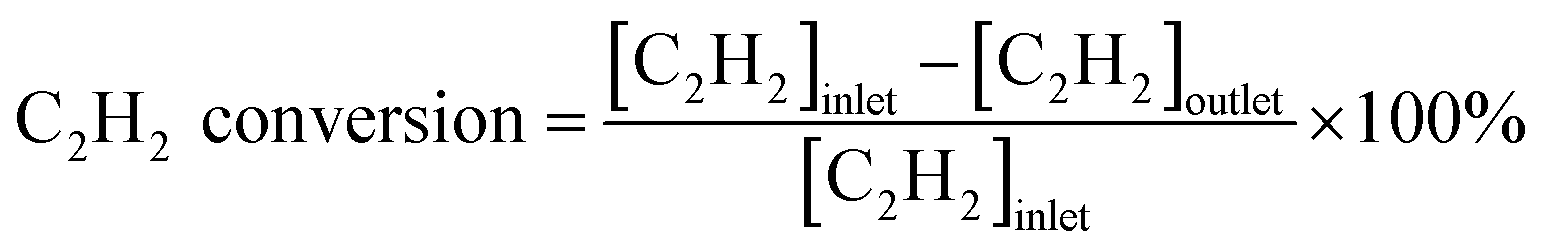DOI:
10.1039/C7RA02266B
(Paper)
RSC Adv., 2017,
7, 18592-18600
Mesoporous Co3O4 supported Pt catalysts for low-temperature oxidation of acetylene
Received
23rd February 2017
, Accepted 22nd March 2017
First published on 28th March 2017
Abstract
Three-dimensionally ordered mesoporous Co3O4 (meso-Co3O4) and its supported Pt catalysts were synthesized for the catalytic oxidation of acetylene. All the catalysts formed mesoporous structures and possessed high surface areas of 110–122 m2 g−1. Meso-Co3O4-supported Pt catalysts exhibited highly catalytic performances, and the 0.6Pt/meso-Co3O4 catalyst gave the lowest temperature of 120 °C for acetylene oxidation. It was concluded that the ordered mesoporous structure, with plenty of structural defects, good low-temperature reducibility and the high concentration of active oxygen species were responsible for the excellent catalytic performance of 0.6Pt/meso-Co3O4.
1. Introduction
Acetylene (C2H2) is mainly emitted from motor vehicle exhausts and petrochemical processing. It enhances the photochemical reaction and promotes the formation of secondary organic aerosols (SOAs) and ozone.1–3 Catalytic oxidation is one of the most effective pathways to eliminate acetylene, in which the key issue is the development of the catalytic materials. However, as far as we know, there are few reports about catalysts for the catalytic oxidation of acetylene. Thus, it is very significant to explore a high performance catalyst for removing acetylene.
Co3O4 shows excellent catalytic performance and is widely used in many reactions, for example, CO oxidation,4 ethylene oxidation,5 formaldehyde oxidation (with6 or without7 noble metal promoters) and the oxidation of other organic molecules.8 It is commonly accepted that the activity of the catalyst is closely related to its morphology and structure. Different structures and morphologies of Co3O4 have been reported, including nano-rod, nano-tube, nano-sheet, nano-sphere and multiporous structures.9,10 Three-dimensional ordered mesoporous metal oxides possess high surface area, large pore volume and controllable pore diameter, which have attracted more and more attentions for their special physicochemical properties. Bai et al.11 prepared nano-Co3O4, 2D-Co3O4, and 3D-Co3O4 catalysts for the catalytic oxidation of formaldehyde. The result showed that 3D-Co3O4 catalyst exhibited the best catalytic performance for formaldehyde oxidation.
Pt particles supported on metal oxides were effective catalysts in the oxidation reaction.12 Zhang et al.13 reported that Pt/TiO2 was the most active catalyst for HCHO oxidation when 1 wt% noble metal (Pt, Rh, Pd and Au) was supported on TiO2. Compared with metal oxide catalysts, oxide-supported Pt catalysts showed higher catalytic activity for the oxidation reaction. Tang et al.14 prepared Pt/MnOx–CeO2 catalysts for the oxidation of formaldehyde. The presence of Pt active sites promoted the adsorption of formaldehyde and the effective activation of oxygen molecule.
Herein, we report the synthesis of meso-Co3O4 and meso-Co3O4-supported Pt catalysts for the oxidation of acetylene. The effects of mesoporous structure, lattice defect, oxygen species and low-temperature reducibility are investigated in terms of acetylene removal.
2. Experimental
2.1. Catalyst preparation
KIT-6 was synthesized according to the strategy reported previously.15 Meso-Co3O4 was prepared using KIT-6 as the hard template. In a typical synthesis process of meso-Co3O4, 1.0 g of KIT-6 was added to a solution of Co(NO3)2·6H2O in ethanol. The samples were evaporated to dryness at 60 °C. The products were calcined at 550 °C for 6 h. The hard template of KIT-6 was removed by NaOH solution (2 mol L−1) at 60 °C. Orderedly meso-Co3O4 was obtained after centrifugation, washing with deionized water and drying at 100 °C. For comparison, the bulk-Co3O4 was prepared via the thermal decomposition of Co(NO3)2·6H2O at 550 °C for 6 h.
The meso-Co3O4 and bulk-Co3O4 supported Pt catalysts were prepared by the means of NaBH4 reduction. In a typical synthesis process, HPtCl4 aqueous solution was mixed with PVP at 35 °C. Then 1 g of meso-Co3O4 or bulk-Co3O4 was added to the solution. The mixed solution was stirred for 4 h. Subsequently, NaBH4 was added under rapidly stirring. The solid was centrifugal separation, washed with deionized water, dried at 100 °C for 8 h and calcined at 350 °C for 4 h. The final catalysts are marked as xPt/meso-Co3O4 or xPt/bulk-Co3O4 (x denotes the weight percentage of Pt element to the related catalyst, x = 0.3, 0.6 and 1.0). The content of Pt were measured by inductively coupled plasma atomic emission spectroscopy (ICP-AES).
2.2. Catalyst characterization
X-ray diffraction (XRD) patterns were tested by a powder meter (Bruker D8 Advance) using Cu Kα radiation (λ = 0.154056 nm), operating at 40 kV and 10 mA. Data were recorded in a 2θ of the low-angle from 0.5 to 3° and wide-angle range of 10–80°. N2 adsorption–desorption isotherms and pore size distributions of the catalysts were tested on an automated gas sorption analyzer (Micrometrics TriStar II 3020). The samples were degassed for 4 h at 300 °C. Transmission electron microscopy (TEM) images were observed using a JEOL JEM 2100 instrument at an accelerating voltage of 200 kV. The catalysts were dispersed in ethanol, and dropped on a copper grid. Scanning electron microscopy (SEM) images measurements were obtained on a SU8010 instruments with the working voltages of 5 kV. Raman spectra were recorded on a Renishaw Laser Raman spectrometer with a laser excitation wavelength of 532 nm under room temperature. The spectra were obtained with resolution of 1 cm−1 and scanned from 100 to 1000 cm−1. X-ray photoelectron spectra (XPS) were recorded on an AxiosmAX spectrometer with an Al Kα X-ray source. The charge-shifted spectra were calibrated using the C 1s photoelectron signal at 284.6 eV. The data were processed by XPS-PEAK software. O2-TPD experiments were tested on a DAS-7000 instrument. 0.05 g of samples were pretreated in the O2 flow (30 mL min−1) at 300 °C for 1 h. The samples were cooled, then the carrier gas changed to pure He. The sample was heated by a flow (30 mL min−1) of pure He from 30 °C to 850 °C at a rate of 10 °C min−1. H2-TPR experiments were also tested on the above instrument. 0.05 g of samples were pretreated in the N2 flow at 300 °C for 1 h. The samples were cooled and reduced by a flow (30 mL min−1) of a 10% H2/90% N2 mixture from 100 to 600 °C with a heating rate of 10 °C min−1. The H2 consumptions of the reduction band were calibrated by a standard CuO (99.998%) powder.
2.3. Catalytic activity test
The catalytic activities of the catalysts were tested in the fixed bed reactor. The samples (100 mg, 40–60 mesh) were placed in the middle of the reaction tube. The concentration of gases was detected by a SP-1000 chromatographic analyzer with a TCD detector. The reaction gas consisted of 50 ppm C2H2, 13 v% O2 and balance gas N2, and the total flow rate of the reactant was 60 mL min−1 (under standard condition). The gas hourly space velocity (GHSV) of the catalytic oxidation was 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. The conversion rate of C2H2 was calculated by the following formula:
000 h−1. The conversion rate of C2H2 was calculated by the following formula:
3. Results and discussion
3.1. Structural and textural analyses
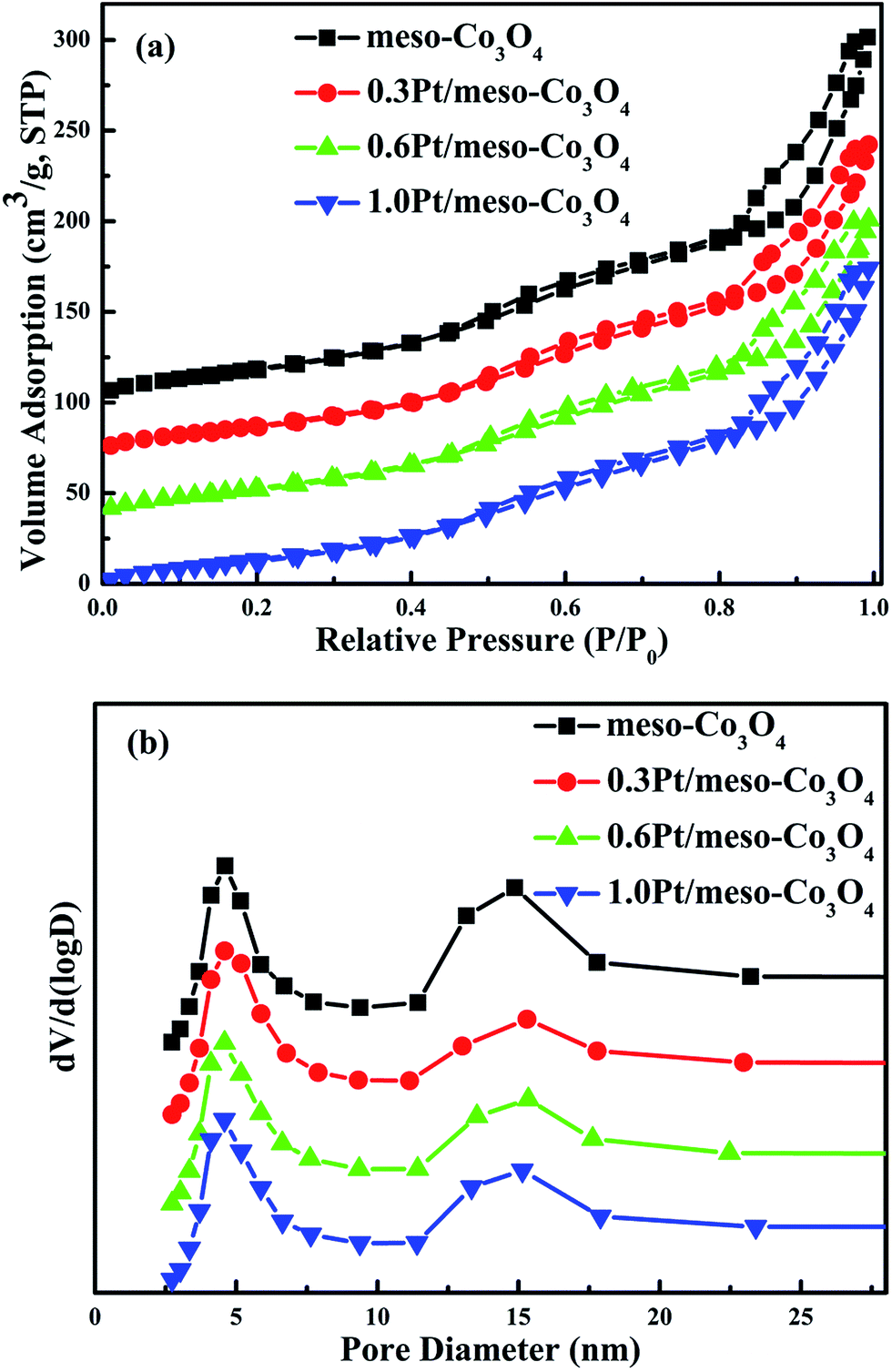
N2 adsorption–desorption isotherms and pore size distributions of the meso-Co3O4 and xPt/meso-Co3O4 are shown in Fig. 1a and b, respectively. All samples show type IV isotherms16 with a H1 hysteresis loop, suggesting the presence of uniform mesopores. It is consistent with the low-angle XRD results (Fig. 2B). BET surface areas, average pore diameters and pore volumes of all samples are displayed in Table 1. All catalysts possess the average pore size of maximum distribution at 5 nm. The broad peak of pore size distribution at 15 nm can be attributed to interspace of the catalysts. The pore diameters, surface areas and pore volumes of xPt/meso-Co3O4 do not significantly reduce. The surface areas of meso-Co3O4 and xPt/meso-Co3O4 (110–122 m2 g−1) are much larger than those of bulk-Co3O4 and 0.6Pt/bulk-Co3O4 (8.1–8.4 m2 g−1). It indicates that most of Pt nanoparticles are dispersed on the surface of the catalysts.
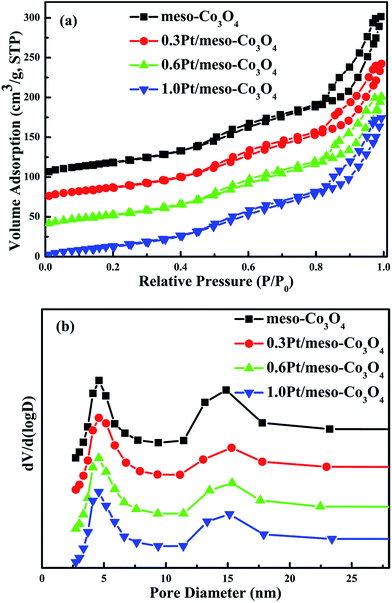 |
| | Fig. 1 (a) N2 adsorption–desorption and (b) pore size distributions of meso-Co3O4, 0.3Pt/meso-Co3O4, 0.6Pt/meso-Co3O4 and 1.0Pt/meso-Co3O4. | |
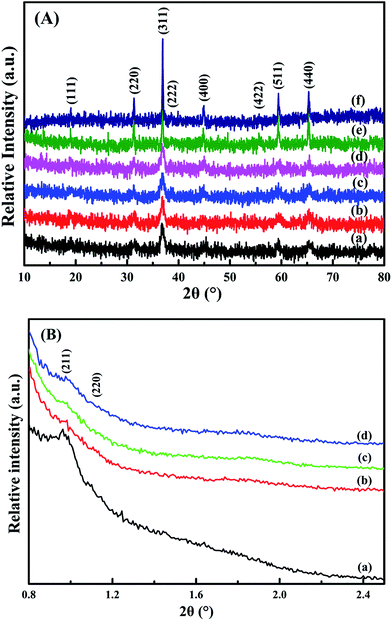 |
| | Fig. 2 (A) Wide-angle and (B) low-angle XRD patterns of (a) meso-Co3O4, (b) 0.3Pt/meso-Co3O4, (c) 0.6Pt/meso-Co3O4, (d) 1.0Pt/meso-Co3O4, (e) bulk-Co3O4 and (f) 0.6Pt/bulk-Co3O4. | |
Table 1 Textural properties of samples
| Sample |
Surface area (m2 g−1) |
Average pore diameter (nm) |
Pore volume (cm3 g−1) |
| Meso-Co3O4 |
122.0 |
8.4 |
0.38 |
| 0.3Pt/meso-Co3O4 |
111.5 |
7.9 |
0.33 |
| 0.6Pt/meso-Co3O4 |
110.9 |
7.8 |
0.32 |
| 1.0Pt/meso-Co3O4 |
110.0 |
8.2 |
0.33 |
| Bulk-Co3O4 |
8.4 |
— |
— |
| 0.6Pt/bulk-Co3O4 |
8.1 |
— |
— |
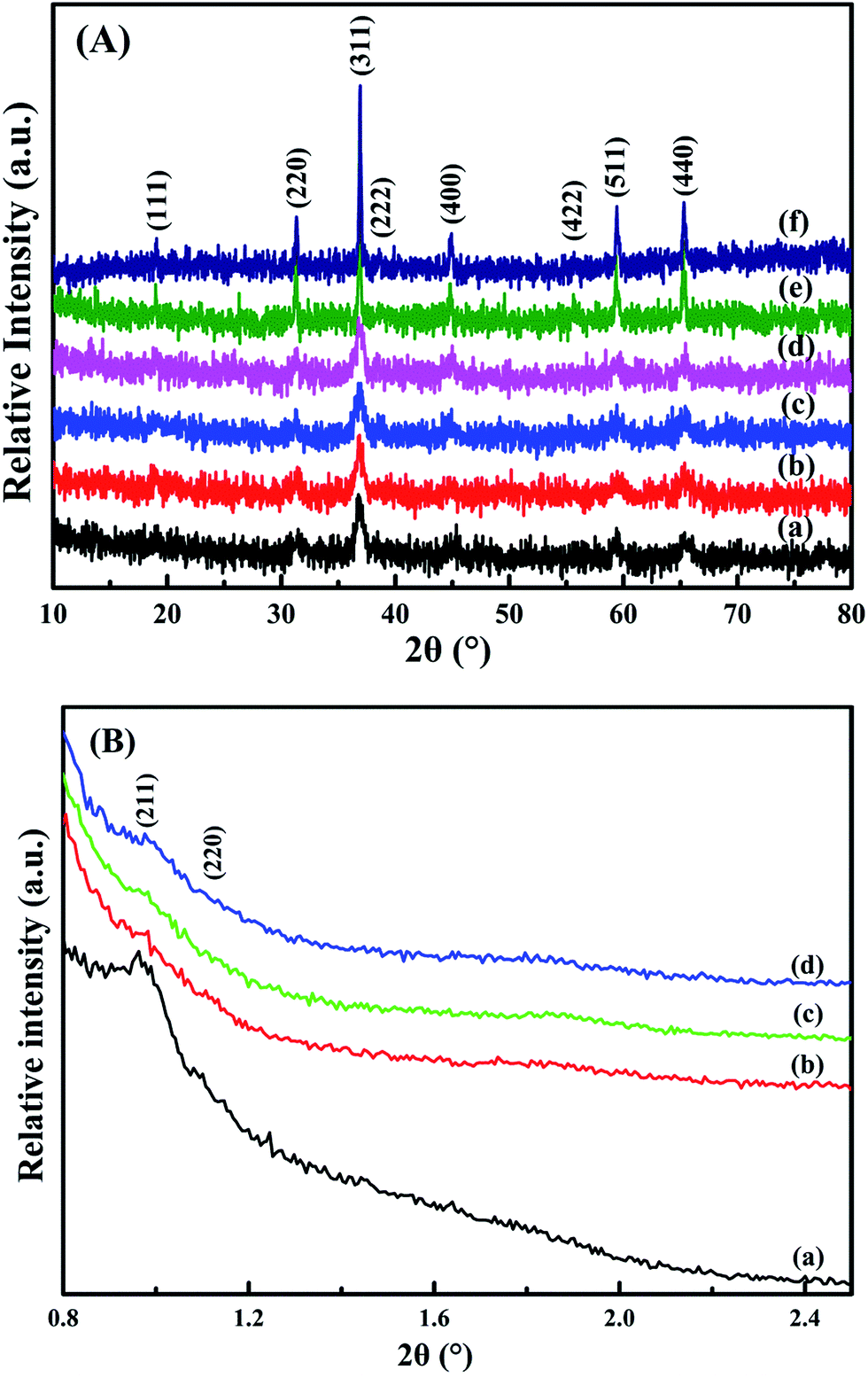
From the wide-angle XRD patterns in Fig. 2A, all the catalysts show diffraction peaks at 19°, 31.3°, 36.9°, 38.2°, 44.5°, 55.6°, 59.4° and 65.3° (2θ), conforming the (111), (220), (311), (222), (400), (422), (511) and (440) planes.17 It is consistent with the standard cobalt oxide spinel structure (JCPDS PDF# 43-1003). The loading of Pt particles rarely affects Co3O4 crystal. Fig. 2B shows the low-angle XRD patterns of the samples. The meso-Co3O4 and xPt/meso-CO3O4 catalysts exhibit (211) and (220) diffraction peaks at 2θ angle 1° and 1.14°.5 It indicates that the catalysts possess a three-dimensional orderedly mesoporous structure. The regularity of the mesoporous structure decreases after Pt loading.
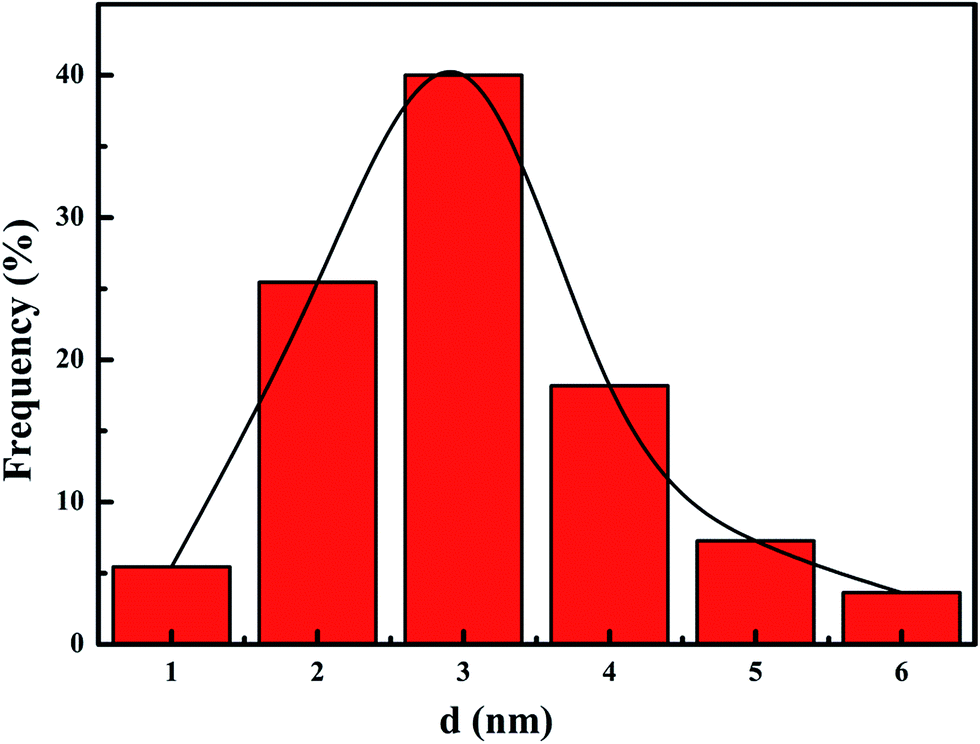
Fig. 3 presents TEM images of the meso-Co3O4 and xPt/meso-Co3O4. All samples display orderedly mesoporous structure. Pt particles are dispersed on the surface of meso-Co3O4. Dispersive Pt particles may provide more active sites for the catalytic oxidation of acetylene. The loading of Pt does not change the mesoporous structure. The orderedly mesoporous structure can facilitate the transport and diffusion of reactant and product molecules. Meanwhile, such a mesoporous structure can prevent Pt particles from leaching and aggregation because of the limitation of the special channels. By making a statistic analysis on the sizes of more than 200 Pt particles in the TEM images of the 1.0Pt/meso-Co3O4 sample, the particle size distribution of Pt particles are shown in Fig. 4. The mean diameter of Pt particles is about 2.9 nm.
 |
| | Fig. 3 TEM images of (a) meso-Co3O4, (b) 0.3Pt/meso-Co3O4, (c and d) 0.6Pt/meso-Co3O4 and (e and f) 1.0Pt/meso-Co3O4. | |
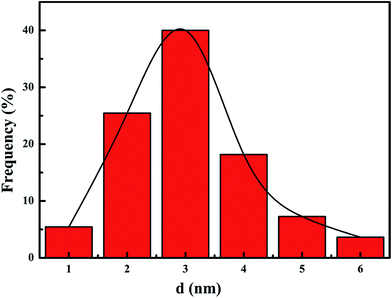 |
| | Fig. 4 Pt particles size distribution of 1.0Pt/meso-Co3O4. | |
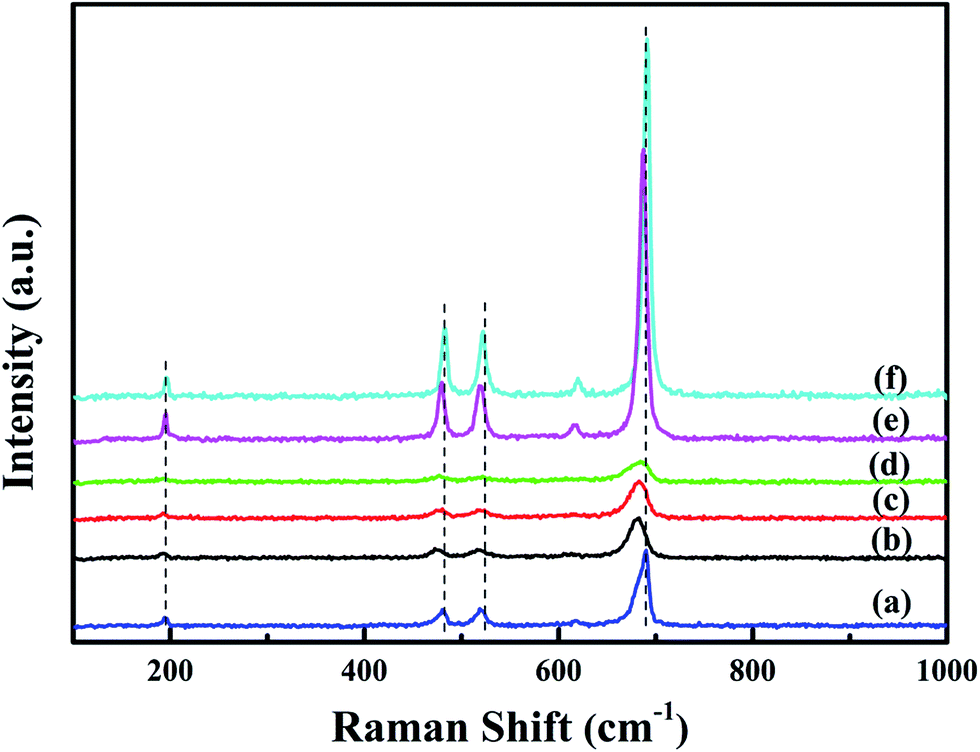
Raman spectra of all the samples are shown in Fig. 5. Raman peaks at 195, 480, 518, 615 and 689 cm−1 correspond to the F2g(1), Eg, F2g(2), F2g(3) and A1g symmetries of crystalline Co3O4, respectively.18 The peak at 689 cm−1 can be ascribed to Raman vibration of Co3+–O2−.7 The overall enhancement of peak intensity in bulk-Co3O4 and 0.6Pt/bulk-Co3O4 samples should be due to the influence of crystallinity and the orientation effect during the Raman test. With the loading of Pt, Raman peak at 689 cm−1 shows an obvious shift to lower frequencies and the peak intensity decreases. The red shift indicates that xPt/meso-Co3O4 catalysts have a lattice defect or residual stress of the spinel structure.19 The existence of lattice defect is beneficial for the formation of the oxygen vacancies.20 The oxygen vacancies can activate, adsorb oxygen and provide the lattice sites of oxygen migration, resulting in the formation of highly active electrophilic oxygen species.21 The decrease of the peak intensities is due to the decrease of Co3+. It can be concluded that there is an electron transfer from Pt0 to Co3+.
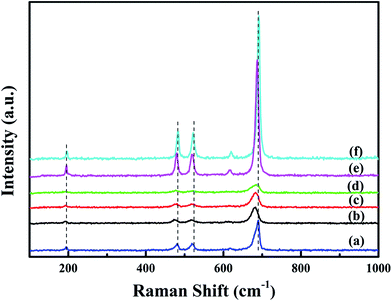 |
| | Fig. 5 Raman spectra of (a) meso-Co3O4, (b) 0.3Pt/meso-Co3O4, (c) 0.6Pt/meso-Co3O4, (d) 1.0Pt/meso-Co3O4, (e) 0.6Pt/bulk-Co3O4 and (f) bulk-Co3O4. | |
3.2. The results of XPS
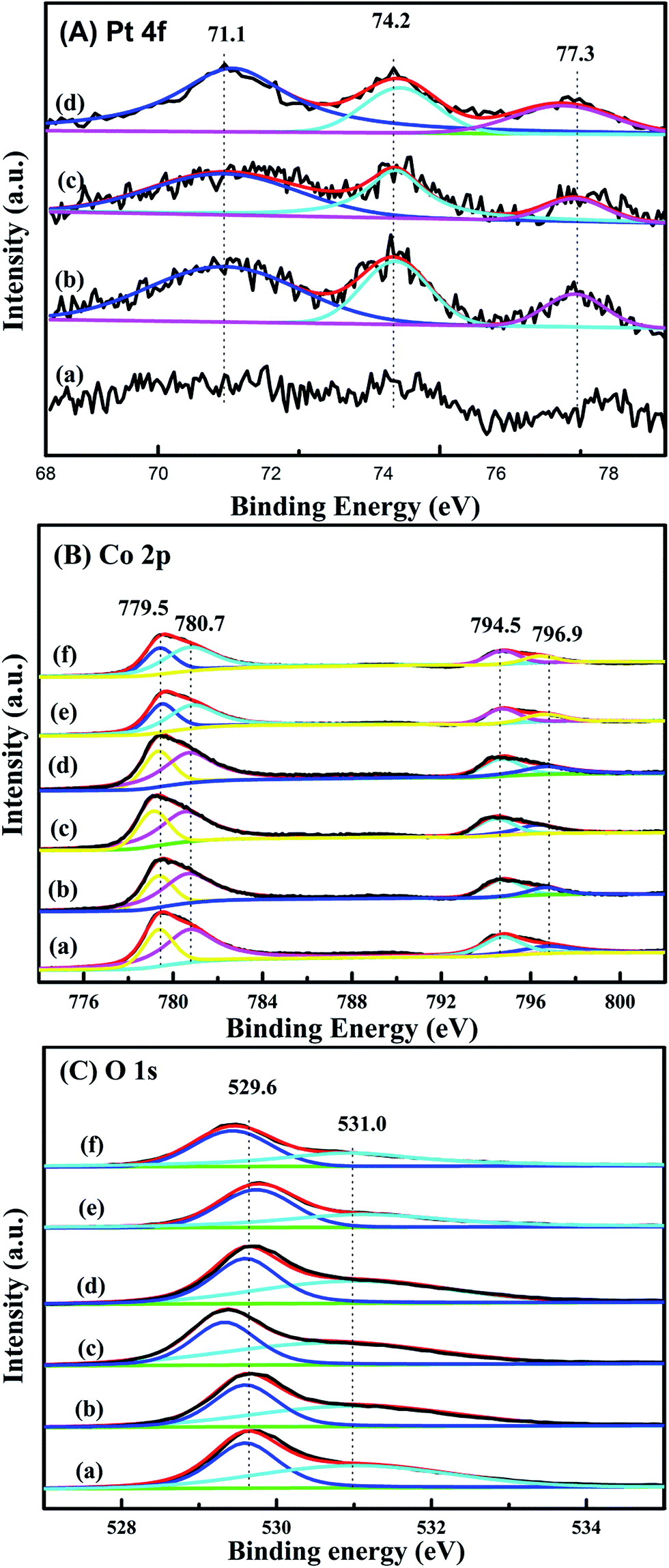
XPS is performed in order to gain the information of surface element compositions, chemical states and adsorbed oxygen species of catalysts. Pt 4f XPS spectra of xPt/meso-Co3O4 and 0.6Pt/bulk-Co3O4 are shown in Fig. 6A. The binding energies of 71.1, 74.2 and 77.3 eV can be assigned to Pt0, Pt2+ and Pt4+ species, respectively.22 This result indicates that both Pt0 and Ptδ+ (Pt2+ and Pt4+) species exist on the surface of Co3O4 support. Ptδ+ species is generally active species rather than metallic Pt in oxidation reaction.23 Table 2 lists the relative amounts of Pt0, Pt2+ and Pt4+ species on the surface of 0.6Pt/meso-Co3O4, 1.0Pt/meso-Co3O4 and 0.6Pt/bulk-Co3O4 by the means of XPS-peak-differentiation-imitating analysis. It is hard to calculate the content of the Pt species for 0.3Pt/meso-Co3O4. The Ptδ+ concentrations over 0.6Pt/meso-Co3O4, 1.0Pt/meso-Co3O4 and 0.6Pt/bulk-Co3O4 catalysts are 42.9%, 37.5% and 35.9%. It is attributed to the dispersive Pt particles on the Co3O4 and the formation of Pt–O–Co bond. Therefore, it is concluded that there is an electron transfer from Pt to Co3+ after the loading of Pt particles. It is consistent with the result of Raman characterization. 0.6Pt/meso-Co3O4 possesses higher concentrations of Ptδ+ than 1.0Pt/meso-Co3O4, and thus increases the density of active oxygen species and Co2+ species of 0.6Pt/meso-Co3O4. Meanwhile, 0.6Pt/meso-Co3O4 shows higher concentrations of Ptδ+ in the meso-Co3O4 surface than 0.6Pt/bulk-Co3O4. It should be due to the order mesoporous structure of meso-Co3O4. Co 2p shows four peaks at BE = 779.5, 780.7, 794.5 and 796.9 eV, which corresponds to Co3+ 2p3/2, Co2+ 2p3/2, Co3+ 2p1/2 and Co2+ 2p1/2 (ref. 11) in Fig. 6B. Table 3 lists the surface Co3+/Co2+ molar ratios of the catalysts. The existence of surface oxygen vacancies is further confirmed by the XPS. The surface Co3+/Co2+ molar ratios of bulk-Co3O4 are higher than meso-Co3O4. Compared to the meso-Co3O4 and bulk-Co3O4, the Co3+/Co2+ molar ratios of xPt/meso-Co3O4 and 0.6Pt/bulk-Co3O4 catalysts decrease, indicating that the surface Co2+ concentration increases after Pt loading. xPt/meso-Co3O4 possess a higher oxygen vacancy concentration than meso-Co3O4. It should be due to the electron transfer from Pt to Co3+ through the reaction of Pt0 + Co3+ → Ptδ+ +Co2+. 0.6Pt/meso-Co3O4 gives the minimum value of the surface Co3+/Co2+ molar ratio and the most surface oxygen vacancy. From Fig. 6C, O 1s exhibits two peaks at 529.6 and 531.0 eV. The former can be assigned to the surface lattice oxygen (Olatt), and the latter corresponds to surface adsorbed oxygen (Oads).24 It is widely recognized that the Oads species play an important role in the deep oxidation of the organics.25 Combined with the results from Table 3, the Oads/Olatt molar ratio (0.98) of bulk-Co3O4 is lower than those of meso-Co3O4 (1.20) and of xPt/meso-Co3O4 (1.25–1.44), indicating that meso-Co3O4 possess more adsorbed oxygen species. The highest Oads/Olatt molar ratio of 0.6Pt/meso-Co3O4 is 1.44, and thus 0.6Pt/meso-Co3O4 has the most surface absorbed oxygen species.
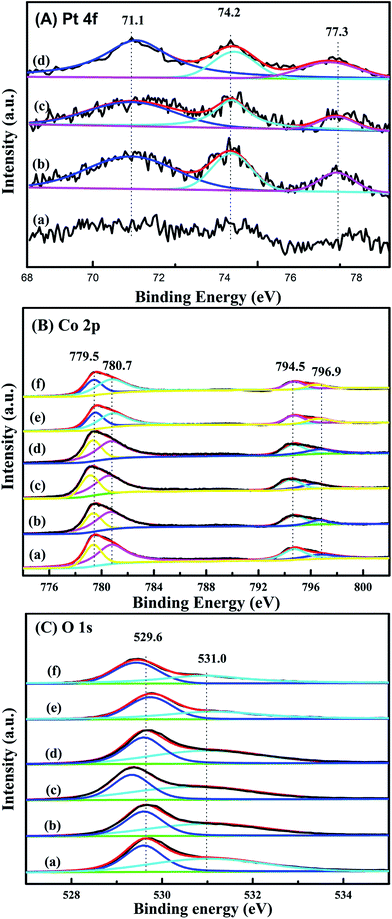 |
| | Fig. 6 (A) Pt 4f XPS spectra of (a) 0.3Pt/meso-Co3O4, (b) 0.6Pt/meso-Co3O4, (c) 1.0Pt/meso-Co3O4 and 0.6Pt/bulk-Co3O4. (B) O 1s and (C) Co 2p XPS spectra of (a) meso-Co3O4, (b) 0.3Pt/meso-Co3O4, (c) 0.6Pt/meso-Co3O4, (d) 1.0Pt/meso-Co3O4, (e) bulk-Co3O4 and (f) 0.6Pt/bulk-Co3O4. | |
Table 2 Surface states of Pt species derived from XPS analyses
| Catalyst |
Pt0 (%) |
Pt2+ (%) |
Pt4+ (%) |
| 0.6Pt/meso-Co3O4 |
57.1 |
29.7 |
13.2 |
| 1.0Pt/meso-Co3O4 |
62.5 |
27.4 |
10.1 |
| 0.6Pt/bulk-Co3O4 |
64.2 |
19.4 |
16.5 |
Table 3 Surface compositions and apparent activation energies of the bulk-Co3O4, 0.6Pt/bulk-Co3O4, meso-Co3O4 and xPt/meso-Co3O4 samples for acetylene oxidation
| Catalyst |
Co3+/Co2+ molar ratio |
Oads/Olatt molar ratio |
H2 consumption (mmol g−1) |
Ea (kJ mol−1) |
| Meso-Co3O4 |
0.69 |
1.20 |
16.1 |
77 |
| 0.3Pt/meso-Co3O4 |
0.65 |
1.31 |
15.4 |
50 |
| 0.6Pt/meso-Co3O4 |
0.60 |
1.44 |
14.6 |
46 |
| 1.0Pt/meso-Co3O4 |
0.66 |
1.25 |
15.6 |
48 |
| Bulk-Co3O4 |
0.83 |
0.98 |
16.9 |
99 |
| 0.6Pt/bulk-Co3O4 |
0.73 |
1.07 |
16.0 |
68 |
3.3. The results of O2-TPD
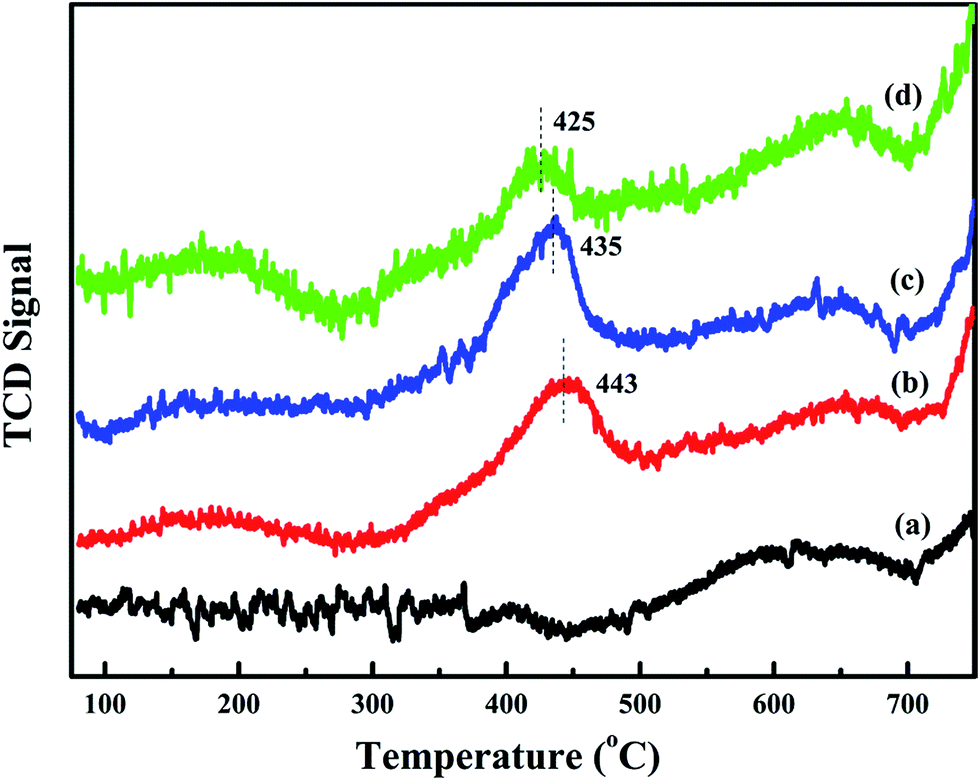
To further study the effect of activated oxygen species for acetylene oxidation, O2-TPD experiments are carried out. The result is shown in Fig. 7. The desorption peak that are less than 450 °C can be ascribed to surface active oxygen, such as O2− and O−, and the desorption temperature of bulk phase lattice oxygen are higher than 700 °C.11 Compared with meso-Co3O4, xPt/meso-Co3O4 presents higher intensities of desorption peaks below 450 °C. This phenomenon indicates that xPt/meso-Co3O4 surface contains more abundant surface active oxygen species than meso-Co3O4. It is in according with the result of XPS. For 0.3Pt/meso-Co3O4, the desorption peak is centered at 443 °C. For 1.0Pt/meso-Co3O4, the desorption peak shifts to lower temperature range, starting at 300 °C and is centered at 425 °C. Among xPt/meso-Co3O4, 0.6Pt/meso-Co3O4 exhibits the lowest beginning oxygen desorption temperature and highest intensities of desorption peak. The desorption temperature and intensity of surface-active oxygen species are connected to the catalytic ability for oxidation reaction. Lower beginning oxygen desorption temperatures and larger intensities of desorption peaks lead to better catalytic ability.11 It indicates that the adding of Pt is beneficial to increasing the surface activity oxygen species of xPt/meso-Co3O4, which can enhance the catalytic performance of acetylene oxidation.
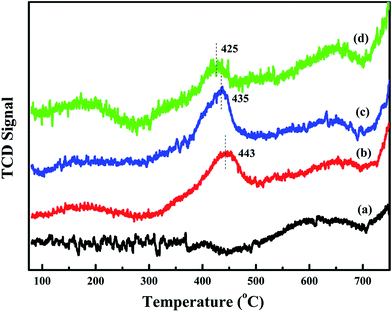 |
| | Fig. 7 O2-TPD profiles of (a) meso-Co3O4, (b) 0.3Pt/meso-Co3O4, (c) 0.6Pt/meso-Co3O4 and (d) 1.0Pt/meso-Co3O4. | |
3.4. The results of H2-TPR
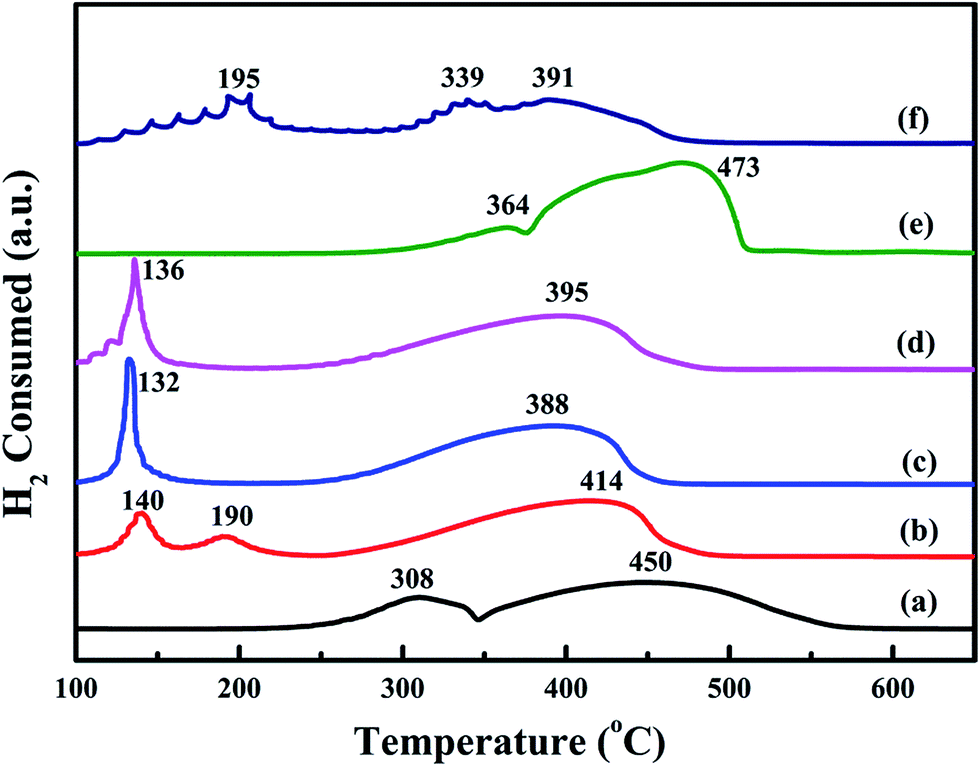
Low-temperature reducibility is an important factor in oxidation reactions. H2-TPR experiments are carried to investigate the reducibility of the samples. The results are shown in Fig. 8. For the bulk-Co3O4, there are two reduction peaks at 364 °C and 473 °C. For the meso-Co3O4, there are two reduction peaks at 308 °C and 450 °C. The former is due to the reduction of Co3O4 to CoO, whereas the latter is attributed to the reduction of CoO to Co0. Comparing the reduction profile of bulk-Co3O4, meso-Co3O4 is more reducible at lower temperatures. Therefore, meso-Co3O4 with high surface area displays a more amount of reducible sites. After the adding of Pt, there is a new reduction peak at 140 °C for 0.3Pt/meso-Co3O4. It should be due to the reduction of Ptδ+. Meanwhile, the reduction peaks of Co3O4 to CoO and CoO to Co0 shift to lower temperatures. For 0.6Pt/meso-Co3O4 and 1.0Pt/meso-Co3O4, the reduction peaks of Ptδ+ to Pt0 and Co3+ to Co2+ are overlapped. 0.6Pt/meso-Co3O4 presents the lowest reduction peak at 132 °C. It indicates that the adding of Pt gives rise to the improvement in low-temperature reducibility of xPt/meso-Co3O4, which can enhance the catalytic performance of acetylene oxidation. Meanwhile, 0.6Pt/meso-Co3O4 displays the highest ability of low-temperature reducibility. By quantitatively analyzing the reduction peaks in H2-TPR profiles, the H2 consumption of the samples are shown in Table 3. The H2 consumption of meso-Co3O4, 0.3Pt/meso-Co3O4, 0.6Pt/meso-Co3O4, 1.0Pt/meso-Co3O4, bulk-Co3O4 and 0.6Pt/bulk-Co3O4 is 16.1, 15.4, 14.6, 15.6, 16.9 and 16.0 mmol g−1, respectively. Due to the presence of the partially overlapping reduction of Co3+, Co2+ and Ptδ+ ions, it is hard to convert the H2 consumption to the Co3+/Co2+ ratios and the content of the Ptδ+. However, it can conclude that there are more amounts of oxygen vacancies in xPt/Co3O4 than in pure Co3O4. It is in good agreement with their Raman and XPS results.
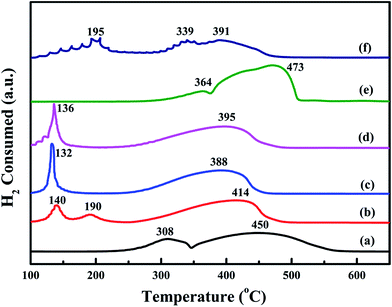 |
| | Fig. 8 H2-TPR profiles of (a) meso-Co3O4, (b) 0.3Pt/meso-Co3O4, (c) 0.6Pt/meso-Co3O4, (d) 1.0Pt/meso-Co3O4, (e) bulk-Co3O4 and (f) 0.6Pt/bulk-Co3O4. | |
3.5. Catalytic performance
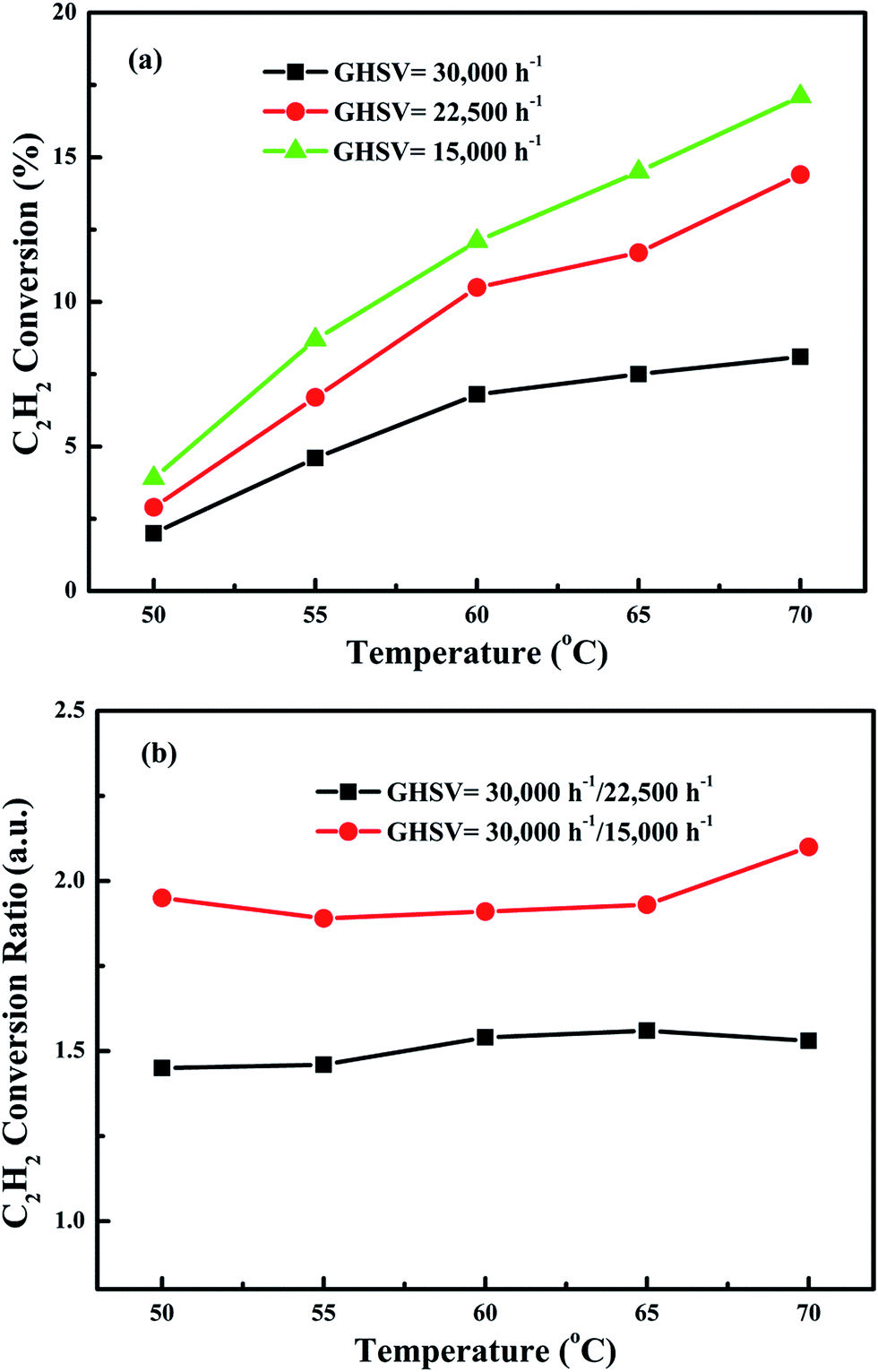
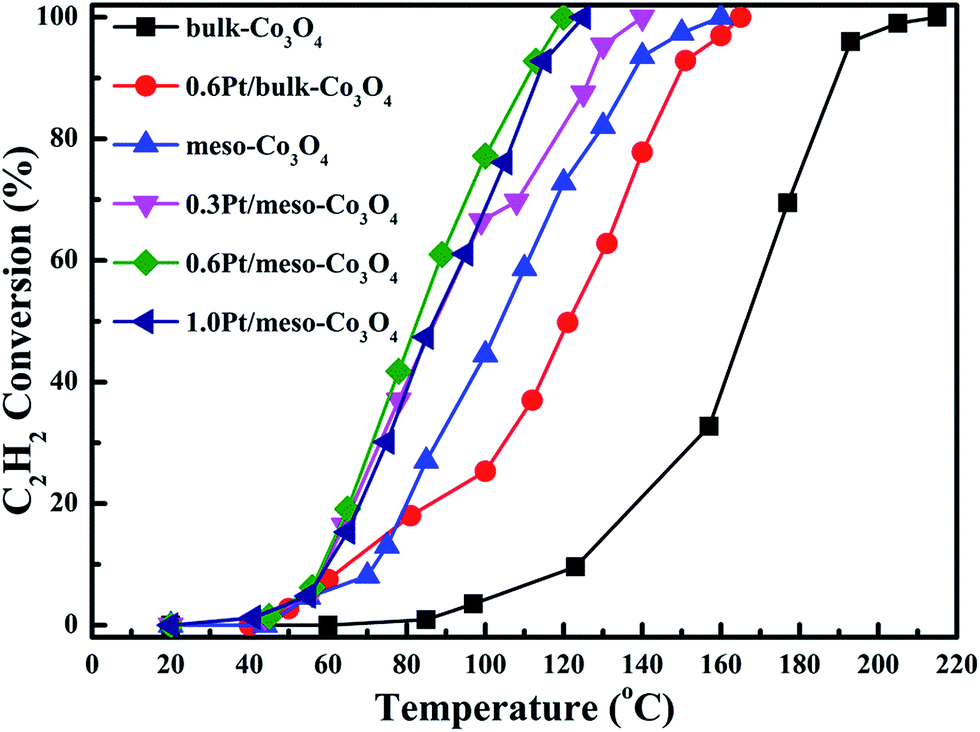
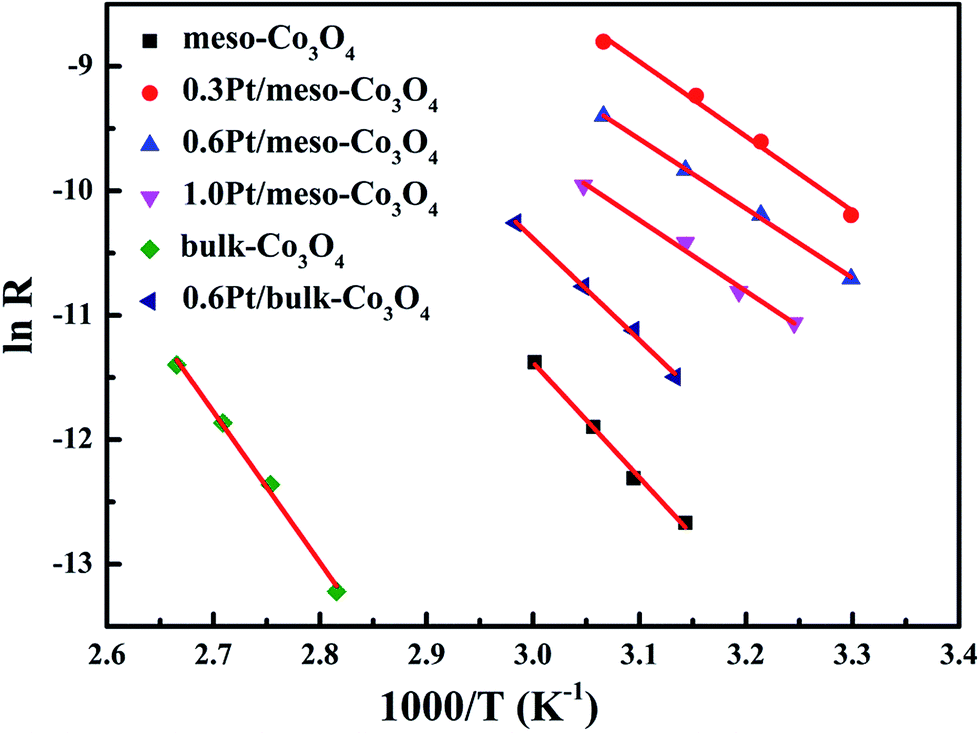
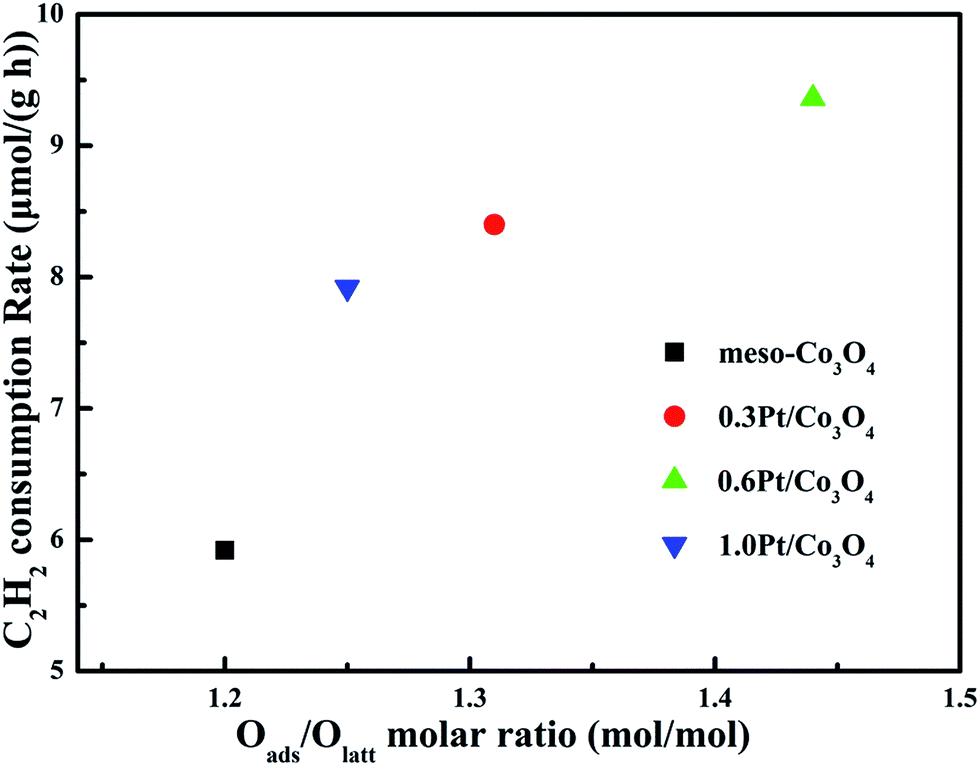
In order to eliminate influence of diffusion, the kinetics research of acetylene oxidation is first carried out. Fig. 9a shows the reaction results for meso-Co3O4 catalyst at various space velocities. In the absence of mass-transfer limitations, acetylene conversion is excepted to increase by n-fold as the space velocity decrease by n-fold. Acetylene conversion ratios (Fig. 9b) suggest that meso-Co3O4 should decrease the influence of diffusion under 30000 h−1 space velocity. Fig. 10 shows the catalytic performances of the catalysts for the oxidation of acetylene. It is evident that the most effective catalyst for acetylene oxidation is 0.6Pt/meso-Co3O4, while the worst active catalyst is bulk-Co3O4. The order of the catalytic activity is 0.6Pt/meso-Co3O4 > 1.0Pt/meso-Co3O4 > 0.3Pt/meso-Co3O4 > meso-Co3O4 > bulk-Co3O4, in good agreement with the reducibility sequences of these catalysts. The bulk-Co3O4 catalyst displays the worst catalytic activity, and the complete conversion temperature of acetylene is at 220 °C. The meso-Co3O4 catalyst shows better oxidation activity, completely converting acetylene at 160 °C. Among xPt/meso-Co3O4 catalysts, 0.6Pt/meso-Co3O4 catalyst exhibits the best catalytic performance, which can completely oxidize acetylene at 120 °C. Fig. 11 shows the Arrhenius plots for acetylene oxidation over the Co3O4 and xPt/Co3O4, and the calculated apparent activation energies (Ea) are summarized in Table 3. It is shown that the Ea value for acetylene oxidation decreased in the sequence of bulk-Co3O4 > meso-Co3O4 > 0.6Pt/bulk-Co3O4 > 0.3Pt/meso-Co3O4 > 1.0Pt/meso-Co3O4 > 0.6Pt/meso-Co3O4. The apparent activation energy of 0.6Pt/meso-Co3O4 is the lowest. The oxidation of acetylene might be more easily over the mesoporous Co3O4 supported Pt catalysts. For the xPt/meso-Co3O4 samples, Pt particles are dispersed on the surface of meso-Co3O4, and more oxygen vacancies are present in xPt/meso-Co3O4. Oads is a beneficial role with respect to catalytic oxidation of acetylene. To further study the effect of Oads in acetylene oxidation. Then, a test is carried out at low conversions under a kinetically controlled regime at 60 °C. As shown in Fig. 12, the acetylene consumption rate increases with the rising of Oads/Olatt molar ratio. That is to say, a higher Oads concentration is beneficial for the enhancement in catalytic oxidation of acetylene.
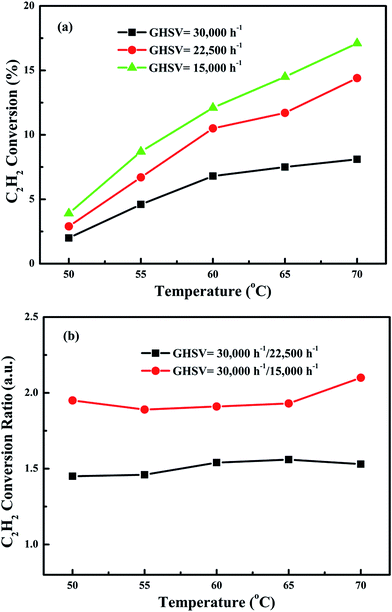 |
| | Fig. 9 (a) Acetylene conversion as a function of reaction temperature over meso-Co3O4 at space velocities of 15000 h−1, 22500 h−1 and 30000 h−1 and (b) acetylene conversion ratios for 15000 h−1, 22500 h−1 and 30000 h−1 as a function of reaction temperature, using date from (a). | |
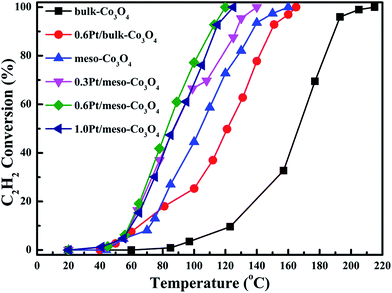 |
| | Fig. 10 Catalytic performance as a function of reaction temperature over bulk-Co3O4, 0.6Pt/bulk-Co3O4, meso-Co3O4, 0.3Pt/mso-Co3O4, 0.6Pt/meso-Co3O4 and 1.0Pt/meso-Co3O4 catalysts for the oxidation of C2H2 under the conditions: [C2H2] = 50 ppm, [O2] = 13 vol%, N2 balance and GHSV = 30000 h−1. | |
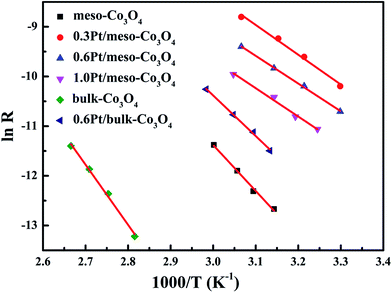 |
| | Fig. 11 Arrhenius plots in acetylene oxidation of meso-Co3O4, 0.3Pt/meso-Co3O4, 0.6Pt/meso-Co3O4, 1.0Pt/meso-Co3O4, bulk-Co3O4 and 0.6Pt/bulk-Co3O4. Reaction gas composition: [C2H2] = 50 ppm, [O2] = 13 vol%, N2 balance and GHSV = 30000 h−1. | |
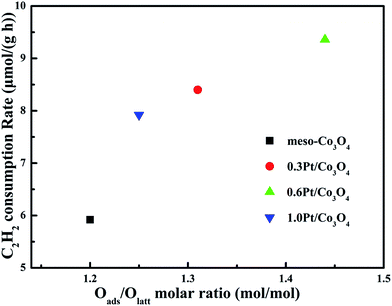 |
| | Fig. 12 Acetylene consumption rates at 60 °C as a function of Oads/Olatt molar ration of meso-Co3O4, 0.3Pt/meso-Co3O4, 0.6Pt/meso-Co3O4 and 1.0Pt/meso-Co3O4. Reaction gas composition: [C2H2] = 50 ppm, [O2] = 13 vol%, N2 balance and GHSV = 30000 h−1. | |
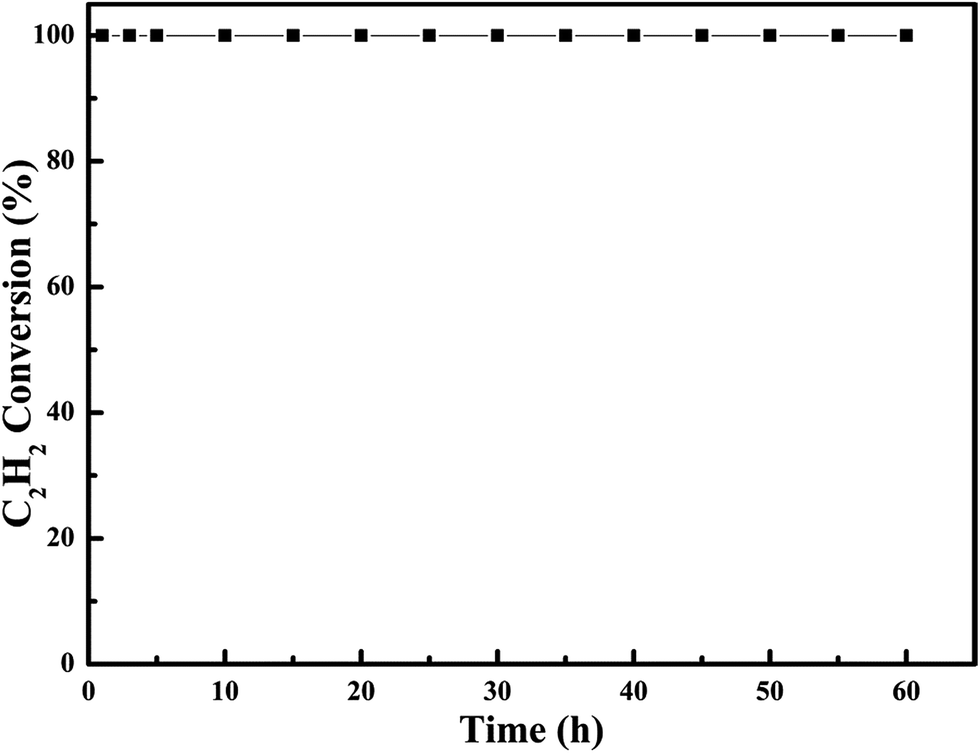
In order to examine the chemical and mechanical stability of the catalysts, the on-steam reaction experiments were carried out over the most effective 0.6Pt/meso-Co3O4 at 120 °C for acetylene oxidation, as shown in Fig. 13. There is no significant loss in catalytic activity. It indicates that 0.6Pt/meso-Co3O4 catalyst is chemical stability for acetylene oxidation. SEM images of the recovered catalyst are shown in Fig. 14. The morphology of the catalyst did not show significant change. Therefore, 0.6Pt/meso-Co3O4 may be a chemical and mechanical durable catalyst for acetylene oxidation under the adopted conditions.
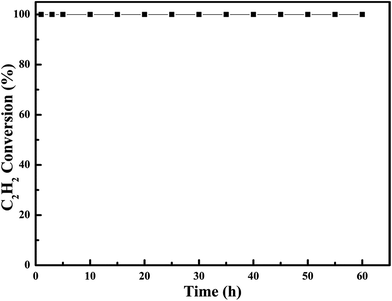 |
| | Fig. 13 Durability test on 0.6Pt/meso-Co3O4 catalyst. Reaction gas composition: [C2H2] = 50 ppm, [O2] = 13 vol%, N2 balance and GHSV = 30000 h−1. | |
 |
| | Fig. 14 SEM images of (a and b) 0.6Pt/meso-Co3O4 catalyst and (c and d) the used 0.6Pt/meso-Co3O4. | |
It is generally known that the porous network of catalysts can provide more contact sites and promote the diffusion of reactant and product molecules. The mesoporous structure is beneficial to enhancing the catalytic oxidation of acetylene. The big surface area of meso-Co3O4 (122.0 m2 g−1) is highly conductive to acetylene oxidation. xPt/meso-Co3O4 also possess the high surface area (111.0–111.5 m2 g−1). After the loading of Pt, xPt/meso-Co3O4 catalysts exhibit higher conversion at low temperature than meso-Co3O4 catalyst. The reaction mechanism of C2H4 oxidation over the Co3O4 has been reported.26 It was reported that the oxidation of hydrocarbons on transition metal oxides follows a Mars-Van Krevelen mechanism.27 The surface oxygen species will participate in hydrocarbons oxidation process and recovery by the gas-phase oxygen molecules. Thus, the reaction mechanism of acetylene oxidation over the catalysts may be related to surface oxygen species. Acetylene molecule can be absorbed on the surface of xPt/meso-Co3O4 to form an adsorbed intermediate. The adsorbed acetylene species are further oxidized to CO2 and H2O under the effect of surface active oxygen. From the results of XPS and O2-TPD, xPt/meso-Co3O4 display more surface absorbed oxygen species than meso-Co3O4. The existence of lattice defect is beneficial for the formation of the oxygen vacancies. The oxygen vacancies can be favorable for the adsorption of oxygen and hence enhancing the catalytic oxidation of acetylene. xPt/meso-Co3O4 exhibit more lattice defect than meso-Co3O4, which can be demonstrated from the result of Raman, XPS and H2-TPR. It has been reported that better catalytic activity is related to low-temperature reducibility. Better reducibility means that oxygen species are easily activated. From the result of H2-TPR, xPt/meso-Co3O4 show better low-temperature reducibility than meso-Co3O4. Among all xPt/meso-Co3O4 catalysts, 0.6Pt/meso-Co3O4 catalyst exhibits the best catalytic performance, which can completely oxidize acetylene at 120 °C. 0.6Pt/meso-Co3O4 shows orderedly mesoporous structure, sufficient oxygen vacancies, more adsorbed oxygen species and better low-temperature reducibility. Therefore, the highest catalytic performance of 0.6Pt/meso-Co3O4 for the oxidation of acetylene was obtained.
4. Conclusions
The meso-Co3O4 with high surface area are synthesized by using the KIT-6 hard template, and xPt/meso-Co3O4 catalysts are prepared by the means of NaBH4 reduction. The activity for acetylene oxidation over different catalysts decreases in the following order: 0.6Pt/meso-Co3O4 > 1.0Pt/meso-Co3O4 > 0.3Pt/meso-Co3O4 > meso-Co3O4 > bulk-Co3O4. 0.6Pt/meso-Co3O4 exhibits the highest catalytic activity of acetylene oxidation at 120 °C and good stability for at least 60 h. The excellent catalytic performance of 0.6Pt/meso-Co3O4 can be associated with its orderedly mesoporous structure, plenty of adsorption oxygen species and better low-temperature reducibility.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (U1662103, 21673290 and 21376261); the National Hi-Tech Research and Development Program (863) of China (2015AA034603); and the China National Offshore Oil Corporation Fund (LHYJYKJSA20160002).
References
- K. Wang, L. Du and M. F. Ge, J. Environ. Sci., 2009, 21, 137–141 CrossRef CAS.
- R. Volkamer, P. J. Ziemann and M. J. Molina, Atmos. Chem. Phys., 2009, 9, 1907–1928 CrossRef CAS.
- W. P. L. Carter, Air Waste, 1994, 44, 881–899 CAS.
- Y. Yu, T. Takei, H. Ohashi, H. He, X. Zhang and M. Haruta, J. Catal., 2009, 267, 121–128 CrossRef CAS.
- C. Y. Ma, Z. Mu, J. J. Li, Y. G. Jin, J. Cheng, G. Q. Lu, Z. P. Hao and S. Z. Qiao, J. Am. Chem. Soc., 2010, 132, 2608–2613 CrossRef CAS PubMed.
- C. Ma, D. Wang, W. Xue, B. Dou, H. Wang and Z. Hao, Environ. Sci. Technol., 2011, 45, 3628–3634 CrossRef CAS PubMed.
- Z. Wang, W. Wang, L. Zhang and D. Jiang, Catal. Sci. Technol., 2016, 6, 3845–3853 CAS.
- Y. Xia, H. Dai, H. Jiang and L. Zhang, Catal. Commun., 2010, 11, 1171–1175 CrossRef CAS.
- J. Yang and T. Sasaki, Cryst. Growth Des., 2010, 10, 1233–1236 CAS.
- L. Hu, Q. Peng and Y. Li, J. Am. Chem. Soc., 2008, 130, 16136–16137 CrossRef CAS PubMed.
- B. Bai, H. Arandiyan and J. Li, Appl. Catal., B, 2013, 142–143, 677–683 CrossRef CAS.
- C. Zhang, F. Liu, Y. Zhai, H. Ariga, N. Yi, Y. Liu, K. Asakura, M. Flytzani-Stephanopoulos and H. He, Angew. Chem., Int. Ed., 2012, 51, 9628–9632 CrossRef CAS PubMed.
- C. Zhang and H. He, Catal. Today, 2007, 126, 345–350 CrossRef CAS.
- X. Tang, J. Chen, X. Huang, Y. Xu and W. Shen, Appl. Catal., B, 2008, 81, 115–121 CrossRef CAS.
- F. Kleitz, S. H. Choi and R. Ryoo, Chem. Commun., 2003, 2136–2137 RSC.
- A. Rumplecker, F. Kleitz, E. L. Salabas and F. Schüth, Chem. Mater., 2007, 19, 485–496 CrossRef CAS.
- M. Casas-Cabanas, G. Binotto, D. Larcher, A. Lecup, V. Giordani and J.-M. Tarascon, Chem. Mater., 2009, 21, 1939–1947 CrossRef CAS.
- Q. Liu, L. C. Wang, M. Chen, Y. Cao, H. Y. He and K. N. Fan, J. Catal., 2009, 263, 104–113 CrossRef CAS.
- K. R. Zhu, M. S. Zhang, Q. Chen and Z. Yin, Phys. Lett. A, 2005, 340, 220–227 CrossRef CAS.
- Y. Liu, H. Dai, J. Deng, S. Xie, H. Yang, W. Tan, W. Han, Y. Jiang and G. Guo, J. Catal., 2014, 309, 408–418 CrossRef CAS.
- M. M. Schubert, S. Hackenberg, A. C. van Veen, M. Muhler, V. Plzak and R. J. Behm, J. Catal., 2001, 197, 113–122 CrossRef CAS.
- Y. Li, Y. Du, Y. Wei, Z. Zhao, B. Jin, X. Zhang and J. Liu, Catal. Sci. Technol., 2017, 7, 968–981 CAS.
- W. Tang, Z. Hu, M. Wang, G. D. Stucky, H. Metiu and E. W. Mcfarland, J. Catal., 2010, 273, 125–137 CrossRef CAS.
- B. Bai and J. Li, ACS Catal., 2014, 4, 2753–2762 CrossRef CAS.
- S. Rousseau, S. Loridant, P. Delichere, A. Boreave, J. P. Deloume and P. Vernoux, Appl. Catal., B, 2009, 88, 438–447 CrossRef CAS.
- H. F. Feng, D. Wang, X. H. Liu, Y. L. Guo, G. Z. Lu and P. J. Hu, ACS Catal., 2016, 6, 5393–5398 CrossRef.
- S. Scirè and L. F. Liotta, Appl. Catal., B, 2012, 125, 222–246 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Yongheng Li,
Zhen Zhao
*,
Yongheng Li,
Zhen Zhao