NPs. The sharp aromatic C–N peak at 1350 cm−1 and the N–H out of plane vibration at 700 cm−1 further confirmed the anchorage of FA at the periphery of Fe3O4@P(MEO2MAx-OEGMAy) NPs.
 | ||
| Fig. 2 FT-IR spectra of (a) Fe3O4@MEO2MA-co-OEGMA (b) Fe3O4@MEO2MA-co-OEGMA–FA NPs. | ||
 | ||
| Fig. 3 TEM micrographs of (a) Fe3O4@silane NPs, (b) Fe3O4@MEO2MA-co-OEGMA and (c) Fe3O4@MEO2MA-co-OEGMA–FA NPs. | ||
 | ||
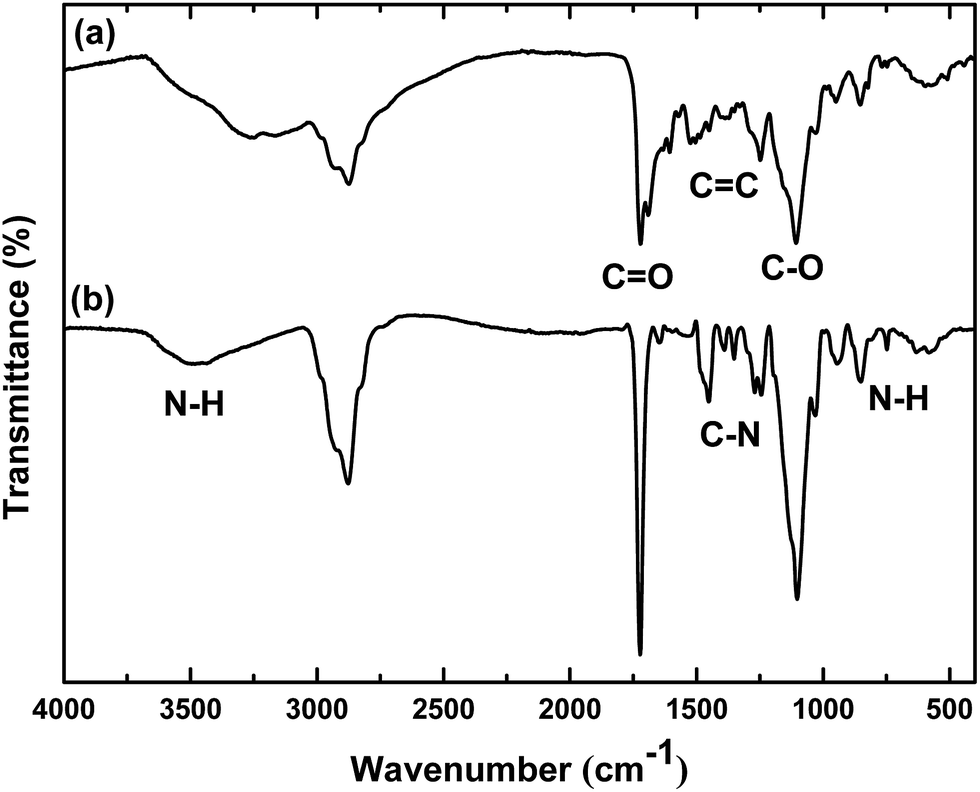
| Fig. 4 (A) Evolution of Fe3O4@P(MEO2MA80-co-OEGMA20)–FA (black), Fe3O4@P(MEO2MA65-co-OEGMA35)–FA (red) and Fe3O4@P(MEO2MA60-co-OEGMA40)–FA (blue) diameter and aggregation with temperature. The solutions of the NPs dispersed in water below the and above the LCST are illustrated in (A) and (B) respectively. | ||
The responsive behavior and the starting temperature at which the NPs started to aggregate was also monitored by DLS. As depicted in Fig. 4, when a solution of Fe3O4@P(MEO2MA80-co-OEGMA20)–FA NPs is heated from 25 to 50 °C, their hydrodynamic diameter started to increase at 30 °C, which corresponds to the LCST of P(MEO2MA60-OEGMA40)–FA in PBS. For Fe3O4@P(MEO2MA60-OEGMA40)–FA and for Fe3O4@P(MEO2MA65-OEGMA35)–FA NPs, the measured hydrodynamic diameter of the NPs started to increase at 38 and 44 °C, respectively. The shift of the LCST towards higher temperatures when increasing the amount of OEGMA, from 30 °C for the Fe3O4@P(MEO2MA80-OEGMA20)–FA NPs to 44 °C for Fe3O4@P(MEO2MA65-OEGMA35)–FA NPs, indicates that the thermal properties of the core/shell NPs are completely driven by the macromolecular properties of the copolymer chains (the same evolution of LCST was observed for the samples without FA as shown in Fig. S3†). Finally, to prove that the processes are fully reversible, five successive cycles of heating (T > LCST) and cooling (T < LCST) were conducted. All the results are presented in Fig. S4.† A reversible behavior in physiological media upon heating and cooling was demonstrated for all the samples. This behavior is in perfect agreement with the high hydration capacity of the ethylene glycol groups below the collapse temperature and the chains shrinking above the latter. No influence of FA was detected on the collapse temperature of the polymer chains.
 | ||
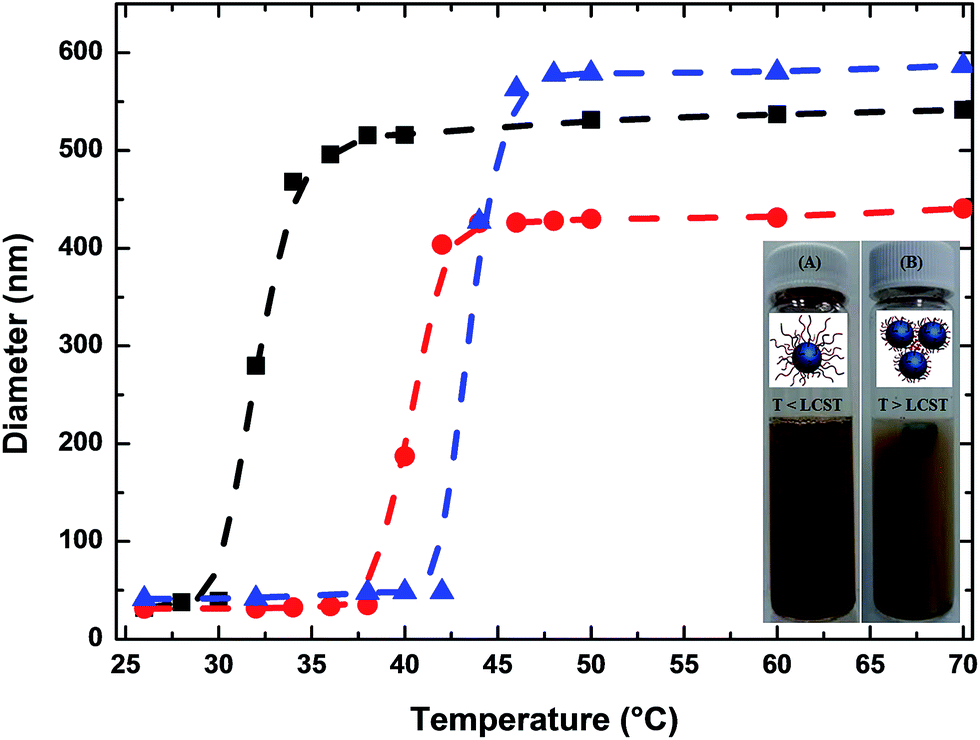
| Fig. 5 Magnetization curves of the Fe3O4@P(MEO2MA80-co-OEGMA20)–FA (black), Fe3O4@P(MEO2MA65-co-OEGMA35)–FA (red) and Fe3O4@P(MEO2MA60-co-OEGMA40)–FA (blue) magnetic NPs. | ||
 | ||
| Fig. 6 Temperature variation of ZFC and FC of Fe3O4@P(MEO2MA60-co-OEGMA40), B = 750 Oe. | ||
Fig. 7 presents the evolution of the cumulative drug release vs. temperature for Fe3O4@P(MEO2MA65-OEGMA35)–FA and Fe3O4@P(MEO2MA60-OEGMA40)–FA NPs. These samples were selected as they display an LCST in PBS close to 40 °C (based on the DLS results presented in Fig. 4), which is important for further applications as drug carrier. Surprisingly, almost no drug was released for both samples below the LCST (Fig. 7). DOX is indeed only stored in the carrier by supramolecular interactions that can be weakened in media with high ionic strength. By increasing the temperature, we could observe a change in the slope of the curve as soon as the LCST is reached. The release was expected to occur at 38 and 44 °C for Fe3O4@P(MEO2MA60-OEGMA40)–FA and Fe3O4@P(MEO2MA65-OEGMA35)–FA NPs, respectively, as shown by the LCST measurements. However, once DOX associated with the NPs, the starting temperature at which DOX is released, which is also the temperature at which the copolymer chain is supposed to start to collapse, is shifted by 5 °C for both samples. DOX incorporated within the polymer shell seems to act as a kosmotrope molecule which is able to induce an early collapsing of copolymer chains.14,35,36 Moreover, as seen in Fig. 7, the temperature range of the DOX release is larger (about 5 °C) for both samples compared to media without DOX, which is the indication that the NPs gradually release DOX. Interestingly, the evolution of the drug release rate is also influenced by the OEGMA amount within the polymer backbone.
 | ||
| Fig. 7 Cumulative DOX release by the core/shell NPs. | ||
For core/shell NPs containing a high amount of OEGMA, i.e. Fe3O4@P(MEO2MA60-OEGMA40)–FA NPs, the temperature range between the starting of the drug release and its complete release is increased compared to Fe3O4@P(MEO2MA65-OEGMA35)–FA NPs. Nevertheless, only 50% of the total amount of the DOX was released within this very short time lapse (30 s), thus confirming the high diffusion of DOX within the copolymer shell34 and indicating that longer times are necessary to release the total amount of the drug from the NPs.
Despite of the short time lapse, we showed that the loading of DOX within these NPs generate a more gradual collapse of the polymer chains. Such a phenomenon is accentuated when the macromolecules are more sterically hindered. Nevertheless, the controlled release of DOX from polymer-coated SPIONPs was obtained. For a good control of the drug release properties of those systems, the kosmotrope nature of DOX has to be taken into consideration when those systems will be considered as carrier for this anti-cancer molecule.
4. Conclusions
In this study, core/shell magnetic NPs covered by polymers allowing cancer targeting and macrophage evasion were successfully prepared. Fe3O4 NPs have excellent dispersibility and stability in aqueous medium due to the surface coating by MEO2MA and OEGMA. The LCST of the copolymer could be easily tuned by varying the initial monomer molar ratio. The combination of temperature-triggering release properties and the possibility of FA targeting would allow to control where and when the drug will released but longer time than 30 s have to be considered to release the total amount of the drug. We are currently investigating the drug release mechanism to further improve our systems for fast and efficient drug release properties. Thus, by virtue of these advantages, Fe3O4@P(MEO2MAx-co-OEGMAy)–FA NPs can potentially be a good candidate for enhancing the targeting efficiency of cancer cells in bioimaging, biodetection, and other in vivo medical applications.Acknowledgements
The financial support was given by the Centre National de Recherche Scientifique (CNRS). The authors are thankful to Jaafar Ghanbaja for the TEM characterizations.References
- D. Thassu, M. Deleers and Y. V. Pathak, Nanoparticulate Drug Delivery Systems, CRC Press, 2007 Search PubMed.
- H. M. Duvernoy, The Human Brain: Surface, Three-Dimensional Sectional Anatomy with MRI, and Blood Supply, Springer Science & Business Media, 2012 Search PubMed.
- L. Li, W. Jiang, K. Luo, H. Song, F. Lan, Y. Wu and Z. Gu, Theranostics, 2013, 3, 595–615 CrossRef PubMed.
- P. B. Santhosh and N. P. Ulrih, Cancer Lett., 2013, 336, 8–17 CrossRef CAS PubMed.
- R. Ghosh Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef CAS PubMed.
- N. Lee, D. Yoo, D. Ling, M. H. Cho, T. Hyeon and J. Cheon, Chem. Rev., 2015, 115, 10637–10689 CrossRef CAS PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- P. Kucheryavy, J. He, V. T. John, P. Maharjan, L. Spinu, G. Z. Goloverda and V. L. Kolesnichenko, Langmuir, 2013, 29, 710–716 CrossRef CAS PubMed.
- M. Mahmoudi, H. Hofmann, B. Rothen-Rutishauser and A. Petri-Fink, Chem. Rev., 2012, 112, 2323–2338 CrossRef CAS PubMed.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- I. Y. Galaev and B. Mattiasson, Trends Biotechnol., 1999, 17, 335–340 CrossRef CAS PubMed.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- E. Wischerhoff, K. Uhlig, A. Lankenau, H. G. Börner, A. Laschewsky, C. Duschl and J.-F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 5666–5668 CrossRef CAS PubMed.
- M. Chanana, S. Jahn, R. Georgieva, J.-F. Lutz, H. Bäumler and D. Wang, Chem. Mater., 2009, 21, 1906–1914 CrossRef CAS.
- A. M. Jonas, K. Glinel, R. Oren, B. Nysten and W. T. S. Huck, Macromolecules, 2007, 40, 4403–4405 CrossRef CAS.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS PubMed.
- S. Edmondson, V. L. Osborne and W. T. S. Huck, Chem. Soc. Rev., 2004, 33, 14–22 RSC.
- E. J. A. Dine, Z. Ferjaoui, T. Roques-Carmes, A. Schjen, A. Meftah, T. Hamieh, J. Toufaily, R. Schneider, E. Gaffet and H. Alem, Nanotechnology, 2017, 28, 125601 CrossRef PubMed.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309–15314 CrossRef CAS PubMed.
- K. Matyjaszewski, H. Dong, W. Jakubowski, J. Pietrasik and A. Kusumo, Langmuir, 2007, 23, 4528–4531 CrossRef CAS PubMed.
- H. Alem, A. Schejn, T. Roques-Carmes, J. Ghanbaja and R. Schneider, Nanotechnology, 2015, 26, 335605 CrossRef PubMed.
- V. M. Gaspar, E. C. Costa, J. A. Queiroz, C. Pichon, F. Sousa and I. J. Correia, Pharm. Res., 2015, 32, 562–577 CrossRef CAS PubMed.
- N. Li, T. Li, C. Liu, S. Ye, J. Liang and H. Han, J. Biomed. Nanotechnol., 2016, 12, 878–893 CrossRef CAS PubMed.
- B. Sahoo, K. S. P. Devi, R. Banerjee, T. K. Maiti, P. Pramanik and D. Dhara, ACS Appl. Mater. Interfaces, 2013, 5, 3884–3893 CAS.
- H. Maeda, K. Tsukigawa and J. Fang, Microcirculation, 2016, 23, 173–182 CrossRef CAS PubMed.
- H. Maeda, Adv. Drug Delivery Rev., 2015, 91, 3–6 CrossRef CAS PubMed.
- Y.-X. J. Wang, S. M. Hussain and G. P. Krestin, Eur. Radiol., 2001, 11, 2319–2331 CrossRef CAS PubMed.
- A. Gloria, T. Russo, U. D'Amora, S. Zeppetelli, T. D'Alessandro, M. Sandri, M. Bañobre-López, Y. Piñeiro-Redondo, M. Uhlarz, A. Tampieri, J. Rivas, T. Herrmannsdörfer, V. A. Dediu, L. Ambrosio and R. D. Santis, J. R. Soc., Interface, 2013, 10, 20120833 CrossRef CAS PubMed.
- K. Hagane, T. Akera and J. R. Berlin, J. Pharmacol. Exp. Ther., 1988, 246, 655–661 CAS.
- A. Hervault, A. E. Dunn, M. Lim, C. Boyer, D. Mott, S. Maenosono and N. T. K. Thanh, Nanoscale, 2016, 8, 12152–12161 RSC.
- D. Missirlis, R. Kawamura, N. Tirelli and J. A. Hubbell, Eur. J. Pharm. Sci., 2006, 29, 120–129 CrossRef CAS PubMed.
- S. Louguet, B. Rousseau, R. Epherre, N. Guidolin, G. Goglio, S. Mornet, E. Duguet, S. Lecommandoux and C. Schatz, Polym. Chem., 2012, 3, 1408–1417 RSC.
- S. Z. Moghaddam and E. Thormann, RSC Adv., 2016, 6, 27969–27973 RSC.
- J. Iwahara, A. Esadze and L. Zandarashvili, Biomolecules, 2015, 5, 2435–2463 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02437a |
| This journal is © The Royal Society of Chemistry 2017 |