
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIonothermally synthesized hierarchical porous Schiff-base-type polymeric networks with ultrahigh specific surface area for supercapacitors†
Yuhang Zhaoa,
Ping Liu *b,
Xiaodong Zhuangb,
Dongqing Wub,
Fan Zhangb and
Yuezeng Su*c
*b,
Xiaodong Zhuangb,
Dongqing Wub,
Fan Zhangb and
Yuezeng Su*c
aSchool of Aeronautics and Astronautics, Shanghai Jiao Tong University, 800 Dongchuan RD, Shanghai 200240, P. R. China
bSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, 800 Dongchuan RD, Shanghai 200240, P. R. China. E-mail: liupingsjj@sjtu.edu.cn
cSchool of Electronic Information and Electrical Engineering, Shanghai Jiao Tong University, 800 Dongchuan RD, Shanghai 200240, P. R. China. E-mail: yzsu@sjtu.edu.cn
First published on 5th April 2017
Abstract
A hierarchical porous polymeric network (HPPN) with ultrahigh specific surface area up to 2870 m2 g−1 was synthesized via a one-step ionothermal synthesis method without using templates. As the electrode material of supercapacitors, HPPN-400-30 exhibits a specific capacitance of 129 F g−1 at 10 A g−1 and demonstrates an excellent cycling stability with 99.93% capacitance retention after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 10 A g−1. Furthermore, an all-solid-state supercapacitor (ASSS) based on HPPN-400-30 exhibits an energy density of 8.1 W h kg−1 at the high power density of 5027 W kg−1.
000 cycles at 10 A g−1. Furthermore, an all-solid-state supercapacitor (ASSS) based on HPPN-400-30 exhibits an energy density of 8.1 W h kg−1 at the high power density of 5027 W kg−1.
Elaborately designed new carbonaceous skeleton polymeric networks with optimized structures and functionality have been intensively studied as their architectures continue to be of interest for many established and emerging applications, ranging from storage of hydrogen, sensing, and catalysis to energy storage and conversion.1 It is well known that many materials display their function only when they exist as or are assembled into a certain structure or morphology.2 Since the capacitance of an electrochemical double layer capacitor (EDLC) is strongly dependent on the interface between its porous electrode material and electrolyte,3,4 the introduction of pores into carbonaceous materials with high surface area, large pore volume and the proper pore distribution will efficiently increase their effective surface area for the formation of electric double layer and enhance their electrochemical properties as electrode materials of EDLCs. Meanwhile, the introduction of heteroatom will influence the electron distribution in carbonaceous materials. For instance, the nitrogen doping will improve the wettability of carbonaceous materials and provide more active sites for the ion accumulation in the electrode materials.5,6 Many efforts have been done to increase the effective surface area while adjusting the pore distribution,7,8 to introduce heteroatoms into the carbon skeleton9,10 and to develop new materials with new modalities.4,11
However, the heteroatom doping in most of common electrode materials are uncontrollable for the limit of doping methods.12,13 Schiff-base chemistry is one of the most popular choices to address this problem by constructing porous organic materials like crystalline COFs and amorphous organic frameworks to introduce heteroatoms through the condensation reaction in a reversible fashion.14–16 Heteroatoms, nitrogen in special, could be controllably doped into the polymeric structure by choosing variable monomers. Nevertheless, from the most of previous reports, Schiff-base-type materials were constructed by conventional organic synthesis methodology at low temperatures which usually close a majority of the very fine pores of the networks. So, most of Schiff-base-type materials have a majority of micropores with relative small pore volumes and exhibit relatively low conductivity as well. Consequently, the high surface areas with proper porosities (surface area ≥ 2000–3000 m2 g−1, pore volume ≥ 2 cm3 g−1) and decent conductivity are still the challenges for Schiff-base-type porous materials. Furthermore, some drawbacks in traditional organic synthesis methods cannot be neglected, such as multiple synthetic steps, long reaction time (mostly 3–7 days) and expensive raw materials.17–19
Ionothermal condensation, a one-step synthesis for porous polymeric networks under catalysts, has attracted intensive attention for its simple synthesizing procedure20 and controllable porosity construction strategies.21 Given that Schiff-base chemistry is an efficient linkage mean in polymerization to control the chemical structure by using diverse monomers. So, the ionothermal approach was utilized for the construction of Schiff-base-type HPPNs. In this work, HPPNs were ionothermally synthesized through a condensation reaction between melamine, a large-scale industrial chemical, and anthraquinone at the presence of aluminium chloride (see details in ESI†). The synthesized nitrogen-enriched HPPNs with high nitrogen content, controllable hierarchical porous architecture and ultrahigh specific surface exhibited high specific capacitance at low current density and an excellent cycling stability at high current density. Moreover, an all solid state supercapacitor (ASSS) based on HPPN-400-30 demonstrated high energy density and power density simultaneously.
Preparation of HPPNs
To ensure that the basic structure of this material can be resembled as that shown in Scheme 1. The polymerization was carried out at a moderate temperature of 400 °C for varied reaction time. Melamine, anthraquinone and aluminium chloride were mixed in a 25 mL Pyrex ampoule evacuated by vacuum, sealed and then heated to 400 °C for 15, 30, 60 and 90 hours, respectively. HPPN-400-ts (t: 15, 30, 60 and 90) were obtained as black powder (see details in ESI†). In order to investigate the temperature effect on the polymeric network structure, a serious of HPPN-Ts (T: 300, 400, 500 and 600) samples were fabricated through 15 hours polymerization at different temperature from 300, 400, 500 to 600 °C in the same way.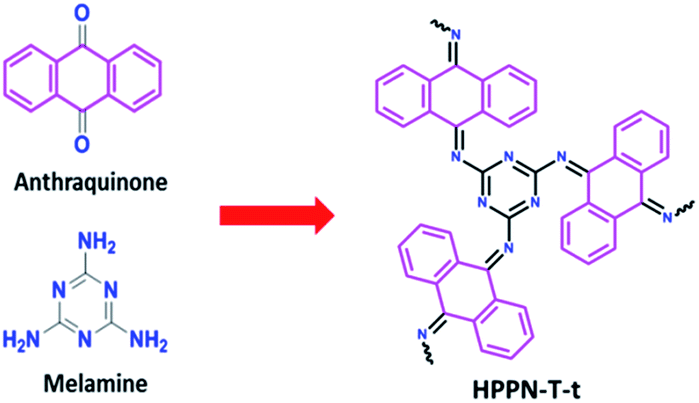 | ||
| Scheme 1 Ionothermal synthesis of a nitrogen-enriched Schiff-base type hierarchical porous polymeric networks (HPPNs) from anthraquinone and melamine in presence of AlCl3. | ||
Results and discussion
The Fourier transform infrared spectroscopy (FT-IR), 13C CP-MAS NMR spectroscopy, XPS and Raman spectra were measured to characterize the chemical structure of HPPNs. The high conversion rate of functional groups initially presented in the monomers and the build-up of polymeric networks in HPPNs are confirmed by the greatly attenuation of the bands attributed to the primary amine group of melamine at 3470, 3420 cm−1 (–NH2 stretching) and 1650 cm−1 (–NH2 deformation) and even absence of the band ascribed to the carbonyl function of anthraquinone at 1675 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) in FT-IR of HPPNs (Fig. S3a and c in ESI†).4,22 The distinct bands can be found at ∼1582 cm−1 and ∼1220 cm−1 in FT-IR of HPPNs, which can be assigned to the imine linkages (CN stretching) formed in the polymeric networks.3,23,24 HPPNs consist of five types of carbon atoms, which are experimentally identified by 13C CP-MAS NMR spectroscopy (see Fig. S4a and b in ESI†). Based on XPS analysis, the nitrogen contents are calculated to be 11.1%, 10.1%, 9.3% and 8.7% in HPPN-400-15, HPPN-400-30, HPPN-400-60 and HPPN-400-90 (Fig. S5e in ESI†), indicating that the nitrogen from melamine was high efficiently integrated into the polymeric networks. The gradual decline of total nitrogen content in HPPNs synthesized through the relatively longer polymerization processes can be ascribed to the further rearrangement or decomposition reactions of the aromatic frameworks in HPPN-400-ts.4,10 The more obvious decline of nitrogen content from 11.4%, 11.1%, and 10.6% to 5.4% was found in HPPN-300, HPPN-400, HPPN-500 and HPPN-600 as shown in Fig. S5f (in ESI†). The fact indicates the more decomposition reactions took placed in the aromatic polymeric networks at 600 °C. Furthermore, the deconvolution peaks in their high resolution N 1s XPS spectra located at ∼398.1, 400.1 and 401.1 eV could be properly ascribed to the pyridinic N (N1), pyrrolic N (N2) and quaternary N (N3), respectively (see Fig. S5c and d in ESI†),23 which suggest the formation of graphitized fragments.25 Remarkably, the quantitative XPS analysis demonstrates that the percentage of quaternary N obviously increased from 6.2% in HPPN-400-15 to 19.4% in HPPN-400-30 with the prolongation of the polymerization time from 15 to 30 hours, which could be probably ascribed to the occurring of further cross-linking and rearrangement reactions. However, the further increase of polymerization time to 60 and 90 hours leaded to a decline of the quaternary N content to 12.3% in HPPN-400-60 and to 10.2% in HPPN-400-90. This may suggest that more defects emerged on the formed graphitized fragments with the increasing polymerization time at 400 °C (Fig. S5c and Table S1†). In contrast, a continuing ascent of the quaternary N content from 0.8%, 6.2%, and 17.9% to 27.2% was observed in HPPN-300, HPPN-400, HPPN-500 and HPPN-600, which also indicates the occurring of the further cross-linking/rearrangement reactions and formation of increased graphitized fragments under the higher polymerization temperatures (Fig. S5d and Table S1†).3,26,27 The above analysis about the graphitization degree based on varied quaternary N content in HPPNs were experimentally confirmed by their Raman spectra. HPPN-400-30 and HPPN-600 have the relative lower ID/IG ratios in comparison with other HPPNs (see Fig. S4c and d in ESI†).
O stretching) in FT-IR of HPPNs (Fig. S3a and c in ESI†).4,22 The distinct bands can be found at ∼1582 cm−1 and ∼1220 cm−1 in FT-IR of HPPNs, which can be assigned to the imine linkages (CN stretching) formed in the polymeric networks.3,23,24 HPPNs consist of five types of carbon atoms, which are experimentally identified by 13C CP-MAS NMR spectroscopy (see Fig. S4a and b in ESI†). Based on XPS analysis, the nitrogen contents are calculated to be 11.1%, 10.1%, 9.3% and 8.7% in HPPN-400-15, HPPN-400-30, HPPN-400-60 and HPPN-400-90 (Fig. S5e in ESI†), indicating that the nitrogen from melamine was high efficiently integrated into the polymeric networks. The gradual decline of total nitrogen content in HPPNs synthesized through the relatively longer polymerization processes can be ascribed to the further rearrangement or decomposition reactions of the aromatic frameworks in HPPN-400-ts.4,10 The more obvious decline of nitrogen content from 11.4%, 11.1%, and 10.6% to 5.4% was found in HPPN-300, HPPN-400, HPPN-500 and HPPN-600 as shown in Fig. S5f (in ESI†). The fact indicates the more decomposition reactions took placed in the aromatic polymeric networks at 600 °C. Furthermore, the deconvolution peaks in their high resolution N 1s XPS spectra located at ∼398.1, 400.1 and 401.1 eV could be properly ascribed to the pyridinic N (N1), pyrrolic N (N2) and quaternary N (N3), respectively (see Fig. S5c and d in ESI†),23 which suggest the formation of graphitized fragments.25 Remarkably, the quantitative XPS analysis demonstrates that the percentage of quaternary N obviously increased from 6.2% in HPPN-400-15 to 19.4% in HPPN-400-30 with the prolongation of the polymerization time from 15 to 30 hours, which could be probably ascribed to the occurring of further cross-linking and rearrangement reactions. However, the further increase of polymerization time to 60 and 90 hours leaded to a decline of the quaternary N content to 12.3% in HPPN-400-60 and to 10.2% in HPPN-400-90. This may suggest that more defects emerged on the formed graphitized fragments with the increasing polymerization time at 400 °C (Fig. S5c and Table S1†). In contrast, a continuing ascent of the quaternary N content from 0.8%, 6.2%, and 17.9% to 27.2% was observed in HPPN-300, HPPN-400, HPPN-500 and HPPN-600, which also indicates the occurring of the further cross-linking/rearrangement reactions and formation of increased graphitized fragments under the higher polymerization temperatures (Fig. S5d and Table S1†).3,26,27 The above analysis about the graphitization degree based on varied quaternary N content in HPPNs were experimentally confirmed by their Raman spectra. HPPN-400-30 and HPPN-600 have the relative lower ID/IG ratios in comparison with other HPPNs (see Fig. S4c and d in ESI†).
The XRD patterns of HPPNs reveal their typical amorphous structures as shown in Fig. S3b and d (in ESI†). The TEM images in Fig. 1b and S2 (in ESI†) demonstrate the emerging of extremely abundant pores, while the elemental mapping images disclose the homogeneous distribution of nitrogen and carbon atoms in the whole sample (Fig. 1c and d). The N2 adsorption–desorption measurements at 77 K display the type IV isotherms for HPPN-400-ts (t: 15, 30, 60 and 90) and for HPPN-Ts (T: 300, 400, 500 and 600) in Fig. S6a and c (in ESI†), suggesting the appearance of an abundant of mesopores besides a large quantity of micropores in these HPPNs. The pore size distribution (Fig. S6b and d in ESI†) evaluated from the adsorption isotherms based on the density functional theory (DFT) furtherly confirms this finding. The high specific surface areas of 1508, 2870, 1958 and 1397 m2 g−1 were achieved for HPPN-400-15, HPPN-400-30, HPPN-400-60 and HPPN-400-90, respectively. A jump in specific surface area from 1508 m2 g−1 for HPPN-400-15 to 2870 m2 g−1 for HPPN-400-30 might be ascribed to the occurring of the further cross-linking/rearrangement in the Schiff-base-type porous aromatic polymeric networks during 30 hours polymerization. In comparison with HPPN-400-15, HPPN-400-30 exhibited a rapid growth of mesopore with size between 2 and 10 nm (see Fig. S6d in ESI†). Reversely, the further prolongation of polymerization reaction to 60 and 90 hours resulted in a shrinkage of specific surface area from 2870 m2 g−1 to 1958 m2 g−1 for HPPN-400-60 and 1397 m2 g−1 for HPPN-400-90. This phenomenon could be probably attributed to the collapse of porous structures in polymeric networks,11 which brings the lower specific surface area. The finding is verified by the pore size distribution and total pore volume based on nitrogen sorption/desorption isotherms of the HPPN-400-ts (Fig. S6d and Table S2†), while an obvious decline of mesopore distribution can be found clearly for HPPN-400-60 and HPPN-400-90. Contrastively, a jump of specific surface area from 578 m2 g−1, 1508 m2 g−1 and 2051 m2 g−1 to 2384 m2 g−1 was achieved with the rising polymerization temperature from 300 °C to 600 °C for HPPN-300, HPPN-400, HPPN-500 and HPPN-600. The fact manifests the occurring of further cross-linking and rearrangement in the porous frameworks of HPPN-Ts under the increasing temperature,3 which is confirmed by the pore size distribution and total pore volume based on nitrogen isotherms of these HPPN-Ts (Fig. S6b and Table S2†).
 | ||
| Fig. 1 Morphology and elemental mapping of the as-prepared HPPNs: (a) SEM image of HPPN-400-30; (b) TEM image of HPPN-400-30; (c and d) elemental mapping of HPPN-400-30 ((c) carbon, (d) nitrogen). | ||
All above characterizations demonstrate that the HPPNs maintain their inherent nitrogen-doping carbonaceous skeleton, controllable hierarchical porous structures, high surface areas as well as high pore volume. It can be envisioned that the HPPNs with great potential hold the promise for the application in electrochemical capacitors as active electrode materials.28 The electrochemical performance of HPPNs based electrodes was examined in a 1 M H2SO4 aqueous electrolyte. Both a conventional cell with a three-electrode configuration and an all solid state supercapacitor (ASSS) device with a two-electrode symmetric structure were employed to investigate the electrochemical characters of HPPNs in this work.
The supercapacitive performance was firstly tested with cyclic voltammetry (CV) measurement and galvanostatic charge–discharge (GCD) cycling experiment. The symmetric CV curves and reversible voltage-dependent current loops were obtained for HPPNs in the potential range from −0.3 to 0.7 V at scan rates from 5 to 100 mV s−1 versus AgCl/Ag (Fig. 2a and S8 in ESI†), which demonstrate a typical performance of the supercapacitive energy storage.29 GCD experiments were then carried out at different current densities from 0.1 to 10 A g−1 to further characterize the performances of HPPNs based electrodes. The specific capacitance calculated from the discharge curves demonstrates that the HPPN-Ts (T: 300, 400, 500 and 600) show the specific capacitance dependency on the synthesis temperature of HPPN-Ts as illustrated in Fig. S10a and Table S3 (in ESI†). For instance, HPPN-300, HPPN-400 and HPPN-500 exhibited an ascend of specific capacitance from 185.8, 231.8 to 251.8 F g−1 at the current density of 0.1 A g−1 in Table S3,† which might be attributed to the rapid growth of specific surface area (SSA) from 587, 1508 to 2051 m2 g−1 and of total pore volume from 0.556, 0.855 to 1.268 cm3 g−1 for HPPN-300, HPPN-400 and HPPN-500 when they have similar nitrogen content (Fig. S6e and Table S2 in ESI†). Furthermore, Fig. S6b† clearly displays that the amount of the mesopores with the pore size between 2 and 10 nm is obviously multiplied with the rising synthetic temperature from 300 to 600 °C for HPPN-Ts. Based on the basic principle of electrochemical double layer capacitor, the specific capacitance is largely depends on the effective SSA with the pore size larger than the ion size of electrolyte.3 Nevertheless, an obvious decline of specific capacitance was observed for the HPPN-600 as illustrated in Fig. S9a† even though it has the highest SSA of 2384 m2 g−1. Remarkably, the nitrogen content rapid declines from above 10% for other three HPPN-Ts (T: 300, 400 and 500) to 5.4% for HPPN-600 as indicated in Table S1.† It is believed that the nitrogen-doping can effectively change the electron distribution in the carbonaceous skeleton of polymeric networks which enable the dipolar interaction between polymeric networks surface and electrolyte cations. It further improve their electrochemical performance by enhancing the wettability and the electro-active SSA of the electrode materials, and finally provide more active sites for the ion accumulation.5,23 Additionally, it is believed that the capacitive performance of the nitrogen-containing carbonaceous materials is originated not only from the double electric layer, but also from the pseudocapacitance through the redox reactions.30–32 Consequently, a sharp fall of nitrogen content in HPPN-600 might be the dominant factor causing the drop of its capacitive performance.
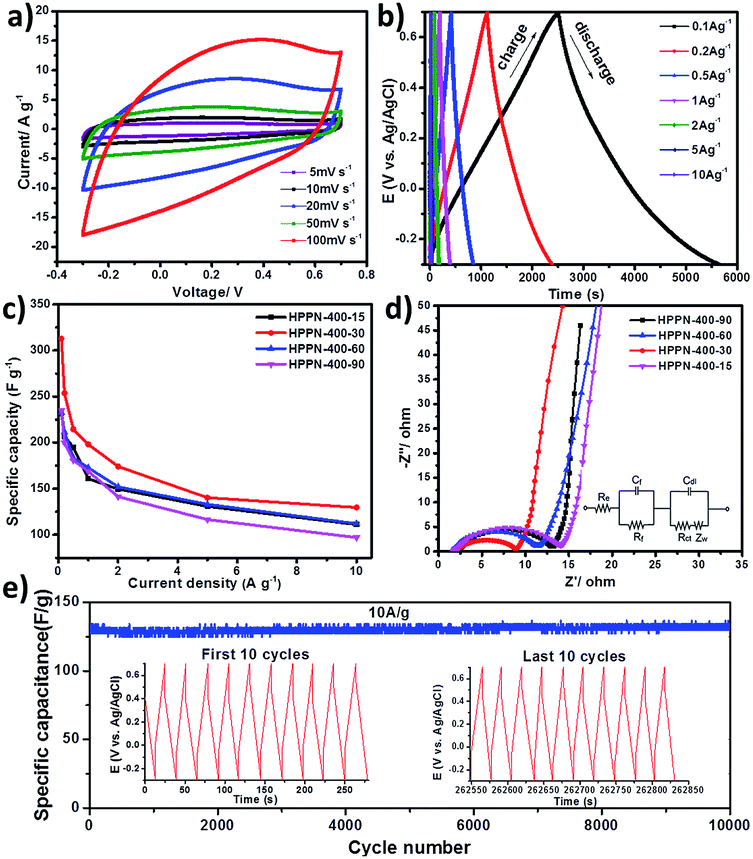 | ||
| Fig. 2 Supercapacitor performance of as-synthesized porous polymeric networks: (a) cyclic voltammograms of HPPN-400-30 at different scan rates; (b) galvanostatic charge–discharge curves of HPPN-400-30 at different current densities; (c) specific capacitance plots at different current densities for HPPN-400-ts; (d) electrochemical impedance spectroscopy (EIS) profiles of HPPN-400-ts measured in the frequency range from 0.01 Hz to 100 kHz; (e) cycle performance of the supercapacitor based on HPPN-400-30 for a 10000-cycle charge–discharge test at a current density of 10 A g−1, the insets show the initial and last 10 cycles to confirm the stability of the supercapacitor. | ||
Furthermore, the electrochemical performances of HPPN-400-ts synthesized at a moderate temperature 400 °C for 15, 30, 60 and 90 hours were investigated. The specific capacitances evaluated from the discharge curves demonstrate that the HPPN-400-30 performed outstanding specific capacitances at the observed current densities from 0.1 to 10 A g−1 as manifested in Fig. 2c and Table S3 (in ESI†). Notably, HPPN-400-30 exhibited the highest capacitance of 312.7 F g−1 at 0.1 A g−1 and it can be operated at high current density of 10 A g−1 to achieve the specific capacitance of 129 A g−1, which allows a rapid charge and power supply. The excellent performance might be assigned to its extremely high SSA (2870 m2 g−1), a relative high nitrogen content (10.1%) and an exceptionally high total pore volume (2.75 cm3 g−1), which can provide the high electro-active SSA, the sufficient charge storage space, efficient ion transfer path and the high mass transfer capability of the electrode.3,5 Both HPPN-400-15 and HPPN-400-60 also demonstrated relatively high specific capacitance of above 110 F g−1 at a high current density of 10 A g−1. However, HPPN-400-90 could not retain the high specific capacitance especially at 10 A g−1, which might ascribed to its relatively lower SSA, less nitrogen content and a reduced pore volume among the four HPPN-400-ts (see Table S3 in ESI†).
The excellent performances of HPPNs are confirmed by the cycling stability tests in Fig. 2e. HPPN-400-30 exhibited extraordinarily high cycling stability with a high capacitance retention of 99.93% at 10 A g−1 after 10000 repeated charge–discharge processes. This is also confirmed by its excellent thermal stability demonstrated in TGA measurement in Fig. S7 (see ESI†), no obvious decomposition was observed until 600 °C for the HPPN-400-30. Additionally, the capability of ion diffusion/transfer within the hierarchical porous architecture can be estimated roughly from the nearly isosceles-triangular shaped charge–discharge profiles of HPPN-400-30 in the inset of Fig. 2e. Such a long term stability of HPPN-400-30 can be attributed to its high mesoporosity ratio, large pore volume and high N-doping content,33,34 which will guarantee the rapid diffusion/transfer of electron and electrolyte ions in its porous skeleton.25
For further understanding the kinetic behaviour of HPPNs based electrodes, electrochemical impedance spectroscopy (EIS) was performed in the frequency range from 100 kHz to 10 mHz as illustrated in Fig. 2d and S11a (in ESI†). HPPN-400-30 has a smaller semicircle in high frequency region and a more vertical straight line in low frequency region as shown in Fig. 2d, which indicates its lower resistance to mass transfer and ion diffusion within its porous networks,9,35,36 which insurances its good rate capacity indicated by the symmetric CV curve at 100 mV s−1 in Fig. 2a. The highest quaternary N content of 19.4% from XPS analysis and the lowest ID/IG ratio from Raman spectra have confirmed the formation of more graphitized fragments, which brings HPPN-400-30 based supercapacitor the better conductivity and lower resistance to transfer/diffusion of electrolyte ions.
The remarkable electrochemical performance of HPPN-400-30 at high current density encouraged us to further assemble an all-solid-state supercapacitors (ASSS) with HPPN-400-30 as active electrode material and the mixed gel of polyvinyl alcohol (PVA)/H2SO4 as electrolyte. Fig. 3a depicts the CV curves of this ASSS device at different scan rates in a potential window from −0.3 to 0.7 V. The CV profiles exhibit the completely symmetric loops even at the high scan rate of 100 mV s−1. All GCD curves show the very regular, triangular shape (Fig. 3b) at the current densities from 0.1 to 10 A g−1, indicating the occurring of fast charge and ion transfer/diffusion. The specific capacitances of this ASSS device at different current densities are illustrated in the inset of Fig. 3c. A specific capacitance of 96.8 F g−1 was achieved at the current density of 0.1 A g−1. Ragone plot is a useful indicator of the performance by connecting the power and energy density of supercapacitor devices.37–39 Calculated by integrating the CV curves at different scan rates, the ASSS device manifests the maximum energy density of 13.4 W h kg−1 at 0.1 A g−1 and the maximum power density of 5027.6 W kg−1 for a reduced energy density of 8.1 W h kg−1 as displayed in Fig. 3c and Table S4 (in ESI†). More importantly, the outstanding performance of this ASSS device is comparable to or even higher than state of the art supercapacitors based on carbonaceous skeleton polymeric networks.37–39
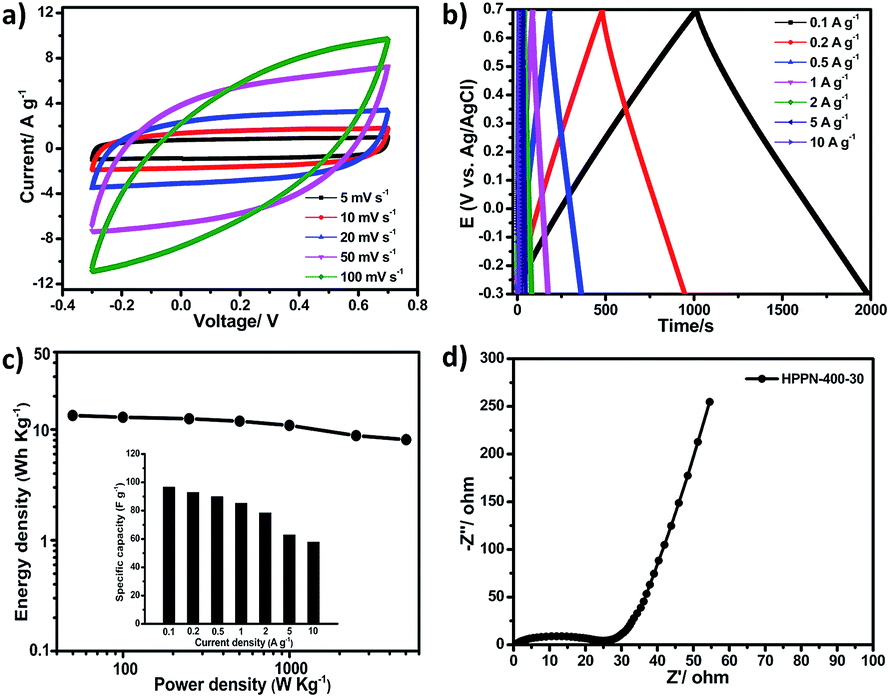 | ||
| Fig. 3 (a) CV curves of the all-solid-state supercapacitor (ASSS) with HPPN-400-30 as electrode at 5–100 mV s−1; (b) galvanostatic charge–discharge profiles of the ASSS with HPPN-400-30 as electrode under different current densities; (c) Ragone plot for the ASSS device with different specific capacitances shown in the insert; (d) electrochemical impedance spectroscopy (EIS) profile of the ASSS measured in the frequency range from 0.01 Hz to 100 kHz. | ||
In summary, we have demonstrated that the one-step ionothermal synthesis is an efficient way to fabricate the hierarchical porous Schiff-base-type polymeric networks with ultrahigh specific surface area, large pore volume and decent conductivity at moderate temperature without using template. A three-electrode system based on HPPN-400-30 demonstrates high specific capacitance and exceptionally high cycling stability. Moreover, a HPPN-400-30 based ASSS device exhibits a high energy density and a large power density simultaneously. The hierarchical porous architecture of HPPNs with high specific surface area, large pore volume, a large fraction of mesopores and high nitrogen content can provide massive active sites for charge accumulation and enhance the rapid diffusion and transfer of electron and ion during the charge/discharge processes. The result outlines a promising strategy towards improved carbonaceous hierarchical porous polymeric networks for high performance energy storage and conversion devices.
Acknowledgements
Financial support from National Natural Science Foundation of China (51403126, 61306018), the Science and Technology Commission of Shanghai Municipal (16JC1400703) are gratefully acknowledged. A lot of thanks to the Instrumental Analysis Centre of Shanghai Jiao Tong University for the characterization of materials.References
- D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959–4015 CrossRef CAS PubMed.
- A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS PubMed.
- L. Hao, J. Ning, B. Luo, B. Wang, Y. Zhang, Z. Tang, J. Yang, A. Thomas and L. Zhi, J. Am. Chem. Soc., 2015, 137, 219–225 CrossRef CAS PubMed.
- X. Zhuang, F. Zhang, D. Wu and X. Feng, Adv. Mater., 2014, 26, 3081–3086 CrossRef CAS PubMed.
- P. J. Hall, M. Mirzaeian, S. I. Fletcher, F. B. Sillars, A. J. R. Rennie, G. O. Shitta-Bey, G. Wilson, A. Cruden and R. Carter, Energy Environ. Sci., 2010, 3, 1238 CAS.
- J. Wei, D. Zhou, Z. Sun, Y. Deng, Y. Xia and D. Zhao, Adv. Funct. Mater., 2013, 23, 2322–2328 CrossRef CAS.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS PubMed.
- W. Tian, X. Mao, P. Brown, G. C. Rutledge and T. A. Hatton, Adv. Funct. Mater., 2015, 25, 4803–4813 CrossRef CAS.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839 CAS.
- N. P. Wickramaratne, J. Xu, M. Wang, L. Zhu, L. Dai and M. Jaroniec, Chem. Mater., 2014, 26, 2820–2828 CrossRef CAS.
- N. Jung, S. Kwon, D. Lee, D. M. Yoon, Y. M. Park, A. Benayad, J. Y. Choi and J. S. Park, Adv. Mater., 2013, 25, 6854–6858 CrossRef CAS PubMed.
- J. Han, L. L. Zhang, S. Lee, J. Oh, K. S. Lee, J. R. Potts, J. Ji, X. Zhao, R. S. Ruoff and S. Park, ACS Nano, 2013, 7, 19–26 CrossRef CAS PubMed.
- P. Karthika, N. Rajalakshmi and K. S. Dhathathreyan, J. Nanosci. Nanotechnol., 2013, 13, 1746–1751 CrossRef CAS PubMed.
- M. Mastalerz, M. W. Schneider, I. M. Oppel and O. Presly, Angew. Chem., Int. Ed., 2011, 50, 1046–1051 CrossRef CAS PubMed.
- S. Wan, F. Gandara, A. Asano, H. Furukawa, A. Saeki, S. K. Dey, L. Liao, M. W. Ambrogio, Y. Y. Botros, X. F. Duan, S. Seki, J. F. Stoddart and O. M. Yaghi, Chem. Mater., 2011, 23, 4094–4097 CrossRef CAS.
- Y. Zeng, R. Zou, Z. Luo, H. Zhang, X. Yao, X. Ma, R. Zou and Y. Zhao, J. Am. Chem. Soc., 2015, 137, 1020–1023 CrossRef CAS PubMed.
- S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed.
- N. Huang, X. Chen, R. Krishna and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 2986–2990 CrossRef CAS PubMed.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- X. X. Lin, B. Tan, L. Peng, Z. F. Wu and Z. L. Xie, J. Mater. Chem. A, 2016, 4, 4497–4505 CAS.
- P. Kuhn, A. Thomas and M. Antonietti, Macromolecules, 2009, 42, 319–326 CrossRef CAS.
- J. Wu, X. Rui, G. Long, W. Chen, Q. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 7354–7358 CrossRef CAS PubMed.
- Y. Kou, Y. Xu, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 8753–8757 CrossRef CAS PubMed.
- Y. B. Zhang, J. Su, H. Furukawa, Y. Yun, F. Gándara, A. Duong, X. Zou and O. M. Yaghi, J. Am. Chem. Soc., 2013, 135, 16336–16339 CrossRef CAS PubMed.
- Y. Liu, X. Wang, Y. Dong, Z. Wang, Z. Zhao and J. Qiu, J. Mater. Chem. A, 2014, 2, 16832–16835 CAS.
- D. Hulicova, J. Yamashita, Y. Soneda, H. Hatori and M. Kodama, Chem. Mater., 2005, 17, 1241–1247 CrossRef CAS.
- H. M. Jeong, J. W. Lee, W. H. Shin, Y. J. Choi, H. J. Shin, J. K. Kang and J. W. Choi, Nano Lett., 2011, 11, 2472–2477 CrossRef CAS PubMed.
- L. Lai, H. Yang, L. Wang, B. K. Teh, J. Zhong, H. Chou, L. Chen, W. Chen, Z. Shen, R. S. Ruoff and J. Lin, ACS Nano, 2012, 6, 5941–5951 CrossRef CAS PubMed.
- L. Zhang, F. Zhang, X. Yang, G. Long, Y. Wu, T. Zhang, K. Leng, Y. Huang, Y. Ma, A. Yu and Y. Chen, Sci. Rep., 2013, 3, 1408 Search PubMed.
- D. Hulicova-Jurcakova, M. Kodama, S. Shiraishi, H. Hatori, Z. H. Zhu and G. Q. Lu, Adv. Funct. Mater., 2009, 19, 1800–1809 CrossRef CAS.
- Y. H. Lee, K. H. Chang and C. C. Hu, J. Power Sources, 2013, 227, 300–308 CrossRef CAS.
- R. Liu, L. Pan, J. Jiang, X. Xi, X. Liu and D. Wu, Sci. Rep., 2016, 6, 21750 CrossRef CAS PubMed.
- D. K. Singh, K. S. Krishna, S. Harish, S. Sampath and M. Eswaramoorthy, Angew. Chem., Int. Ed., 2016, 55, 2032–2036 CrossRef CAS PubMed.
- D. Zhu, Y. Wang, L. Gan, M. Liu, K. Cheng, Y. Zhao, X. Deng and D. Sun, Electrochim. Acta, 2015, 158, 166–174 CrossRef CAS.
- Z. Niu, W. Zhou, J. Chen, G. Feng, H. Li, W. Ma, J. Li, H. Dong, Y. Ren, D. Zhao and S. Xie, Energy Environ. Sci., 2011, 4, 1440 CAS.
- Z. Niu, W. Zhou, X. Chen, J. Chen and S. Xie, Adv. Mater., 2015, 27, 6002–6008 CrossRef CAS PubMed.
- F. Ran, X. Zhang, Y. Liu, K. Shen, X. Niu, Y. Tan, L. Kong, L. Kang, C. Xu and S. Chen, RSC Adv., 2015, 5, 87077–87083 RSC.
- Z. S. Wu, A. Winter, L. Chen, Y. Sun, A. Turchanin, X. Feng and K. Mullen, Adv. Mater., 2012, 24, 5130–5135 CrossRef CAS PubMed.
- K. Yuan, Y. Xu, J. Uihlein, G. Brunklaus, L. Shi, R. Heiderhoff, M. Que, M. Forster, T. Chasse, T. Pichler, T. Riedl, Y. Chen and U. Scherf, Adv. Mater., 2015, 27, 6714–6721 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, FT-IR, XRD, XPS, BET, TGA analysis profiles and additional electrochemical data are included. See DOI: 10.1039/c7ra01203a |
| This journal is © The Royal Society of Chemistry 2017 |