
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceToward multifunctional hybrid platforms for tissue engineering based on chitosan(PEO) nanofibers functionalized by bare laser-synthesized Au and Si nanoparticles
Ahmed Al-Kattan *a,
Viraj P. Nirwanb,
Emilie Munnierc,
Igor Chourpac,
Amir Fahmib and
Andrei V. Kabashina
*a,
Viraj P. Nirwanb,
Emilie Munnierc,
Igor Chourpac,
Amir Fahmib and
Andrei V. Kabashina
aAix-Marseille University, CNRS, LP3 UMR 7341, Campus de Luminy, Case 917, 13288, Marseille cedex 9, France. E-mail: ahmed.al-kattan@univ-amu.fr
bRhine-waal University of Applied Sciences, Faculty of Technology and Bionics, Marie-Curie-straße 1, 47533 Kleve, Germany
cFrançois-Rabelais University, EA6295 “Nanomédicaments et Nanosondes”, 37200 Tours, France
First published on 21st June 2017
Abstract
Exhibiting a variety of unique optical, structural and physicochemical properties, laser-synthesized nanomaterials have become increasingly popular during recent years in a variety of biomedical, catalytic, photovoltaic and other applications. Here, we explore the use of bare laser-synthesized gold and silicon nanoparticles (AuNPs and SiNPs) as additives to functionalize electrospun chitosan(PEO) nanofibers and then assess the potential of such hybrid structures as multifunctional platforms for tissue engineering. We demonstrate that bare AuNPs and SiNPs can be easily grafted on the surface of the chitosan(PEO) nanofibers without any interference, via electrostatic interaction between a strong negative surface charge of NPs and the polycationic surface of the fibers. We also show that the nanofibers functionalized with nanoparticles can affect the morphology and physico-chemical characteristics of the resulting nanostructures. As an example, the functionalization of nanofibers by SiNPs led to quite different thicknesses of fibers (386 ± 80 nm and 632 ± 170 nm), suggesting a potential improvement of fibre surface reactivity. Finally, biological toxicity of the nanofibers was assessed through preliminary viability tests conducted on HaCaT cells. After 24 h of incubation time, no adverse effects were observed confirming satisfactory biocompatibilty of the hybrid nanofiber structures. The proposed concept promises exciting perspectives in the development of innovative multifunctional scaffolds structures gathering new properties for tissue engineering.
Introduction
The electrospinning technique has gained a lot of interest within recent years due to its flexibility in the fabrication of (nano)fibrous matrixes for regenerative medicine and tissue engineering.1,2 This technique offers several advantages over conventional techniques such as template synthesis or mechanical drawing,3,4 including a much better flexibility in the choice of operational synthetic or natural polymers,5,6 and a better efficiency in the control of nanofiber size (from submicron to nanometer scale) and porosity.7 In particular, significant efforts were applied to the development of biologically-derived polymers such as chitosan.8 Its conversion into electrospun nanofibers is particularly challenging due to hydrogen bonding interactions between polysaccharide chains, leading to high crystallinity and poor solubility in common solvents. To overcome this problem, numerous formulations of chitosan nanofibers were developed by employing biocompatible co-spinning agents such as poly(ethylene oxide) (PEO) to facilitate its electrospinnability.9 Chitosan is by far one of the most exploited polymers due to its biocompatibility, biodegradability and bioactivity (anti-microbial, scar reduction, wound healing, etc.).10–12 Furthermore, the presence of amino groups on chitosan surface can enable interactions with a variety of biological species (e.g. lipids, proteins, DNA, etc.), making chitosan(PEO) nanofibers promising host structure for tissue engineering.One of the promising strategies to further improve physicochemical, biological and/or therapeutic characteristics of polymer nanofibers is based on the incorporation of various additives into the nanofiber structure.13 As an example, inorganic nanomaterials with desired properties can be used as additives. Silver (Ag) nanoparticles present one of the most prominent examples, which can offer outstanding antibacterial activity and thus extend the application area of nanofibers compositions toward wound healing and tissue engineering applications.14,15 Gold (Au) nanoparticles is another important example, as they are often used to improve mechanical properties of polymer fibers, promote proteins adsorption and cell adhesion, and finally reduce bacterial colonization.16–18 Mesoporous silica nanoparticles present another promising class as nanocarriers for drug delivery in nanofibers system.19 However, the dominating majority of currently used nanoparticles are obtained by conventional chemical methods, which normally use hazardous raw materials such as nitrate salts or citrate ligands, provoking nanoparticle surface contamination (e.g., residual anions) and leading to residual toxicity of final products.18 In addition, chemically-synthesized nanomaterials are not always adapted for operation in specific conditions (e.g. protection from day-light), which makes their application prospects complicated.
Laser ablation in liquid ambience has recently emerged as a “green” alternative to chemical nanomaterial synthesis as it can avoid the use of toxic by-products.20–26 As an example, profiting from ultrashort pulsed laser ablation in deionized water,23,24 we recently managed to fabricate stable solutions of bare Au and Si nanoparticles, which do not contain any protective ligands on the surface and are not contaminated by any toxic substance.25–27 The cleanness of laser synthesis opens up opportunities for novel efficient applications of these nanomaterials in cancer theranostics,26–30 electrocatalysis,31 etc. In addition, the presence of high oxidations states (e.g. AuO−, SiOx−) in surface composition of laser-synthesized nanomaterials27,32 promises their efficient interactions with many substances such as polymer fibers.33 When linked to polycationic nanofiber polymers such as chitosan(PEO), such nanomaterials are expected to create additional adhesion/interaction sites with biological matrix (e.g. cells, proteins, DNA, etc.) in tissue engineering applications. Finally, bare-NPs can serve as sensitizers of therapeutics modalities such as radiofrequency radiation-induced hyperthermia,27,28 which promises their use as local drug/gene delivery vehicles activated either by external (e.g., optical stimuli, radio frequency) or internal (e.g., pH, temperature) stimuli.34,35
This work is conceived as attempt to elaborate novel hierarchical hybrid nanofibers based-on chitosan(PEO) functionalized with bare laser-synthetized AuNPs and SiNPs. AuNPs and SiNPs were prepared by laser-ablative synthesis and their structural properties (size-distribution, charge surface) were tested by HR-TEM and zeta measurements before electrospinning. Chitosan/PEO and NPs were then electrospun at appropriate ratio. The obtained nanofibers were characterized by a panel of physicochemical methods (SEM-EDX, FTIR, TGA and DSC), and the role of NPs on the nanofibers properties was assessed. Finally first successful MTT tests were presented.
Materials and methods
Materials
Commercial chitosan powder (Mw = 200 kD) with 75% of deacetylation was used (Sigma-Aldrich). Poly(ethylene oxide) (PEO) powder (Mw = 300 kD) and acetic acid (ReagentPlus®, 99%) solution were purchased respectively from Sigma Aldrich and Normapur (PROLABO). Gold solid target (99.99%) purchased from Sigma Aldrich was used to synthesize AuNPs. Raw-silicon micro-particles powder was prepared as described by Blandin et al.26 Cell culture reagents were purchased from Fisher bioblock (France).Preparation of electrospun nanofibers
Chitosan and PEO powder were added at chitosan/PEO ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 in 90 wt% of aqueous acetic acid. The solutes were then mixed under magnetic stirring for 5 hours complete the dissolution. AuNPs and SiNPs were introduced at increasing percentage 0, 10 and 30 wt% (0 wt% is the reference). The prepared solutions were degasified for 1 hour and kept in a sealed container at room temperature for electrospinning. All experiments were carried out using fresh solutions.
:4 in 90 wt% of aqueous acetic acid. The solutes were then mixed under magnetic stirring for 5 hours complete the dissolution. AuNPs and SiNPs were introduced at increasing percentage 0, 10 and 30 wt% (0 wt% is the reference). The prepared solutions were degasified for 1 hour and kept in a sealed container at room temperature for electrospinning. All experiments were carried out using fresh solutions.
Electrospinning of hybrid chitosan(PEO)/NPs blend nanofibers
Climate-controlled electrospinning platform from IME technologies was employed to elaborate the nanofibers. Electrospinning process was performed using a horizontal setup. Briefly, the prepared solution was transferred into a 3 ml syringe (IME technologies) with Luer-Lock connected via a PTFE tube to flat-needles. The syringe was mounted on a programmable pumps module to ensure a steady electrospinning flow and connected to a rotating target collector. The flow rate was fixed at 0.3 ml h−1 for all experiments. Nanofibers were collected on an aluminum foil placed around a rotating target collector equipped with nozzle stage. The speed of the rotating collector and the nozzle translation were 80 rpm and 10 mm s−1, respectively. Voltages applied to the solution and the collector was fixed at 12 kV and −1 kV, respectively. All experiments were conducted under normal atmospheric conditions at 28 °C temperature and a relative humidity of 30%.Laser-ablative synthesis of bare Au and Si nanoparticles
AuNPs and SiNPs were prepared using femtosecond radiation from a Yb:KGW laser (Amplitude Systems, 1025 nm, 480 fs, 1 kHz) under ambient atmosphere conditions. In the case of AuNPs, radiation was focused with the help of a 75 mm lens on the surface of target (1 cm × 1 cm), which was continually moved at the speed of 0.5 mm s−1. The concentration of AuNPs was determined by the calculation of the weight loss of the target during the ablation process. The size distribution of the nanoparticles was measured by TEM. In the case of SiNPs, initial Si-micropowder was preliminary prepared by mechanical milling of a Si wafer. The powder was then transferred into a glass cuvette at 0.35 g l−1 and dispersed in deionized water using a sonication bath for 30 minutes. Then, the dispersed Si-microparticles were fragmented under laser irradiation for one hour. The laser beam was focused at 0.5 cm in the middle of glass cuvette filled with the NPs solution, while the solution was continually homogenized by a magnetic stirrer. The formed SiNPs solution was then transferred into centrifugal plastic tubes (Cellestar®tubes) and centrifuged at 3500 rpm for 20 minutes to remove possible unfragmented Si-micropowder. The concentration of the SiNPs in the solution was measured by induced coupled plasma mass spectroscopy (ICP-MS) method.Fourier transforms infrared (FTIR) spectroscopy
Chemical characteristics were evaluated by FTIR spectrometer (Perkin-Elmer) equipped with a universal attenuated total reflection (ATR) sampling accessory. All analyses were performed at room temperature on chitosan/PEO (reference), chitosan/PEO/AuNPs and chitosan/PEO/SiNPs blend nanofibers. In addition, two references spectra of pure chitosan and PEO dissolved in 10 ml of aqueous acetic acid solution (90%) were measured. The measurements were recorded at operating wavelengths in the range of 4000 to 400 cm−1. Each measurement was composed of 32 scans to ensure a high signal-to-noise ratio with a resolution of 4 cm−1.Thermal analysis
Thermal decomposition profiles of the blend nanofibers were characterized by TGA analysis performed on Perkin-Elmer system (TGA 4000). For each composition, 12 mg of obtained nanofibers were scratched from the aluminum foil and placed into the TGA with a nitrogen flow rate of 20 ml min−1 and at heating rate of 10 °C min−1 in a temperature range from 25 °C to 800 °C. Differential scanning calorimetry (DSC) was also performed on the elaborated nanofibers. The specimens were equilibrated to 30 °C and then heated up to 250 °C. The measurements were repeated three times.Characterization of bare Au and Si nanoparticles prepared by laser process
A high-resolution transmission electron microscopy (HR-TEM) system (JEOL JEM 3010) was employed in imaging and diffraction modes to characterize size distribution and crystalline structure of NPs. ζ-Potential of NPs was also measured by dynamic light scattering (DLS) using a Zetasizer Nano ZS (Malvern, UK).Scanning electron microscopy (SEM)
The size and the surface morphologies of the blend nanofibers were characterized by a scanning microscope using a DSM 982 Gemini (Zeiss, Germany) at accelerating voltage of 15 kV. The observations were conducted on piece of aluminum foil cut from the mat and placed on an aluminum stub. For high magnification, a Joel JSM-6320F microscope operating at 0.5–30 kV was used, coupled with energy dispersive X-ray analysis (EDX) to detect AuNPs and SiNPs.Cell viability assay
Toxicity of hybrid nanofibers was assessed via in vitro tests on HaCaT cells (human keratinocytes obtained from adult skin) purchased from AddexBio. HaCaT cells were grown in DMEM enriched with 10% FBS and 1% antibiotics, at 37 °C and 5% CO2. A sample of 10 mg of hybrid nanofibers was incubated in cell culture medium for 24 h. The preparation was then centrifugated (3000 × g, 15 min) and the supernatant was kept. MTT assay was performed in 96 multiwell plates. HaCaT cells were seeded at 5000 cells per well and grown for 72 h in complete culture medium. Culture medium was discarded and replaced by 200 μl of the supernatant at different concentrations in culture medium. After 24 h, the solution was discarded and cells were incubated for 4 h with 100 μl of MTT solution at 0.05%. Cell viability was evaluated by using microplate reader working at 540 nm. Incubations and measurements were repeated in triplicate.Results and discussion
Five formulations of chitosan(PEO)/NPs blend nanofibers were prepared with increasing concentrations 0, 10 and 30 wt% of nanoparticles (NPs). Nomenclature and compositions used are summarized in Table 1. Nanofibers were prepared from optimized chitosan/PEO ratio of 0.25. Polymers were then dissolved in 10 ml of 90% acetic acid solution. Electrospinning of pure chitosan dissolved in the range 0.5–2 wt% was unsuccessful, resulting in the formation of nanobeads instead of nanofibers. Several studies reported the difficulty to electrospun pure chitosan solution mainly because of its polycationic nature and rigid chemical structure due to the formation of specific inter and intra-molecular interactions (strong hydrogen bonds) preventing free movements of polymeric chain segments leading to explosion-like behavior of the jet (intermittent spinning).36,37 On the other hand, chitosan nanofibers were obtained by incorporating poly(ethylene oxide) PEO as co-spinning agent. A continuous jet was thus observed leading to the formation of uniform nanofibers on the collector. PEO can act as plasticizer by breaking down inter and intra molecular interactions of chitosan chains.38,39 The interaction of PEO with chitosan chains through hydrogen bonds ensure the formation of nanofibrous matrix based-on chitosan(PEO).| Nomenclature sample | Reference | Polymer composition wt (%) | |||
|---|---|---|---|---|---|
| Chitosan | PEO | AuNPs | SiNPs | ||
| Chitosan(PEO) | Ch-0 | 1 | 4 | — | — |
| Chitosan(PEO)–AuNPs | Ch-10Au | 1 | 4 | 10 | — |
| Chitosan(PEO)–AuNPs | Ch-30Au | 1 | 4 | 30 | — |
| Chitosan(PEO)–SiNPs | Ch-10Si | 1 | 4 | — | 10 |
| Chitosan(PEO)–SiNPs | Ch-30Si | 1 | 4 | — | 30 |
Before mixing with chitosan/PEO solution and electrospinning, bare NPs were characterized by HR-TEM. As shown in Fig. 1 NPs were typically spherical with mean size of 40 ± 0.89 nm and 32 ± 0.97 nm for AuNPs and SiNPs, respectively. Crystallographic structures of NPs were examined by electron diffraction measurements, which revealed characteristic orientation plane of Au (Fig. 1a) and Si (Fig. 1b). ζ-Potential measurements, were carried out revealing negative surface charge of NPs (−23.1 ± 2.61 mV for AuNPs, 45 ± 1.5 mV for SiNPs) due to partial oxidation of their surface.
 | ||
| Fig. 1 Typical HR-TEM images of AuNPs (a) and SiNPs (b) prepared by laser ablation in water, with corresponding size distribution and electron diffraction patterns. | ||
SEM observations showed that the nanofibers exhibit cylindrical morphology with diameter of 687 ± 120 nm forming a dense network with no trace of beads (Fig. 2a). The functionalization of chitosan(PEO) fibers with AuNPs and SiNPs was done at 10 and 30 wt% while all the electrospinning parameters were constant (Fig. 2b–e). The addition of NPs had no effect on the chitosan(PEO) nanofiber morphology, while the presence of spherical NPs on the surface of the nanofibers were clearly visible. Precise size measurements revealed that NPs had diameters close to 44 ± 3.07 nm (Fig. 2b and c) and 37 ± 2.75 nm (Fig. 2d and e), corresponding to the mean sizes observed by HR-TEM for AuNPs and SiNPs, respectively. Elemental analyses (EDX) confirmed the presence of the NPs where Au (Fig. 3 Ch-30Au) and Si (Fig. 3 Ch-30Si) were detected in addition to the main elements of the fiber (C, O) and the substrate foil (Al). Other morphological observations revealed that the diameter of the nanofibers elaborated in presence of AuNPs was unchanged (632 ± 170 nm) (Fig. 2b and c) while the fibers co-electrospun with SiNPs were twice thinner (386 ± 80 nm) (Fig. 2d and e). These observations evidenced electrostatic interactions between positive group (NH3+) on the surface of the nanofibrous and the negative oxidized surface of NPs (Au–O−/Au–OH− and SiOx−);26,27 and suggested better affinity of interaction between SiNPs and chitosan(PEO) due to higher ζ-potential of SiNPs (−45 ± 1.5 mV) than of AuNPs (23.1 ± 2.61 mV).
 | ||
| Fig. 2 SEM of hybrid chitosan(PEO) nanofibers prepared with different content of NPs: 0 wt% (a) of NPs, 10 and 30 wt% of AuNPs (b and c), 10 and 30 wt% of SiNPs (d and e). | ||
 | ||
| Fig. 3 EDX spectra of Ch-30AuNPs and Ch-30SiNPs hybrid nanofibers. | ||
Chemical compositions of the nanofibers were analyzed by FTIR and results are presented in Fig. 4. Chitosan(PEO) nanofibers (Ch-0) exhibited characteristics bands of chitosan and PEO. Thus, a broad band was observed in the region of 3400–3000 cm−1 encompassing stretching modes of NH2 and OH groups attributed to chitosan.37,40,41 In addition, the band observed at 1560 cm−1 for pure chitosan attributed to amine group was shifted by almost 40 cm−1 for chitosan(PEO), associated with the formation of hydrogen bonds between polyether oxygen and amino hydrogen of PEO and chitosan, respectively.42,43 Another band of chitosan as C–O stretching was observed at 1029 cm−1, but overlapped with intense PEO bands 1145, 1095 and 1059 cm−1 assigned to a C–O–C stretching vibration.40 Finally intense PEO band at 2883 cm−1 was observed and attributed to CH2 stretching for chitosan(PEO).44 The addition of AuNPs (Fig. 4 Ch-10Au and Ch-30Au) and SiNPs (Fig. 4 Ch10Si and Ch30Si) into the formulation had no observable effect on the chemical nature of chitosan(PEO) nanofibers. No additional bands were observed to evidence any formation of second phase.
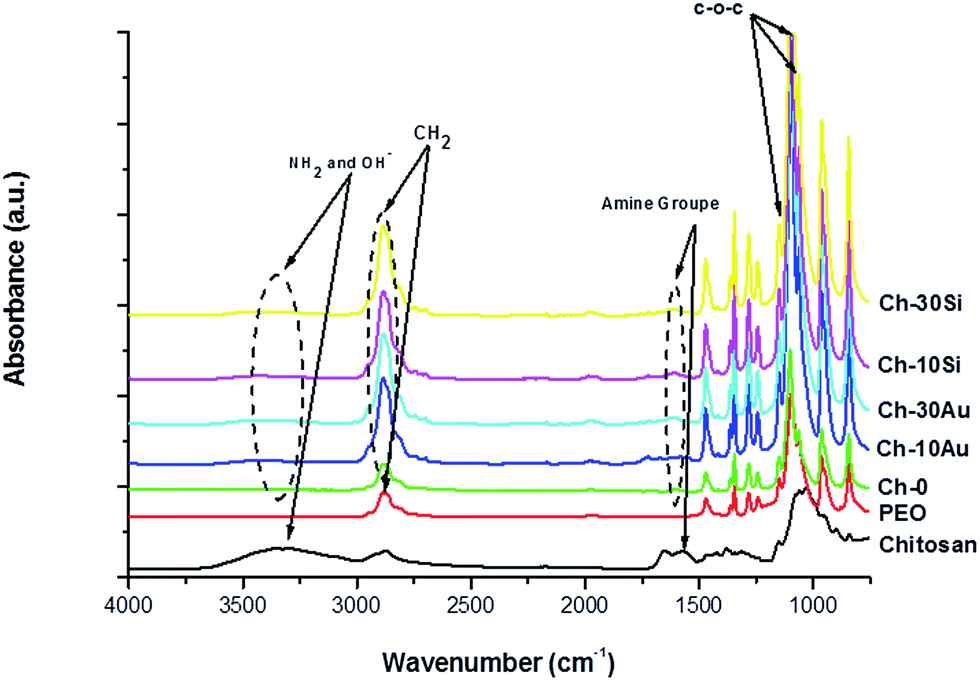 | ||
| Fig. 4 FTIR of pure chitosan, pure PEO, and hybrid chitosan(PEO) nanofibers prepared with different amounts 0, 10 and 30 wt% of bare-NPs. | ||
TGA characterization of nanofibers conjugated with AuNPs and SiNPs is presented in Fig. 5. The measurements were performed at heating rate of 10 °C min−1 in 25–800 °C interval. Chitosan(PEO) (Ch-0) displayed one weight loss region at 383 °C corresponding to 95.44% of initial mass loss and attributed to the degradation of the polymer molecules.38
 | ||
| Fig. 5 TGA thermogram curves of hybrid chitosan(PEO) nanofibers blended with AuNPs and SiNPs. | ||
In addition, around the region of 100 °C, no significant weight loss was observed, which was probably due to the absence of water molecules in the fiber composition. The functionalization of the nanofibers with AuNPs had no influence on the weight loss estimated as 95.15% and 95.47% for Ch-10Au and Ch-30Au, respectively. For SiNPs, similar thermic events were observed at 417 °C and 425 °C, respectively for Ch-10Si and Ch-30Si respectively, corresponding to the degradation of polymer molecules. It is interesting to note that a slight shift of curve Ch-30Si to higher temperature suggests a better thermal stability, which is probably due to crystalline structure of NPs as it explored in literature for AuNPs.23,26 Based-on this hypothesis it will be interesting to elaborate hybrid chitosan(PEO) with higher content of NPs to highlight this aspect. Finally, the absence of supplementary thermal event confirmed for AuNPs and SiNPs suggests that NPs are cross-linked to the backbone polymer fiber and did not form a secondary phase in agreement with microscopy and FTIR analyses.
Thermal gravimetric analysis was also complemented by differential scanning calorimetry (DSC). These results are presented in Fig. 6. A classical DSC curve was observed with two events for Ch-0 nanofibers. The first one occurred at 56.4 °C corresponding to latent heat of melting attributed in the literature to favorable interaction between crystalline PEO and amorphous chitosan chain to form strong intermolecular hydrogen bonds between the ether groups of PEO and the amino/hydroxyl groups of chitosan.6,45 At higher temperature around 125 °C, a second predominant event related to a degradation of the polymer matrix takes place. The nonappearance of thermal episode at 100 °C might be related to the absence of water molecules signaled by TGA measurements.
 | ||
| Fig. 6 DSC thermogram curves of hybrid chitosan(PEO) nanofibers blended with AuNPs (a) and SiNPs (b). | ||
The addition of AuNPs (Ch-10Au and Ch-30Au) into the initial formulation has no significant influence on the first endothermic peak. Here, the first peak is slightly shifted to 59.14 °C and 55.73 °C for Ch-10Au and Ch-30Au, respectively, compared to 56.4 °C for Ch-0. However, the intensity of the peaks is decreased when concentration of AuNPs is increased, which can be explained by a lower mass loss which results in better thermal stability. Such a supposition is confirmed by the increase of endothermic enthalpy value ranging from 185.81 J g−1 for Ch-0 to 433.41 J g−1 and 1170.77 J g−1 for Ch-10Au and Ch-30Au, respectively. We suppose that the same increase of enthalpy is responsible for the shift of the second endothermic peak to higher temperatures at increased AuNPs concentration (132.33 °C and 160 °C for Ch-10Au and Ch-30Au, respectively, compared to 125 °C for Ch-0). Similar results were observed with SiNPs (Fig. 6b). Here, only slight shifts of positions of the first endothermic peak was recorded (62.21 °C and 55.02 °C for Ch-10Si and Ch-30Si, respectively, compared to 56.4 °C for Ch-0). Similar to observations in presence of AuNPs, the intensity of the peak decreased as the SiNPs concentration was increased. However, the deviation of curves for Ch-10Si and Ch-30Si is relatively small compared to Ch-10Au and Ch-30Au, which is obviously explained by a much weaker increase of endothermic enthalpy values while passing from Ch-10Si (364.87 J g−1) to Ch-30Au (433.44 J g−1).
As hybrid nanofibers are intended to be used as biomaterials in tissue engineering, their toxicity was evaluated by in vitro tests on human keratinocytes (HaCaT), the main cells present in the skin. Cells were incubated for 24 h with different concentrations of nanofibers dispersed in cell culture medium (from 0.1 μg l−1 to 1 mg l−1). The tests were performed at highest concentration of 30 wt% of NPs. The results are shown on Fig. 7. In the experimental conditions, the hybrid nanofibers did not show any significant toxicity on HaCaT cells when the NPs were incorporated into nanofibers formulations.
 | ||
| Fig. 7 Cell viability of HaCaT cells as function of the concentration of hybrid nanofibers loaded with 30 wt% of AuNPs (red curve) or SiNPs (black curve). | ||
Indeed, the viability holds around 100% for a concentration up to 100 μg l−1. Furthermore, at higher nanofiber concentrations (1 mg l−1), the cell viability drops only slightly to 80%. Therefore, we evidence in these experimental conditions that the functionalization of the chitosan(PEO) nanofibers by AuNPs or SiNPs does not create toxicity. This result looks very encouraging, but has to be confirmed by further studies (in progress) to assess (i) the biocompatibility at longer time, and (ii) the ability of the NPs to stimulate the attachment and proliferation of cells.
At this point of the work, we demonstrated that AuNPs and SiNPs can be potentially used as relevant additives/key modules for extended modalities for tissue engineering applications. Indeed, two points can be reminded: (i) AuNPs and SiNPs can easily interact via electrostatic interactions without interference with chemical composition of the nanofibers. Furthermore, the introduction of SiNPs reduces the size of the nanofibers which can potentially promote the proliferation and attachment of cells in accordance to the literature. Indeed, Chen et al. found that fibroblast attachment on 428 nm polycaprolactone nanofibrous scaffolds was significantly better than fibers having micronic size diameter.46 In another study, chondrocyte proliferation was also positively influenced by reducing the size of fibers below 500 nm.47 Thus, we can reasonably conclude that the fiber size reduction observed during the incorporation of SiNPs into chitosan(PEO) can promote the intrinsic ability of chitosan(PEO) to interact with tissues. To check this hypothesis, additional tests will be performed.
Another important finding is related to the capability of AuNPs and SiNPs to delay the degradation of the nanofibers, as it was observed in DSC thermal tests. This effect is probably due to the ability of bare-NPs to absorb heating energy leading to lower transfer of thermal energy to nanofibers and thus the increase thermal stability at higher temperatures. We believe that this property can be explored for advanced therapy modalities (e.g., radio frequency, hyperthermia).40
Conclusions
We demonstrated the possibility for electrospinning of some promising nanofiber composites based on chitosan(PEO) functionalized with bare (ligand-free) AuNPs and SiNPs synthesized by laser ablation. Structural analysis showed that the nanofibers are cylindrical and uniform, coated with NPs via electrostatic interactions to offer additional anchoring biological sites. In addition, we observed a decrease of fiber size under their functionalization with SiNPs, which can contribute to higher surface efficiency in biological immobilizations. Spectroscopic and thermal analysis established that the addition of AuNPs and SiNPs into the nanofibers formulation had no significant effect on the chemical composition, but improved the thermal stability, opening exciting perspectives for advanced therapeutics modalities. Finally, preliminary cytotoxicity tests on HaCaT skin cells revealed that NPs-blended nanofibers has no great influence on the cell viability. As the main objective of this study, we present the demonstration of conceptual possibility to fabricate chitosan(PEO) nanofiber blended with ultra-clean inorganic NPs. We also reveal some promising properties of such nanofibers, which can be important for potential tissue engineering applications.Acknowledgements
The authors express their thanks to DAAD program (German Exchange Academic Service), “LASERNANOCANCER” (No. PC201420), GRAVITY projects of the ITMO “Plan Cancer 2014–2019” INSERM program and Hynanodend project (BMBF). The authors would like to acknowledge Dr Duygu Disci-Zayed for her contribution for SEM-EDX and the improvement of the manuscript.Notes and references
- J. Quirós, K. Boltes and R. Rosal, Polym. Rev., 2016, 56, 631–667 CrossRef.
- J. Cheng, Y. Jun, J. Qin and S. H. Lee, Biomaterials, 2017, 114, 121–143 CrossRef CAS PubMed.
- S. L. Tao and T. A. Desai, Nano Lett., 2007, 7, 1463–1468 CrossRef CAS PubMed.
- X. Xing, Y. Wang and B. Li, Opt. Express, 2008, 16, 10815–10822 CrossRef CAS PubMed.
- Z. Ma, M. Kotaki, T. Yong, W. He and S. Ramakrishna, Biomaterials, 2005, 26, 2527–2536 CrossRef CAS PubMed.
- Z. Chen, C. He and H. Wang, Carbohydr. Polym., 2008, 72, 410–418 CrossRef CAS.
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670–5703 CrossRef CAS PubMed.
- K. Sun and Z. H. Li, eXPRESS Polym. Lett., 2011, 5, 342–361 CrossRef CAS.
- N. Bhattarai, D. Edmondson, O. Veiseh, F. A. Matsen and M. Zhang, Biomaterials, 2005, 26, 6176–6184 CrossRef CAS PubMed.
- F. L. Mi, Y. C. Tan, H. F. Liang and H. W. Sung, Biomaterials, 2002, 23, 181–191 CrossRef CAS PubMed.
- M. Z. Elsabee, H. F. Naguib and R. Elsayed, Mater. Sci. Eng., C, 2012, 32, 1711–1726 CrossRef CAS.
- C. Mahoney, M. B. Mccullough, J. Sankar and N. Bhattarai, J. Nanomed. Biother. Discovery, 2012, 2, 1–9 Search PubMed.
- F. E. Ahmed, B. S. Lalia and R. Hashaikeh, Desalination, 2015, 356, 15–30 CrossRef CAS.
- D. Annur, Z. K. Wang, J. Der Liao and C. Kuo, Biomacromolecules, 2015, 16, 3248–3255 CrossRef CAS PubMed.
- S. W. Ali, S. Rajendran and M. Joshi, Carbohydr. Polym., 2011, 83, 438–446 CrossRef CAS.
- C. R. Deeken, D. B. Fox, S. L. Bachman, B. J. Ramshaw and S. A. Grant, J. Biomed. Mater. Res., Part B, 2011, 97, 334–344 CrossRef PubMed.
- S. A. Grant, C. S. Spradling, D. N. Grant, D. B. Fox, L. Jimenez, D. A. Grant and R. J. Rone, J. Biomed. Mater. Res., Part A, 2014, 102, 332–339 CrossRef PubMed.
- Q. Cui, A. Yashchenok, L. Zhang, L. Li, A. Masic, G. Wienskol, H. Möhwald and M. Bargheer, ACS Appl. Mater. Interfaces, 2014, 6, 1999–2002 CAS.
- J. Jalvandi, M. White, Y. Gao, Y. B. Truong, R. Padhye and I. L. Kyratis, Int. J. Polym. Mater., 2017, 66, 507–513 CrossRef CAS.
- A. Fojtik and A. Henglein, Ber. Bunsen-Ges. Phys. Chem., 1993, 97, 1493–1496 CrossRef CAS.
- M. S. Sibbald, G. Chumanov and T. M. Cotton, J. Phys. Chem., 1996, 100, 4672–4678 CrossRef CAS.
- F. Mafune, J. Kohno, Y. Takeda and T. Kondow, J. Phys. Chem. B, 2002, 106, 7575–7577 CrossRef CAS.
- A. V. Kabashin and M. Meunier, J. Appl. Phys., 2003, 94, 7941–7943 CrossRef CAS.
- K. Maximova, A. I. Aristov, M. Sentis and A. V. Kabashin, Nanotechnology, 2015, 26, 065601 CrossRef CAS PubMed.
- D. Rioux, M. Laferrière, A. Douplik, D. Shah, L. Lilge, A. V. Kabashin and M. M. Meunier, J. Biomed. Opt., 2009, 14, 021010 CrossRef PubMed.
- P. Blandin, K. A. Maximova, M. B. Gongalsky, J. F. Sanchez-Royo, V. S. Chirvony, M. Sentis, V. Yu. Timoshenko and A. V. Kabashin, J. Mater. Chem. B, 2013, 1, 2489–2495 RSC.
- A. Al-Kattan, Y. V. Ryabchikov, T. Baati, V. Chirvony, J. F. Sanchez-Royo, M.-A. Estève, M. Sentis, V. Yu. Timoshenko, D. Braguer and A. V. Kabashin, J. Mater. Chem. B, 2016, 4, 7852–7858 RSC.
- A. V. Kabashin and V. Yu. Timoshenko, Nanomedicine, 2016, 11, 2247–2250 CrossRef CAS PubMed.
- K. P. Tamarov, L. A. Osminkina, S. V. Zinovyev, K. A. Maximova, J. V. Kargina, M. B. Gongalsky, Y. Ryabchikov, A. Al-Kattan, A. P. Sviridov, M. Sentis, A. V. Ivanov, V. N. Nikiforov, A. V. Kabashin and V. Y. Timoshenko, Sci. Rep., 2014, 4, 7034 CrossRef CAS PubMed.
- T. Baati, A. Al-kattan, M. Esteve, L. Njim, Y. Ryabchikov, F. Chaspoul, M. Hammami, M. Sentis, A. V. Kabashin and D. Braguer, Sci. Rep., 2016, 6, 25400 CrossRef CAS PubMed.
- S. Hebie, Y. Holade, K. Maximova, M. Sentis, P. Delaporte, K. Kokoh, T. Napporn and A. V. Kabashin, ACS Catal., 2015, 5, 6489–6496 CrossRef CAS.
- J. P. Sylvestre, S. Poulin, A. V. Kabashin, E. Sacher, M. Meunier and J. H. T. Luong, J. Phys. Chem. B, 2004, 108, 16864–16869 CrossRef CAS.
- A. E. Deniz, H. A. Vural, B. Ortaç and T. Uyar, Mater. Lett., 2011, 65, 2941–2943 CrossRef CAS.
- E. Y. Kim, D. Kumar, G. Khang and D.-K. Lim, J. Mater. Chem. B, 2015, 3, 8433–8444 RSC.
- L. Tian, L. Lu, Y. Qiao, S. Ravi, F. Salatan and M. P. Melancon, J. Funct. Biomater., 2016, 7, 19 CrossRef PubMed.
- K. Desai, K. Kit, J. Li and S. Zivanovic, Biomacromolecules, 2008, 9, 1000–1006 CrossRef CAS PubMed.
- L. Li and Y. L. Hsieh, Carbohydr. Res., 2006, 341, 374–381 CrossRef CAS PubMed.
- J. Deitzel, J. Kleinmeyer, D. Harris and N. Beck Tan, Polymer, 2001, 42, 261–272 CrossRef CAS.
- J. An, H. Zhang, J. Zhang, Y. Zhao and X. Yuan, Colloid Polym. Sci., 2009, 287, 1425–1434 CAS.
- Y. Wan, H. Wu, A. Yu and D. Wen, Biomacromolecules, 2006, 7, 1362–1372 CrossRef CAS PubMed.
- I. Slowing, B. G. Trewyn and V. S.-Y. Lin, J. Am. Chem. Soc., 2006, 128, 14792–14793 CrossRef CAS PubMed.
- L. Guo, H. Sato, T. Hashimoto and Y. Ozaki, Macromolecules, 2010, 43, 3897–3902 CrossRef CAS.
- K. Deyao, P. Tao, M. F. a. Goosen, J. M. Min and Y. Y. He, J. Appl. Polym. Sci., 1993, 48, 343–354 CrossRef.
- S. Zivanovic, J. Li, P. M. Davidson and K. Kit, Biomacromolecules, 2007, 1505–1510 CrossRef CAS PubMed.
- B. Duan, C. Dong and X. Yuang, J. Biomater. Sci., Polym. Ed., 2004, 15, 797–811 CrossRef CAS PubMed.
- M. Chen, P. K. Patra, S. B. Warner and S. Bhowmick, Tissue Eng., 2007, 13, 579–587 CrossRef CAS PubMed.
- S. E. Noriega, G. I. Hasanova, M. J. Schnieder, G. F. Larsen and A. Subramanian Chen, Cells Tissues Organs, 2012, 195, 207–221 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |