
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stabilizing silica nanoparticles in hydrogels: impact on storage and polydispersity†
Giorgia Giovanninia,
Filip Kunca,
Carmen C. Pirasa,
Ondrej Stranikb,
Alison A. Edwardsa,
Andrew J. Halla and
Vladimir Gubala *a
*a
aMedway School of Pharmacy, University of Kent, Central Avenue, Chatham, ME44TB, UK. E-mail: V.Gubala@kent.ac.uk
bThe Leibniz Institute of Photonic Technology (IPHT), Albert-Einstein-Straße 9, 07745 Jena, Germany
First published on 5th April 2017
Abstract
For successful nanomedicine, it is important that the unique, size-dependent physico-chemical properties of the nanomaterial remain predictably constant during both the storage and the manipulation of the material. Here a novel approach to preserve the colloidal stability and degradation of NPs is described. The concept is simple: (a) a solution of monodisperse particles is formulated into a responsive water- or PBS-based hydrogel; (b) the gel can be reversibly turned into a solution after long term storage by shaking it by hand; (c) the NP can be diluted and used in any desired application without the need for excessive manipulation. The differences between the physico-chemical properties of NPs stored in solution and in gel are compared. Two types of NPs were involved in this study: silica NPs of ∼100 nm and Au-NPs of 30 and 80 nm in diameter. The key findings are: the fibrous matrix of the hydrogel limits the NP mobility, significantly reduces NP aggregation and conserves the NP morphology; both the hydrogelator and the NPs show negligible toxicity towards the model U937 human hematopoietic cell line; undesired leaching of cargo material loaded inside the particles is reduced, which could be an important feature for drug delivery systems.
Introduction
The majority of the nanomedicine products that promised to revolutionize healthcare have been formulated as intravenous injectable samples, typically in the form of a solution or a suspension.1 The simple argument is that it is easier to retain the unique, highly size-dependent properties of a nanomaterial in a liquid as opposed to aerosols2 or solids (e.g. powders, tablets or capsules).3 However, controlling the colloidal and chemical stability of nanoparticles (NP) in solution/suspension for extended periods of time may still represent quite an onerous task. The difficulties in controlling the dispersion of nanoparticles in liquid media have contributed to the poor translation of these exciting materials into routine clinical use.The colloidal stability of NPs in solution is mostly governed by forces related to the particles size, density and charge. For example, Brownian motion4 and electrostatic repulsions5 (if the NP surface is charged) keep the particles in constant movement thus controlling aggregation. On the other hand, the gravitational force causes particles to sediment and it is strongly correlated with their size and density.6 Storing nanoparticles in solutions with high salt concentration such as buffers and cell culture media diminishes the effects of the electrostatic repulsions and increases the probability of aggregation. Number of theoretical (e.g. the DLVO theory7,8) and experimental approaches to stabilise NPs in liquid media have been proposed. Kamiya and Iijima comprehensively reviewed range of post-synthesis and in situ surface modification techniques for dispersion of nanoparticles in various solvents.9 For example, Sapra et al. improved water-dispersed nanoparticle suspension for at least 30 days by surface modification of nanoparticles during aerosol or gas-phase synthesis, followed by direct transfer into liquid media.10 And of course significant amount of research has been done to stabilise silica (SiNP) and gold (AuNP)11 nanoparticles, two good examples of widely used nanomaterial in biological applications. For AuNPs, the most common stabilisation methods rely on the use of ionic and non-ionic surfactants.12,13 For SiNPs, one of the most elegant solution was proposed by Bagwe.5 It relies on electrostatic repulsions between NPs, which is done by simple, post-synthetic introduction of organosilanes with charged and chemically “inert” functional groups on the SiNP surface. SiNP also attracted much attention because it is possible to incorporate various hydrophobic and hydrophilic cargo inside the silica matrix,14 which made them popular material to be studied as drug delivery systems (DDS).15 It is easy to store non-functionalised SiNPs in organic solvent such as ethanol for long term, however functionalized SiNP (e.g. equipped with antibodies or sensitive, biologically active cargo) cannot be stored in organic solvents and are known to aggregate in enriched aqueous solutions such as buffers,16 cell culture media or serum.17 Once nanoparticles aggregate in solution, the most common manipulation to redisperse the sample involves the use of ultrasound to ‘break’ the aggregates into a monodispersed suspension. However, access to ultrasonic bath or probe is not always possible and some of the mechanical energy used to move and vibrate NPs in solution could also be transferred to another undesired processes associated with a decomposition of the material, premature leaching of the drug load, denaturation of proteins (if used) or in case of SiNPs to the acceleration of a process called Ostwald ripening.18–21 Therefore, a new method for formulation and storage of NPs would be welcome. Ideally, the alternative method should be straightforward, and extremely simple for the end user to do. The requirement for simplicity of the NP-sample manipulation stems from the fact that the end user often isn't the material chemist that prepared the material but someone from quite different scientific discipline (e.g. biological and environmental sciences, medicine etc.). In our previous work, we have explored the possibility of formulating silica nanoparticles (NPs) in freeze-dried solid forms upon addition of cryoprotectants.22 While this method was relatively simple, it was also time consuming and limited to material that could tolerate freezing and dehydration stress. Here we report on a new, more generic approach by formulating silica nanoparticles into responsive hydrogels.
The combination of hydrogels and nanoparticles to enhance the properties of the individual components for various applications has been well studied. The concepts, design and applications of nanoparticle–hydrogel composites were comprehensively reviewed by Thoniyot et al.23 Stimuli-responsive AuNP-gel and SiNP-gel hybrids showed promising efficacy in surface plasmon resonance (SPR) based sensors,24 anti-bacterial applications,25 as medicated dressings to prevent infection in chronic wounds,26 in thermoreversible27 and temperature sensitive28 hydrogels for drug delivery,29 for improved tissue adhesion,30 in electrochemical applications and optoelectronic devices31 among others. However, simple approach of a well-designed, mechanically responsive hydrogel that would be a great alternative for the standard, liquid formulation of nanoparticles, while preserving their colloidal stability is still missing.
In this work, we trapped nanoparticles in a reversible hydrogel prepared from low-molecular-weight gelator and studied the effect of this formulation on their colloidal stability. To demonstrate that this method was not exclusive only to preserve the colloidal stability of SiNPs, we have also used commercially available AuNPs (30 and 80 nm in diameter). The concept is illustrated in Fig. 1a. The critical prerequisites of our new vehicle for nanoparticle storage were: (1) the gelator must be inert to the nanomaterial; (2) nanoparticles and gel must be easily separable without any chemical alteration; (3) the hydrogel must be able to withstand certain level of mechanical stress but also be able to turn back into monodispersed suspension with minimum user manipulation (e.g. by shaking the sample by hand) and (4) once the hydrogel is turned back to solution, any remaining gel-fibre debris must be biocompatible and non-toxic. Considering these conditions, an amphiphilic molecule, Fmoc-galactosamine (Fmoc-gal) was chosen as the best candidate to form a responsive hydrogel when added to the NP suspension. This derivative is well-studied, chemically inert when in contact with nanoparticles and its various structural analogues were already explored for instance in the preparation of cell culture scaffolds.32 The mechanically responsive hydrogel was generated by the self-assembly of Fmoc-gal (at 2 mg mL−1), which was mixed with the aqueous NP suspension. We have tested this concept on several types of in-house made SiNPs and commercially sourced 30 nm and 80 nm gold nanoparticles.
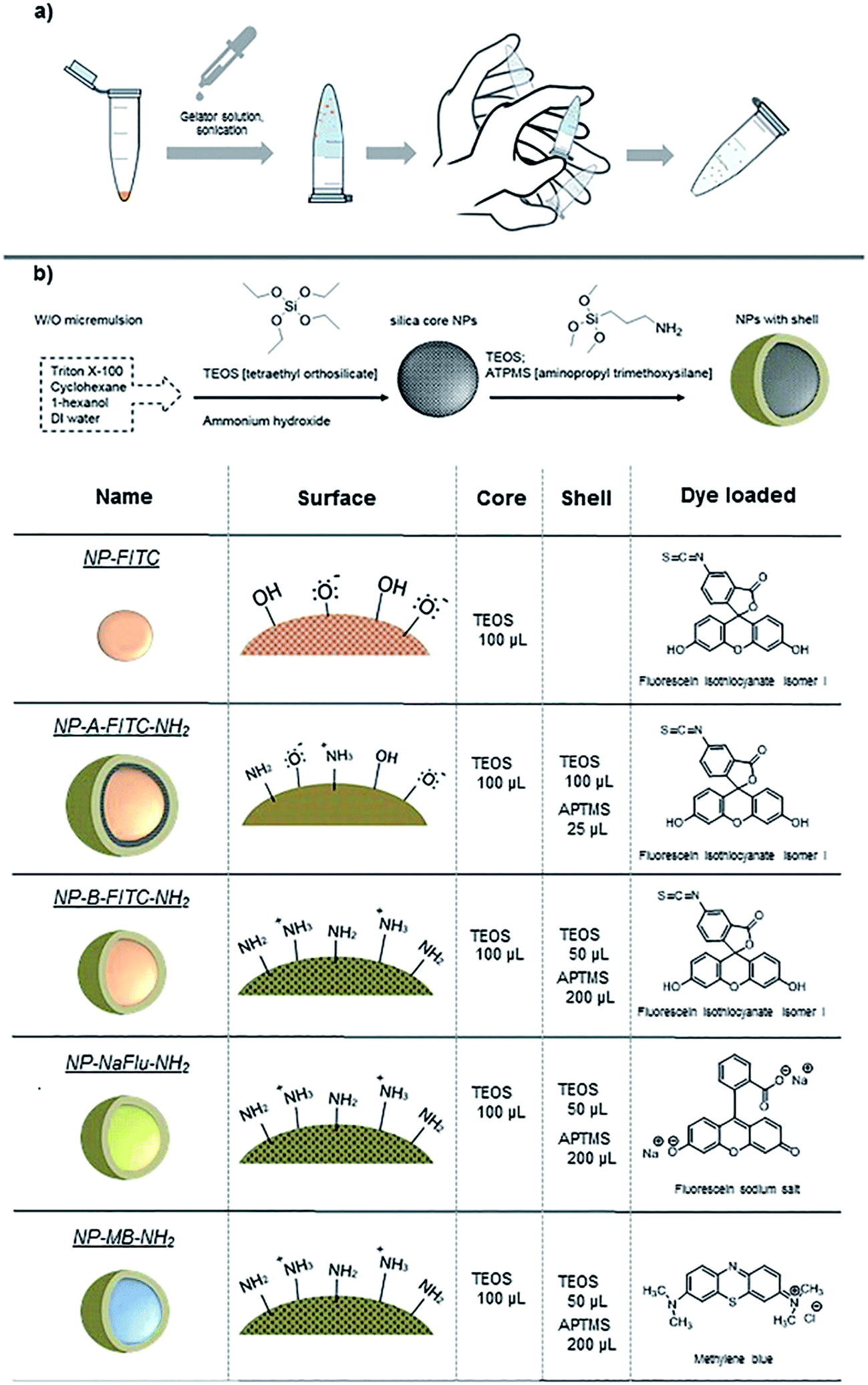 | ||
| Fig. 1 (a) Illustration of the general concept, (b) scheme showing the synthesis of SiNP; the types and specifications of nanomaterial studied in this work. | ||
For SiNPs, we have compared the longer term colloidal stability of dye doped silica nanoparticles prepared by microemulsion method both with net negative charge and with net positive charge suspended in water, phosphate buffer saline (PBS) and also when formulated in a supramolecular hydrogel. The specifics of the SiNPs used in this study are summarized in Fig. 1b. Because SiNPs are good models for DDS, we studied the SiNP ability to retain their hydrophobic and hydrophilic cargo while being stored over longer period of time in the gel. In a model experiment, we have compared the effect of the dye-leaching phenomenon (i.e. analogous to controlled drug release) between three formulations: (i) NPs in organic suspension, (ii) in aqueous suspension and (iii) in the hydrogel. Three different dyes (as drug surrogates) with ranging hydrophobicity – hydrophilicity were incorporated inside the NP core to provide insights into the stability of the silica matrix when formulated into the gel. Some of the engineered nanomaterial, especially the ones intended to be used in medicine will be equipped with often light-sensitive drugs, organic/inorganic dyes or functionalised on their surface with biorelevant material such as antibodies or oligonucleotides. For such reasons, the NPs in this study were stored in the dark, at 4 °C. The analyses were performed at room temperature and the model drug release experiment was conducted at 37 °C to mimic the physiological temperature.
Results and discussion
NP preparation and formulation
While the AuNPs used in this study were obtained from commercial sources, the SiNPs were prepared in-house by reverse microemulsion method.33 The size and surface charge of the prepared SiNPs was measured by dynamic light scattering both in water and ethanol. The size, the monodispersity and the fact that the SiNPs were of spherical shape was then corroborated by transmission electron microscope as illustrated in Fig. 2. As a reference, we have firstly prepared plain fluorescein isothiocyanate-doped silica NPs (NP-FITC), which exhibit surface negative charge due to the presence of silanols. The FITC molecules were covalently incorporated inside the silica matrix using aminated organosilane (APTMS) as a cross-linker, following a method described by Tan et al. previously.33 Summarized in Table 1, the plain SiNPs had a diameter of 60 nm/72 nm and zeta potential of −30/−34.4 mV in ethanol and water respectively. | ||
| Fig. 2 DLS and TEM data showing the size, charge and the morphology of the prepared SiNPs. Right column shows the SiNP suspension and the SiNPs in gel. | ||
| Ethanol | Water | Vacuum | |||||
|---|---|---|---|---|---|---|---|
| Ø DLS [nm] | PDI | ζ [mV] | Ø DLS [nm] | PDI | ζ [mV] | Ø TEM [nm] | |
| NP-FITC | 64 ± 24 | 0.382 | −30.0 | 72 ± 6 | 0.374 | −34.4 | 38 ± 6 |
| NP-A-FITC-NH2 | 157 ± 13 | 0.373 | −18.1 | 101 ± 8 | 0.249 | −22.5 | 68 ± 14 |
| NP-B-FITC-NH2 | 378 ± 31 | 0.918 | 46.0 | 712 ± 1 | 0.754 | 3.6 | 89 ± 10 |
| NP-NaFlu-NH2 | 342 ± 1 | 0.112 | 17.7 | 53 ± 5 | 0.148 | 12.2 | 62 ± 11 |
| NP-MB-NH2 | 297 ± 44 | 0.179 | 1.4 | 84 ± 19 | 0.286 | 11.4 | 80 ± 17 |
The size of the individual SiNPs measured by TEM was 38 nm. The discrepancy between the two techniques were attributed to the fact that the TEM images were acquired in vacuum from SiNPs dried on the copper grid. In an attempt to prepare SiNPs with less negative surface charge, we synthesized FITC-doped silica NPs with an outer shell composed of combination of the negatively charged silanols and limited amount of positively charged amines (TEOS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :APTMS = 4:1 by volume). This type of SiNPs were labelled as NP-A-FITC-NH2.
:APTMS = 4:1 by volume). This type of SiNPs were labelled as NP-A-FITC-NH2.
The thick layer of TEOS grafted on the SiNP surface caused the NP-A-FITC-NH2 grow to diameter of 157 nm/101 nm with surface charge of −18/−22.5 mV in ethanol and water respectively (Table 1). Interestingly, the presence of the shell resulted in NPs with much brighter yellow/orange colour when compared to the NP-FITC without the shell (Fig. 2, right column). This visible ‘brightness’ increase is presumably attributed to entrapping FITC molecules in the different environments of the SiNPs with and without the shell. The final three types of SiNPs involved in this study were synthesized with identical surface chemistry, the only difference was the dye used. Fluorescein isothiocyanate (FITC), highly water soluble sodium salt of fluorescein (NaFlu) and water insoluble methylene blue (MB) were doped inside SiNPs (Fig. 1) and modified on the surface with relatively thick shell consisting of polymerized TEOS:APTMS molecules in a ratio of 1:4 (by volume). Positively charged, APTMS-coated silica nanoparticles (with higher density of –NH2/–NH3+groups) are known to aggregate readily in liquids, which created a great opportunity for us to test the ability of the hydrogel to stabilize the colloidally ‘highly unstable’ SiNPs.
The fact that the positively charged APTMS-coated SiNPs tend to aggregate in solution is evident from Fig. 2. Although the TEM pictures show that the size of the individual SiNPs is still in the range of 62–89 nm, DLS data indicate significant aggregation with average size in the range of 297–378 nm in ethanol and even larger range in water. Importantly, as shown in Fig. 2, we were able to formulate each prepared SiNP sample into a suspension and also into a gel.
Colloidal stability – long term storage
The in-house prepared SiNP and two commercially sourced AuNPs samples were formulated into an aqueous suspension (in water) and hydrogels at the concentration of 1 mg of NP per mL, which corresponds to approximately ∼1 × 1012 NPs per mL. The concentration of the Fmoc-gal gelator was 2 mg mL−1. Both the aqueous and hydrogel formulations were stored at 4 °C for either several days or weeks. After storing the NPs for set amount of weeks, both the aqueous and hydrogel NP samples were gently shaken by hand for the hydrogel to convert back to liquid and then diluted to 0.5 mg of NP per mL. This represents an optimal concentration required for accurate analysis of the size and polydispersity by Dynamic Light Scattering (DLS), Nanoparticle Tracking Analysis (NTA) and Transmission Electron Microscopy (TEM). Importantly, we have previously reported on the use of silica NPs as drug delivery carriers and demonstrated that the concentration of NPs for in vitro and in vivo experiments is optimal in the range of 1–0.001 mg of NP per mL.18 Qualitatively, as seen on Fig. S1 and S2,† it was possible to observe the aggregation of both AuNPs and SiNPs stored in a solution by a naked eye as early as after 1 week of storage. Storing AuNPs in solution for one week resulted in significant aggregation, evident by the high polydispersity and particle average size of ∼175 nm. On the other hand, the AuNPs stored in the hydrogel formulations appeared to have no visible aggregates. Quantitative analysis of the particle size inside the hydrogel or when the AuNPs were dispersed to suspension by ‘breaking’ the gels proved to be more challenging due to the presence of gel fibre debris. Therefore, the DLS data were corroborated by NTA, a technique that is not affected by the presence of gel fibre debris. The analysis of NTA data, shown on Fig. S3,† indicate that the size distribution of AuNPs stored in hydrogel is significantly improved when compared to the AuNPs stored in solution.SiNPs have also high tendency to aggregate in solution, however in this case the silica matrix is also prone to hydrolysis, which leads to dissolution of the SiNPs. In order to study the colloidal stability of SiNPs and their changes in morphology, we stored SiNPs both in solution and hydrogel for 30 days and used TEM for analysis (Fig. 3). TEM provided clearer picture of the various SiNP morphologies, the distinct populations present in the mixture and the presence of the gel-fibre debris was not detrimental to the analysis and interpretation.
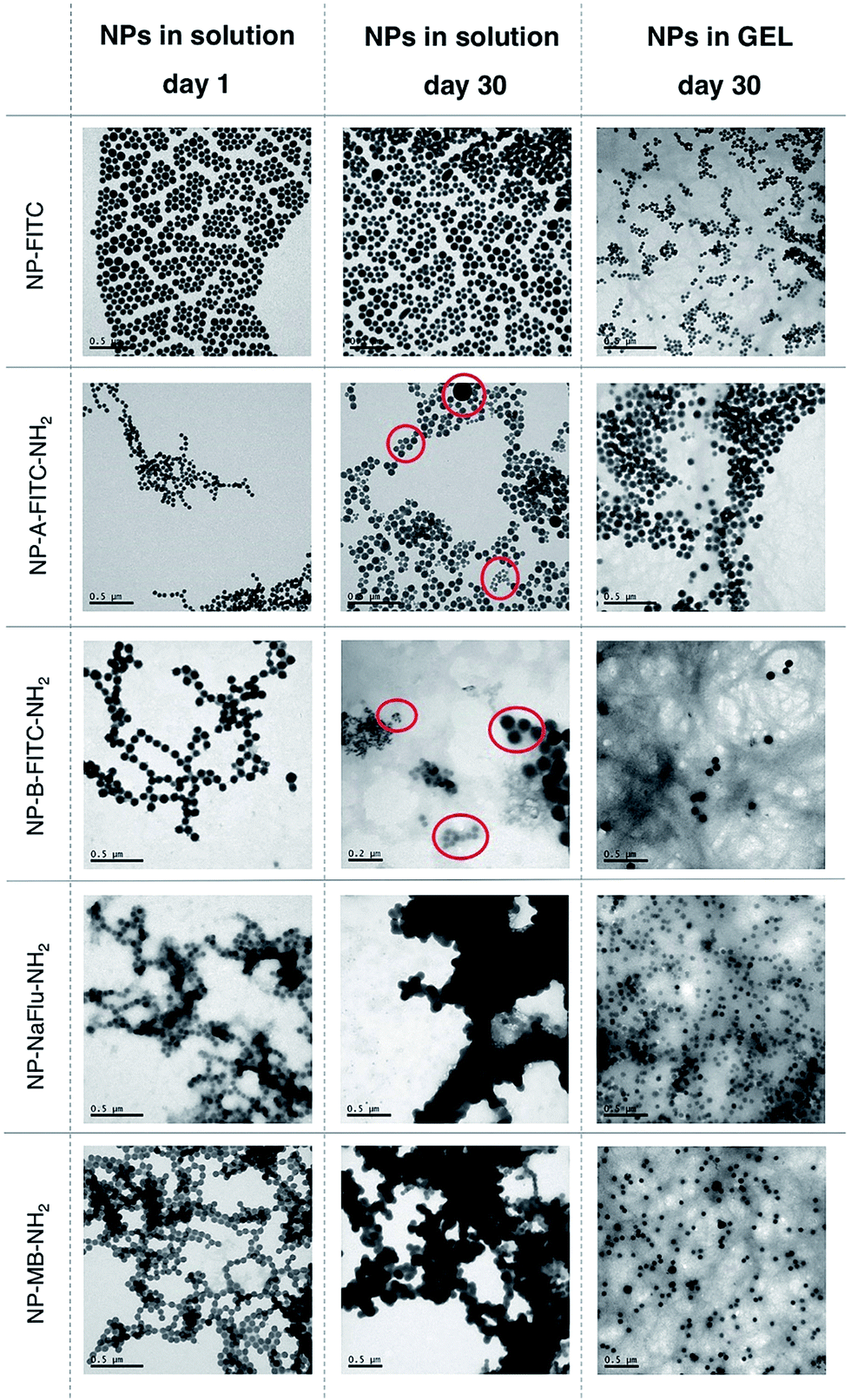 | ||
| Fig. 3 TEM images showing the evolution of the SiNP morphologies and size distribution when stored in water for 1 day and 30 days (left and middle column) and in gel for 30 days (right column). The red circles highlight the evolution of various SiNP populations over time, which clearly demonstrates that the silica NPs are behaving as a dynamic system in water. On the other hand, when stored for 30 days in gel, the morphology and uniformity of the SiNPs is extremely monodispersed. The 0.2 μm scale for NP-B-FITC-NH2 was used on purpose to illustrate the various NP populations. Storing NP-NaFlu-NH2 and NP-MB-NH2 in solution for 30 days resulted in significant NP-polymerisation/aggregation, which means that this material should no longer be classified as nanomaterial. | ||
As seen on Fig. 3 (with Gaussian distribution on Fig. S4†), when all five SiNP samples were synthesized and placed in water, they were of uniform size with relatively narrow size distribution. No visible aggregation was observed, although DLS data (Table 1) suggest some degree of NP–NP interactions for the positively charged SiNPs (NP-B-FITC-NH2, NP-NaFlu-NH2 and NP-MB-NH2). After storing the SiNPs in solution for 30 days, clear indication of polydispersity was evident across all tested samples. In case of NP-A-FITC-NH2 and NP-B-FITC-NH2, two or even three very distinct populations of SiNPs were observed (Fig. 3, red circles). Apart from the original ∼75 nm particles, we have observed a population of some ultrasmall, ∼20 nm spherical SiNPs and also some very large, ∼200 nm species.
This phenomenon could be explained by a process known as Ostwald ripening, in which monomers dissolved from smaller silica particles contribute to the growth of larger ones and the particle size distribution constantly evolves towards an equilibrium.19 This process is mainly observed during the formation of particles when the catalyst is present, however it unavoidably continues when SiNPs are stored in solutions too, since silica condensation is reversible.20 Importantly, the evolution of various populations of silica NPs is not observed in the hydrogel samples. We assume that the mobility of SiNPs stored in gel is inhibited by the gel fibers, hence both the dissolution of silica matrix and Ostwald ripening are retarded. A net results of storing SiNPs in such less dynamic system is that the structural integrity of SiNPs and their content can be secured for longer period of time. NP-NaFlu-NH2 and NP-MB-NH2 samples were highly aggregated when stored in solution for 30 days, with visible precipitates suggesting a very unstable colloidal system (Fig. 3, middle column). In fact, the precipitates were clearly visible in the solution by a naked eye even after 1 week of storage (Fig. S1†).
These findings were in stark contrast with the images obtained from the hydrogel samples. Although the debris from the gel fibres were still visible under the TEM, the SiNPs were highly monodispersed, with retained shape and extremely narrow size distribution. We have observed no deformation of the SiNPs, no evidence of extensive hydrolysis, no nanoparticle-bridging and no visible precipitates. The forces that usually govern the motion and aggregation of the particles in solution were restricted inside the hydrogel, which translated to a highly stable SiNP sample after storing it for 30 days.
Dye leaching – silica degradation
A critical feature for any successful drug delivery systems (DDS) is the ability to undergo biodegradation (or self-degradation) but also to retain the clinically relevant concentration of the drug while in storage. Firstly, we have studied the capacity of NP-B-FITC-NH2, NP-NaFlu-NH2 and NP-MB-NH2 to retain their cargo when stored in ethanol and water for 3 weeks. The amount of released dye from the silica NPs was tracked by measuring the fluorescence of the SiNPs and the supernatant at different time intervals while keeping the samples in suspension for 21 days (Fig. S5†). The differences in dye-leaching between the studied solvents were quite remarkable. All three studied dyes were leaching relatively slowly when the SiNPs were stored in EtOH but the rate of leaching was significantly accelerated in water. While the water-soluble salt of fluorescein in NP-NaFlu-NH2 is only physically adsorbed inside the silica pores, the FITC dyes in NP-B-FITC-NH2 are covalently incorporated into the silica matrix via organosilane linkage. However, because the leaching rate of both dyes (covalently and non-covalently linked to silica matrix) from the SiNPs in aqueous environment is equally fast, this suggests rapid degradation of the silica matrix in water. Although this might be beneficial for drug delivery as it satisfied the requirement of degradability of the nanomaterial, it represents a challenge for the storage of silica based DDS.To gain more insight on how fast silica degradation could be under a model physiological conditions, we have monitored the dye leaching of NP-B-FITC-NH2, NP-NaFlu-NH2 and NP-MB-NH2 when shaken in an isotonic medium (0.01 M PBS pH 7.4 with 0.137 M NaCl) at 37 °C for 8 hours. For reference purposes, we have performed identical experiments in ethanol and water. The results are summarized in Fig. S6.† The dye release, as a consequence of silica matrix hydrolysis was very prominent both in water and PBS in the case of NP-B-FITC-NH2 and NP-NaFlu-NH2. Significant accumulation of FITC and the water-soluble fluorescein analogue (Na+Flu) in the supernatant was evident immediately after several minutes. On the other hand, methylene blue, as the most hydrophobic dye was not released into the aqueous environment readily. In fact, the release of methylene blue from NP-MB-NH2 was slow in both water and ethanol and only a small degree of accelerated release of methylene blue was observed in PBS after 6 hours. These simple experiments highlighted two important requirements: (1) it is preferable to store silica NPs at 4 °C and (2) it is necessary to formulate the silica NPs into a preparation, where the dye leaching effect could be minimized.
Dye leaching – long term storage
To demonstrate the advantage of the hydrogel approach, NP-B-FITC-NH2, NP-NaFlu-NH2 and NP-MB-NH2 were stored for 28 days at 4 °C in water-based Fmoc-Gal hydrogel and also PBS-based Fmoc-Gal hydrogel. On the 28th day, all samples were gently shaken by hand, the resulting suspension was diluted to 0.5 mg of SiNP per mL, centrifuged to separate the supernatant, which was analysed by fluorescence (absorbance in the case of methylene blue). As a reference, identical procedure was followed with SiNPs stored in water and PBS in the absence of the gelator. The results are presented in Fig. 4 and S7.† | ||
| Fig. 4 Leaching of the dye from SiNPs to the supernatant while being stored in solution or in gel. (a) FITC covalently linked to silica matrix – the leaching was partially reduced when stored in gel (excitation/emission wavelengths: 471/525 nm). (b) FITC-sodium salt physically trapped in the silica matrix – the amount of dye leached from the NPs is dramatically reduced when stored in gel (excitation/emission wavelengths: 456/518 nm). (c) Methylene blue physically trapped in the silica matrix – very slow leaching of the hydrophobic dye in both formulations but very dramatic difference in colloidal stability of the NP-B-FITC-NH2 when stored in water and gel (absorbance: 664 nm). | ||
The hydrogel had stabilizing effect on both the dye leaching and the colloidal stability of the measured NPs. The most notable differences between the NPs' capacity to retain their cargo when stored in solution and gel was observed in case of NP-NaFlu-NH2. The degradation of the SiNPs and the subsequent release of the dye was three times slower in the gel (for both water-based gel and PBS-based gel) when compared to the corresponding solutions. Less dramatic effect was observed in the case of NP-B-FITC-NH2. Remarkably, despite that the hydrophobic methylene blue leached out of the NPs very slowly in both formulations, the differences in the colloidal stability of NP-MB-NH2 between water and gel were enormous (Fig. 4C).
NP-hydrogel stability
So far it was established that the prepared hydrogel is very stable while being stored ‘still’ (i.e. without shaking or strong vibrations) for long time and that it is possible to ‘break’ it into a solution by simply shaking it in hand. However, we recognized the need to test the ability of this new formulation to retain the protective, gel-like properties as a function of time and external vibrations, and how this ability correlates with some intrinsic factors of the SiNPs such as charge and the nature of the cargo inside the NPs. To do this, all SiNPs were formulated into gels and exposed to strong vibrations using a simple orbital shaker. The conversion of the hydrogel into a solution as a function of shaking speed and time was then qualitatively compared.Some interesting trends were observed (Fig. S8–S12†). Firstly, the gel stability positively correlated with increased zeta potential. Secondly, the more hydrophobic the dye was, the more stable the gel appeared to be. The first trend is clearly showed on Fig. 5. The NP-A-FITC-NH2 (ζ = −22.5 mV) were evidently converted from the gel to a suspension after 29 hours and 30 minutes of shaking at 1200 rpm, while the NP-B-FITC-NH2 (ζ = +3.6 mV) retained its self-supporting gel characteristic even after and 30 hours of shaking at 1200 rpm. Similarly, the highly negatively charged NP-FITC (ζ = −34.4 mV) were found to convert to a suspension at 800 rpm and after more than 7 hours of shaking. The second trend is best illustrated in Fig. S8 and S9.† The NP-MB-NH2 with the hydrophobic methylene blue had very positive effect on the gel stability, while the NP-NaFlu-NH2 with the highly soluble NaFlu dye had detrimental effect on the integrity of the gel. Presumably NPs with highly water soluble cargo will need to be stored under conditions avoiding strong vibrations or they will require to be formulated in a gel with higher stiffness than the one reported here.
 | ||
| Fig. 5 NP-A-FITC-NH2 and NP-A-FITC-NH2 gel formulations shaken by orbital shaker for up to 30 hours with increasing speed. | ||
NP-hydrogel in vitro cytotoxicity
Silica nanoparticles have been studied in several biomedical applications, both in vitro and in vivo and their low cytotoxicity is relatively well documented18,21,22 Once formulated in a hydrogel however, even basic toxicology data are not available. The typical concentration of silica NPs used either in animal studies or in vitro work ranges between 1–0.001 mg of NP per mL. In this work, we have observed that once the NP-gel formulation was converted back to suspension and diluted to 0.5 mg of NP per mL, some of the hydrogel fibres remained in the suspension and they were clearly visible under TEM (Fig. 3). Although further dilutions could dissolve the gel fibres completely, the cytotoxic effect of either the diluted Fmoc-Gal or some of the remaining nano-sized gel fibres needed to be investigated. This was done by performing an MTS Cell Proliferation Assay on human glioblastoma astrocytoma cell line (U87MG). This assay allow to evaluate the cell viability determining their metabolic activity. If cells are alive and metabolically active, they produce mitochondrial dehydrogenase. This enzyme reduces MTS's reagent (tetrazolium salt), forming a product that absorbs light at 490 nm. U87MG cells have been chosen because they adhere well on surfaces. This property allows to remove the analysed sample after incubation with cell, limiting interferences between the sample and MTS's reagent. To obtain basic cytotoxicity data, three formulations were studied: SiNP in hydrogel, SiNP in solution and Fmoc-Gal hydrogel without SiNPs. All formulations were allowed to incubate with U87MG cells for 1, 2 and 4 hours in a 96-well plate format in a cell culture medium. The assay has been performed in triplicate and the results are shown in Fig. 6. The cytotoxic effect either of the gel or the silica nanoparticles was minimal. In fact, the proportion of live cells was marginally increasing over time, suggesting that the interaction of Fmoc-Gal, SiNPs or both with U87MG cells did not have detrimental effect on the cell life. Additional in vivo nanotoxicology studies are currently being conducted to corroborate these findings. | ||
| Fig. 6 Cell viability measured by MTS assay with U937 human hematopoietic cell line incubated with silica nanoparticles (red bars), hydrogel (blue bars) and SiNPs in hydrogel (green lines). | ||
Conclusion
In summary, we have presented a simple and perhaps the most universal solution to storing silica NPs without significant effects to their colloidal stability and degradation. The elegant hydrogel restricts the movement of the individual nanoparticles and slows down any undesired ongoing reactions of the silica matrix or collisions of particles. This in turns presents obvious benefits in terms of nanoparticle aggregation and drug release. An initial cytotoxicity data showed negligible effect on cell growth, however additional in vivo toxicology tests are currently in progress in our laboratory to fully elucidate the impact of these findings on live biological systems. Because of its simplicity and versatility, our method has the real potential to be exploited by scientists working with various other types of NPs. We're confident that our findings could be very significant especially for the material scientists active in the field of nanomedicine. We hope that this could finally accelerate the progress towards the development of real, commercially available products that will revolutionize healthcare.Experimental section
Materials
Cyclohexane (anhydrous, 99.5%), 1-hexanol (anhydrous, ≥99%), Triton® X-100, aminopropyl trimethoxysilane [APTMS] (97%), tetraethyl orthosilicate [TEOS] (99.99%), ammonium hydroxide solution (28% w/v in water, ≥99.99%), fluorescein isothiocyanate isomer I, fluorescein sodium salt, sodium pyruvate solution. All mentioned were purchased from Sigma Aldrich. Methylene blue was purchased from Acros Organics. DMEM, antibiotic–antimycotic (containing 10000 units per mL of penicillin, 10000 μg mL−1 of streptomycin and 25 μg mL−1 of FungizoneTM) and fetal bovine serum (FBS) were purchased from GIBCO (Life Technologies). The model cell line U937 was sourced from European collection of cell cultures. Gold nanoparticles 30 nm and 80 nm diameter were purchased from BBI solutions. CellTiter 96®Aqueous One was purchased from Promega. Absolute ethanol, phosphate buffer saline tablets (one tablet dissolved in 200 mL DI water yields 0.01 M phosphate buffer, pH 7.4) were purchased from Fisher Scientific. Thereafter, the use of ‘PBS’ refers to 0.01 M PBS, pH 7.4.
Method
Dye precursor formation. In a dried glass vial fluorescent dye FITC (2.5 mg) was dissolved in 2 mL of 1-hexanol and APTMS (5.6 μL) was added. The reaction was allowed to proceed over 2 hours under nitrogen atmosphere. After that 0.666 mL of mixture was collected and added into prepared microemulsion.
Formation of microemulsion. All nanoparticles were formed in a microemulsion prepared by combining cyclohexane (7.5 mL), 1-hexanol (1.133 mL), Triton® X-100 (1.894 g), DI water (0.48 mL) in a 30 mL plastic bottle under constant stirring.
Synthesis of NP-FITC. Firstly, the microemulsion was prepared as described above. Into this 100 μL of TEOS was added followed by 0.666 mL dye precursor solution. After 30 min 40 μL of ammonium hydroxide was added to trigger polymerisation. The mixture was stirred for further 24 hours. After that the microemulsion was broken by adding 30 mL ethanol. Formed silica cores were purified by centrifugation (14
000 rpm, 10 min) and re-dispersed in 30 mL ethanol (3×). After purification, the nanoparticles were stored in ethanol solution at 4 °C.
Synthesis of NP-A-FITC-NH2. Silica nanoparticle core was synthesised similarly to NP-FITC protocol, silica nanoparticle core was synthesised similarly to NP-FITC protocol. After 24 h stirring two 50 μL portions of TEOS were added to the mixture 30 min apart of each other. 30 min later APTMS (25 μL) was added, the mixture was stirred for further 24 hours and purified as mentioned above.
Synthesis of NP-B-FITC-NH2. Similar to the protocol above, after 24 h of stirring, one portion of 50 μL of TEOS was added to the microemulsion. After additional 30 min, ATPMS (200 μL) was added and the mixture was stirred for further 24 h and purified as mentioned above.
Synthesis of NP-MB-NH2 and NP-NaFlu-NH2. Methylene blue and fluorescein sodium salt dyes were encapsulated in nanoparticle cores by using 0.48 mL of their aqueous solutions (6.25 mM) as the water phase in the microemulsion. The rest of the procedure was similar to NP-B-FITC-NH2.
Sample preparation
Characterisation
Analysis
000 rpm), 600 μL of the supernatant from the top-part of the tube was collected and the amount of dye was quantified. This was done to avoid contamination by any un-centrifuged nanoparticles. The remaining supernatant was discarded; the pellet of isolated SiNPs was re-dispersed in equal amount of solvent (2 mL). 200 μL of each solution were pipetted into 96-well plate and fluorescence and absorbance values were recorded.Alternatively 1 mg of NPs was suspended into 2 mL of water, or ethanol and shaken at 37 °C. The signal of the dye leached from the NPs to the solvent was detected every 2 hours for 8 hours, following the same procedure mentioned above.
All three samples were shaken by hand (to break the gel) and vortexed for 1 min at 500 rpm. They were then diluted up to by factor of 4, using the cell culture medium (RPMI).
The cells were plated at a density of 3 × 103 per well in a 96-well plate. In each well the cells were treated with 20 μL of each samples, reaching a concentration of 50 μg of NPs per well. The total volume of 100 μL per well was reached by adding the cell culture medium. All plates were incubated at 37 °C with 5% CO2 for up to 4 hours. After 1 h, 2 h and 4 h, 20 μL of MTS solution was added to the each well. The plate was then incubated for further 2 hours to allow MTS reagent to react. The absorbance was read at 490 nm to determine the cell viability. For the calculation of cell viability, a calibration curve was done measuring the absorbance of different cell-concentrations after 2 hours incubation with MTS.
Acknowledgements
G. Giovannini, F. Kunc and C. C. Piras are thankful to Medway School of Pharmacy, University of Kent Faculty of Sciences Research fund for providing funding for their postgraduate studies.References
- D. Pooja, H. Kulhari, D. J. Adams and R. Sistla, Expert Opin. Ther. Pat., 2016, 26, 745–749 CrossRef CAS PubMed
.
- A. Costa, M. Pinheiro, J. Magalhaes, R. Ribeiro, V. Seabra, S. Reis and B. Sarmento, Adv. Drug Delivery Rev., 2016, 102, 102–115 CrossRef CAS PubMed
- K. Yoncheva, B. Tzankov, M. Popova, V. Petrova and N. Lambov, J. Dispersion Sci. Technol., 2016, 37, 113–118 CrossRef CAS
- K. Petersson, D. Ilver, C. Johansson and A. Krozer, Anal. Chim. Acta, 2006, 573, 138–146 CrossRef PubMed
- R. P. Bagwe, L. R. Hilliard and W. H. Tan, Langmuir, 2006, 22, 4357–4362 CrossRef CAS PubMed
- T. L. Moore, L. Rodriguez-Lorenzo, V. Hirsch, S. Balog, D. Urban, C. Jud, B. Rothen-Rutishauser, M. Lattuada and A. Petri-Fink, Chem. Soc. Rev., 2015, 44, 6287–6305 RSC
- E. J. W. Verwey, J. Phys. Colloid Chem., 1947, 51, 631–636 CrossRef CAS PubMed
- B. V. Derjaguin, Y. I. Rabinovich and N. V. Churaev, Nature, 1978, 272, 313–318 CrossRef CAS
- H. Kamiya and M. Iijima, Sci. Technol. Adv. Mater., 2010, 11, 044304 CrossRef PubMed
- M. Sapra, A. A. Pawar and C. Venkataraman, J. Colloid Interface Sci., 2016, 464, 167–174 CrossRef CAS PubMed
- C. K. Kim, P. Ghosh and V. M. Rotello, Nanoscale, 2009, 1, 61–67 RSC
- A. Das, R. Chadha, N. Maiti and S. Kapoor, J. Nanopart., 2014, 2014, 7 Search PubMed
- Y. Zhao, Z. Wang, W. Zhang and X. Jiang, Nanoscale, 2010, 2, 2114–2119 RSC
- R. I. Nooney, E. McCormack and C. McDonagh, Anal. Bioanal. Chem., 2012, 404, 2807–2818 CrossRef CAS PubMed
- A. Burns, H. Ow and U. Wiesner, Chem. Soc. Rev., 2006, 35, 1028–1042 RSC
- L. S. de Lara, V. A. Rigo and C. R. Miranda, Eur. Phys. J. B, 2015, 88, 261–271 CrossRef
- Y. S. Lin, N. Abadeer and C. L. Haynes, Chem. Commun., 2011, 47, 532–534 RSC
- C. Fang, N. Bhattarai, C. Sun and M. Zhang, Small, 2009, 5, 1637–1641 CrossRef CAS PubMed
- S. W. Bae, W. Tan and J.-I. Hong, Chem. Commun., 2012, 48, 2270–2282 RSC
- M. X. Yu and J. Zheng, ACS Nano, 2015, 9, 6655–6674 CrossRef CAS PubMed
- J. L. Provis and D. G. Vlachos, J. Phys. Chem. B, 2006, 110, 3098–3108 CrossRef CAS PubMed
- C. J. Moore, H. Monton, R. O'Kennedy, D. E. Williams, C. Nogues, C. Crean and V. Gubala, J. Mater. Chem. B, 2015, 3, 2043–2055 RSC
- P. Thoniyot, M. J. Tan, A. A. Karim, D. J. Young and X. J. Loh, Adv. Sci., 2015, 2, 13 Search PubMed
- J. Wang, S. Banerji, N. Menegazzo, W. Peng, Q. Zou and K. S. Booksh, Talanta, 2011, 86, 133–141 CrossRef CAS PubMed
- E. Marsich, A. Travan, I. Donati, A. Di Luca, M. Benincasa, M. Crosera and S. Paoletti, Colloids Surf., B, 2011, 83, 331–339 CrossRef CAS PubMed
- G. S. Alvarez, C. Helary, A. M. Mebert, X. L. Wang, T. Coradin and M. F. Desimone, J. Mater. Chem. B, 2014, 2, 4660–4670 RSC
- C. P. Silveira, L. M. Apolinário, W. J. Fávaro, A. J. Paula and N. Durán, ACS Biomater. Sci. Eng., 2016, 2, 1190–1199 CrossRef CAS
- S. R. Sershen, S. L. Westcott, N. J. Halas and J. L. West, J. Biomed. Mater. Res., 2000, 51, 293–298 CrossRef CAS PubMed
- X. Hu, X. Hao, Y. Wu, J. Zhang, X. Zhang, P. C. Wang, G. Zou and X.-J. Liang, J. Mater. Chem. B, 2013, 1, 1109–1118 RSC
- Y. Liu, H. Meng, S. Konst, R. Sarmiento, R. Rajachar and B. P. Lee, ACS Appl. Mater. Interfaces, 2014, 6, 16982–16992 CAS
- H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 Search PubMed
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS PubMed
- R. P. Bagwe, C. Yang, L. R. Hilliard and W. Tan, Langmuir, 2004, 20, 8336–8342 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02427d |
| This journal is © The Royal Society of Chemistry 2017 |