
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe influence of the constituent elements on the corrosion mechanisms of silver alloys in sulphide environments: the case of sterling silver
I. Tissot *a,
O. C. Monteirob,
M. A. Barreirosc,
J. Correiab and
M. F. Guerrad
*a,
O. C. Monteirob,
M. A. Barreirosc,
J. Correiab and
M. F. Guerrad
aLIBPhys – UNL 2829-516 Caparica, Portugal & Faculdade de Ciências, Universidade de Lisboa, Campo Grande, 1749-016, Lisboa, Portugal. E-mail: isabel.tissot@archeofactu.pt
bCentro de Química e Bioquímica, Faculdade de Ciências, Universidade de Lisboa, Campo Grande, 1749-016 Lisboa, Portugal. E-mail: ocmonteiro@fc.ul.pt; jmcorreia@fc.ul.pt
cLaboratório Nacional de Energia e Geologia, LEN/UER, Estrada do Paço do Lumiar, 22, 1649-038 Lisboa, Portugal. E-mail: alexandra.barreiros@lneg.pt
dArchAm – UMR 8096 CNRS, MAE, 21 allée de l’Université, 92023 Nanterre, France. E-mail: maria.guerra@cnrs.fr
First published on 31st May 2017
Abstract
The corrosion of copper and silver in sulphide environments was assessed by several analytical techniques to investigate the influence of each one on the corrosion of sterling silver. The surface colour changes with the immersion time due to the formation of corrosion products composed of particles with distinct sizes and shapes and the consequent layer thickening. Ag2S is the main corrosion product of silver. At early stages of corrosion, Cu develops Cu2O and Cu2S, and later Cu2O again. After the corrosion of sterling silver, pure Cu and Ag are compared, and the predominant influence of Cu at the first stages may be suggested whilst Ag mainly contributes to longer corrosion stages. The layer-by-layer corrosion structure observed for the sulphidation of sterling silver was not observed for its constituent elements.
1. Introduction
When exposed to high sulphide containing atmospheres, cultural heritage silver objects corrode.1–9 To establish adequate conservation treatments, it is fundamental to identify the corrosion products formed at the surface and to fully describe the corrosion mechanisms. Although there is significant research in this area, the influence of the constituent elements of the silver alloy on the corrosion mechanism is far away from being clearly identified.10–14 In previous work, the corrosion of sterling silver (92.5 wt% of Ag and 7.5 wt% of Cu), one of the main silver alloys used in jewellery making,4,15,16 in sulphide environments was investigated.10 In this study, it was suggested that the surface colour variation (from yellow to violet, blue grey and dark grey) is related to both the formation of distinct corrosion products and the corrosion layer thickness. At the early stages of corrosion, the corrosion products are mainly composed of Cu compounds and at longer corrosion times by either Ag2S or AgCuS. The corroded surface morphology suggests the development of a layer-by-layer film. The corroded layer structure is the result of overlapped layers with distinct compositions, continuously growing at different rates. The corrosion of Ag and Cu in high sulphide environments has been widely investigated,11,13,17–32 however, the influence of the constituent elements on the corrosion mechanism of sterling silver was, from our knowledge, not studied yet.It was demonstrated that α-Ag2S (acanthite) is the main corrosion product of pure Ag. The formation can occur either by direct conversion of Ag to Ag2S, by an oxidative dissolution of Ag to Ag+ followed by the formation of Ag2S, or by the formation of Ag nanoparticles that are converted into Ag2S.6–11 These different corrosion mechanisms are dependent on the environment characteristics. The colour of the corroded surfaces depends on the size and shape of the Ag2S particles.33 The oxidation of Ag and Ag2S may lead to the formation of silver oxides (AgO and Ag2O), but it has also been reported the formation of Ag2S over a Ag2O film layer.34
Copper sulphides are the resulting compounds of pure Cu corrosion in sulphide environments. There are several stoichiometric phases of copper sulphides, from copper-rich Cu2S (chalcocite) to copper-poor CuS (covellite) phases.35,36 The corrosion mechanism involves the nucleation and growth of CuxS particles at the surface. Thicker films are composed of copper-rich phases whereas thinner films are constituted by copper-poor phases.35 Depending on the relative humidity level, a first layer of Cu2O is formed on the Cu surface. The CuxS growths over this oxide layer, firstly following a linear rate and then, when a minimum thickness is reached, following a parabolic rate.37–40 The colour of the corroded copper surfaces depends on the Cu2S layer thickness.27 However, apart from studies on natural patinas, the colour variation of the copper corroded surface is rarely concerned in sulphidation mechanism investigations.41,42
The overlapped layers with distinct compositions of the silver-copper alloys corroded layer structure10 should depend on the pure elements (Ag and Cu) corrosion mechanisms. The present work is a continuation of the study on corrosion of silver alloy in sulphide environments10 and aims to compare the corrosion mechanisms of sterling silver with those of its constituent metals. The colour of the corroded layers depending on the composition and thickness of the corrosion layers, and the contribution of each pure element to the colour variation of the corroded silver alloy were also investigated.
Samples of pure Ag and Cu employed by jewellers to fabricate sterling silver were corroded in a sulphide environment and the surfaces characterised using the analytical strategy applied for the previous study of sterling silver corrosion by Tissot et al.10 The surfaces were optically characterised by ultraviolet-visible (UV-Vis) spectrophotometry, the surface morphology investigated by scanning electron microscopy with a field emission gun (SEM-FEG) and the surface elemental composition achieved by energy dispersive X-ray spectrometry (EDS) and by X-ray diffraction (XRD). The corrosion thickness was determined by ellipsometry.
2. Material and methods
2.1 Silver and copper samples and accelerated ageing test conditions
Sheets of Cu and Ag were acquired from a local jewellery supplier (Lima & Teixeira, Lisbon), to test the pure metals employed by jewellers to fabricate sterling silver. A set of 11 samples of 1.5 cm2 area and 1 mm thickness were cut from the sheets and then abraded with SiC-paper with grits up to 1000 mesh, using the method described in Tissot et al.10 To remove grinding residues, the samples were washed with deionised water in an ultrasonic bath.The Ag and Cu samples were corroded by immersion in a 0.1 M Na2S aqueous solution during the same immersion time as the sterling silver samples published in Tissot et al.10: 1, 3, 5, 7, 15, 30, 60, 120, 240, 480 and 1020 minutes.
2.2 Surface characterisation
The analytical protocol described in Tissot et al.10 was applied to characterise the Ag and Cu samples surface. The methods and the analytical techniques are briefly summarised below.The optical characterisation was carried out by UV-Vis spectrophotometry using a Shimadzu UV-2450PC spectrometer. The diffuse reflectance spectra (DRS) were recorded with a detecting wavelength ranging from 220 to 1400 nm, using bare Ag and bare Cu, prepared with the same surface finishing of the samples, as reference to the baselines. Absorption data (α/Λ) were calculated from reflectance data with Kubelka−Munk equations: α/Λ = (1 − R)2/(2R), where R is the reflectance and α and Λ are the absorption and scattering coefficients, respectively.
Surface morphology was observed under a SEM-EDS Philips XL 30 FEG model operated with acceleration voltage from 10 to 25 kV. Semi-quantitative elemental composition was obtained with an EDS (EDAX) system equipped with a Si(Li) detector with a 3 μm super ultra-thin window (SUTW), allowing detection of light elements. The X-ray spectra were collected in spot mode analysis for 300 s acquisition time.
Crystalline corrosion products were identified by XRD using a Philips X-ray diffractometer (PW 1730) with automatic data acquisition (APD Philips v3.6B) with a Cu Kα radiation working at 40 kV/30 mA. Data were collected with a 0.020 step size and an acquisition time of 200 s deg−1. A selection of different ranges of 2θ was made to avoid the Ag and Cu diffraction peaks. Powder X-ray diffraction files were obtained from the Pearson's Crystal Database (PCD).
The corrosion layer thickness was estimated by ex situ ellipsometric measurements using a Sentech SE400 Ellipsometer operating in PSA mode at a constant wavelength of 632.8 nm. The ellipsometric parameters azimuthal angle (Ψ) and phase shift (Δ), retrieved from the change of polarization state of the light beam upon reflection on the sample, were measured at two distinct angles of incidence (φ = 65° and 70° for Ag and φ = 60° and 70° for Cu). Due to the significant light absorption and increasing thickness of the corrosion layer with time, it was only possible to acquire the ellipsometric data for the Ag samples immersed until 30 min and for the Cu samples immersed until 60 min. For longer immersion times, the reflected radiation intensity was too low to be analysed, like in the case of sterling silver.10
3. Results and discussion
3.1 Corroded silver surfaces characterisation
Artificially aged Ag surfaces present colour variations that depend on the immersion time. Fig. 1 shows this colour variation that goes from light brown (1 and 3 min) to blue (5 and 7 min), blue-grey (15, 30, 60 and 120 min) and grey (240 min onwards). Contrary to what has been suggested,33 the colours of the corroded Ag surfaces are different from those reported for corroded sterling silver. The latter, under the same conditions, are characterised by a colour variation with different tones of yellow, red, violet, blue and grey.10 | ||
| Fig. 1 Surface colour of the corroded Ag samples immersed during 1, 3, 5, 7, 15, 30, 60, 120, 240, 480 and 1020 min in a 0.1 M Na2S aqueous solution. | ||
Fig. 2A presents the UV-Vis absorption spectra for the samples immersed during 1, 3, 5 and 7 min. All these samples show an absorption band increase around 370 nm, that can be ascribe to the presence of Ag nanoparticles.43 The sample immersed during 1 min has a broad band with a maximum absorption at 484 nm, which can be attributed to the nucleation of amorphous Ag2S with a particle size inferior to 10 nm.44 The sample immersed during 3 min has a well-defined absorption band with a maximum at 528 nm, corresponding to an estimated band gap energy of 1.2 eV, value reported for both Ag2O and Ag2S band gap energies.45,46
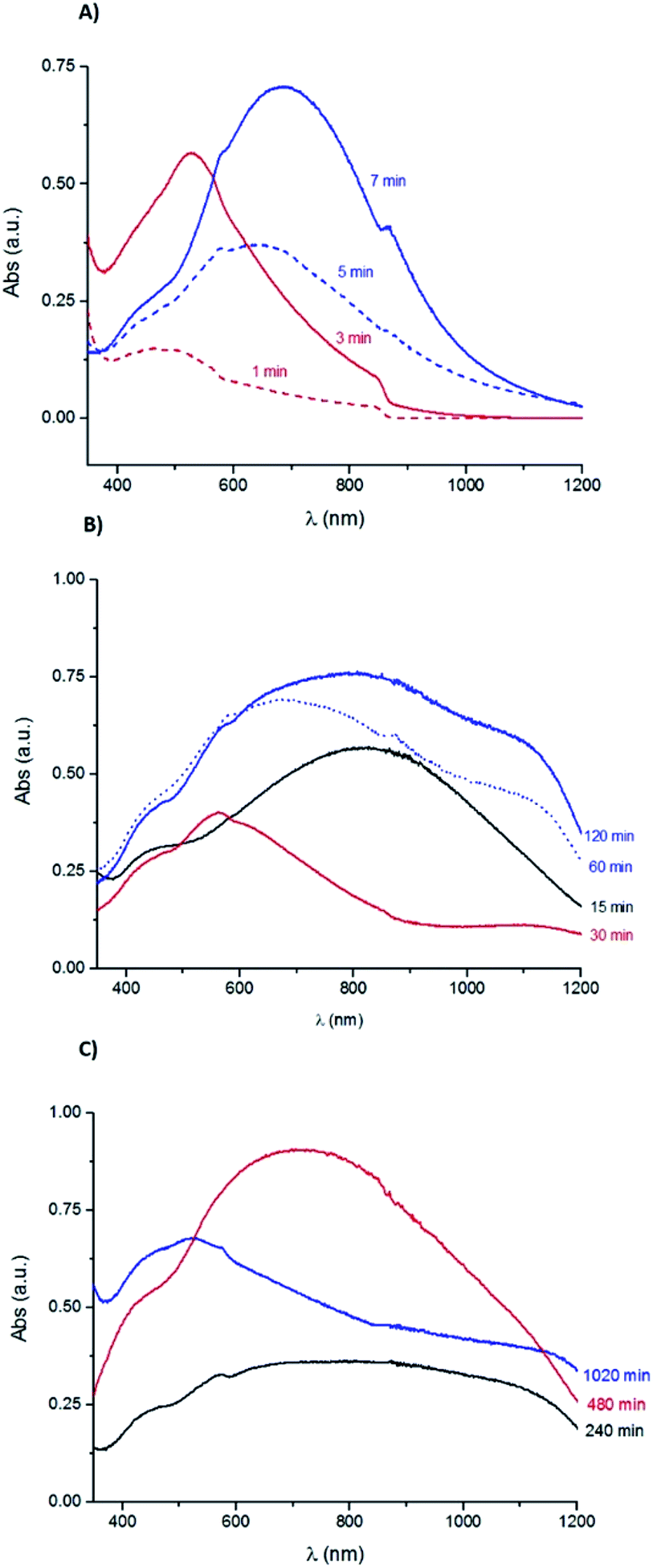 | ||
| Fig. 2 UV-Vis absorption spectra obtained for the Ag samples corroded during (A) 1, 3, 5 and 7 min; (B) 15, 30, 60 and 120 min; (C) 240, 480 and 1020 min in a 0.1 M Na2S aqueous solution. | ||
As shown in Fig. 2B, the UV-Vis absorption spectra of the samples with immersion time longer than 15 min have a broad band between 370 and 480 nm, similar to the one observed for the sample immersed during 1 min. The UV-Vis spectra of samples immersed during 5, 7 and 15 minutes show another band which broadens with the immersion time increase and shifts towards higher wavelengths with a maximum absorbance at 646 nm (5 min), 689 nm (7 min) and 820 nm (15 min), respectively. This band can be ascribed to the formation of Ag2S particles, which size increases with the immersion time.44 The band energy gap estimated for the 7 min sample is 1.1 eV that could correspond to the presence of Ag2S particles.47
The UV-Vis spectra of the samples immersed during 60 and 120 minutes show a slight bump at 566 nm followed by a wide band with a maximum absorbance at 679 nm (60 min) and at 839 nm (120 min). At higher wavelengths, it is observed a slight increase with a maximum absorbance at 1141 nm (60 min) and at 1123 nm (120 min). Fig. 2C shows the data obtained for the samples with immersion times of 240, 480 and 1020 minutes. The absorption spectrum of the 240 min sample has a small bump at 569 nm followed by a wide band with an absorbance decrease at around 1000 nm. The sample immersed during 480 min has a wide band with a maximum of absorbance at 708 nm. The 1020 min sample has a broad band with a slight bump at 575 nm followed by an absorbance decrease and then by another accentuated decrease at 1150 nm. The bands broadening can be related to the corrosion layer increase.
The UV-Vis absorption data suggest the coexistence of Ag2S and/or Ag2O particles with distinct sizes. For the samples immersed during short periods of time (1 to 3 minutes) there is a possible nucleation of small particles that increase in size with the immersion time, followed by the nucleation of new particles in a continuous corrosion process.
The surface morphology and the elemental composition of the compounds formed during corrosion were investigated by SEM-EDS. Fig. 3A shows the bare Ag surface before accelerated ageing. The grid marks are visible on the surfaces of samples immersed until 120 min. The surface of the samples immersed during 1, 3 and 5 minutes are characterised by the presence of small near-spherical particles (Fig. 3C). The 7 and 15 minutes corroded surfaces present a uniform layer, composed of spherical particles which sizes increase with time. The layer has localised hollows resulting from the sulphidation process.47 The corrosion layer of the sample immersed during 30 min, Fig. 3F, that appears thicker contains particles with well-defined boundaries. From 240 min onwards, the shape of the formed structures resemble crystallites. Fig. 3H shows Ag protrusions caused by the electron beam acceleration voltage as reported by Morales-Masis et al.48
 | ||
| Fig. 3 SEM-secondary electron (SE) images of (A) Ag and corroded Ag by immersion in a 0.1 M Na2S aqueous solution at: (B) 3 min, (C) 5 min, (D) 7 min, (E) 15 min, (F) 30 min, (G) 60 min, (H) 240 min and (I) 1020 min (scale bar is 2 μm for (A to H) and 5 μm for (I)). | ||
Table 1 summarises the EDS data obtained for the Ag corroded samples. With the immersion time increase, the Ag content decreases and the S and O values increase. The increase of S content is more pronounced than the increase of O. Keast et al.11 attribute the increase of S to an Ag2S corrosion layer thickening. For samples immersed more than 120 min, the Ag/S ratio in at% is consistent with the presence of Ag2S, based on the stoichiometry of this compound.
| Sample ID | Composition in wt% | ||
|---|---|---|---|
| Ag | S | O | |
| Lα | Kα | Kα | |
| 1 min | 97.7 | 1.5 | 0.8 |
| 3 min | 97.7 | 1.2 | 1.2 |
| 5 min | 97.6 | 1.5 | 0.6 |
| 7 min | 97.3 | 1.5 | 0.9 |
| 15 min | 97.8 | 1.5 | 0.7 |
| 30 min | 96.7 | 3.0 | 0.3 |
| 60 min | 91.2 | 7.2 | 1.6 |
| 120 min | 88.2 | 10.7 | 1.2 |
| 240 min | 88.3 | 10.4 | 1.3 |
| 480 min | 87.2 | 11.5 | 1.3 |
| 1020 min | 86.9 | 11.8 | 1.3 |
XRD analysis allowed to identify α-Ag2S for all the samples except for the sample immersed during 1 min. As shown in Fig. 4, the intensity of the different Ag2S diffraction peaks increases with the immersion time, which can be justified either by an increase of the compound quantity or by an increase of the degree of crystallisation of the corrosion products.
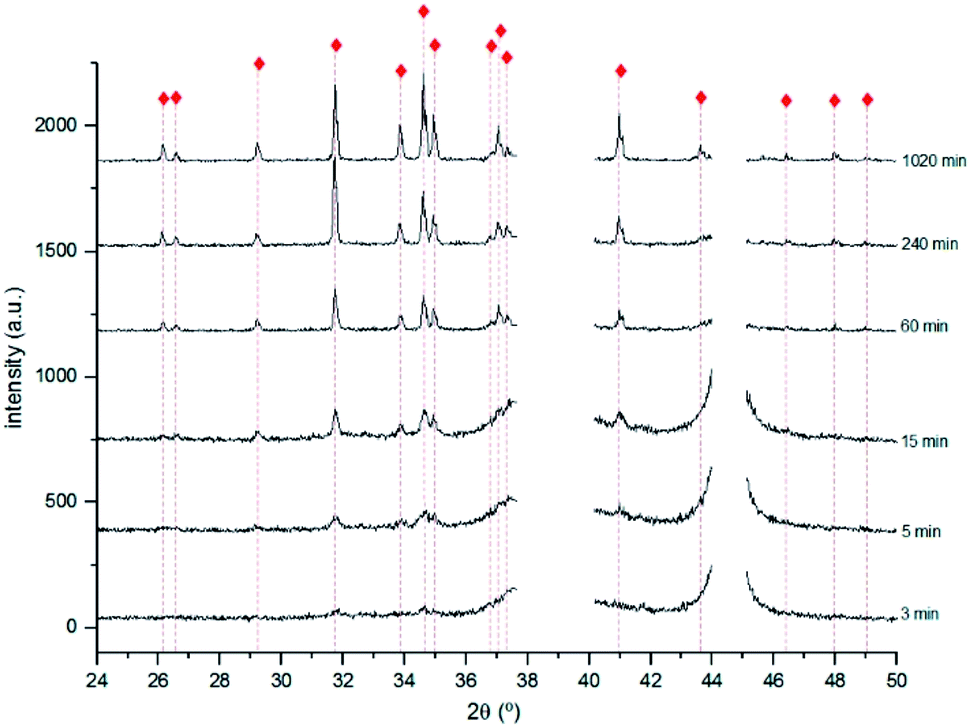 | ||
Fig. 4 X-ray diffractograms obtained for the 3, 5, 15, 60, 240 and 1020 min corroded pure Ag with the identification of the main diffraction peaks of Ag2S  (PCD file no. 00-014-0072). (PCD file no. 00-014-0072). | ||
For immersion time inferior to 240 min, the most intense diffraction peaks are distinct from those reported for α-Ag2S.
Fig. 5 shows the diffractogram obtained for the 240 min sample. The highest intensity peak corresponds to the (103) diffraction plane of Ag2S instead of the expected (121). This difference can be justified by a distinct crystal orientation acquired during the growing process.48 The presence of Ag2O suggested by the UV-Vis spectra obtained for the 3 min sample was not confirmed by XRD. This can be justified either by its absence or presence at too low quantities, or by a non-crystalline structure. Data indicates the formation of Ag2S only.
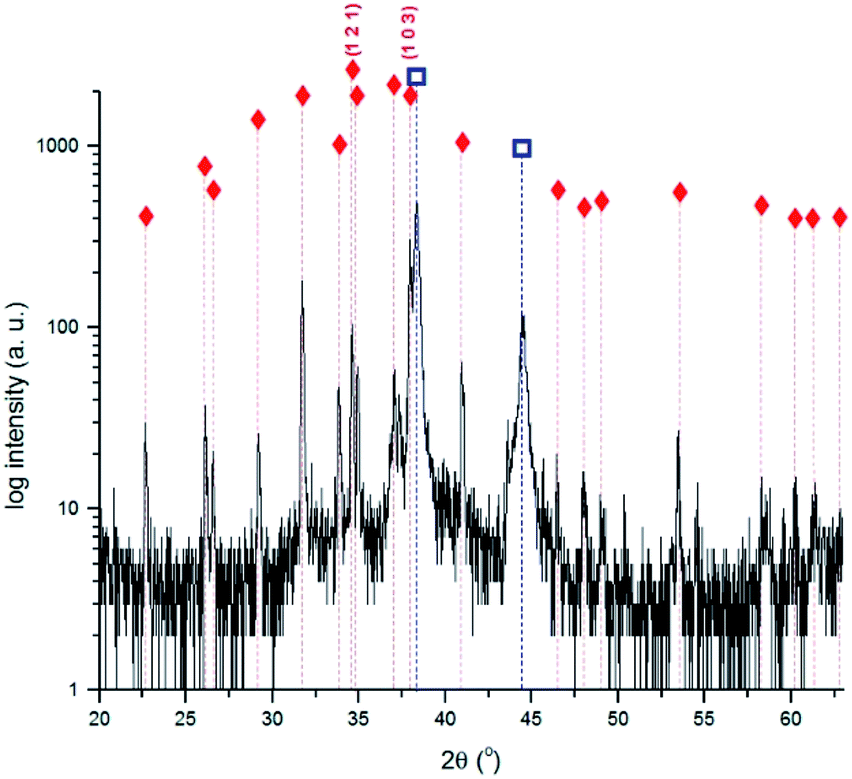 | ||
Fig. 5 X-ray diffractogram obtained for the 240 min corroded Ag sample with the identification of the main diffraction peaks of Ag and Ag2S.  Ag (PCD file no. 00-004-0783); Ag (PCD file no. 00-004-0783);  Ag2S (PCD file no. 00-014-0072). Ag2S (PCD file no. 00-014-0072). | ||
The Ag corrosion layer thicknesses were assessed by ex situ ellipsometry. For each sample, the ellipsometric parameters, azimuthal angle (Ψ) and phase shift (Δ) were measured at 65° and 70° and the data fitted with a two-layer model in a multiangle approach. This model considers one homogeneous and isotropic layer on a semi-infinite homogeneous substrate, and allows the estimation of the thickness of the corrosion layer, being known the optical properties of the substrate. The obtained dielectric constants, refractive index (n) and extinction coefficient (k) are effective values (in the restrict sense of the optical outcome) and not necessarily characteristic of a single compound. Table 2 presents the complex refractive index and the computed thickness of the corrosion films for the samples immersed until 30 min. The thickness values obtained are higher than those reported for Ag2S layer formed in natural environments49,50 and by artificial corrosion.34,51
| Sample ID | ñ | d (nm) |
|---|---|---|
| 1 min | 3.0–0.1i | 118 ± 0.6 |
| 3 min | 3.0–0.1i | 126 ± 1.3 |
| 5 min | 3.0–0.4i | 127 ± 2.6 |
| 7 min | 3.0–0.4i | 129 ± 0.5 |
| 15 min | 3.0–0.4i | 214 ± 0.5 |
| 30 min | 3.0–0.3i | 222 ± 0.4 |
The obtained data indicate that α-Ag2S is, as expected, the main corrosion product of pure Ag immersed in the sulphide solution. The formation of this compound occurs in a continuous process. At the early stages of corrosion, there is the nucleation of small particles of Ag2S that increase in size with the immersion time until they join until a continuous film develops.39 With immersion time increases, the layer becomes uniform, containing crystalline Ag2S. Based on the composition and morphology of the corroded surface, it can be suggested that the surface colour variation is related to the size and shape of the Ag2S particles and to the film thickness.
3.2 Copper corroded surfaces characterisation
Fig. 6 shows the colour variation of the Cu corroded surfaces with the immersion time. Alike the colour variation reported for pure Cu corroded in Na2S solutions,27,42 the surface of samples change from violet (1 min) to blue-green (3 min), green (5 and 7 min), violet (15 min), dark violet (30 min), blue-grey (60 to 240 min) and then grey-black (480 and 1020 min). A violet tone is observed for samples immersed during 1 and 15 minutes whilst those immersed 3, 5 and 7 minutes have green tones. | ||
| Fig. 6 Surface colour of the corroded Cu samples immersed during 1, 3, 5, 7, 15, 30, 60, 120, 240, 480 and 1020 min in a 0.1 M Na2S aqueous solution. | ||
The UV-Vis absorption spectrum shown in Fig. 7, obtained for the sample corroded during 1 min, has a well-defined band. This band has a maximum absorbance at 560 nm corresponding to a 1.78 eV band gap energy, which can be ascribed to the presence of Cu2O.52 For the sample immersed during 3 min, two bands are observed: one in the same wavelength range as in the previous sample, but with lower intensity, and a well-defined band with a maximum at 670 nm. The latter band may correspond to an increase of the particles size.
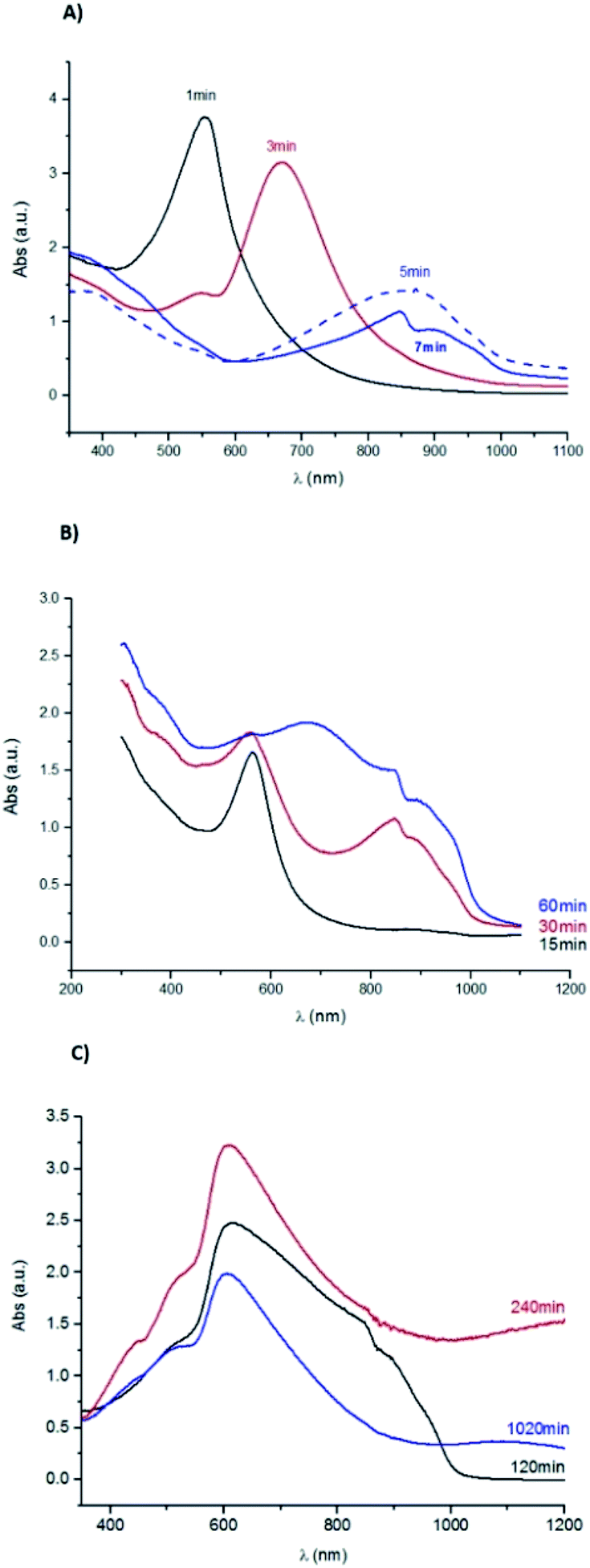 | ||
| Fig. 7 UV-Vis absorption spectra for the Cu samples corroded during (A) 1, 3, 5 and 7 minutes; (B) 15, 30 and 60 min; (C) 120, 240 and 1020 min. | ||
For samples immersed during 5 and 7 minutes, the second band shifts towards higher wavelengths attaining a maximum of absorbance at 856 nm and 848 nm, respectively. These bands correspond to 1.1 eV and 1.2 eV band gap energies, the latter value is reported for the formation of both Cu2S53 and Cu2O particles.54
This band shifts to lower wavelengths for the sample immersed during 15 min. The maximum absorbance reached, around 560 nm, is identical to the one observed in the spectrum of the sample immersed during 1 min. This shift can be related to the nucleation of new particles of distinct size, similar to those formed on the 1 min sample surface. Since the optical properties also depend on the size and shape of the formed particles,53 this result supports the colour similarity cited above. For the samples immersed during 30 min onwards, the bands broadening should be related to the corrosion layer thickening.
SEM observation of the sample immersed during 3 min (Fig. 8B) revealed the presence of a uniform layer composed of small round particles. The layer morphology reminds the one reported for Cu2S films.42 The corrosion layer thickens with the immersion time increase and, from an immersion time of 5 min onwards, it covers the grit marks (Fig. 8C).
 | ||
| Fig. 8 SEM-SE images of (A) pure Cu and corroded Cu by immersion in a 0.1 M Na2S aqueous solution at: (B) 3 min, (C) 5 min, (D) 7 min, (E) 15 min, (F) 30 min, (G) 60 min, (H) 240 min and (I) 1020 min (scale bar is 2 μm for (A to G) and 1 μm for (H and I)). | ||
Round particles of different sizes can be seen in the corrosion layer developed on the sample immersed 15 min (Fig. 8E). For longer immersion times, the particles change to an undefined shape that is characteristic of the layer growth mechanism.55–57 On samples immersed during times longer than 60 min, the particles have either a round or a laminar shape (Fig. 8F–I). The latter can be ascribed to Cu2S.56,57
Table 3 summarises the EDS data obtained for the corroded samples. With the immersion time increase, the S and O contents increase and the Cu content decreases. The accentuated increase of S on the 60 min sample surface can be related to the formation of corrosion products with sulphur. The increase of O is more pronounced in the case of samples immersed during 240 and 1020 min, suggesting the formation of copper oxide.
| Sample ID | Composition in wt% | ||
|---|---|---|---|
| Cu | S | O | |
| Kα | Kα | Kα | |
| 1 min | 97.3 | 1.2 | 1.5 |
| 3 min | 96.7 | 1.5 | 1.8 |
| 5 min | 93.6 | 4.9 | 1.5 |
| 7 min | 94.5 | 3.5 | 2.1 |
| 15 min | 95.1 | 3.2 | 1.6 |
| 30 min | 92.1 | 5.5 | 2.4 |
| 60 min | 82.8 | 14.8 | 2.4 |
| 120 min | 88.4 | 8.0 | 3.6 |
| 240 min | 87.7 | 4.6 | 7.7 |
| 480 min | 86.3 | 10.2 | 3.4 |
| 1020 min | 83.8 | 7.1 | 9.1 |
The presence of Cu2S was identified by XRD for all the samples with immersion times longer than 15 min.
As shown in Fig. 9, Cu2O was identified by XRD for the 120, 240 and 1020 minutes samples suggesting the formation of two different compounds for immersion times longer than 60 min. The highest intensity peaks of Cu2S are different from those reported in the PCD file no. 00-033-0490 showing that it grows with a distinct crystal orientation. The formation of Cu2O could result from the Cu2S oxidation. With the corrosion layer thickening, cracks and defects appear allowing S to react with Cu, to form Cu2S.
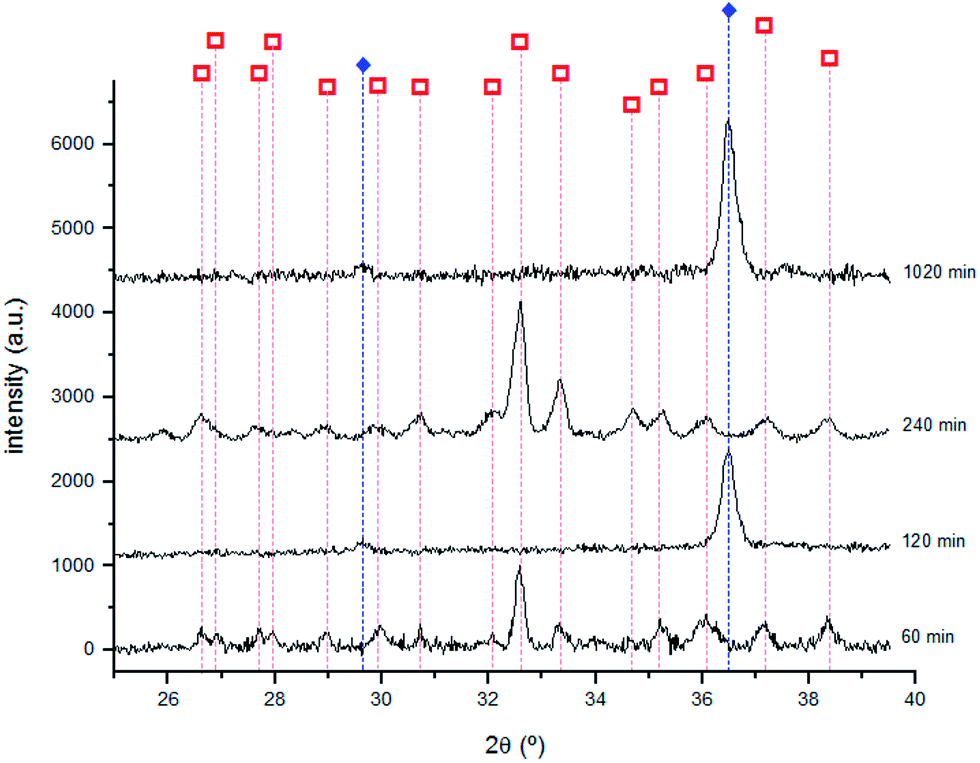 | ||
Fig. 9 X-ray diffractograms obtained for the samples immersed 60, 120, 240 and 1020 min with the identification of main diffraction peaks of Cu2S and Cu2O. Cu2S  (PCD file no. 00-033-0490); Cu2O (PCD file no. 00-033-0490); Cu2O  (PCD file no. 00-077-0199). (PCD file no. 00-077-0199). | ||
The corrosion layer thickness was also assessed for the Cu samples by ex situ ellipsometry using a multiangle analysis (70° and 60°) and the two-layer model. Table 4 presents the complex refractive index and the estimated corrosion film thickness for the samples corroded until 60 min. The values obtained for the sample immersed during 1 min are in accordance with those reported for Cu2S films with violet colour surfaces.42
| Sample ID | ñ | d (nm) |
|---|---|---|
| 1 min | 3.0–0.13i | 129 ± 0.3 |
| 3 min | 3.0–0.11i | 135 ± 0.6 |
| 5 min | 3.0–0.11i | 209 ± 0.5 |
| 7 min | 3.0–0.11i | 300 ± 0.4 |
| 15 min | 3.0–0.11i | 332 ± 0.8 |
| 30 min | 3.0–0.28i | 326 ± 0.6 |
| 60 min | 3.0–0.23i | 340 ± 0.6 |
Other information, such as the corrosion layer structural composition, can be retrieved from the dielectric constants. The composition was estimated by modelling the corrosion film structure using the Bruggeman Effective Medium Approximation (BEMA) theory.58 In this model, the corrosion layer structure is considered as a mixture of known compounds.59 Based on the elemental composition obtained by EDS (Table 3) that identified the presence of S and O, the corrosion layer could be modelled as a combination of Cu2S and Cu2O, using reported n and k values.60,61 For samples corroded until 5 min, a mixture of Cu2O/Cu2S with relative proportions of 85 ± 5/15 ± 5 vol% was found. For longer immersion times (until 60 min), it prevails the Cu2S component with a concentration ranging from 70 to 80 vol%. The latter result is in agreement with the XRD data.
The 15 min sample could not be fitted by the BEMA model. The similar violet surface colour and UV-Vis spectra obtained for the samples corroded during 1 and 15 minutes (Fig. 6 and 7) suggested the presence of similar species at the surface. However, the estimated corrosion layer for the 15 min sample is, as expected, higher than the thickness of the 1 min corrosion layer. For this reason, the existence of a multilayered corrosion film was considered, and data was fitted using the three-layer model. The results of this theoretical model indicate the presence of a 160 nm inner-layer composed of 80 vol% of Cu2O and 20 vol% Cu2S and a 230 nm outer layer composed exclusively of Cu2O. These values are in agreement with the calculated corrosion layer thickness (332 nm by the multiangle ellipsometry approach) and with the UV-Vis data.
The results obtained for the corrosion of Cu in sulphide solution indicate that the process follows the reported copper sulphidation mechanisms.37–40 At early corrosion stages, occurs the formation of Cu2O followed by the formation of Cu2S. In fact, the surface characterisation of the samples immersed until 15 minutes indicates the coexistence of Cu2O and Cu2S. For immersion times between 15 and 60 minutes, it was shown the prevalence of Cu2S, with a layer thickening with the immersion time. For longer immersion times, the Cu2S is interspersed with the formation of Cu2O, possibly due to the oxidation of the Cu2S layer. Like in the case of Ag, the different colours of the corroded surface are a result of the size and shape of the corrosion products particles and of the corrosion layer thickness.
3.3 Ag and Cu corrosion versus sterling silver corrosion
When the colour of corroded pure Ag is compared with the colour of corroded sterling silver, it can be seen that at the early stages of corrosion the colour variation of pure Cu and sterling silver are in appearance alike. For longer immersion times the colour variation of sterling silver approaches that of pure Ag. The corrosion films of pure Ag and Cu mainly contain Ag2S and Cu2S respectively, suggesting that the surface colour variation depends on the corrosion film thickness. The colour variation of sterling silver is instead due not only to the film thickness but also to the formation of different compounds. At the early stages of corrosion, based on the corroded surface composition and on the surface colour variation, we can suggest a significant influence of the Cu on the corrosion mechanism of sterling silver. The formation of Cu compounds on the surface of sterling silver for early stages of corrosion was suggested by Tissot et al.10 Nevertheless, these new data on the pure elements provide evidence on the predominant role of Cu even when this element is present at low concentrations (7.5 wt% Cu in the sterling silver).The morphology of the corroded surfaces of the pure elements and the alloy were compared. In the case of Ag, it was shown the nucleation and growth of the same species, but with sizes and shapes that change with the immersion time. Cu presents identical morphologies until 60 min of immersion time, but for longer times the formation of particles with laminar and round shapes was observed. At the early stages of corrosion, the surface morphology of the corroded sterling silver has similarities with those of Ag and Cu. For longer corrosion times, the morphology of the sterling silver corrosion evidently approaches the morphology of the Ag corrosion. The corrosion layer was found to be thicker for pure elements. When all the samples immersed during 1 min are compared, the estimated corrosion layer thickness is about 90 nm higher for the pure elements than for sterling silver.
Ag is the main constituent element of the sterling silver alloy. It would be expected a major influence of this element on the corrosion mechanisms of the alloy. Nonetheless, data (colour, composition and morphology) suggest that the corrosion mechanism of sterling silver is predominantly influenced by Cu at the early stages of corrosion, and by Ag for longer times. However, it should be highlighted that sterling silver develops a layer-by-layer corrosion structure. This type of structure was not observed for any of the pure metals, suggesting a distinct corrosion mechanism. Fig. 10 summarises this discussion.
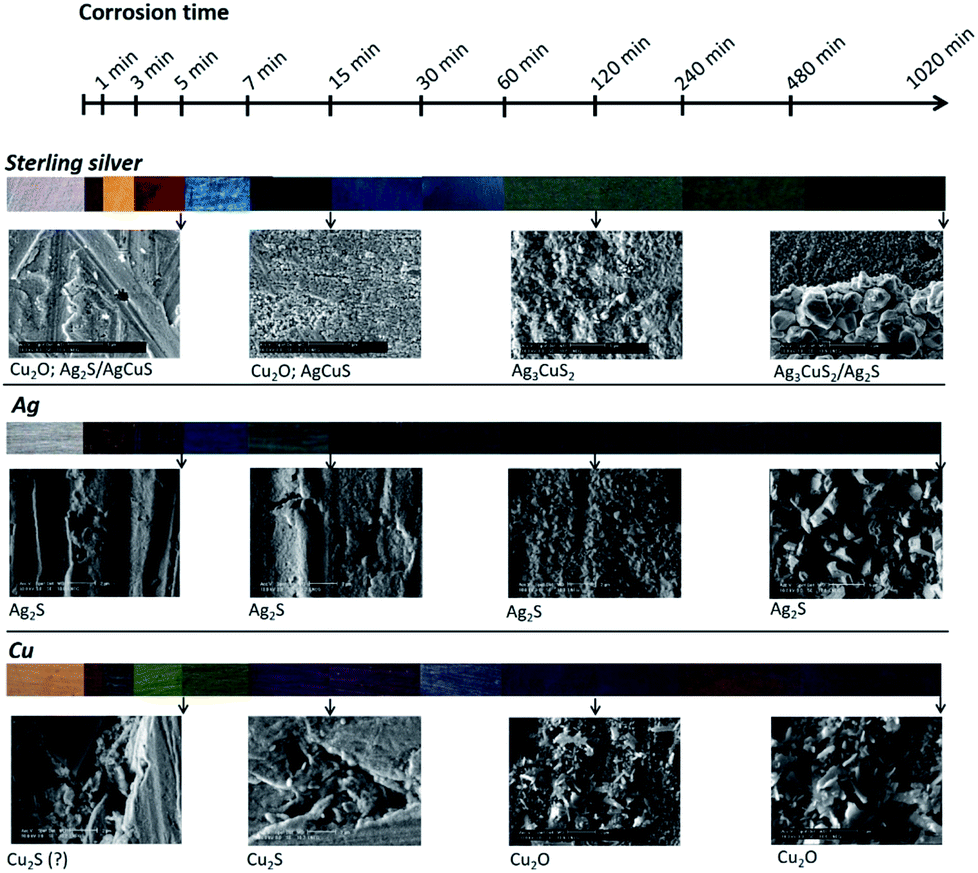 | ||
| Fig. 10 Schema of sterling silver,10 copper and silver surface colour, morphology and composition variation with the corrosion time increase in a 0.1 M Na2S aqueous solution. | ||
4. Conclusions
In this work, sheets of pure Ag and pure Cu used by jewellers to fabricate sterling silver were corroded by immersion in an aqueous Na2S solution. The corroded surfaces were characterised with different analytical techniques. The results obtained for these pure metals were compared to data previously published for sterling silver, and their contribution to the alloy corrosion mechanism was analysed and discussed.α-Ag2S is the main corrosion product of Ag. For early corrosion stages, UV-Vis spectrophotometry and SEM indicated the formation of Ag2S particles of different shapes and sizes, which increase in number and size with the immersion time. For longer immersion times, the corrosion layer becomes uniform and composed of crystalline Ag2S, identified by XRD. The corrosion layer thickness was estimated to about 222 nm for samples immersed until 30 min.
The characterisation by UV-Vis, ellipsometry and XRD of Cu corroded surfaces showed for early corrosion stages the coexistence of Cu2O and Cu2S. For longer immersion times, it was revealed the prevalence of Cu2S, and the surface morphology observed by SEM showed a layer thickening with time. For longer immersion times, Cu2S and Cu2O were identified by XRD, suggesting the Cu2S oxidation. The corrosion layer thickness was estimated to about 340 nm for samples immersed until 60 min.
The different colours of the Ag and Cu corroded surfaces are a result of the distinct size and shape of the corrosion products particles and of the corrosion layer thickness.
Based on the corroded surface composition it is possible to propose that the corrosion mechanisms of Ag and Cu are different from the corrosion mechanism of sterling silver. At the early stages of corrosion, the influence of Cu predominates whilst for longer corrosion times it is Ag that assumes the major role. In addition, at the early corrosion stages the colours of corroded sterling silver and corroded pure Cu are similar, while for longer corrosion times sterling silver acquires a colour close to that of pure Ag. The layer-by-layer structure observed for sterling silver was not observed for the pure metals, and therefore should be characteristic of this alloy corrosion mechanism.
Acknowledgements
This research was partially supported by the Fundação para a Ciência e Tecnologia (SFRH/BDE/51439/2011 and ID/MULTI/00612/2013).References
- T. Palomar, B. R. Barat, E. García and E. Cano, J. Cult. Herit., 2016, 17, 20 CrossRef.
- A. Marquardt, E. M. Breitung, T. Drayman-Weisser, G. Gates and R. J. Phaneuf, Heritage Sci., 2015, 3, 37 CrossRef.
- J. C. D. de Figueiredo Junior, S. S. Asevedo and J. H. R. Barbosa, Appl. Surf. Sci., 2014, 317, 67 CrossRef.
- J. Novakovic, P. Vassiliou and E. Georgiza, Int. J. Electrochem. Sci., 2013, 8, 7223 CAS.
- C. Liang, C. Yang and N. Huang, Surf. Coat. Technol., 2009, 203, 1034 CrossRef CAS.
- H. Kim and J. H. Payer, Corros. Sci. Technol., 2006, 5, 206 Search PubMed.
- M. Bernard, E. Dauvergne, M. Evesque, M. Keddam and H. Takenouti, Corros. Sci., 2005, 47, 663 CrossRef CAS.
- M. Evesque, M. Keddam and H. Takenouti, Electrochim. Acta, 2004, 49, 2937 CrossRef CAS.
- K. Hallet, D. Thickett, D. S. McPhail and R. J. Chater, Appl. Surf. Sci., 2003, 203–204, 789 CrossRef.
- I. Tissot, O. C. Monteiro, M. A. Barreiros, V. Corregidor, J. Correia and M. F. Guerra, RSC Adv., 2016, 6, 51856 RSC.
- V. J. Keast, T. A. Myles, N. Shahcheraghi and M. B. Cortie, J. Nanopart. Res., 2016, 18, 45 CrossRef.
- J. Hirunyagird, G. Lothongkum and E. Nisaratanaporn, Adv. Mater. Res., 2014, 894, 138 CrossRef.
- C. Kleber, R. Wiesinger, J. Schnöller, U. Hilfrich, H. Hutter and M. Schreiner, Corros. Sci., 2008, 50, 1112 CrossRef CAS.
- A. M. Hacke, C. Carr, A. Brown and D. Howell, J. Mater. Sci., 2003, 38, 3307 CrossRef CAS.
- P. Storme, O. Schalm and R. Wiesinger, Heritage Sci., 2015, 3, 25 CrossRef.
- A. Cusma, M. Sebastiani, D. De Felicis, A. Basso and E. Bemporad, Coatings, 2015, 5, 78 CrossRef CAS.
- C. Leygraf, I. O. Wallinder, J. Tidblad and T. Graedel, in The Atmospheric Corrosion, ed. C. Leygraf, I. O. Wallinder, J. Tidblad and T. Graedel, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2016, pp. 337–347 Search PubMed.
- C. E. Sanders, D. Verreault, G. S. Frankel and H. C. Allen, J. Electrochem. Soc., 2015, 162, C630 CrossRef CAS.
- R. Wiesinger, R. Grayburn, M. Dowsett, P.-J. Sabbe, P. Thompson, A. Adriaens and M. Schreiner, J. Anal. At. Spectrom., 2015, 30, 694 RSC.
- P. Homem, PhD thesis, Faculdade de Letras, Universidade do Porto, 2013.
- T. W. H. Oates, M. Losurdo, S. Noda and K. Hinrichs, J. Phys. D: Appl. Phys., 2013, 46, 145308 CrossRef.
- I. Martina, R. Wiesinger and M. Schreiner, J. Raman Spectrosc., 2013, 44, 770 CrossRef CAS.
- R. Wiesinger, I. Martina, C. Kleber and M. Schreiner, Corros. Sci., 2013, 77, 69 CrossRef CAS.
- I. Martina, R. Wiesinger, D. Jembrih-Simbürger and M. Schreiner, e-Preserv. Sci., 2012, 9, 1 CAS.
- N. Li, PhD thesis, Université Pierre et Marie Curie-Paris VI, 2012.
- C. E. Lemon, PhD thesis, Ohio State University, 2012.
- S. Nakayama and T. Umemoto, SEI Tech. Rev., 2012, 74, 8 Search PubMed.
- C. Kebler, U. Hilfrich and M. Schreiner, Appl. Surf. Sci., 2007, 253, 3712 CrossRef.
- J. L. Elechiguerra, L. Larios-Lopez, C. Liu, D. Garcia-Gutierrez, A. Camacho-Bragado and M. J. Yacaman, Chem. Mater., 2005, 17, 6042 CrossRef CAS.
- H. Kim, Mater. Corros., 2003, 54, 243 CrossRef CAS.
- D. W. Rice, P. Peterson, E. B. Rigby, P. B. P. Phipps, R. J. Cappell and R. Tremoureux, J. Electrochem. Soc., 1981, 128, 275 CrossRef CAS.
- V. I. Birss and G. A. Wright, Electrochim. Acta, 1981, 12, 1809 CrossRef.
- T. Baše, Z. Bastl, V. Havránek, K. Lang, J. Bould, M. G. S. Londesborough, J. Macháček and J. Plešek, Surf. Coat. Technol., 2010, 204, 2639 CrossRef.
- Y. B. Amor, E. Sutter, H. Takenouti, B. Tribollet, M. Boinet, R. Faure, J. Balencie and G. Durieu, Electrochim. Acta, 2014, 131, 89 CrossRef CAS.
- I. Carbone, Q. Zhou, B. Vollbrecht, L. Yang, S. Medling, A. Bezryadina, F. Bridges, G. B. Alers, J. T. Norman and T. Kinmen, J. Vac. Sci. Technol., A, 2011, 29, 051505 Search PubMed.
- N. Taniguchi and M. Kawasaki, J. Nucl. Mater., 2008, 379, 154 CrossRef CAS.
- M. Reid, J. Punch, C. Ryan, L. F. Garfias, S. Belochapkine, J. P. Franey, G. E. Derkits Jr and W. D. Reents Jr, J. Electrochem. Soc., 2007, 154, C209 CrossRef CAS.
- J. C. Barbour, J. W. Braithwaite and A. F. Wright, Nucl. Instrum. Methods Phys. Res., Sect. B, 2001, 175, 382 CrossRef.
- T. E. Graedel, J. P. Franey, G. J. Gualtieri, G. W. Kammlott and D. L. Malm, Corros. Sci., 1985, 25, 1163 CrossRef CAS.
- S. P. Sharma, J. Electrochem. Soc., 1980, 127, 21 CrossRef CAS.
- K. P. FitzGerald, J. Nairn, G. Skennerton and A. Atrens, Corros. Sci., 2006, 48, 2480 CrossRef CAS.
- I. Grozdanov and M. Najdoski, J. Solid State Chem., 1995, 114, 469 CrossRef CAS.
- M. Valodkar, S. Modi, A. Pal and S. Thakore, Mater. Res. Bull., 2011, 46, 384 CrossRef CAS.
- R. Chen, N. T. Nuhfer, L. Moussa, H. R. Morris and P. M. Whitmore, Nanotechnology, 2008, 19, 1 Search PubMed.
- X. Wang, S. Li, H. Yu, J. Yu and S. Liu, Chem.–Eur. J., 2011, 17, 7777 CrossRef CAS PubMed.
- S. Kashida, N. Watanabe, T. Hasegawa, H. Iida, M. Mori and S. Savrasov, Solid State Ionics, 2003, 158, 167 CrossRef CAS.
- P. Chattopadhyay and S. Guha Roy, Indian J. Pure Appl. Phys., 2013, 51, 98 CAS.
- M. Morales-Masis, S. J. van der Molen, W. T. Fu, M. B. Hesselberth and J. M. van Ruitenbeek, Nanotechnology, 2009, 20, 095710 CrossRef CAS PubMed.
- H. Gil, C. P. Buitrago and A. Echavarría, J. Solid State Electrochem., 2015, 19, 1817 CrossRef CAS.
- H. Águas, R. J. C. Silva, M. Viegas, L. Pereira, E. Fortunato and R. Martins, Phys. Status Solidi C, 2008, 5, 1215 CrossRef.
- T. Tamai, IEEE Trans. Compon., Hybrids, Manuf. Technol., 1989, 12, 43 CrossRef CAS.
- Z. Wu, M. Shao, W. Zhang and Y. Ni, J. Cryst. Growth, 2003, 260, 490 CrossRef.
- K. Itoh, T. Kuzuya and K. Sumiyama, Mater. Trans., 2006, 47, 1953 CrossRef CAS.
- M. R. Johan, M. S. M. Suan, N. L. Hawari and H. A. Ching, Int. J. Electrochem. Sci., 2011, 6, 6094 CAS.
- M. Ramya and S. Ganesan, Iran. J. Sci. Technol., 2013, 37, 293 Search PubMed.
- T. Martino, R. Partovi-Nia, J. Chen, Z. Qin and D. W. Shoesmith, Electrochim. Acta, 2014, 127, 439 CrossRef CAS.
- J. Chen, Z. Qin and D. W. Shoesmith, Mater. Res. Soc. Symp. Proc., 2012, 1475, 465 CrossRef.
- S. Berthier, Optique des milieux composites, Polytechnica, Paris, 1993 Search PubMed.
- A. Szekeres, S. Bakalova, S. Grigorescu, A. Cziraki, G. Socol, C. Ristoscu and I. N. Mihailescu, Appl. Surf. Sci., 2009, 255, 5271 CrossRef CAS.
- N. K. Abbas and N. J. Ghdeeb, Iraqi Journal of Physics, 2015, 13, 121 Search PubMed.
- Handbook of Optical Constants of Solids, ed. E. D. Palik, Academic Press, Boston, vol. 2, 1991 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |