
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous quantification of coenzyme A and its salvage pathway intermediates in in vitro and whole cell-sourced samples
R. Goosen and
E. Strauss *
*
Department of Biochemistry, Stellenbosch University, Stellenbosch, South Africa. E-mail: estrauss@sun.ac.za
First published on 4th April 2017
Abstract
We report a method for the simultaneous quantification of the essential metabolic cofactor coenzyme A (CoA) and its thiol-bearing precursors—including 4′-phosphopantetheine, which was recently shown to play a potentially important role as nexus metabolite in CoA biology—with pmol sensitivity. This sensitivity is gained by making use of an established thiol-derivatisation agent that produces a fluorophore upon labeling, which is subsequently separated by HPLC and quantified by fluorescence detection. While previous reports have made use of a similar strategy to quantify CoA, very few have extended the method to the CoA biosynthetic intermediates (some of which occur at levels much lower than CoA) or have critically evaluated its analytical performance. In this study we addressed these shortcomings, and also overcame the difficulty associated with the independent confirmation of the concentrations of the analytical standards used for quantification. The method's utility is showcased through time-course analyses of in vitro reconstituted enzyme reactions and by analysis of extracts from Escherichia coli and Staphylococcus aureus, demonstrating its potential in advancing studies of CoA biosynthesis and CoA-dependent biology in a wide range of systems.
Introduction
Coenzyme A (CoA, 1) is an essential enzyme cofactor involved in the transfer and activation of acyl groups, and plays a key role in central energy and fatty acid metabolism.1 However, is also important in other contexts.2 First, the N-substituted pantothenamide antimicrobials that are active against Staphylococcus aureus and the malaria parasite Plasmodium falciparum, most likely target CoA biosynthesis—which usually occurs in five steps from pantothenic acid (Pan, 2, vitamin B5) (Scheme 1)—and CoA utilization.3,4 These compounds are metabolically activated by the truncated CoA salvage pathway that starts from the CoA degradation product pantetheine (PantSH, 5) and only requires three of the five biosynthetic steps (Scheme 1).5 Second, the first CoA biosynthetic enzyme pantothenate kinase (PanK) is implicated in PKAN (pantothenate kinase-associated neurodegeneration) a neurodegenerative disease.6,7 Recently it was shown that extracellular 4′-phospho-pantetheine (P-PantSH, 6) can also serve as a precursor for CoA biosynthesis in eukaryotic cells, bypassing the PanK step and opening new treatment options for PKAN.8 The ability to simultaneously analyse and quantify CoA and its biosynthetic intermediates is a key requirement for advancing our knowledge of both these systems.9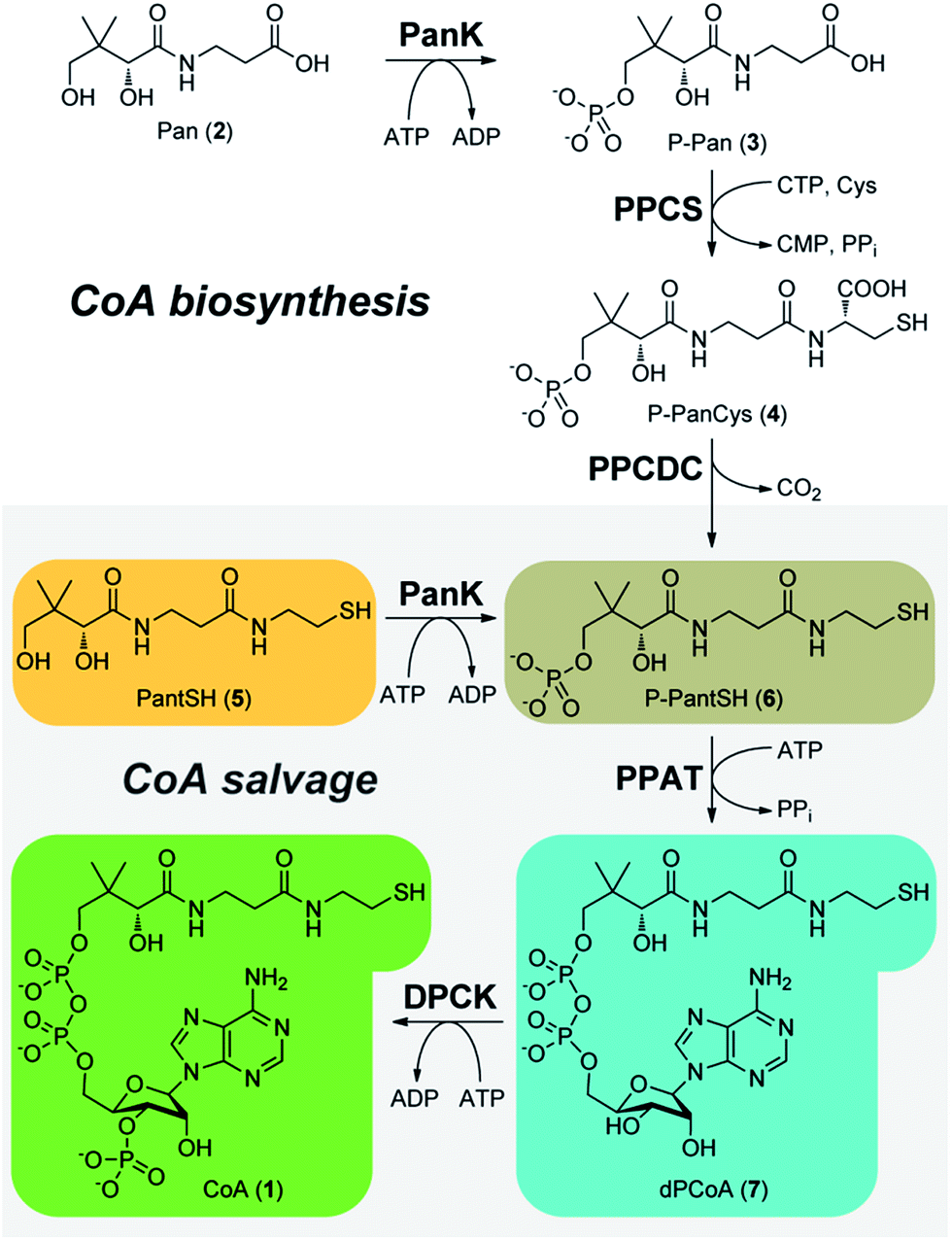 | ||
| Scheme 1 The five-step CoA (1) biosynthetic pathway catalysed by the sequential transformation of pantothenic acid (Pan, 2) by PanK (pantothenate kinase), PPCS (phosphopantothenoylcysteine synthetase), PPCDC (phosphopantothenoyl-cysteine decarboxylase), PPAT (phosphopantetheine adenylyltransferase) and DPCK (dephospho-CoA kinase). CoA salvage (grey box) takes place by means of a truncated pathway that starts with phosphorylation of pantetheine (PantSH, 5) by PanK. | ||
The quantification of CoA in biological samples has received wide attention as highlighted by a recent review.10 The most practical of the CoA analysis methods usually involve separation by HPLC followed by UV- or MS-based detection, or fluorescence detection following pre- or post-column derivatisation with a fluorophore. However, these methods cannot all be applied with equal success to the CoA biosynthetic intermediates, and consequently the analytical performance of these methods has not been critically evaluated with these compounds. Careful sample handling during analysis is also crucial, as previous studies have reported that CoA in cell extracts were found to be unstable.11 In this study we therefore set out to develop a practical method for the simultaneous detection of CoA and its salvage pathway intermediates PantSH, P-PantSH, dephospho-coenzyme A (dPCoA, 7) with pmol sensitivity, to validate its analytical performance, and to showcase its utility in the analysis of actual samples: in vitro enzyme reaction mixtures, and extracts from different types of bacterial cells.
Methods
General materials and methods
All chemicals and solvents were purchased from Sigma-Aldrich and were of the highest purity. Proteins were purified on an ӒKTAprime protein purification system. Protein determinations were performed with the Quick Start Bradford Protein Assay Kit (Bio-Rad) and a bovine serum albumin standard set. Chromatograms from the high performance liquid chromatography (HPLC) analyses were traced, generated, integrated and analysed using Chemstation for LC, Rev. A.10.02[1757] (Agilent). All graphical and statistical analyses were performed with SigmaPlot 12.0 (Systat Software, Inc.).Overexpression and purification of E. coli CoA salvage enzymes
Pure E. coli PanK, PPAT and DPCK enzymes were obtained by overexpression from previously prepared pET-28a(+)-based expression vectors and by purification as previously reported.12 These plasmids are available from Addgene (plasmid # 50386, 50388 and 50390). Fractions containing pure protein (as judged by SDS-PAGE analysis) were pooled, and glycerol added to a final concentration of 5% before being stocked and stored at −80 °C in 50 μL aliquots.Reconstituting E. coli CoA salvage reactions in vitro
Reconstituting the CoA salvage pathway in vitro entailed the addition of PantSH to a reaction mixture containing PanK, PPAT and DPCK enzymes along with an excess of ATP. A 10 mM PantSH stock solution was prepared by reducing a 5 mM pantethine (PantSH disulfide, obtained from Sigma-Aldrich) solution with 7.5 mM tris(2-carboxyethyl)phosphine (TCEP) for a period of 5 min. The CoA salvage pathway reaction mix contained 50 mM Tris–HCl buffer (pH 7.6), 10 mM MgCl2, 20 mM KCl, 5 mM ATP, 1.75 mM TCEP, 0.04 U μL−1 pyruvate kinase, 1.8 mM phosphoenolpyruvate (PEP) and 0.1 μg μL−1 each of PanK, PPAT and DPCK respectively in a total reaction volume of 1 mL. The reaction was initiated by the addition of PantSH from the stock solution to a final concentration of ∼900 μM. The reaction mixture was incubated at 37 °C for 60 minutes on a rotary mixer.Standardization of analyte standards
The concentration of PantSH, dPCoA and CoA standards prepared from commercially-obtained material were determined according to the method of Lukesh et al.13 In brief, analyte standards were first treated with the reducing agent dithiobutylamine (DTBA) to reduce any disulfides. The reduction reaction contained approximately 500 μM of the standard and 500 μM DTBA in a total volume of 280 μL made up with assay buffer (0.1 M sodium phosphate buffer, pH 8 and 1 mM EDTA). The reaction was incubated at room temperature for 15 minutes to allow for complete disulfide reduction. Oxidized and residual reduced DTBA were removed by the addition of 7 mg Dowex 50WX4-400 cation exchange resin (previously washed with deionized water) to the reduction reaction. The mixture was swirled for 5 minutes before the resin was removed by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 1 minute. Thiol content in the metabolite standard was subsequently determined by treatment with 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB); 25 μL of the reduced sample was immediately added to the DTNB assay mixture (250 μL assay buffer and 5 μL DTNB reagent solution, the latter prepared by the addition of 4 mg DTNB to 1 mL assay buffer) and incubated at room temperature for 15 minutes. The amount of yellow 2-nitro-5-thiobenzoate chromophore produced by the reaction of DTNB with free thiols was measured spectrophotometrically at 412 nm and quantified using an extinction coefficient of 14150 M−1 cm−1.14
000 × g for 1 minute. Thiol content in the metabolite standard was subsequently determined by treatment with 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB); 25 μL of the reduced sample was immediately added to the DTNB assay mixture (250 μL assay buffer and 5 μL DTNB reagent solution, the latter prepared by the addition of 4 mg DTNB to 1 mL assay buffer) and incubated at room temperature for 15 minutes. The amount of yellow 2-nitro-5-thiobenzoate chromophore produced by the reaction of DTNB with free thiols was measured spectrophotometrically at 412 nm and quantified using an extinction coefficient of 14150 M−1 cm−1.14
P-PantSH standards were prepared by the phosphorylation of a standardized solution of PantSH using E. coli PanK, followed by dilution of the resulting P-PantSH to the appropriate standard concentrations. Reaction mixtures contained 900 μM PantSH, 5 mM ATP, 1 mM TCEP, 0.1 μg μL−1 PanK in a total volume of 1 mL and were incubated for 20 minutes. Complete conversion of PantSH to P-PantSH was confirmed by HPLC analysis.
Sampling, derivatisation and HPLC analysis
To perform metabolite analysis of in vitro reactions, 50 μL aliquots were removed from the reaction mixture at set intervals. Each aliquot was immediately added to 10 μL trichloroacetic acid (TCA) (90% w/v) to precipitate the enzymes and quench the reaction. The 60 μL precipitation mixture, containing 15% TCA, was incubated for 10 min at 4 °C. Following acid precipitation, the mixture was neutralised by the addition of 40 μL of 2.75 M ammonium acetate. The precipitated proteins were removed by centrifugation at 10000 × g for 10 min. An aliquot (7 μL) of the resulting supernatant was removed for derivatisation with 7-diethylamino-3-(4-maleimidophenyl)-4-methyl-coumarin (CPM). The derivatisation reaction contained 21 μL acetonitrile, 1.3 μL CPM (10 mM in acetonitrile) and the 7 μL supernatant, made up to 70 μL with H2O. The derivatisation mix was incubated overnight at room temperature to ensure complete hydrolysis of the CPM derivatives, after which 5 μL was analysed by HPLC. Note that it is important to at all times maintain a CPM concentration that is at least 1.5 times the sum of the concentration of the thiolated metabolites and TCEP present in the sample; the amount of CPM should therefore be adjusted based on the total amount of thiolated metabolites and TCEP.
HPLC analyses were performed on an Agilent 1100 series system equipped with an in-line fluorescence detector, using a Supelcosil LC-DP (250 × 4.60 mm, 5 μm particle size) reverse phase column protected by a Supelcosil LC-DP Supelguard cartridge (Sigma-Aldrich). Samples (5 μL) were analysed at a flow rate of 1 mL min−1 using a ternary solvent system that consisted of 50 mM potassium phosphate buffer, pH 6.8 (solvent A), 60% acetonitrile (solvent B) and 100% acetonitrile (solvent C). The method started with 50% A and 50% B; the following elution protocol was followed to achieve separation: 5 min isocratic (50% A and 50% B); 20 min linear gradient, ending at 40% A and 60% B; 1 min linear gradient, ending at 40% A and 60% C; 6 min isocratic (40% A and 60% C); 3 min linear gradient ending at 10% A and 90% C; 6 min isocratic (10% A and 90% C); 10 min linear gradient back to initial conditions (50% A and 50% B); 5 min isocratic (50% A and 50% B) (total analysis time of 57 minutes).
Separation was monitored using fluorescence (Em 465 nm, Ex 387 nm). Peaks of interest were integrated to determine the peak area, which was used for quantification by comparison to the calibration curves obtained as described below.
Obtaining calibration curves of analyte standards
To obtain calibration curves for each metabolite, the standardized analyte solutions were used to prepare a dilution series (in triplicate) covering the concentration range of 2.5 to 45 μM. Solutions were sampled, derivatised and analysed by HPLC as described above (including the acid precipitation and neutralisation steps), to ensure that calibration curves were representative of analytes in actual samples. A typical set of calibration curves are shown in Fig. 1, the corresponding linear regression parameters are shown in Table 1.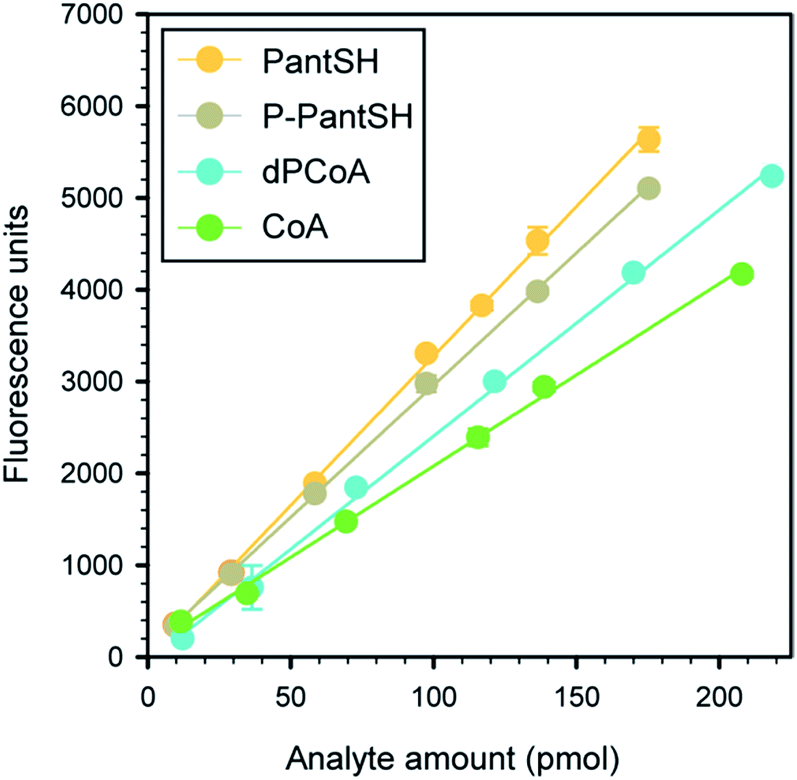 | ||
| Fig. 1 Typical calibration curves obtained for CoA and its thiolated biosynthetic intermediates. All data points represent the mean of triplicate values, with the error bars denoting the standard deviation. Where errors are not visible they are smaller than the symbol. Linear regression parameters are given in Table 1. | ||
| Analyte | Calibration equations (y = mx + c) | |
|---|---|---|
| m | c | |
| a Errors denote the standard deviation in the regression parameters determined for at least six different concentrations, each measured in triplicate. | ||
| PantSH | 32.58 ± 0.39 | 21.91 ± 40.41 |
| P-PantSH | 28.81 ± 0.23 | 81.34 ± 24.05 |
| dPCoA | 24.64 ± 0.43 | 58.34 ± 54.22 |
| CoA | 19.88 ± 0.30 | 91.14 ± 35.50 |
Calculation of calibration data and analytical figures of merit
Variation in retention time (Rt) for each analyte was determined by calculating the standard deviation (SD) in retention time for at least three different concentrations (each analysed in triplicate). The linearity of the response was evaluated by linear regression analysis, which showed that in all cases the calibration curves gave excellent R2 values in the range 0.995–0.999.The analytical figures of merit were determined according to International Conference on Harmonisation (ICH) guidelines.15 The limits of detection (LOD) and limits of quantification (LOQ) were calculated by considering σ, which is the residual SD of the regression line of the calibration curve. Based on a 95% confidence level, the LOD was calculated as 3.3σ divided by the slope of the regression line of the calibration curve, while LOQ was calculated as 10σ divided by the same slope.
Extraction and derivatisation of CoA salvage intermediates in E. coli and S. aureus
The extraction of CoA salvage intermediates was performed similarly for both S. aureus RN4220, E. coli K12 and E. coli JTG10 (ref. 16) (obtained from CGSC, stock # 6926) with the exception that S. aureus was cultured in 500 mL tryptic soy broth (TSB) and E. coli in 500 mL Luria-Bertani (LB) medium, both at 37 °C with shaking. Cells were harvested in stationary phase (OD600 ∼ 1.75) by the removal of 100 mL culture from the main culture, which was then split into two 50 mL samples. One of these was used for dry cell weight determination, while the other was used for metabolite extraction and analysis.Both 50 mL culture samples were centrifuged at 4500 × g and 4 °C for 20 minutes to remove media. Cells were washed twice by resuspension in 50 mL ice cold PBS followed by centrifugation under the same conditions. The cell pellet used for dry cell weight determination was dried on a SpeedVac system until constant weight was recorded, normally after 18–20 hours. Cells earmarked for metabolite extraction were resuspended in ice cold extraction solution consisting of 80% acetonitrile and 0.1 M formic acid in water using a ratio of 50 μL extraction solution per 20 mg wet cells harvested. The extraction mixture was shaken at 4 °C for 30 minutes to complete cell lysis and metabolite extraction. Next, cell debris was removed from the cell extract by centrifugation at 4 °C and 10000 × g for 15 minutes. The supernatant was then filtered by centrifugation through Nanosep 3K membrane filtration tubes (Pall) for 10 min at 10000 × g. The filtered cell extracts were neutralized by taking 44.62 μL cell extract and adding 14 μL Tris buffer (500 mM, pH 7.6) and 65.62 μL H2O. Any disulfides present were reduced by addition of 9.46 μL of TCEP (1 mM). After 10 minutes, 6.3 μL CPM (10 mM in 100% acetonitrile) was added to bring the total volume up to 140 μL. The total acetonitrile content of a sample prepared in this manner is 30%, the same value as found in the analysis of the standards used to prepare the calibration curves.
Results and discussion
Choosing an analysis method: advantages of derivatisation with the thiol-reactive reagent CPM
We decided that the derivatisation with a fluorescent reagent, followed by HPLC separation and post-column fluorescence detection, presented the best strategy for developing a method for the simultaneous detection of CoA and its salvage pathway intermediates. Our choice of derivatisation over one of the several MS-based detection methods that have gained popularity was based on our desire to maintain a similar sensitivity for quantification of all the analytes in spite of the variation in their structure and ionizing potential, and to use instrumentation that would be readily available for the analysis of large numbers of samples. Additionally, to our knowledge there have been no reports of successful MS-based quantification of PantSH or P-PantSH in cell extracts.In choosing a thiol-labeling agent we decided against the widely used monobromobimane (mBBr) as it has been reported to have several undesirable characteristics, including background fluorescence (of unreacted mBBr); non-specific binding to amines, carboxylates and phosphates; photodegradation of mBBr derivatisation products (requiring derivatised samples to be protected against light until analysis); and non-baseline separation of the mBBr-derivatives of PantSH and P-PantSH in some cases.17
Instead, 7-diethylamino-3-(4-maleimidophenyl)-4-methyl-coumarin (CPM, 8) was chosen as fluorescent derivatisation agent, as several studies18,19 have showed that it is highly specific and reactive towards thiols and that the derivatisation products are stable with a high quantum yield, allowing for the sensitive detection of low molecular weight thiols in cell extracts (Scheme 2). Ideally, it is also only weakly fluorescent until bound to a thiolated analyte. Other reagents such as the fluoro-benzoxadiazoles ABD-F and SBD-F would most likely also be appropriate choices (SBD-F was in fact recently used to analyse P-PantSH and CoA levels in various extracts8).
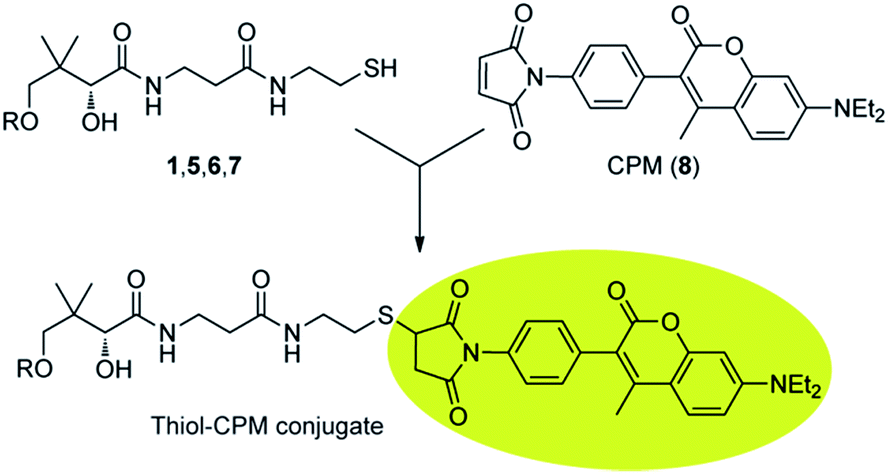 | ||
| Scheme 2 Derivatisation of CoA and its thiolated biosynthetic intermediates with 7-diethylamino-3-(4-maleimidophenyl)-4-methyl-coumarin (CPM, 8). | ||
Reactivity and specificity of CPM as labelling reagent, and stability of the resulting adducts
Although CPM was chosen as the preferred derivatisation reagent, various factors regarding its reactivity and specificity and the stability of the adducts that forms need to be taken into account.In regards to reactivity, the pH of samples to be derivatised was found to be of critical importance to ensure complete derivatisation and accurate quantification. CPM does not react with free thiols at an acidic pH but requires a near neutral environment for optimal reactivity. At higher pH levels it has been reported that hydrolysis of the maleimide moiety can occur before derivatisation takes place.20 Hence the pH has to be closely controlled in samples to be derivatised with CPM by the incorporation of a neutralisation step into the sample preparation workflow. This is especially important in cases (such as in our method) where acid is used for the rapid quenching of in vitro reactions, as well as in biological extraction solutions.
In regards to specificity, we were surprised to find that CPM also formed fluorescent products with tris(2-carboxyethyl)phosphine (TCEP), the reducing agent used to ensure that the thiol groups of the analytes are in the free, reduced form. At least two different adducts were found to be formed (see below). However, the separation method was developed in such a manner that these peaks did not interfere with the analysis of the analytes of interest.
Finally, in regards to stability, it is known that the maleimide moiety of CPM-thiol adducts are unstable, undergoing hydrolysis to form two ring-cleaved fluorophores at the N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O position.21 While no effect on the fluorescence quantum yield of the coumarin signalling group has been reported following maleimide hydrolysis, the presence of multiple adduct forms will lead to more than one peak being observed for the metabolite–CPM adduct in HPLC analysis. Prolonged incubation of derivatisation reactions were found to ensure complete hydrolysis and elution of CPM derivatives in a single peak.
O position.21 While no effect on the fluorescence quantum yield of the coumarin signalling group has been reported following maleimide hydrolysis, the presence of multiple adduct forms will lead to more than one peak being observed for the metabolite–CPM adduct in HPLC analysis. Prolonged incubation of derivatisation reactions were found to ensure complete hydrolysis and elution of CPM derivatives in a single peak.
Sample preparation workflow and HPLC analysis method
To establish the proposed method, we devised a sample preparation workflow as schematically summarised in Fig. 2A. First, samples are prepared from the source of interest in the presence of the thiol reducing agent TCEP. Next, the samples are acidified either with 15% trichloroacetic acid (TCA) (for in vitro enzyme reaction mixes) or formic acid in acetonitrile (for bacterial cell samples). This quenches any enzymatic reactions by precipitating the proteins, and aids with extraction of the analytes of interest. Third, the acidified samples are neutralised prior to derivatisation with excess CPM in the fourth step. Finally, the derivatised samples were analysed by HPLC with fluorescent detection.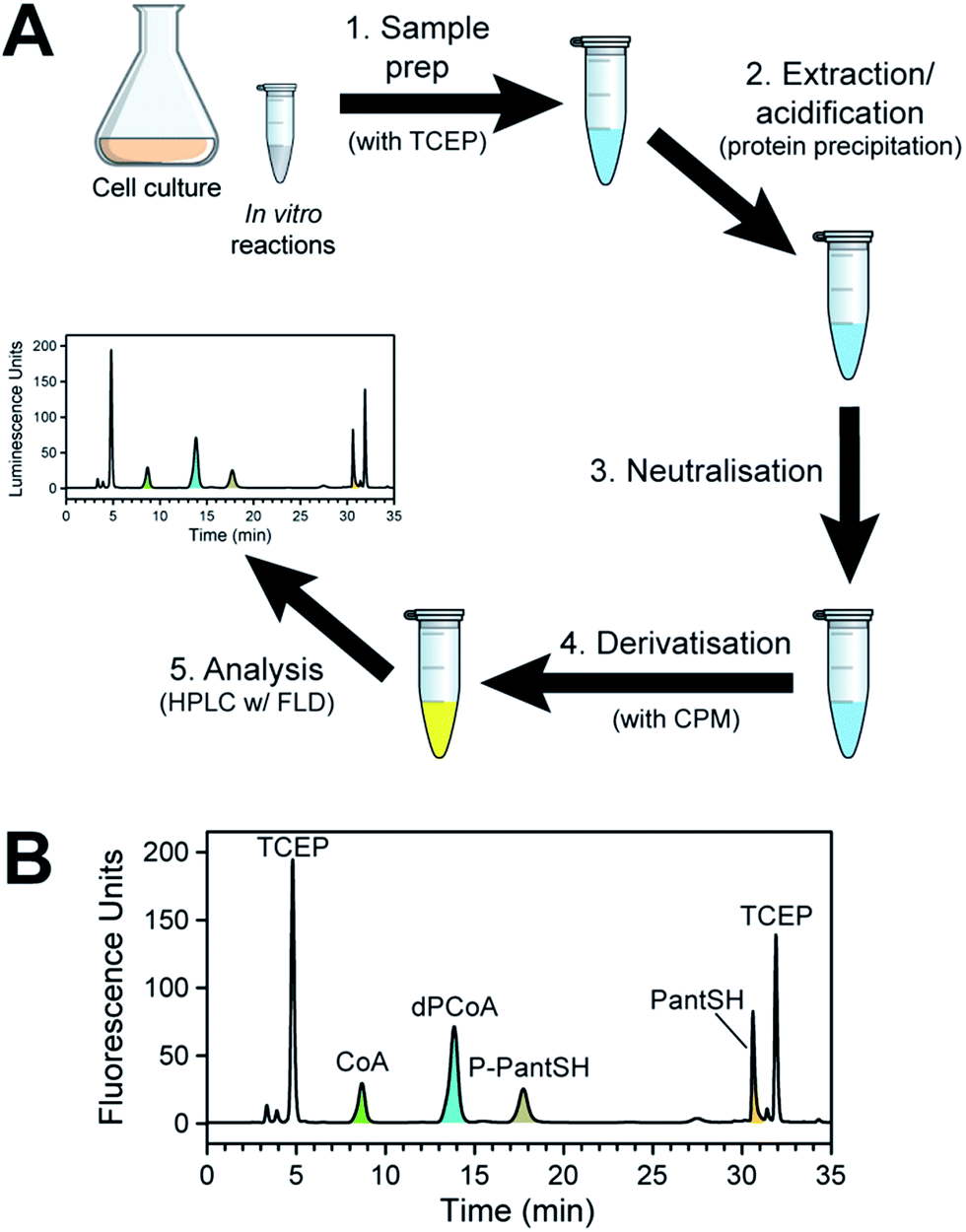 | ||
| Fig. 2 Method for the analysis of CoA and its salvage pathway precursors by HPLC with fluorescence detection (FLD). (A) Workflow for the preparation of samples (from cell cultures or from in vitro enzyme reactions). (B) Typical chromatogram obtained in the analysis of CoA and its salvage pathway metabolites using fluorescent detection (Em 465 nm; Ex 387 nm), with the relevant peaks indicated and coloured according to the legend used in Scheme 1. Note the two peaks that represent CPM–TCEP adducts (at 4.8 and 31.9 min). | ||
A typical chromatogram of a sample analysed using the optimised LC method is shown in Fig. 2B. We found that the baseline separation of the various analyte and reagent peaks could be obtained using a reverse phase di-phenyl column and a gradient method with increasing amounts of acetonitrile in a potassium phosphate buffer at pH 6.8. The total run time per sample was less than 1 h, including the time required to re-equilibrate the column to initial conditions.
Analyte quantification: determining the concentration of analyte standards in an independent manner
Quantification of the analytes presented a much bigger challenge than originally anticipated. Although three of the four analytes of interest are commercially available (P-PantSH was obtained by enzymatic phosphorylation of PantSH using existing protocols), they are not provided as analytical standards. The concentrations of the analyte standards therefore had to be determined by an independent method. Towards this end the well-known thiol-reactive reagent 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) was used for quantification of the thiol standards after their reduction using dithiobutylamine (DTBA).13 Since it has an ionisable amino group, both reduced and oxidised DTBA can be removed by acidic cation exchange prior to analysis with DTNB.Evaluating the analytical performance of the method
To determine the analytical performance of the method, samples of the analyte standards in a concentration range of ∼10–220 pmol were derivatised according to the standard protocol (Fig. 2) and analysed in triplicate to obtain calibration curves (Fig. 1 and Table 1). From these data the calibration data and analytical figures of merit could be obtained; Table 2 summarises these values for a typical calibration curve. This shows that the variation in retention time (Rt) was less than 0.15 min in all cases except for dPCoA, while all the calibration curves gave excellent R2 values of 0.995–0.999.| Parametera | PantSH | P-PantSH | dPCoA | CoA |
|---|---|---|---|---|
| a See text for detailed description of parameters and how they were obtained. | ||||
| Rt SD (min) | 0.12 | 0.09 | 0.51 | 0.08 |
| Linearity (R2) | 0.997 | 0.999 | 0.995 | 0.997 |
| LOD (pmol) | 9.40 | 6.33 | 16.44 | 12.30 |
| LOQ (pmol) | 28.49 | 19.19 | 49.82 | 37.28 |
| Precision (repeatability CV%) | 1.90 | 1.43 | 1.26 | 1.11 |
| Accuracy (recovery %) | 99.91 | 98.47 | 100.59 | 99.57 |
The limit of detection (LOD) and limit of quantification (LOQ) values were both found to be in similar ranges at low pmol levels, the lowest LOD being that for P-PantSH at 6.33 pmol. These values compare very favourably with those obtained for analysis of CoA (the only analyte in this study for which such parameters have been published) by other methods.10 For example, CoA has been quantified by HPLC following derivatization with SBD-F with a reported LOD of 120 pmol,22 and with an LOD of 1.3 nmol using an LC/MS-based technique.23 Only commercially-available ELISA kits claim lower LODs (as low as 18.3 fmol) for the quantification of free CoA.10 Interestingly, the quantification of the more stable acyl-CoAs by LC-MS is similarly sensitive, with an LOD of 10 fmol reported in one study.24 This again serves to highlight the difficulty in measuring the levels of thiolated metabolites with the analytical robustness of the method described here. Finally, the precision and accuracy values were also found to be in the range required for metabolite quantification.
Evaluating the robustness of the analytical method: time-course analysis of in vitro CoA biosynthesis
Next, a set of experiments were conducted to evaluate the utility and robustness of the analytical method under conditions where the analytes are all present, but in varying concentrations. This was done by following the time-dependent changes in the levels of the biosynthetic intermediates of an in vitro reconstituted CoA salvage pathway, i.e. in which PanSH is converted to CoA by purified E. coli PanK, PPAT and DPCK with excess ATP present. Reactions were initiated by the addition of reduced PanSH, followed by removal of aliquots at various time points and analysis according to the standard protocol (Fig. 2). The analyte concentrations were determined from the calibration curves to obtain a time course-profile (Fig. 3). In a robust method, in which the quantification of all the analytes can be done with equal accuracy, precision and sensitivity, one would expect the total amount of analyte measured to always add up to the amount of PanSH introduced at the start. The profile clearly showcases the analytical capabilities of our method; across the time interval the total concentration measured averaged 95.2 ± 4.6% of the initial amount of 900 μM PanSH added. This also demonstrates that our method is an excellent tool for studying the progress of CoA salvage biosynthesis.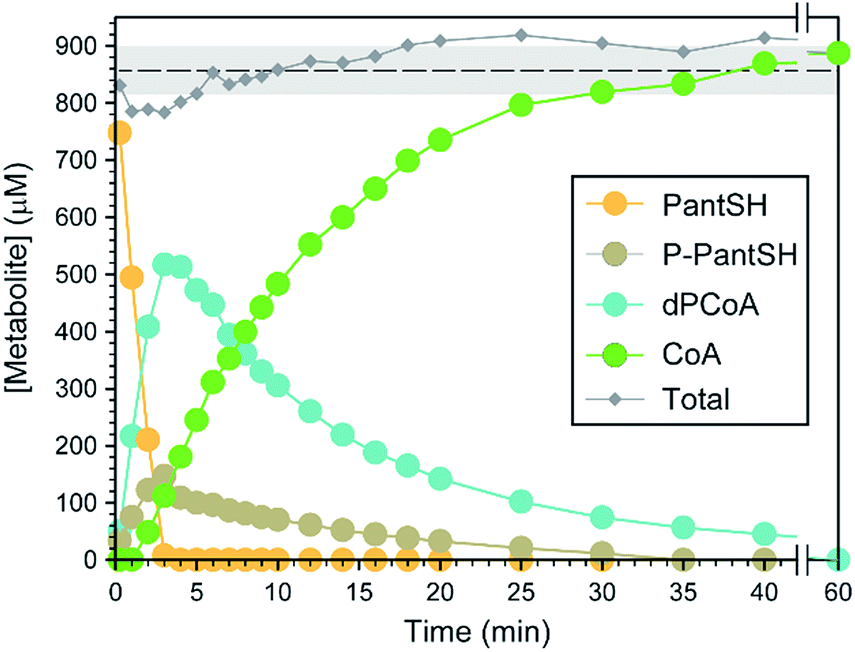 | ||
| Fig. 3 Time-course analysis of the conversion of 900 μM PantSH to CoA by an in vitro reconstituted CoA salvage pathway (i.e. PanK, PPAT and DPCK) from E. coli at 37 °C. Symbols represent the concentration measured for each metabolite at a time point using the standard protocol; the total metabolite concentration at each time point is also indicated. The dashed line shows the average total metabolite concentration, with the grey shaded area representing one standard deviation from that value. | ||
Analysis of intracellular CoA and CoA intermediate concentrations in E. coli and S. aureus
To demonstrate the practical utility of the method in analysing cellular extracts, the intracellular concentration of CoA and its biosynthetic intermediates were measured in extracts from two bacteria, Escherichia coli and Staphylococcus aureus, representing Gram-negative and Gram-positive organisms respectively. Cells were harvested in stationary phase, followed by addition of cold acidic acetonitrile to cause lysis and to quench metabolism by precipitation of proteins. Samples were subsequently treated as per the standard derivatisation protocol (Fig. 2). Due to the large variation in the intracellular concentration among analytes, different volumes of the derivatised extract were analysed to ensure accurate quantification of each analyte in the linear range of its calibration curve. The resulting chromatograms are shown in Fig. 4.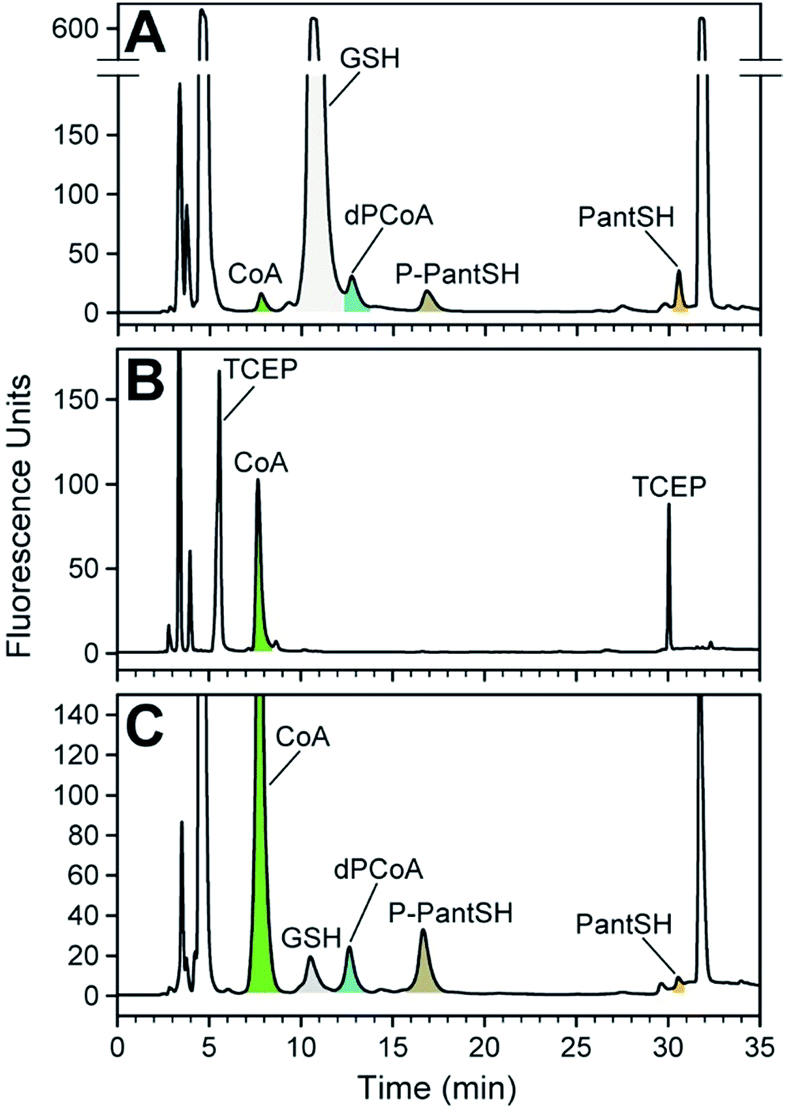 | ||
| Fig. 4 Analysis of CoA and its biosynthetic intermediates in bacterial cell extracts. (A) Analysis of the intracellular thiol metabolites of E. coli K12 using CPM-derivatisation, with metabolite peaks indicated and coloured as in A. The identity of the GSH peak was confirmed by comparison with a standard. (B) As for A, but for E. coli JTG10, a strain that is deficient in producing GSH. (C) As for A, but for S. aureus RN4220. Note the peak corresponding to GSH. Note the differences in the y-axis scale for each of the chromatograms. | ||
The analyte levels determined in this manner are reported in amount per gram cell dry weight (cdw) (Table 3). However, to allow comparison to previous reports of studies in E. coli in which concentration were used, the values were converted to micromolar by using the ratio of cell volume to cell dry weight that has previously been established for this organism.25 This shows that the CoA concentration of ∼127 μM determined in E. coli K12 (Fig. 4A) compares well to the range of between 56 and 108 μM determined for exponentially growing E. coli cultured with different carbon sources (glucose, acetate and glycerol) using radiolabelled precursors;26 it is also similar to the value determined using HPLC analysis using another fluorescent label.27 However, these values are lower than the range of 1.37 mM and 4.54 mM reported in a study using LC-MS and isotopically labeled internal standards for quantification;25 this highlights the importance of independently verifying the concentrations of the standards used when performing metabolite analyses (as done in this study by using DTNB) to ensure confidence in the analyte values that are finally reported.
| Analyte | E. coli K12 | E. coli JTG10b | S. aureus RN4220 |
|---|---|---|---|
| a Analyte levels are given in nmol per gram cell dry weight (cdw); for E. coli, these values were converted to concentrations using a ratio of 0.0023 L g−1 (a value determined in a previous study25) to correlate cdw with cell volume.b A strain incapable of producing GSH.16c For S. aureus, values represent the mean of triplicate analyses with the error representing the standard deviation.d ND, not determined due to interference from GSH peak (see Fig. 4A). | |||
| PantSH | Value < LOQ | Value < LOD | 44.5 ± 3.9c |
| P-PantSH | 151.2 (∼66 μM) | Value < LOD | 146.4 ± 5.4 |
| dPCoA | NDd | Value < LOD | 155.6 ± 1.9 |
| CoA | 293.1 (∼127 μM) | 548.1 (∼238 μM) | 658.2 ± 4.3 |
We found that the known major low molecular weight thiol in E. coli, glutathione (GSH), predominates in the analysis of the K12 strain; in fact, its levels are so high that it prevented the accurate quantification of dPCoA (Fig. 4A).† We therefore repeated the analysis with a mutant E. coli strain16 that is incapable of producing GSH (Fig. 4B). Interestingly, in its case the concentration of CoA was increased, and no CoA biosynthetic intermediates could be detected. This is most likely caused by the CoA biosynthetic flux in the stationary phase E. coli JGT10 being modified in such a manner that it increases CoA levels, which apparently results in the steady state of its precursors to fall below detectable levels. The reason for the higher than normal requirement for CoA is unclear, but the finding raises important questions about the role of CoA as a possible redox buffer when GSH is not present. The fact that the measured CoA levels are similar to those found in S. aureus, which also does not have the ability to produce GSH de novo, supports such a hypothesis (Table 3).
Extracts of S. aureus were analysed without any interference, allowing for the detection and quantification of all the analytes of interest (Fig. 4C). Importantly, the amounts of PanSH, P-PantSH and dPCoA in stationary phase S. aureus cultures are reported here for the first time. The detection of PanSH at levels above the LOQ is a notable finding, as a recent study showed that this metabolite inhibits S. aureus's PanK.5 The measured CoA levels was in a similar range to that reported for other S. aureus strains,27 but more than double that found in E. coli K12—again lending support to a potential role for CoA in redox balance maintenance in the absence of GSH.
We were surprised to find a thiol unrelated to CoA present in measurable amounts in the S. aureus extracts; this was identified as GSH (Rt 11.7 min) by comparison to known standards. The identification of GSH was unexpected given that it is not produced by S. aureus. However, Pöther et al. had previously shown that intracellular accumulation of GSH occurs in S. aureus in the post-exponential phase with increasing levels in the stationary phase when grown in media (such as the tryptic soy broth used in this study) that contain GSH.28 Nonetheless, the GSH levels measured here were lower than all the analytes of interest except PanSH, and whether its presence is biologically relevant remains unknown at this stage.
Conclusions
We have presented here a new analytical method for the accurate and sensitive measurement of CoA—and the first such method also applicable to its biosynthetic intermediates PantSH, P-PantSH and dPCoA—in in vitro enzyme reactions and in whole cell extracts. This method bridges many of the difficulties previously encountered with quantification of these metabolites in biological samples. As such, it should prove to be particularly useful in studies aimed at increasing our understanding of CoA biosynthesis and its regulation in various systems and in various contexts—including the development of new antimicrobial and PKAN treatment options.Acknowledgements
This work was supported by the South African National Research Foundation (NRF). RG received an NRF-DAAD doctoral scholarship. We thank Konrad Mostert for technical assistance.References
- E. Strauss, in Comprehensive Natural Products II Chemistry and Biology, ed. L. Mander and H.-W. Liu, Elsevier, Oxford, 2010, pp. 351–410 Search PubMed.
- B. Srinivasan and O. C. M. Sibon, Biochem. Soc. Trans., 2014, 42, 1075–1079 CrossRef CAS PubMed.
- W. J. A. Moolman, M. de Villiers and E. Strauss, Biochem. Soc. Trans., 2014, 42, 1080–1086 CrossRef CAS PubMed.
- K. J. Saliba and C. Spry, Biochem. Soc. Trans., 2014, 42, 1087–1093 CrossRef CAS PubMed.
- M. de Villiers, L. Barnard, L. Koekemoer, J. L. Snoep and E. Strauss, FEBS J., 2014, 281, 4731–4753 CrossRef CAS PubMed.
- S. J. Hayflick, Biochem. Soc. Trans., 2014, 42, 1063–1068 CrossRef CAS PubMed.
- P. Venco, S. Dusi, L. Valletta and V. Tiranti, Biochem. Soc. Trans., 2014, 42, 1069–1074 CrossRef CAS PubMed.
- B. Srinivasan, M. Baratashvili, M. van der Zwaag, B. Kanon, C. Colombelli, R. A. Lambrechts, O. Schaap, E. A. Nollen, A. Podgorsek, G. Kosec, H. Petkovic, S. Hayflick, V. Tiranti, D.-J. Reijngoud, N. A. Grzeschik and O. C. M. Sibon, Nat. Chem. Biol., 2015, 11, 784–792 CrossRef CAS PubMed.
- O. C. M. Sibon and E. Strauss, Nat. Rev. Mol. Cell Biol., 2016, 17, 605–606 CrossRef CAS PubMed.
- Y. Tsuchiya, U. Pham and I. Gout, Biochem. Soc. Trans., 2014, 42, 1107–1111 CrossRef CAS PubMed.
- S. U. Bajad, W. Lu, E. H. Kimball, J. Yuan, C. Peterson and J. D. Rabinowitz, J. Chromatogr. A, 2006, 1125, 76–88 CrossRef CAS PubMed.
- E. Strauss and T. P. Begley, J. Biol. Chem., 2002, 277, 48205–48209 CrossRef CAS PubMed.
- J. C. Lukesh, M. J. Palte and R. T. Raines, J. Am. Chem. Soc., 2012, 134, 4057–4059 CrossRef CAS PubMed.
- P. W. Riddles, R. L. Blakeley and B. Zerner, Anal. Biochem., 1979, 94, 75–81 CrossRef CAS PubMed.
- ICH, in International Conference on Harmonization, ed. ICH, ICH, 2012 Search PubMed.
- J. T. Greenberg and B. Demple, J. Bacteriol., 1986, 168, 1026–1029 CrossRef CAS PubMed.
- R. C. Fahey and G. L. Newton, Methods Enzymol., 1987, 143, 85–96 CAS.
- D. J. Steenkamp, Biochem. J., 1993, 292, 295–301 CrossRef CAS PubMed.
- C. C. Chung, K. Ohwaki, J. E. Schneeweis, E. Stec, J. P. Varnerin, P. N. Goudreau, A. Chang, J. Cassaday, L. Yang, T. Yamakawa, O. Kornienko, P. Hodder, J. Inglese, M. Ferrer, B. Strulovici, J. Kusunoki, M. R. Tota and T. Takagi, Assay Drug Dev. Technol., 2008, 6, 361–374 CrossRef CAS PubMed.
- M. N. Khan, J. Pharm. Sci., 1984, 73, 1767–1771 CrossRef CAS PubMed.
- K. Shimada and K. Mitamura, J. Chromatogr. B: Biomed. Sci. Appl., 1994, 659, 227–241 CrossRef CAS PubMed.
- K. Imai, T. Toyo'oka and Y. Watanabe, Anal. Biochem., 1983, 128, 471–473 CrossRef CAS PubMed.
- L. Coulier, R. Bas, S. Jespersen, E. Verheij, M. J. van der Werf and T. Hankemeier, Anal. Chem., 2006, 78, 6573–6582 CrossRef CAS PubMed.
- C. A. Haynes, J. C. Allegood, K. Sims, E. W. Wang, M. C. Sullards and A. H. Merrill, J. Lipid Res., 2008, 49, 1113–1125 CrossRef CAS PubMed.
- B. D. Bennett, E. H. Kimball, M. Gao, R. Osterhout, S. J. Van Dien and J. D. Rabinowitz, Nat. Chem. Biol., 2009, 5, 593–599 CrossRef CAS PubMed.
- D. S. Vallari, S. Jackowski and C. O. Rock, J. Biol. Chem., 1987, 262, 2468–2471 CAS.
- G. L. Newton, K. Arnold, M. S. Price, C. Sherrill, S. B. Delcardayre, Y. Aharonowitz, G. Cohen, J. Davies, R. C. Fahey and C. Davis, J. Bacteriol., 1996, 178, 1990–1995 CrossRef CAS PubMed.
- D.-C. Pöther, P. Gierok, M. Harms, J. Mostertz, F. Hochgräfe, H. Antelmann, C. J. Hamilton, I. Borovok, M. Lalk, Y. Aharonowitz and M. Hecker, Int. J. Med. Microbiol., 2013, 303, 114–123 CrossRef PubMed.
- Y. Awasthi, H. Garg, D. Dao, C. Partridge and S. Srivastava, Blood, 1981, 58, 733–738 CAS.
Footnote |
| † For analysis by the method presented here, samples that are expected to contain large amounts of GSH should be pre-treated to remove or reduce the GSH present; this can be achieved by using glutathione S-transferase to conjugate chloro-2,4-dinitro-benzene to the thiol of GSH to prevent its reaction with CPM.29 |
| This journal is © The Royal Society of Chemistry 2017 |