
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe synthesis of two long-chain N-hydroxy amino coumarin compounds and their applications in the analysis of aldehydes†
Zhaobing Guan‡
,
Manman Ding‡,
Yao Sun,
Sisi Yu,
Ao Zhang,
Shuguang Xia,
Xiaosong Hu * and
Yawei Lin*
* and
Yawei Lin*
Department of Chemistry, School of Chemistry, Chemical Engineering and Life Sciences, Wuhan University of Technology, Wuhan, 430070, P. R. China. E-mail: xhu@whut.edu.cn; linyawei2012@whut.edu.cn
First published on 4th April 2017
Abstract
Herein, two N-substituted coumaryl hydroxylamines (compounds 1a and 1b) with long aliphatic chains were prepared in 8 steps with a novel synthetic protocol. They served as derivatization agents for aldehydes by the formation of nitrones under mild conditions, which can be readily analysed by LC-MS with good chromatographic performance, excellent fluorescent response, and high ionization strength. We successfully used compounds 1a and 1b in the analysis of furfuraldehydes in raisins, based on the derivatization reactions of standard furfural (F), 5-methylfurfural (5-MF) and 5-(hydroxymethyl)furfural (5-HMF) samples. It proved that they are excellent agents for the analysis of aldehydes in foodstuffs. The derivatization reaction between compound 1a and hexanal suggested that the applications of our designed derivatization agents can be further extended to the analysis of aliphatic aldehydes. This study demonstrated that the designed N-substituted coumaryl hydroxylamines are excellent probes for the analysis of various aldehydes which are important in multiple research areas.
Introduction
Aldehydes are widely found in foods,1–6 beverages,7–12 the atmosphere,13–16 environmental water17,18 and biological systems.19–24 Concentrations of them have major impact on human health and quality of life. In the household building materials industry, the concentrations of aldehydes in the materials are very important indexes of quality. Certain aldehydes have been classified by the World Health Organization (WHO) as carcinogenic and teratogenic compounds. The concentration of aldehydes in the materials must be low enough for them to be qualified for home building.16 In the food industry, concentrations of aldehydes play important roles in the quality evaluation system. A low abundance of aldehydes may provide the food with pleasant flavour, while a high abundance of them will accelerate the corruption of food and thus affect the freshness.1 Though aldehydes are minor compounds in foodstuffs, they have drawn more and more attention in the detection and analysis of them.1–5 The existence of certain aldehydes can serve as indicators of quality deterioration, microbacterial fermentation and so on.2,5,6 This makes the determination of aldehydes in foodstuffs critical for consumer protection and quality control. Besides the points mentioned above, certain aliphatic aldehydes are key biomarkers for lipid peroxidation in biological organisms, which has a major effect on a variety of diseases including heart disease, diabetes mellitus, et al.22–24 The analysis of aldehydes is thus very helpful for the evaluation of these diseases.The combined liquid chromatography-mass spectrometry (LC-MS) is one of the most widely used technique for the analysis of aldehydes.13,22–24 Derivatization is primarily applied to enhance ionization characteristics in LC-MS analysis of aldehydes based on soft ionization techniques such as electrospray (ESI) and atmospheric pressure chemical ionization (APCI).22 This is because aldehydes and other analytes such as sugars and steroids are poorly ionized by ESI or APCI. The derivatization process usually involves an organic reaction of an analyte molecule with a suitable derivatization reagent. 2,4-Dinitrophenylhydrazine (DNPH) is the most widely used derivatization agent for aldehydes.7,8,13,22,24 It contains a reactive functional group (hydrazine) which selectively reacts with a complementary functional group (carbonyl) in the analytes. DNPH derivatives of aldehydes are subjected to LC-MS analysis using an ultraviolet absorption detector, which can response to the DNPH moiety of the derivatives.
Derivatization agents containing other reactive function groups, such as hydroxylamines, have also been used in the analysis of aldehydes.8,17,18,20 They are used in various applications including chemical synthesis, drug discovery and chemical biology, due to their high nucleophilicity and other specific properties.25 Both O- and N-substituted hydroxylamine derivatives are widely used in these applications (Scheme 1). O-Substituted hydroxylamine derivatives realize their applications through functionalization with O-amino or aminooxy (–ONH2) group, forming oximes in most situations. Oximes usually need to be further reduced for derivatization study.26 The applications of N-substituted hydroxylamine derivatives are carried out through functionalization with N-hydroxy amino (–NHOH) group, usually forming nitrones as products. Though nitrones can be further reduced, the analysis of aldehydes based on N-substituted hydroxylamine can get satisfactory results without reduction step.26 Furthermore, the reactivity of hydroxylamine with aldehyde is high and the reaction only requires mild condition. It thus has been chosen in this study as the reactive function group in the derivatization agents for the aldehyde analysis.
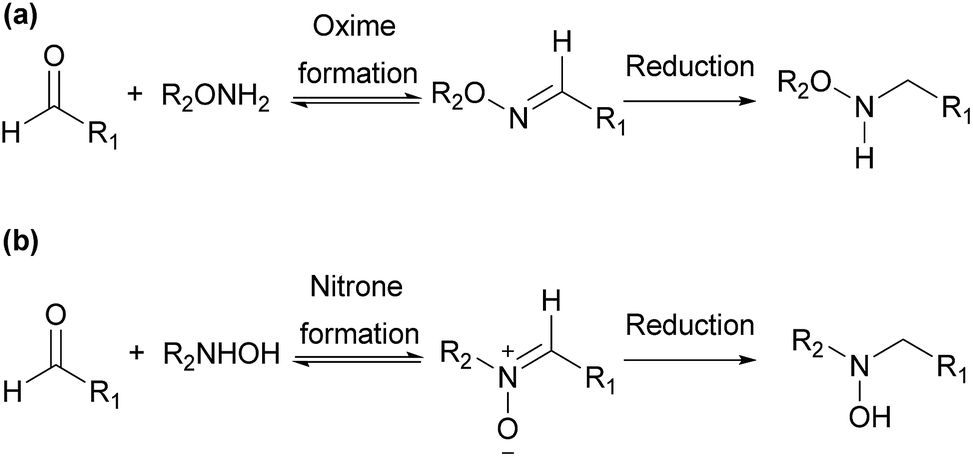 | ||
| Scheme 1 Derivatization of aldehyde using O- and N-substituted hydroxylamine. | ||
Fluorescence spectroscopy is one of the most sensitive optical detection technique used with high-performance liquid chromatography (HPLC). In fluorescence, the intensity of the emission of the sample is usually measured above a low background level, which explains why fluorescence techniques have high sensitivity. Fluorescence of a molecule can occur naturally, or it can be introduced by labelling the molecule with a fluorescent tag through derivatization reaction.27–29 We therefore incorporated fluorophore into the designed derivative agents with hydroxylamine moiety to facilitate the detection during HPLC-MS analysis. The fluorophore we chose is coumarin group, which has been widely used as fluorescent probe in biological studies due to its minimal toxicity, high fluorescence quantum yield, relatively large Stokes shift, small size, ease to synthesize, sensitivity to pH and solvent polarity.30–32 An unnatural amino acid bearing coumarin group was widely used in a variety of biological applications.30,31 Coumarin derivatives containing hydrodxylamine functional group were used in the selective detection of 5-formyl-2′-deoxycytidine in DNA,33 and the structural analysis of glycans.26 In these coumaryl derivatives of O- and N-substituted hydroxylamines, hydroxylamine group is attached to the coumarin ring with a short aliphatic carbon chain. Muddiman et al. found that increasing the length of carbon chain in the alkyl tags made them more hydrophobic, thus improved the chromatographic and electrospray response of the peptide labeled by the tags.34 Encouraged by this founding, we synthesized two novel designed long-chain coumaryl derivatives of hydroxylamine (compound 1a and 1b in Scheme 2), and used them in the structural analysis of aldehydes based on LC-MS analysis. The hydroxylamine group is attached to the coumarin ring at 4-position and separated from the coumarin ring by four carbon atoms. The coumarin ring is assembled via Pechmann condensation,30,31,35 which allows derivatization at other positions on the coumarin ring, and provides applicable method to further increase the chain length between the coumarin and hydroxylamine moieties. Thus more hydroxyamino coumarin derivatization agents with different fluorescent properties can be prepared and applied in aldehyde analysis. These coumaryl hydroxylamine was N-substituted, since it was more convenient for the derivatization as discussed above.
 | ||
| Scheme 2 Two novel designed long-chain coumaryl derivatives of hydroxylamine. | ||
Furfural (F), 5-methylfurfural (5-MF) and 5-(hydroxymethyl)furfural (5-HMF) occur in many foodstuffs and flavorants at certain level.2,5,10,12 The existences and concentrations of these furfuraldehydes are relevant to food safety and flavor, which makes the detection and analysis of them quite important. In this study, we focused on the analysis of them via derivatization reaction with 1a and 1b. The LC-MS results proved they are excellent derivatization agents for furfuraldehydes. Derivatization reaction of hexanal with 1a indicated the application of them can be extended to other aldehydes important in environmental or biological studies.
Results and discussion
The synthesis of compound 1a and 1b is outlined in Scheme 3. The coumarin ring with a 4-bromobutyl group at the 4-position (compound 7) was constructed via Pechmann condensation30,31,35,36 between ethyl 7-bromo-3-oxoheptanoate (compound 5) and resorcinol or 3-methoxyphenol, using sulphuric acid as the condensing agent (7a, 43.3% yield; 7b, 23.5% yield). Compound 7 was then hydrolyzed under reflux in water to afford corresponding alcohol 8, which was further oxidized to aldehyde 9 with pyridinium chlorochromate (PCC)37 in acetone at 65 °C. It then reacted with hydroxylamine hydrochloride to form oxime 10, which was then treated with sodium cyanoborohydride to afford N-substituted coumaryl hydroxylamine 1 as the target compound. | ||
| Scheme 3 The synthetic protocol for the derivatization agents 1a and 1b. | ||
Ethyl 7-bromo-3-oxoheptanoate 5 was a major intermediate compound for the synthesis of the target compound. It was synthesized in three steps, which started with the Reformatsky reaction38 between cyclopentanone 2 and ethyl 2-bromoacetate to form a tertiary alcohol (compound 3). The Reformatsky reaction was chosen to convert 2 into an alcohol, because the enolates formed are less reactive compared with lithium enolates or Grignard reagents, which prevents the occurrence of nucleophilic addition to the ester group. Then under alkaline bromination condition via retro-Reformatsky fragmentation,39 the tertiary alcohol was converted efficiently into α,α,ω-tribromo-β-ketoester (4, 95.3% yield). Compound 4 was reduced using Cu–Zn alloy in saturated ammonium chloride methanol solution at 10–15 °C to afford compound 5 (64.4% yield).
Pechmann condensation was chosen to assemble the coumarin ring. It is an efficient and practical procedure for the synthesis of 4-substituted coumarins. The length of carbon chain attached to the 4-the corresponding carbon chain which is attached to ω-bromo-β-ketoester. This make it relatively easy to adjust the length of the sidechain in the target compounds, which is important for the chromatographic performance and electrospray response of the derived products. Furthermore, derivatives with substituents at the 5-, 6- or 8-position can also be prepared with Pechmann condensation, some of which can serve as potential useful probes with different functionalities or fluorescent properties for a variety of applications. The Pechmann condensation used in this study were performed under mild condition, using sulphuric acid as the condensing agent at ambient temperature. The yield is not very high (7a, 43.3% yield; 7b, 23.5% yield) compared with condensation between resorcinol and ethyl acetoacetate at similar condition. This clearly demonstrates that the substituent attached to the β-keto may have a great effect on the condensation result. The substitution of hydroxy group at 3-position in resorcinol with methoxy group drastically decreased the yield of the condensation reaction, suggesting that the reactivity of 3-methoxyphenol is greatly reduced compared with resorcinol.
The hydrolysis of compound 7 was straight forward, which could be achieved smoothly in a sealed tube in a 115 °C oil bath to afford the corresponding alcohols (8a, 93.0% yield; 8b, 56.4% yield). Compound 7b is less soluble in water compared with 7a, since it has a methoxy group at 7-postion whereas 7a has a more hydrophilic 7-hydroxy group on the coumarin ring. The less solubility of 7b might lead to the lower yield of hydrolysis product. PCC oxidation37 was then carried out to convert compound 8 to 9 in acetone at 65 °C with sodium acetate and diatomite added. It was chosen because it can oxidize the terminal primary alcohol to aldehyde with high selectivity and no over-oxidation occurs to form carboxylic acid. Using manganese dioxide as oxidant didn't provide comparable yields and were less efficient based on our test.
Oxime 10 was prepared by reacting compound 9 with hydroxylamine hydrochloride in a mixture of tetrahydrofuran and water at r.t., while the pH value was adjusted to 4.26 The 13C NMR of 10 indicated that both Z- and E-isomer of oxime existed and one of them was predominating. To determine which isomer is the major product needs further investigation. Finally, oxime 10 was reduced to N-substituted hydroxylamine 1 using sodium cyanoborohydride in a methanol solution of hydrogen chloride at a pH value of 2–3.26 After TLC indicated the complete consumption of the reactant, the solution was directly applied to a RP-HPLC employing a semi-preparative C18 column to afford 1 as a white solid (1a, 63.9% yield; 1b, 70.6% yield). The purity was very high according to 1H and 13C NMR spectra. Sodium cyanoborohydride was chosen as the reductant because it can selectively reduce the oxime to N-substituted hydroxylamine with a high efficiency at relatively low cost.
This is a relatively efficient protocol to synthesize the N-substituted hydroxylamine as designed, which consists of 8 steps of reactions in total. Most steps are relatively straight forward while some steps need more attention. The synthetic protocol provided a practical way to synthesize N-substituted coumaryl hydroxylamine with long aliphatic chain, and the length of it can be varied when proper cyclic ketone with different ring sizes are used as starting reagents. The Pechmann condensation method used to install the coumarin moiety, which makes it possible to introduce substituent groups at the 5-, 6- and 8-position of the coumarin ring. This provides the possibility to introduce more functional groups at each position for further derivatization and may improve the fluorescent properties of the designed probes.
The fluorescent spectra of derivatization agent 1a and 1b were shown in Fig. 1. The excitation and emission wavelengths of compounds 1a are 322 nm and 447 nm, and those of 1b are 320 nm and 390 nm respectively. Stokes shift of 1b is 70 nm, which is much smaller compared with that of 1a (125 nm). The quantum yields of 1a and 1b were 0.61 and 0.72 respectively, which were measured according to the literature40 using quinoline sulfate as standard. This result indicates that the modification of the substituents on the coumarin ring will affect the fluorescent property, which might be important in certain investigations based on the derivatization reaction with these compounds. The structure modification of the derivatization reagents can be realized via Pechmann condensation discussed previously.
 | ||
| Fig. 1 Fluorescence spectra of compounds 1a (a) and 1b (b). | ||
The derivatization reaction was initially tried between compound 1a with furfural to test the feasibility of the aldehyde analysis using the hydroxyamino coumarin compounds. After compound 1a reacted with furfural at 70 °C in an acetic acid/sodium acetate buffer for 1 h, the mixture was analysis by HPLC with a fluorescence detector, the excitation and emission wavelengths of which are set at 322 nm and 447 nm. The result was compared with that of the sample blank of compound 1a in an acetic acid/sodium acetate buffer, as shown in Fig. 2. A new peak with a high intensity showed up around 31 min in the chromatogram of the derivatization mixture. It suggested that compound 1a might have reacted with furfural, forming the nitrone derivative. This was confirmed by collecting the fractions corresponding to the peak showing up at 31 min, and analyzing them by LC-MS.
 | ||
| Fig. 2 HPLC analysis of the furfural derivatization mixture and sample blank of compound 1a. | ||
The chemical equation for the derivatization reaction is shown in Scheme 4 with m/z values of compound 1a and the corresponding nitrone derivative listed below. Mass spectrum in Fig. 3 is obtained from the peak showing up at 31 min in the HPLC chromatogram in Fig. 2. It indicates that the major peak has an m/z value of 328.1180, which corresponds to the singly charged [M + H]+. This confirms the successful derivatization of furfural with compound 1a to form the nitrone. The intensity of peak is pretty high, suggesting that the long aliphatic chain in the derivative can enhance the ionization strength in mass spectroscopy. It also validates that no further reduction reaction is needed in the aldehyde analysis. The HPLC detection limit, linear calibration range, regression equation and correlation coefficient of the 1a-furfural nitrone derivative under our experimental condition was measured and summarized (Table S1 in ESI†). The calibration curves showed good linearity between concentration and peak area with correlation coefficient as 0.9965. The reproducibility of the proposed method, expressed by relative standard deviation (RSD), is 7.23%. The detection limit was 1.6 nM at a signal-to-noise ratio (S/N) of 3. Compared with other analytical methods, the limit of detection LOD of our method is 1.6 nM, which is more sensitive than those of GC-MS method (49 nM) and HPLC with pulsed amperometric detection method (400 nM) according to the ref. 41 and 42 The strong peak at 31 min in Fig. 2 indicates the excellent fluorescent property of the coumarin fluorophore.
 | ||
| Scheme 4 Derivatization reaction of furfural with compound 1a. | ||
 | ||
| Fig. 3 Mass spectrum of the nitrone derivative which was generated from the reaction between furfural and compound 1a. | ||
After the successful derivatization of furfural with compound 1a, we also tested analogous derivatization with compound 1b. A singly charged [M + H]+ peak with an m/z value of 342.1339 proved that compound 1b is also a good derivatization agent for aldehydes (Fig. S35 in ESI†). The peak comes from the corresponding nitrone derivative of furfural which has an m/z value of 341.13 for the most abundant form.
We then carried out more derivatization reactions of aldehydes to include 5-MF and 5-HMF as well. Respective HPLC analysis of F, 5-MF, 5-HMF derivatization mixtures are compared with that of compound 1a as a sample blank, which is shown in Fig. 4. It clearly indicated that all of these three aldehydes can be successfully derived with compound 1a. These three derivatives have distinct retention times and strong fluorescent responses at certain chromatographic condition, which makes compound 1a an excellent derivatization agent for the analysis of them. The analysis of a mixture of F, 5-MF and 5-HMF at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 molar ratio based on derivatization with 1a further confirmed this. After the three aldehydes in the mixture were derived with compound 1a, the mixture was analyzed with HPLC, three derivatives were well separated, which is illustrated in Fig. 5. Each derivative has same retention time as that derived separately and shown in Fig. 4.
:1:1 molar ratio based on derivatization with 1a further confirmed this. After the three aldehydes in the mixture were derived with compound 1a, the mixture was analyzed with HPLC, three derivatives were well separated, which is illustrated in Fig. 5. Each derivative has same retention time as that derived separately and shown in Fig. 4.
 | ||
| Fig. 4 Respective HPLC analysis of F, 5-MF, 5-HMF derivatization mixtures, compared with corresponding HPLC analysis of compound 1a as a sample blank. | ||
 | ||
| Fig. 5 HPLC analysis of a mixture of F, 5-MF, 5-HMF (1:1:1 molar ratio) derived by compound 1a, which is compared with the corresponding HPLC analysis of compound 1a as a sample blank. | ||
With these results in hand, we further applied compound 1a in the analysis of F, 5-MF, 5-HMF in common foods. Using commercially purchased raisin as an example, after it was extracted by a mixture of methanol and ammonium acetate and purified, the supernate was analyzed by compound 1a using previously discussed method. HPLC result indicated the existence of F, 5-MF, 5-HMF (Fig. 6). The peak areas of each corresponding signal suggest that the concentration of 5-HMF is much higher compared with that of F and 5-MF. There is only trace amount of F and 5-MF existing in the raisin since the signals of these two derivatives are very weak. It strongly validates that the designed long-chain hydroxyamino coumarin compounds are excellent agents for the analysis of aldehydes in foodstuff, and it should also work well in the analysis of aldehydes from other sources.
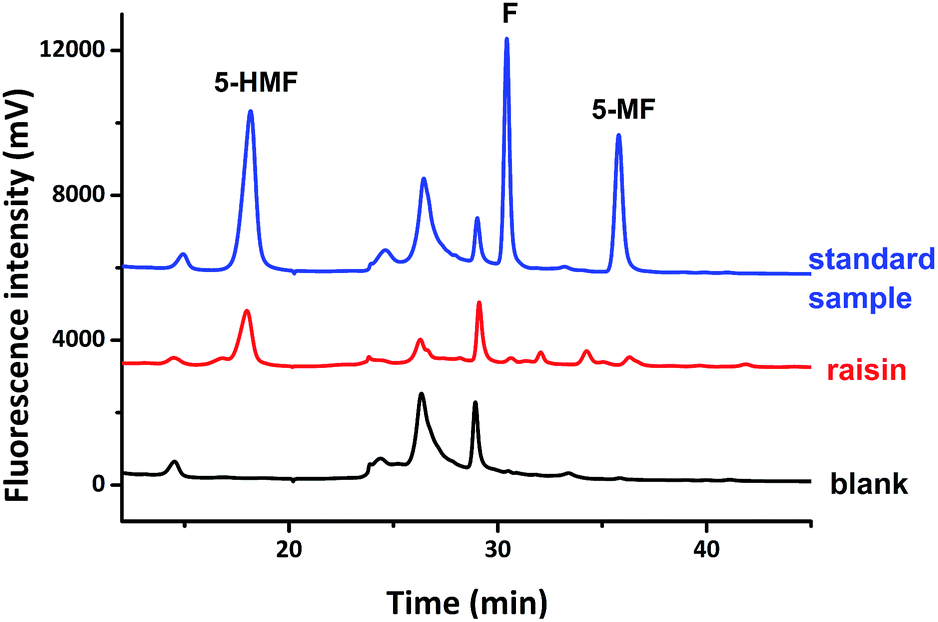 | ||
| Fig. 6 Analysis of aldehydes in raisin based on derivatization reaction using compound 1a, compared with the analysis of compound 1a as a sample blank, as well as that of the derivatives of F, 5-MF and 5-HMF as standard. | ||
Besides the derivatization with furfural related aldehydes, hydroxyamino coumarin compounds can also react with aliphatic aldehydes. We studied the derivatization reaction between compound 1a and hexanal. A singly charged [M + H]+ peak with an m/z value of 332.1858 indicated the successful derivatization of hexanal, since the corresponding nitrone derivative of hexanal has an m/z value of 331.1784 for the most abundant form (Fig. S36 in ESI†). Because aliphatic aldehydes widely exist in nature, the usage of these two long-chain hydroxyamino coumarin agents can be extend to more research areas.
Conclusion
Through a well-designed synthetic protocol, N-substituted coumaryl hydroxylamines with long aliphatic chain (1a and 1b) were prepared and served as derivatization agents for aldehydes. The coumarin rings in the derivatization agents were assembled via Pechmann condensation, which provides a powerful means to change the length of the sidechain. Different substituents at 5-, 6-, 7- or 8-position of the coumarin ring in the derivatization agents can also be introduced via Pechmann condensation to provide more potential useful probes with various functionalities or fluorescent properties. This has been verified by the successful preparation of compound 1a and 1b. These two compounds were prepared with good yield and high purity as indicated by the NMR spectra. Long sidechain in the derivatization agents can improve the chromatographic performance and enhance ionization strength in mass spectrometry, as shown in the analysis of aldehydes through derivatization reaction with 1a and 1b followed by LC-MS analysis. Nitrone can be easily formed by the derivatization reaction between aldehydes and 1a or 1b under mild condition, and it can be readily analyzed without difficulty, which proves the effectiveness of N-substituted hydroxylamine in aldehyde labeling. The excellent fluorescent properties of 1a, 1b and their derived products from reacting with aldehydes validate the correctness of choosing coumarin as the fluorophore in the derivatization agents. Aldehydes at μmol level can be detected and analysed with our agents. Aldehydes chosen in the study are focused on furfuraldehydes including F, 5-MF, and 5-HMF, the existences and concentrations of which are closely relevant to food safety and flavor. We successfully used these N-substituted coumaryl hydroxylamines in the detection of these furfuraldehydes in raisin, based on the derivatization reactions of standard F, 5-MF, and 5-HMF samples. This result proved that they can serve as excellent agents for the analysis of aldehydes in foodstuffs. The derivatization reaction between 1a and hexanal indicated that the usage of them can be further extended to the analysis of aliphatic aldehydes. In the future, we will broaden the application of these two agents to the research areas of environmental and biological sciences, and synthesize more derivatization agents with different sidechain length and substituents on the coumarin ring. It should be able to diversify the applications of N-substituted coumaryl hydroxylamine and further enhance the effectiveness and efficiency of them in the aldehyde analysis.Experimental
Materials and instrumentation
All chemicals were purchased from commercial suppliers and used without further purification. Flash chromatography was carried out with silica gel (200–300 mesh). Analytical TLC was performed with silica gel GF254 plates. Melting points were determined on a SGW X-4 melting point apparatus with microscope (Shanghai Precision & Scientific Instrument Co., Ltd). The H-NMR spectra and C-NMR spectra were recorded at 25 °C on 500 MHz or 125 MHz NMR spectrometer (Bruker Avance III). Chemical shifts (δ) are reported in PPM using Me4Si as an internal standard (δ = 0 ppm), or using the deuterated solvent signal as reference. Spin–spin coupling constants (J) are given in Hz. Mass spectra were measured with an Agilent Technologies 2100 mass spectrometer, or with a TripleTOF 5600 System (AB SCIEX) equipped with a nanospray source. Fluorescence spectra were recorded on a RF-5301 fluorescence spectrophotometer (Shimadzu, Tokyo, Japan). Chromatographic analyses were performed on a LC-20AT HPLC system with a RF-20 fluorescence detector and a chromatography data acquisition system (Shimadzu, Tokyo, Japan). Separations were performed on an Agilent Eclipse 5 μm, XDB-C18 column (USA).Synthetic procedures
:ethyl acetate, 3:1) to afford compound ethyl 2-(1-hydroxycyclopentyl)acetate as a colourless liquid (9.66 g, 56.1 mmol, 93.5% yield). 1H NMR (500 MHz, CDCl3): δ 4.18 (q, 2H, J = 7.1 Hz), 2.60 (s, 1H), 1.90–1.78 (m, 4H), 1.66–1.50 (m, 4H), 1.28 (t, 3H, J = 7.1 Hz). 13C NMR (125 MHz, CDCl3): δ 173.27, 79.48, 60.60, 44.58, 39.67, 23.80, 14.19.:ethyl acetate, 30:1) gave pure ethyl 2,2,7-tribromo-3-oxoheptanoate as a yellow liquid (11.7 g, 28.6 mmol, 95.3% yield). 1H NMR (500 MHz, CDCl3): δ 4.38 (q, 2H, J = 7.1 Hz), 3.43 (t, 2H, J = 6.4 Hz), 2.98 (t, 2H, J = 7.0 Hz), 1.97–1.85 (m, 4H), 1.36 (t, 3H, J = 7.1 Hz). 13C NMR (125 MHz, CDCl3): δ 193.26, 163.73, 64.83, 59.76, 34.96, 32.89, 31.55, 23.45, 13.77.:40, W/W, 12.3 g, 300 mesh). The mixture was then stirred at 10–15 °C for 1.5 h. The TLC indicated the complete consumption of ethyl 2,2,7-tribromo-3-oxoheptanoate. After the reaction mixture was filtered, the organic layer was separated, dried with sodium sulfate and concentrated. Purification by flash chromatography (petroleum ether:ethyl acetate, 30:1) afforded ethyl 7-bromo-3-oxoheptanoate as a red brown liquid (1.94 g, 7.73 mmol, 64.4% yield). 1H NMR (500 MHz, CDCl3): δ 4.20 (q, 2H, J = 7.1 Hz), 3.44 (s, 2H), 3.41 (t, 2H, J = 6.6 Hz), 2.60 (t, 2H, J = 7.1 Hz), 1.95–1.85 (m, 2H), 1.81–1.72 (m, 2H), 1.29 (t, 3H, J = 7.1 Hz). 13C NMR (125 MHz, CDCl3): δ 202.17, 167.15, 61.43, 49.26, 41.81, 33.14, 31.79, 21.94, 14.11.:ethyl acetate, 3:1) to afford 4-(4-bromobutyl)-7-hydroxy-2H-chromen-2-one as a white solid (1.03 g, 3.47 mmol, 43.3% yield). mp: 139–141 °C. 1H NMR (500 MHz, DMSO-d6): δ 10.05 (s, 1H), 7.66 (d, 2H, J = 8.9 Hz), 6.80 (dd, 1H, J1 = 8.9 Hz, J2 = 2.4 Hz), 6.71 (d, 1H, J = 2.4 Hz), 6.10 (s, 1H), 3.59 (t, 2H, J = 6.7 Hz), 2.77 (t, 2H, J = 7.6 Hz), 1.88–1.94 (m, 2H), 1.70–1.76 (m, 2H); 13C NMR (125 MHz, DMSO-d6): δ 161.56, 160.84, 155.62, 126.83, 113.39, 111.60, 109.85, 102.89, 35.12, 32.27, 30.38, 27.03.:ethyl acetate, 3:1) to afford 4-(4-bromobutyl)-7-methoxy-2H-chromen-2-one as a white solid (1.83 g, 5.88 mmol, 23.5%). mp: 71–72 °C. 1H NMR (500 MHz, DMSO-d6): δ 7.75 (d, 1H, J = 8.9 Hz), 7.66 (d, 2H, J1 = 8.9 Hz, J2 = 1.5 Hz), 6.95–6.99 (m, 2H), 6.18 (s, 1H), 3.60 (t, 3H, J = 6.4 Hz), 3.59 (t, 2H, J = 6.7 Hz),2.80 (t, 2H, J = 6.7 Hz), 1.89–1.94 (m, 2H), 1.71–1.76 (m, 2H); 13C NMR (125 MHz, DMSO-d6): δ 162.78, 160.70, 157.02, 155.57, 126.68, 112.64, 110.74, 101.45, 56.39, 35.14, 32.24, 30.36, 26.98.:ethyl acetate, 5:1) to afford another portion of the product. In total, 0.842 g of 4-(4-hydroxybutyl)-7-methoxy-2H-chromen-2-one as white needle-like crystals was produced (3.39 mmol, 56.4% yield). mp: 97–98 °C. 1H NMR (500 MHz, DMSO-d6): δ 7.74 (d, 1H, J = 8.9 Hz), 6.95–6.98 (m, 2H), 6.16 (s, 1H), 4.41 (m, 1H), 3.86 (s, 3H), 3.42–3.46 (m, 2H), 2.75–2.78 (m, 2H), 1.62–1.68 (m, 2H), 1.51–1.55 (m, 2H); 13C NMR (125 MHz, DMSO-d6): δ 162.73, 160.75, 157.54, 155.55, 126.67, 112.63, 110.63, 101.41, 60.76, 56.37, 32.47, 31.19, 25.12.:ethyl acetate, 1:1) to afford 4-(7-hydroxy-2-oxo-2H-chromen-4-yl)butanal as a white powder (141 mg, 0.607 mmol, 47.4% yield). 1H NMR (500 MHz, DMSO-d6): δ 10.50 (s, 1H), 9.69 (s, 1H), 7.67 (d, 1H, J = 8.9 Hz), 6.80 (d, 1H, J1 = 8.6 Hz, J2 = 2.1 Hz), 6.71 (d, 1H, J = 2.1 Hz), 6.09 (s, 1H), 2.74 (m, 2H), 2.58 (m, 2H), 1.83–1.88 (m, 2H); 13C NMR (125 MHz, DMSO-d6): δ 203.52, 161.57, 160.83, 156.93, 155.64, 126.80, 113.42, 111.57, 110.02, 102.91, 42.83, 30.72, 21.18. ESI-MS, m/z: 233 [M + H]+.:ethyl acetate, 1:1) to afford 4-(7-methoxy-2-oxo-2H-chromen-4-yl)butanal as a white powder (286 mg, 1.16 mmol, 61.4% yield). mp: 73–75 °C. 1H NMR (500 MHz, DMSO-d6): δ 9.70 (s, 1H), 7.70 (d, 1H, J = 8.9 Hz), 6.96–6.98 (m, 2H), 6.17 (s, 1H), 3.86 (s, 3H), 2.75–2.78 (m, 2H), 2.57–2.60 (m, 2H), 1.83–1.89 (m, 2H); 13C NMR (125 MHz, DMSO-d6): δ 203.53, 162.78, 160.69, 156.80, 155.58, 126.63, 112.66, 110.90, 101.44, 56.38, 42.80, 30.71, 21.13.:V = 1:1) was added hydroxylamine hydrochloride (45 mg, 0.65 mmol), and the pH value of the solution was adjusted to 4 with NaOH (2 M). It was then stirred at r.t. until TLC indicated the completion of the reaction. The solvent was then removed and the residue was purified by flash chromatography (petroleum ether:ethyl acetate, 1:1) to afford 4-(7-hydroxy-2-oxo-2H-chromen-4-yl)butanal oxime as a white powder (101 mg, 0.408 mmol, 94.8% yield). mp: 136–138 °C. 1H NMR (500 MHz, DMSO-d6): δ 10.49 (s, 1H), 7.64 (dd, 2H, J1 = 8.9 Hz, J2 = 2.2 Hz), 6.80 (m, 1H), 6.71 (d, 1H, J = 2.4 Hz), 6.10 (d, 1H, J = 8.9 Hz), 2.75 (t, 2H, J = 6.7 Hz), 2.77 (t, 2H, J = 7.6 Hz), 1.88–1.94 (m, 2H), 2.18–2.36 (m, 2H), 1.70–1.82 (m, 2H); 13C NMR (125 MHz, DMSO-d6): δ 161.55, 160.85, 157.10, 155.62, 149.50, 126.84, 113.44, 111.61, 109.98, 102.90, 30.85, 29.12, 25.65. ESI-MS, m/z: 248 [M + H]+, 270 [M + Na]+.:V = 1:1) was added hydroxylamine hydrochloride (306 mg, 4.40 mmol), and the pH value of the solution was adjusted to 4 with NaOH (2 M). It was then stirred at r.t. until TLC indicated the completion of the reaction. The solvent was then removed and the residue was purified by flash chromatography (petroleum ether:ethyl acetate, 1:1) to afford 4-(7-methoxy-2-oxo-2H-chromen-4-yl)butanal oxime as a white powder (249 mg, 0.953 mmol, 80.1%). 1H NMR (500 MHz, DMSO-d6): δ 10.44 (s, 1H), 7.75 (d, 1H, J = 8.9 Hz), 7.36–7.38 (m, 1H), 6.95–6.99 (m, 2H), 6.17 (s, 1H), 3.32 (s, 3H), 2.78–2.81 (m, 2H), 2.21–2.25 (m, 2H), 1.75–1.81 (m, 2H); 13C NMR (125 MHz, DMSO-d6): δ 162.77, 160.70, 156.94, 155.57, 149.48, 126.67, 112.67, 111.87, 101.44, 56.38, 30.83, 29.10, 25.60.Fluorescent analysis
The fluorescent spectra of compound 1a and 1b were collected on a Shimadzu RF-5301 fluorescence spectrophotometer. Compound 1a was dissolved in methanol, diluted by 10% aqueous acetonitrile solution to a concentration of 0.5 mM. It was further diluted by acetic acid/sodium acetate buffer (0.1 M, pH = 4) to a final concentration of 0.5 μM and used in the fluorescent analysis. The procedure for compound 1b was similar. The only difference was that the final concentration of 1b used in the fluorescent analysis was 0.8 μM.The quantum yield quantum yields (φF) of compound 1a and 1b were determined using quinoline sulfate solution (2 μM) in sulfuric acid (0.1 M) as a standard (φF = 0.55). The fluorescent quantum yield was calculated with the expression φ1 = φ2 × (F1A2N12)/(F2A1N22), where φ1 and φ2 are the quantum yields of the samples and the standard, F1, F2 are the peak areas under the emission curves of the samples and the standard, A1, A2 are the absorbance of the sample and the standard, N1 and N2 are the solvent refractive factors of the sample and the standard, respectively.
Derivatization with aldehydes
In a typical derivatization procedure for standard aldehyde samples, methanol solution of aldehydes (20 μL, 0.1 mM), derivatization agents (1a or 1b, 20 μL, 0.1 mM), acetic acid/sodium acetate buffer (10 μL, 0.1 M, pH = 3.5) were mixed and allowed to react at 70 °C for 1 h. A similar procedure without aldehydes was carried out as a control. The mixture was then cool to r.t. and directly applied to HPLC analysis with a fluorescence detector.Analysis of raisin sample
Commercially purchased raisin (5.0 g) was put in a 50 mL centrifuge tube, methanol (5 mL) and ammonium acetate (20 mL, 0.02 M) solution were then added. The pH value of the solution was adjusted to 3.5 with acetic acid. The mixture was then sonicated for 30 min. The supernate was further filter by an ultrafiltration tube which has a molecular weight cutoff of 5 kD. Raisin extract (10 μL), acetic acid/sodium acetate buffer (10 μL, 0.1 M, pH = 3.5), compound 1a (20 μL, 1 mM) and aniline solution (10 μL, 20 mM) were mixed in an Eppendorf tube (1.5 mL) and allowed to react at 50 °C for 4 h.Chromatographic separation
Separations were performed by a HPLC system using a C18 column (4.5 × 150 mm) with the temperature of the column set at 30 °C. The flow rate was kept at 1 mL min−1. The excitation and emission wavelengths of the fluorescence detector were set at 322 and 447 nm for the mixture derived from compound 1a, and at 320 and 390 nm for that from compound 1b. Before the analysis, the C18 column was pre-equilibrated with the mobile phase for 30 min.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (21472144 to X. H. and 21405117 to Y. L.), the Fundamental Research Funds for the Central Universities (WUT: 2016-IB-005, 2016-IB-007, 2017-IB-007), and Chutian Scholar Program of the Hubei provincial government (to X. H.) for financial support.References
- J. Schultheiss, D. Jensen and R. Galensa, J. Chromatogr. A, 2000, 880, 233–242 CrossRef CAS PubMed.
- E. M. S. M. Gaspar and A. F. F. Lucena, Food Chem., 2009, 114, 1576–1582 CrossRef CAS.
- M. H. Azarbad and H. Jeleń, Food Anal. Methods, 2014, 8, 1727–1733 CrossRef.
- Q. Zhang, W. Qin, D. Lin, Q. Shen and A. S. Saleh, J. Food Sci. Technol., 2015, 52, 7683–7696 CrossRef CAS PubMed.
- M. Kedzierska-Matysek, M. Florek, A. Wolanciuk, P. Skalecki and A. Litwinczuk, J. Food Sci. Technol., 2016, 53, 2092–2098 CrossRef CAS PubMed.
- M. I. Afzal, C. C. Ariceaga, K. A. Boulahya, M. Jacquot, S. Delaunay and C. Cailliez-Grimal, Crit. Rev. Food Sci. Nutr., 2017, 57, 399–406 CrossRef CAS PubMed.
- D. M. Rodríguez, K. Wrobel and K. Wrobel, Eur. Food Res. Technol., 2005, 221, 798–802 CrossRef.
- J. Ledauphin, D. Barillier and M. Beljean-Leymarie, J. Chromatogr. A, 2006, 1115, 225–232 CrossRef CAS PubMed.
- Z. Yi, T. Feng, H. Zhuang, R. Ye, M. Li and T. Liu, Food Anal. Methods, 2016, 9, 2364–2373 CrossRef.
- P. C. Wietstock, T. Kunz and F. J. Methner, J. Agric. Food Chem., 2016, 64, 8035–8044 CrossRef CAS PubMed.
- V. Pereira, M. Santos, J. Cacho and J. C. Marques, LWT – Food Sci. Technol., 2017, 75, 719–726 CrossRef CAS.
- D. Kahoun, S. Rezkova and J. Kralovsky, Food Chem., 2017, 219, 357–363 CrossRef CAS PubMed.
- Y. Chi, Y. Feng, S. Wen, H. Lu, Z. Yu, W. Zhang, G. Sheng and J. Fu, Talanta, 2007, 72, 539–545 CrossRef CAS PubMed.
- Y. Yu, S. Wen, H. Lu, Y. Feng, X. Wang, G. Sheng and J. Fu, Environ. Monit. Assess., 2008, 137, 275–285 CrossRef CAS PubMed.
- Y. Okada, A. Nakagoshi, M. Tsurukawa, C. Matsumura, J. Eiho and T. Nakano, Environ. Sci. Pollut. Res., 2012, 19, 201–213 CrossRef CAS PubMed.
- E. Hollbacher, C. Rieder-Gradinger, D. Stratev and E. Srebotnik, Holzforschung, 2015, 69, 457–462 CrossRef.
- Q. Liu, Y. Liu, S. Chen and Q. Liu, J. Sep. Sci., 2010, 33, 2376–2382 CrossRef CAS PubMed.
- M. Serrano, M. Silva and M. Gallego, Anal. Chim. Acta, 2013, 784, 77–84 CrossRef CAS PubMed.
- K. Akira, Y. Matsumoto and T. Hashimoto, Clin. Chem. Lab. Med., 2004, 42, 147–153 CrossRef CAS PubMed.
- N. Li, C. Deng, X. Yin, N. Yao, X. Shen and X. Zhang, Anal. Biochem., 2005, 342, 318–326 CrossRef CAS PubMed.
- N. R. Neng, C. A. Cordeiro, A. P. Freire and J. M. Nogueira, J. Chromatogr. A, 2007, 1169, 47–52 CrossRef CAS PubMed.
- M. Eggink, M. Wijtmans, R. Ekkebus, H. Lingeman, I. J. D. Esch, J. Kool, W. M. Niessen and H. Irth, Anal. Chem., 2008, 80, 9042–9051 CrossRef CAS PubMed.
- C. A. Sobsey, J. Han, K. Lin, W. Swardfager, A. Levitt and C. H. Borchers, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1029–1030, 205–212 CrossRef CAS PubMed.
- M. El-Maghrabey, N. Kishikawa and N. Kuroda, J. Chromatogr. A, 2016, 1462, 80–89 CrossRef CAS PubMed.
- N. Chen and J. Xie, Org. Biomol. Chem., 2016, 14, 11028–11047 CAS.
- S. L. Ramsay, C. Freeman, P. B. Grace, J. W. Redmond and J. K. MacLeod, Carbohydr. Res., 2001, 333, 59–71 CrossRef CAS PubMed.
- Y. Ohkura, M. Kai and H. Nohta, J. Chromatogr. B Biomed. Sci. Appl., 1994, 659, 85–107 CrossRef CAS PubMed.
- T. Toyo'oka, J. Chromatogr. B Biomed. Sci. Appl., 1995, 671, 91–112 CrossRef.
- P. G. Rigas, Anal. Bioanal. Chem., 2013, 405, 7957–7992 CrossRef CAS PubMed.
- J. Wang, J. Xie and P. G. Schultz, J. Am. Chem. Soc., 2006, 128, 8738–8739 CrossRef CAS PubMed.
- T. Koopmans, M. van Haren, L. Q. van Ufford, J. M. Beekman and N. I. Martin, Bioorg. Med. Chem., 2013, 21, 553–559 CrossRef CAS PubMed.
- X. Dai, Q. H. Wu, P. C. Wang, J. Tian, Y. Xu, S. Q. Wang, J. Y. Miao and B. X. Zhao, Biosens. Bioelectron., 2014, 59, 35–39 CrossRef CAS PubMed.
- P. Guo, S. Yan, J. Hu, X. Xing, C. Wang, X. Xu, X. Qiu, W. Ma, C. Lu and X. Weng, Org. Lett., 2013, 15, 3266–3269 CrossRef CAS PubMed.
- D. K. Williams, C. W. Meadows, I. D. Bori, A. M. Hawkridge, D. L. Comins and D. C. Muddiman, J. Am. Chem. Soc., 2008, 130, 2122–2123 CrossRef CAS PubMed.
- M. K. Potdar, S. S. Mohile and M. M. Salunkhe, Tetrahedron Lett., 2001, 42, 9285–9287 CrossRef CAS.
- X. Xu, X. Hu and J. Wang, Beilstein J. Org. Chem., 2013, 9, 254–259 CrossRef CAS PubMed.
- E. J. Corey and J. W. Suggs, Tetrahedron Lett., 1975, 16, 2647–2650 CrossRef.
- R. Ocampo and W. R. Dolbier, Tetrahedron, 2004, 60, 9325–9374 CrossRef CAS.
- M.-X. Zhao, M.-X. Wang, C.-Y. Yu, Z.-T. Huang and G. W. Fleet, J. Org. Chem., 2004, 69, 997–1000 CrossRef CAS PubMed.
- L.-W. Cao, H. Wang, X. Liu and H.-S. Zhang, Talanta, 2003, 59, 973–979 CrossRef CAS PubMed.
- N. Moreira, S. Meireles, T. Brandão and P. G. de Pinho, Talanta, 2013, 117, 523–531 CrossRef CAS PubMed.
- J. L. da Silva, M. A. Beluomini and N. R. Stradiotto, J. Sep. Sci., 2015, 38, 3176–3182 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02177a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |