
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel thiohydantoin analogues bearing the 1-hydroxyl-2,2,2-trifluoro-1-ethyl moiety as androgen receptor inhibitors for the potential treatment of castration resistant prostate cancer†
Yingwei Wang‡
 ab,
Yufang Deng‡a,
Xuehai Pangb,
Jiang Yua,
Lei Fanc,
Yuanwei Chen*abc and
Lifeng Zhao*d
ab,
Yufang Deng‡a,
Xuehai Pangb,
Jiang Yua,
Lei Fanc,
Yuanwei Chen*abc and
Lifeng Zhao*d
aLab of YW Chen, Cancer Center, West China Hospital, Sichuan University and Collaborative Innovation Center, Chengdu, 610041, China. E-mail: ywchen@scu.edu.cn; Fax: +86 028 8598 0460; Tel: +86 028 8598 0460
bChengdu Institute of Organic Chemistry, Chinese Academy of Sciences, Chengdu, 610041, China
cHinova Pharmaceuticals Inc., Suite 402, Building B, #5 South KeYuan Road, Chengdu, 610041, China
dChengdu University, Sichuan Industrial Institute of Antibiotics, Chengdu, 610041, China. E-mail: lifengzhao@scu.edu.cn; Fax: +86 028 8505 8465; Tel: +86 158 8241 1095
First published on 21st June 2017
Abstract
Enzalutamide (ENT) is an approved drug for the treatment of castration resistant prostate cancer (CRPC). Despite its success, the duration of response in patients is still limited with drug resistance. More robust CRPC drugs with novel structural motifs are urgently needed. Here, we designed and synthesized a series of 1-hydroxyl or 1-amino-2,2,2-trifluoro-1-ethyl compounds as isosteres to replace the amide group of ENT. Among the compounds prepared and tested, compund 13b is 2-fold more potent than ENT against LNCaP-AR cells. Western blot analysis showed that 13b dose-dependently inhibits the expression of the prostate-specific antigen (PSA). Further in vivo efficacy studies established that 13b has anti-tumor activity with oral administration at 15 mg kg−1 once daily.
1 Introduction
Prostate cancer is the most commonly diagnosed cancer and the third-leading cause of cancer deaths in men.1 Although approximately 75% of these patients will be cured by surgery or radiation when cancer remains within the prostate, the remainder will inevitably progress to a state of metastatic prostate cancer.2 Androgen deprivation therapy or other combination therapy, through lowering serum testosterone or competitively blocking the binding of androgens to the androgen receptor (AR), are initially effective in 90% of patients at this stage.3 Nevertheless, the vast majority of these patients will eventually develop metastatic castration resistant prostate cancer (CRPC) after long term treatments.4–6 Recent research efforts have indicated that CRPC growth is driven by AR signaling as well.7 Furthermore, many possible mechanisms influencing AR signaling have been suggested for the acquired resistance, among which AR mutations, androgen synthesis by prostate cancer cells, and over-expression of AR or AR co-activators have been found to directly or indirectly render AR more sensitive to low androgen concentrations or sometimes turn antagonist responses to agonistic.8Enzalutamide (ENT) was approved by FDA in 2012 for the treatment of CRPC. Unlike the first-generation AR inhibitors, such as flutamide, nilutamide, and bicalutamide (Fig. 1), ENT not only binds to AR, but also reduces nuclear translocation of AR and impairs AR binding to DNA.9 In addition, further studies suggested that ENT possesses a higher binding affinity of AR than the first-generation AR inhibitors. Clinical studies also demonstrated unequivocally its survival benefit in men with CRPC.10,11 Despite its success, the duration of response in patients is still limited. Recently, AR with a specific mutation (F876L) was identified to confer resistance to ENT.12 This disappointing outlook suggests that better compounds with novel structure motif need to be developed urgently.
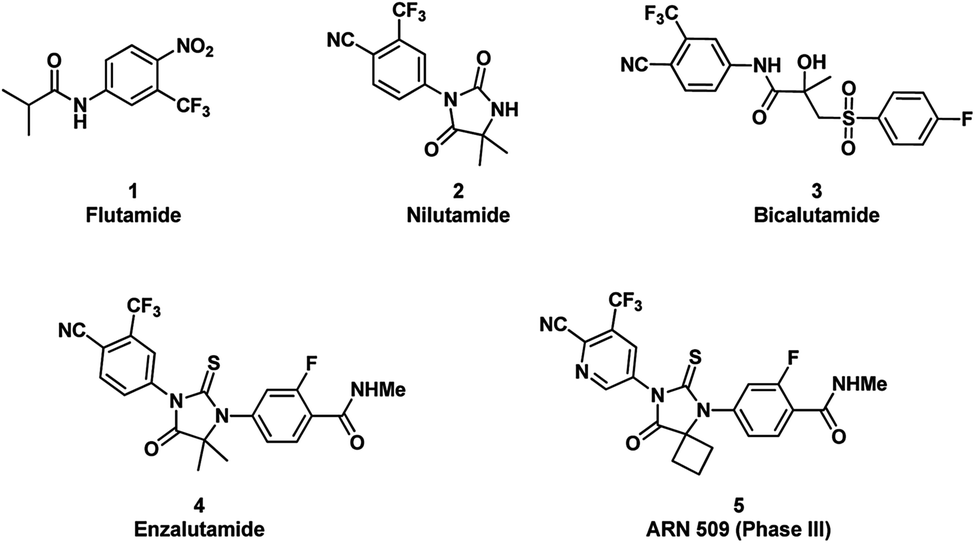 | ||
| Fig. 1 The first and second generation AR inhibitors. | ||
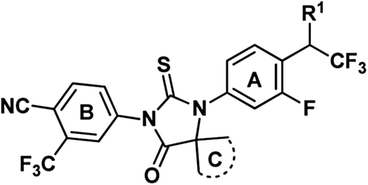
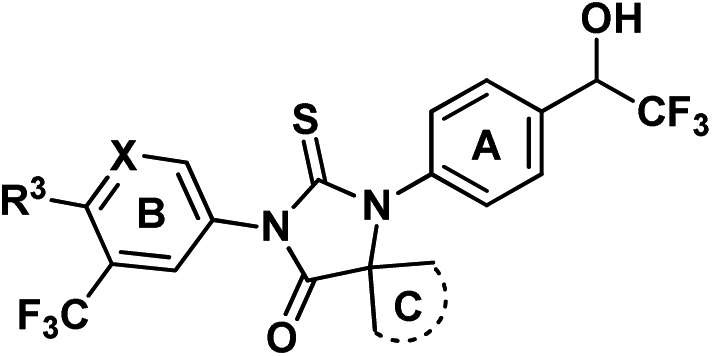
Replacement of amide group by other isosteres is a common practice in medicinal chemistry effort. Black and his colleagues from Merck reported to use trifluoroethylamine group as the isostere of amide in their searching of novel cathepsin K inhibitors.13 After replacement of amide with trifluoroethylamine, the potency and selectivity were improved, also the new function group are stable for P1–P2 cleavage that was observed for other amide inhibitors. Considering the major metabolic path way of ENT is through N-demthylation and amide hydrolysis,14,15 a series of isosteres analogs bearing trifluoroethylamine or trifluoroethanol were prepared as novel AR inhibitors. At the same time, C-ring was also optimized for potentially improvement of drug resistance (Fig. 2). Here we report our progress and also the in vivo and in vitro data of compound 13b which is dose-dependently inhibiting expression of prostate-specific antigen and has anti-tumor activity at oral administration dose of 15 mg kg−1 once daily.
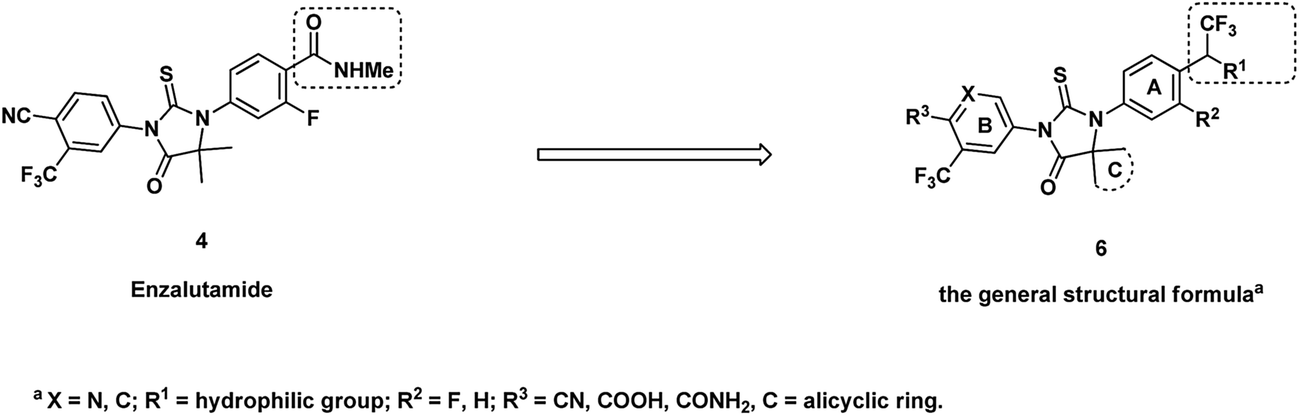 | ||
| Fig. 2 Proposed novel structure as potential AR inhibitors. | ||
2 Results and discussion
2.1 Chemistry
Synthetic routes of evaluated compounds 13a–f were depicted in Scheme 1 as path A and path B, respectively. In path A, compounds 8a–d was obtained by coupling of bromide 7a–b with the appropriated amino-acid under Ullmann coupling reaction conditions. Then, compounds 8a–d were directly subjected to esterification without further purification to afford compounds 9a–d under conditions of K2CO3 and CH3I. Finally, compounds 13a–f were obtained through the reaction between 9a–d and 10a–b.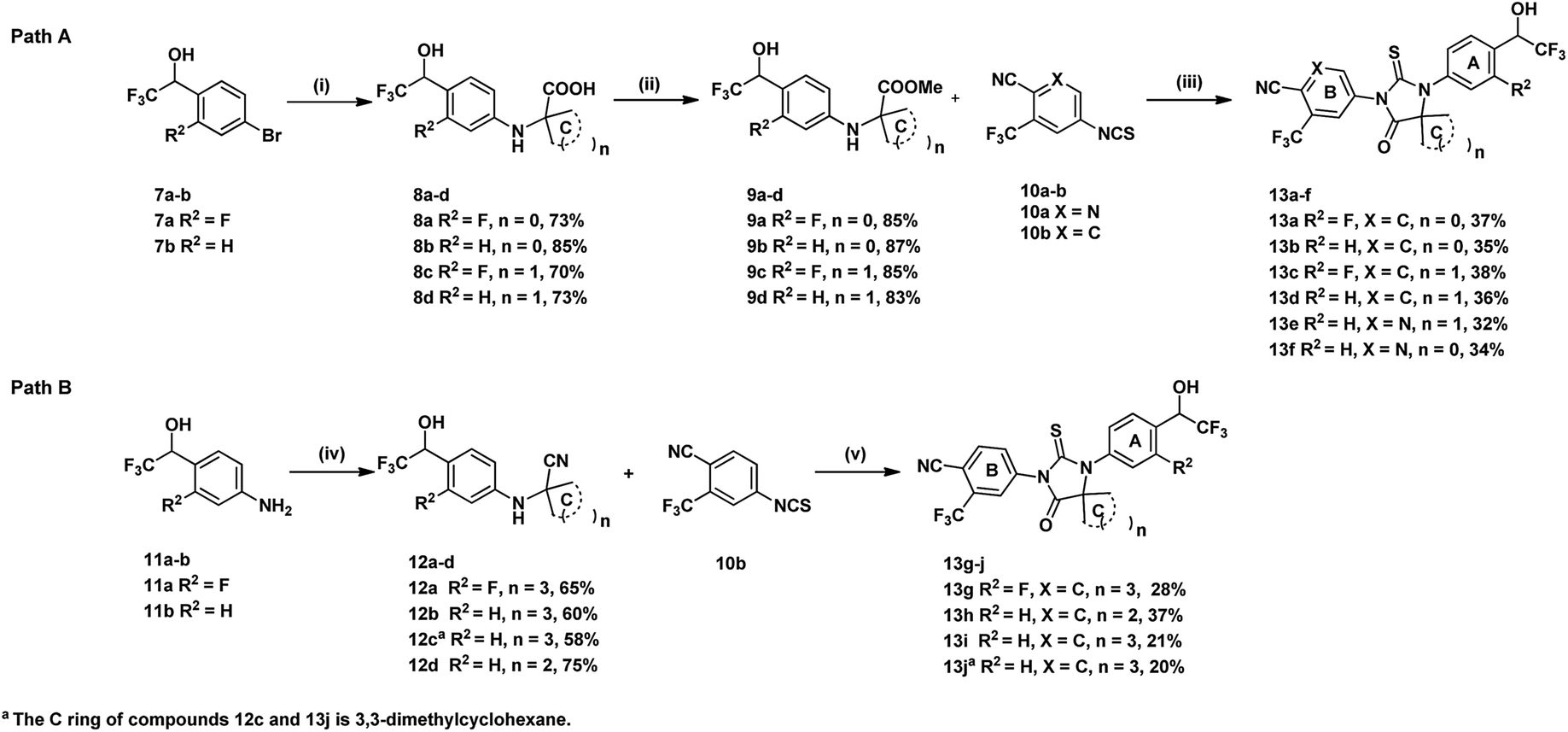 | ||
| Scheme 1 Reagents and conditions: (i) 10% CuI, 2% Cu, 20% N,N-dimethylglycine as ligand, K2CO3, amino-acid, DMF, 80 °C; (ii) K2CO3, CH3I, DMF, rt; (iii) 10a or 10b, DMAc, 80 °C, 24 h; (iv) TMSCN, AcOH, ketone, reflux; (v) 10b, DMAc, 80 °C, 24 h, then MeOH and 1 N HCl, reflux. | ||
In path B, the starting material 11a–b were provided from corresponding benzaldehyde according the reported procedure.15 Compounds 12a–d, then, were prepared under Strecker reaction conditions using compounds 11a–b, TMSCN and corresponding ketone as substrates. Finally, compounds 13g–j were obtained by reacting 12a–d with 10b. Other analogues, 14a–d and 15a–i, were synthesized form 13a–b by substitution, alkylation, oxidation, or hydrolysis reaction as shown in Scheme 2.
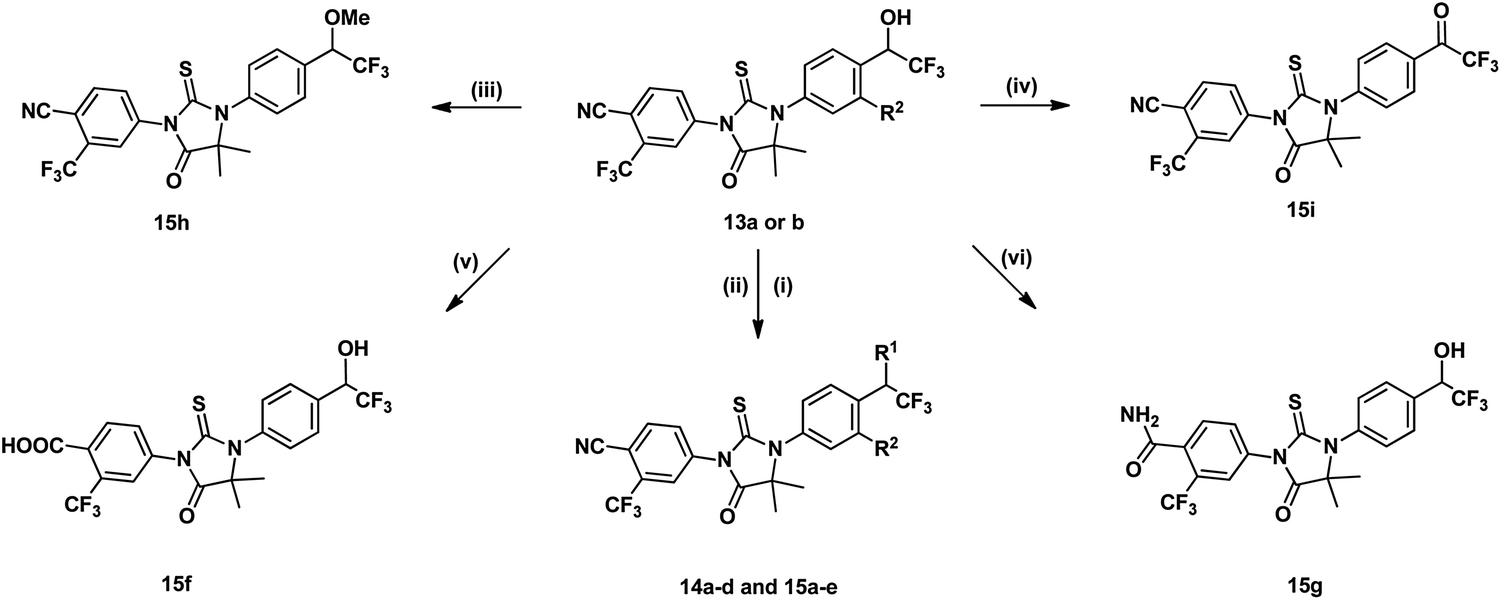 | ||
| Scheme 2 Reagents and conditions: (i) Et3N, MsCl, DCM, 0 °C, 2 h; (ii) amino or amino alcohol in THF, reflux, 12 h; (iii) NaH, MeI, DMF, rt; (iv) IBX, DMSO, rt, 12 h; (v) H2SO4, 100 °C, 24 h; (vi) 4 N NaOH, 30% H2O2, DMSO, 0 °C, 40 min. | ||
2.2 Biology activity
The in vitro anti-proliferation activities were evaluated by using LNCaP/AR cells (prostate cancer cells overexpressing AR). In order to mimic the CRPC state, the cells were cultured in charcoal stripped serum and treated with compounds for 6 days. The results were summarized in Tables 1, 2 and 3.
|
|||
|---|---|---|---|
| ID | R1 | C | IC50 (μM) |
| a IC50 was average of three determinations and deviation from the average was <5% of average value. | |||
| ENT | — | — | 0.25 |
| 13a | 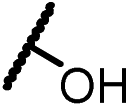 |
 |
0.38 |
| 14a |  |
 |
2.52 |
| 14b | 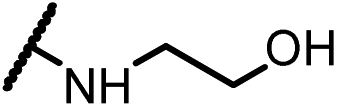 |
 |
0.35 |
| 14c | 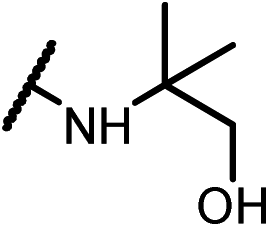 |
 |
0.64 |
| 14d | 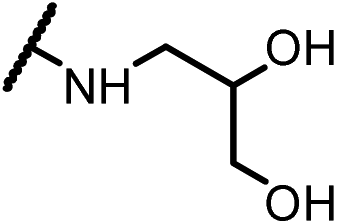 |
 |
0.92 |
| 13c | 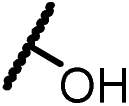 |
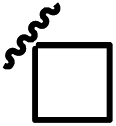 |
0.86 |
| 13g | 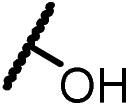 |
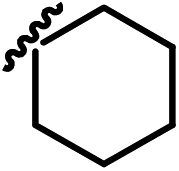 |
0.98 |
|
|||||
|---|---|---|---|---|---|
| ID | R1 | IC50 (μM) | ID | R1 | IC50 (μM) |
| a Control compound (IC50 of ENT is 0.25 ± 0.01 μM) used in all assays; IC50 was average of three determinations and deviation from the average was <5% of average value. | |||||
| 13b | 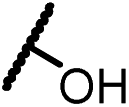 |
0.10 | 15d | 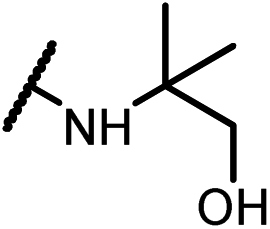 |
0.83 |
| 15a | 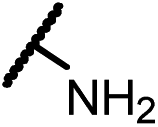 |
0.15 | 15e | 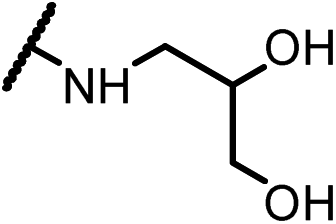 |
1.51 |
| 15b |  |
0.13 | 15h |  |
0.20 |
| 15c | 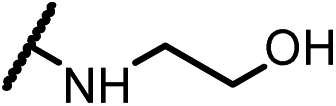 |
0.44 | 15i | 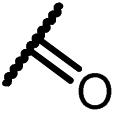 |
0.31 |
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | C | X | R3 | IC50 (μM) | ID | C | X | R3 | IC50 (μM) |
| a Control compound (IC50 of ENT is 0.25 ± 0.01 μM) used in all assays; IC50 was average of three determinations and deviation from the average was <5% of average value. | |||||||||
| 13d | 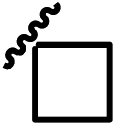 |
C | CN | 9.72 | 13e | 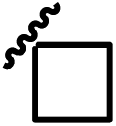 |
N | CN | 0.26 |
| 13h | 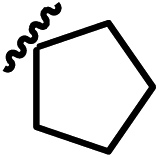 |
C | CN | 8.10 | 13f |  |
N | CN | 5.60 |
| 13i | 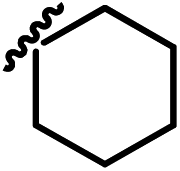 |
C | CN | 7.63 | 15f |  |
C | COOH | >10 |
| 13j | 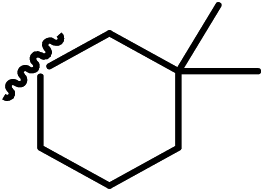 |
C | CN | 5.40 | 15g |  |
C | CONH2 | 0.86 |
The hydroxyl trifluoroethyl analog compound 13a, was first prepared because of its easily synthesis, showed 0.38 μM activity against LNCaP-AR cells compared with ENT of 0.25 μM. Encourage by this result, C-ring analogs of cyclobutyl 13c and cyclohexyl 13g were also prepared and achieved sub-μM activity. However, to our surprise, the closest isostere of amide, N-methylamino trifluoroethyl analog 14a lost the activity by 10-fold. Considering the poor solubility of ENT, more hydroxyl groups were introduced. To our delight, compounds 14b, 14c and 14d were also expressed good activity with IC50 from 0.35 to 0.92 μM.
Analogues with unsubstituted aromatic A-ring were also prepared in order to further study SAR (Table 2). Compound 13b represented an approximate 2-fold improvement over ENT. The second most active compound is 15b, with N–CH3 trifluoroethyl substitutions. Compound 15e with dihydroxyl propyl amino substitution provided 1.51 μM activity against LNCaP-AR. Other compounds 15a–d, 15h–i were found to have moderate to potent activities in the range of 0.15–0.83 μM against LNCaP-AR cell line.
ARN-509, which has substituted pyridine as B-ring and cyclobutyl as C-ring is under phase III trial for CRPC,16 therefore modification of A-ring substitution and C-ring size were also conducted in this studies (Table 3). However, in comparison with compound 13b, expanded the ring size of C-ring (compounds 13d and 13h–j) was detrimental to the activity. When C-ring was cyclobutyl group and B-ring is pyridine, compound 13e regained activity with IC50 of 0.26 μM. To our surprise, the IC50 value of compound 13f is inferior compared with 13e. In addition, replacement of CN group with carboxyl group (compound 15f) totally lost activity and the carboxamide (compound 15g) retained some activity.
To test whether 13b has an effect on prostate-specific antigen (PSA) levels, compound 13b was verified in LNCaP-AR cells by Western blot analysis. The result (Fig. 3) showed that compound 13b inhibited the expression of PSA even at very low doses (2.5 μM) and the inhibition activity was dose-dependent. Interestingly, the result also showed that expression of AR was down-regulated, further investigation is needed to probe the mechanism.
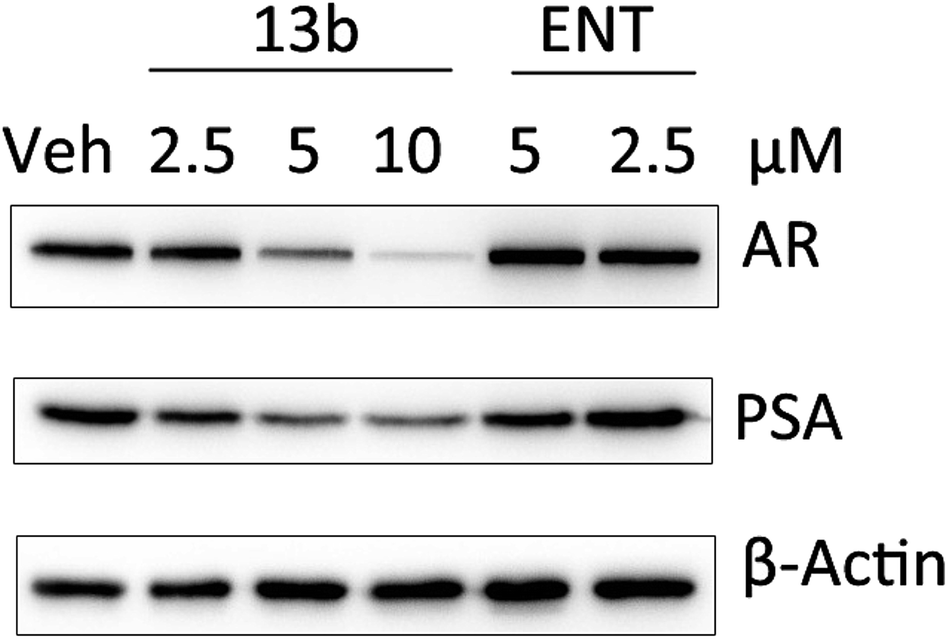 | ||
| Fig. 3 Western blot analysis of PSA expression in LNCaP-AR cells. | ||
The in vivo anti-tumor activity of 13b was evaluated using the CPRC (LNCaP-AR) xenograft model. SCID mice were castrated and subcutaneously inoculated with LNCaP-AR cells. When the tumor volumes reached 100–200 mm3, the mice were randomly grouped (8 animals each group) and oral administrated with compound 13b at 15 mg per kg q.d, 15 mg per kg q.3d, 5 mg per kg q.3d, and for ENT at 15 mg per kg q.d for 27 days. The tumor volumes and weight were measured every 3 days (Fig. 4). Compound 13b at all dose levels inhibited the tumor proliferation compared with the control. 5 mg per kg q.3d of compound 13b have a moderate activity. 15 mg per kg q.d has similar inhibition compared ENT. The tumor inhibition rate of 13b at dose of 5 mg per kg q.3d, 15 mg per kg q.3d, 15 mg per kg q.d were 58.9%, 66.0%, 96.9%, respectively, whereas, the ENT was more than 100% (Fig. 4a). In addition, the weight change 13b, ENT treated group and control group were slightly decreased (Fig. 4b).
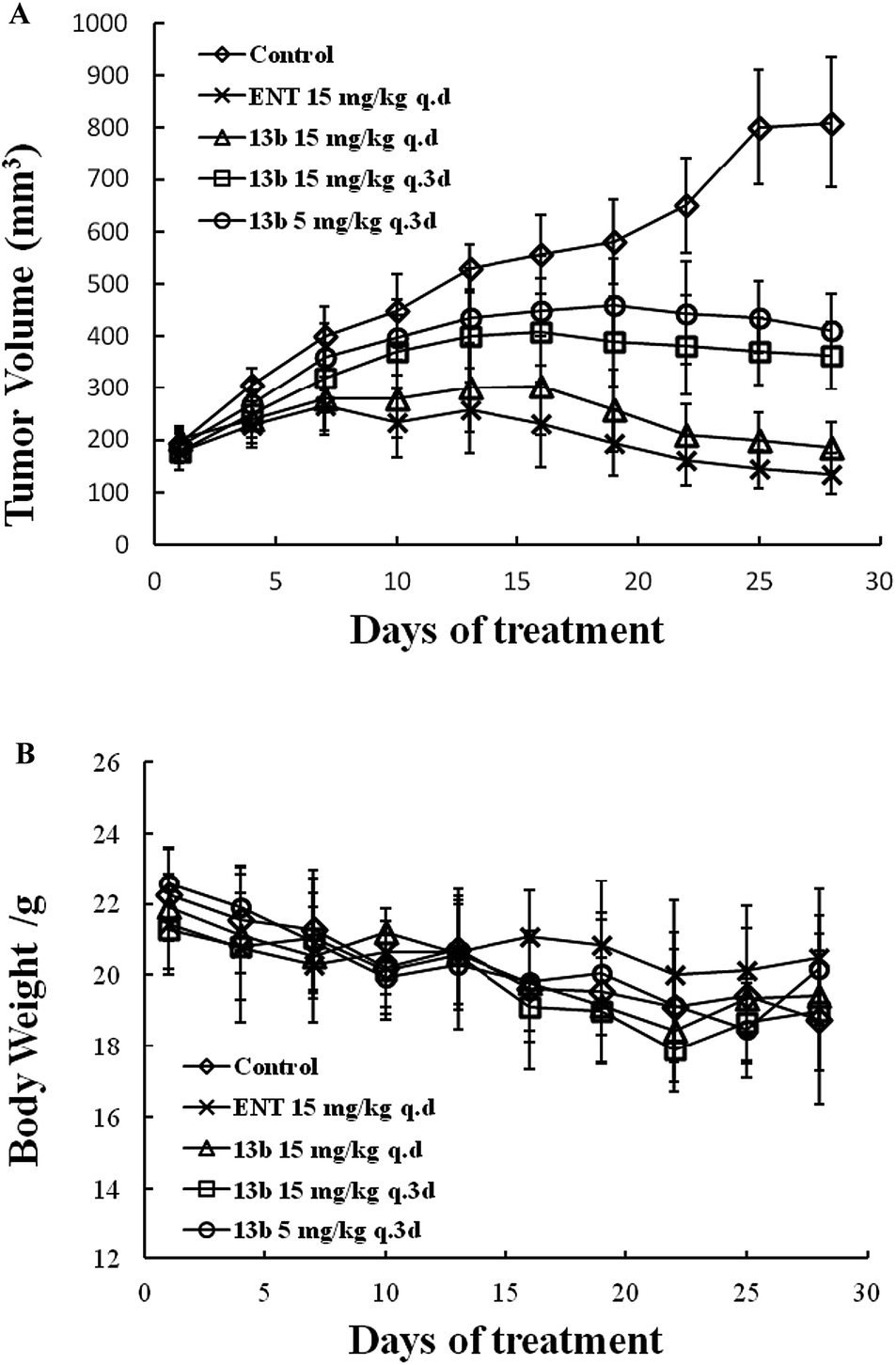 | ||
| Fig. 4 Tumor volume and body weight changes of mice after oral administration of ENT and 13b. | ||
3 Conclusions
In summary, we have synthesized a series of thiohydantoin analogues bearing substituted 2,2,2-trifluoro-1-phenylethanyl moiety. Compound 13b showed good cell activity against CRPC LNCaP-AR cell. Western blot analysis also verified compound 13b inhibited the expression of PSA in LNCaP-AR cells. Further evaluation in xenograft CRPC model showed compound 13b has anti-tumor activity of 96.9% inhibition at 15 mg per kg q.d. Further optimization and relevant works are in progress.4 Experimental
4.1 General information
The 1H and 13C 19F NMR spectra were recorded on Avance DMX 400 MHz NMR spectrometers (Bruker, Germany) in CDCl3 or deuterated DMSO using TMS as internal standard. Spectra are reported as follows: chemical shift δ (ppm), (integral, multiplicity (s = singlet, br s = broad singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, m = multiplet), coupling constant J (Hz), assignment). ESI-HRMS spectra were recorded on a commercial apparatus and methanol was used to dissolve the sample. Reagents were purchased from commercial sources and were used as received unless mentioned otherwise. Reactions were monitored by thin layer chromatography using silica gel GF 254 plates. Column chromatography was performed on silica gel (300–400 mesh).4.2 Experimental section
Compounds 9, 12, 13 were prepared by using a slightly modified literature-known procedure.17–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EA/1:3) to given compounds 9 (yield from 59–76%).:EA/1:2) to give 12 (yield from 58–75%).:EA/1:1) to give a brown solid as crude product, which was purified by preparative chromatography to give compound 13a as a white solid (32 mg, 37% yield).
:EA/1:3) to given compounds 9 (yield from 59–76%).:EA/1:2) to give 12 (yield from 58–75%).:EA/1:1) to give a brown solid as crude product, which was purified by preparative chromatography to give compound 13a as a white solid (32 mg, 37% yield).Compounds 13b–f were synthesized by a similar procedure as described for compound 13a.
4-(3-(3-Fluoro-4-(2,2,2-trifluoro-1-hydroxyethyl)phenyl)-4,4-di-methyl-5-oxo-2-thioxoimidazolidin-1-yl)-2-(trifluoromethyl)ben-zonitrile (13a). 1H NMR (400 MHz, CDCl3) δ 7.97 (dd, J = 9.0, 4.9 Hz, 2H), 7.87–7.76 (m, 2H), 7.21 (dd, J = 8.3, 1.7 Hz, 1H), 7.12 (td, J = 9.7, 1.9 Hz, 1H), 5.43 (q, J = 6.3 Hz, 1H), 3.88–3.45 (m, 1H), 1.61 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 179.85, 174.78, 160.31 (d, J = 248.6 Hz), 137.18 (d, J = 9.2 Hz), 137.00, 135.41, 134.61 (q, J = 33.1 Hz), 132.28, 130.24 (d, J = 4.5 Hz), 127.15 (q, J = 4.8 Hz), 125.94 (d, J = 3.5 Hz), 123.93 (q, J = 281.1 Hz), 123.81 (d, J = 13.2 Hz), 121.87 (q, J = 283.3 Hz), 117.43 (d, J = 23.8 Hz), 114.83, 110.18, 66.75, 65.62 (q, J = 33.5 Hz), 23.69. 19F NMR (376 MHz, CDCl3) δ −62.38, −78.42, −113.83. HRMS (EI) calcd for C21H14F7N3O2S [M + H]+: 506.0768, found: 506.0769.
4-(4,4-Dimethyl-5-oxo-2-thioxo-3-(4-(2,2,2-trifluoro-1-hydroxyeth-yl)phenyl)imidazolidin-1-yl)-2-(trifluoromethyl)benzonitrile (13b). 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 9.2 Hz, 2H), 7.84 (dd, J = 8.3, 1.6 Hz, 1H), 7.68 (d, J = 8.2 Hz, 2H), 7.37 (d, J = 8.4 Hz, 2H), 5.28–5.02 (m, 1H), 2.87 (s, 1H), 1.61 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 179.88, 174.96, 165.29, 137.09, 136.00, 135.77, 135.32, 133.62 (q, J = 34.1 Hz), 132.26, 129.76, 129.07, 127.16 (q, J = 5.1 Hz), 124.04 (q, J = 280.1 Hz), 121.89 (q, J = 273.3 Hz), 114.83, 110.19, 71.97 (q, J = 32.1 Hz), 66.60, 23.73. 19F NMR (376 MHz, CDCl3) δ −61.97, −78.16. HRMS (EI) calcd for C21H15F6N3O2S [M + H]+: 488.0862, found: 488.0869.
4-(5-(3-Fluoro-4-(2,2,2-trifluoro-1-hydroxyethyl)phenyl)-8-oxo-6-thioxo-5,7-diazaspiro[3.4]octan-7-yl)-2-(trifluoromethyl)benzoni-trile (13c). 1H NMR (400 MHz, CDCl3) δ 7.98 (dd, J = 8.4, 5.0 Hz, 2H), 7.93–7.79 (m, 2H), 7.24 (dd, J = 8.4, 1.9 Hz, 1H), 7.14 (dd, J = 9.9, 1.9 Hz, 1H), 5.52 (q, J = 6.3 Hz, 1H), 2.88 (ddd, J = 5.3, 3.5, 2.5 Hz, 1H), 2.70 (ddd, J = 8.9, 6.6, 3.4 Hz, 2H), 2.56 (ddd, J = 20.0, 10.0, 2.4 Hz, 2H), 2.36–2.15 (m, 1H), 1.83–1.64 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 179.86, 174.51, 160.54 (d, J = 252.3 Hz), 137.52 (d, J = 9.6 Hz), 136.88, 135.28, 133.68 (d, J = 33.5 Hz), 132.11, 130.39 (d, J = 4.1 Hz), 127.04 (q, J = 5.0 Hz), 126.43 (d, J = 3.1 Hz), 124.38 (q, J = 280.6 Hz), 123.57 (d, J = 13.1 Hz), 121.73 (q, J = 282.6 Hz), 117.89 (d, J = 23.4 Hz), 114.78, 110.24, 67.51, 65.83 (q, J = 32.1 Hz), 31.69, 13.67. 19F NMR (376 MHz, CDCl3) δ −61.98, −78.38, −113.55. HRMS (EI) calcd for C22H14F7N3O2S [M + H]+: 518.0768, found: 518.0772.
4-(8-Oxo-6-thioxo-5-(4-(2,2,2-trifluoro-1-hydroxyethyl)phenyl)-5,7-diazaspiro[3.4]octan-7-yl)-2-(trifluoromethyl)benzonitri-le (13d). 1H NMR (400 MHz, CDCl3) δ 7.98 (dd, J = 5.1, 3.2 Hz, 2H), 7.90–7.83 (m, 1H), 7.74 (t, J = 9.1 Hz, 2H), 7.43–7.34 (m, 2H), 5.15 (q, J = 6.6 Hz, 1H), 3.11–2.76 (m, 1H), 2.75–2.63 (m, 2H), 2.64–2.50 (m, 2H), 2.34–2.16 (m, 1H), 1.70 (dtt, J = 10.5, 7.7, 5.1 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 179.91, 174.77, 137.04, 136.16, 135.72, 135.23, 133.61 (q, J = 34.1 Hz), 132.16, 130.20, 129.25, 127.07 (q, J = 5.1 Hz), 124.03 (q, J = 280.1 Hz), 121.89 (q, J = 273.3 Hz), 114.84, 110.12, 72.10 (q, J = 32.1 Hz), 67.45, 31.59, 13.70. 19F NMR (376 MHz, CDCl3) δ −61.96, −78.09. HRMS (EI) calcd for C22H15F6N3O2S [M + H]+: 500.0862, found: 500.0866.
5-(8-Oxo-6-thioxo-5-(4-(2,2,2-trifluoro-1-hydroxyethyl)phenyl)-5,7-diazaspiro[3.4]octan-7-yl)-3-(trifluoromethyl)picolino-nitrile (13e). 1H NMR (400 MHz, DMSO) δ 9.23 (s, 1H), 8.77 (d, J = 1.9 Hz, 1H), 7.75 (d, J = 8.3 Hz, 2H), 7.48 (d, J = 8.4 Hz, 2H), 7.03 (s, 1H), 5.49–5.19 (m, 1H), 2.72–2.59 (m, 2H), 2.50–2.38 (m, 2H), 2.07–1.90 (m, 1H), 1.66–1.43 (m, 1H). 13C NMR (101 MHz, DMSO) δ 180.05, 174.96, 154.07, 137.59, 136.19, 136.03 (q, J = 4.2 Hz), 133.82, 130.31, 129.51, 129.24 (q, J = 34.1 Hz), 129.20 (q, J = 2.0 Hz), 125.43 (q, J = 277.1 Hz), 122.04 (q, J = 273.3 Hz), 114.73, 70.41 (q, J = 31.5 Hz), 67.98, 31.49, 13.86. 19F NMR (376 MHz, DMSO) δ −60.83, −76.56. HRMS (EI) calcd for C21H14F6N4O2S [M + H]+: 501.0814, found: 501.0815.
5-(4,4-Dimethyl-5-oxo-2-thioxo-3-(4-(2,2,2-trifluoro-1-hydro-xyeth-yl)phenyl)imidazolidin-1-yl)-3-(trifluoromethyl)picolinonitrile (13f). 1H NMR (400 MHz, CDCl3) δ 9.08 (dd, J = 12.6, 2.2 Hz, 1H), 8.37 (t, J = 3.9 Hz, 1H), 7.67 (dd, J = 18.2, 8.3 Hz, 2H), 7.45–7.31 (m, 2H), 5.13 (q, J = 6.6 Hz, 1H), 1.63 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 179.08, 174.67, 152.34, 135.96, 135.72, 134.16 (q, J = 4.2 Hz), 132.41, 130.54 (q, J = 34.3 Hz), 129.96, 129.67, 129.15, 124.04 (q, J = 283.1 Hz), 121.26 (q, J = 270.9 Hz), 113.75, 71.99 (q, J = 26.3 Hz), 66.81, 23.76. 19F NMR (376 MHz, CDCl3) δ −61.87, −78.12. HRMS (EI) calcd for C20H14F6N4O2S+ [M + H]+: 489.0814, found: 489.0815.
:EA/1:1) to give a brown solid as crude product, which was purified by preparative chromatography to give compound 13g as a white solid (26 mg, 28% yield).Compounds 13h–j were synthesized by a similar procedure as described for compound 13g.
4-(1-(3-Fluoro-4-(2,2,2-trifluoro-1-hydroxyethyl)phenyl)-4-oxo-2-thioxo-1,3-diazaspiro[4.5]decan-3-yl)-2-(trifluoromethyl)benzo-nitrile (13g). 1H NMR (400 MHz, CDCl3) δ 7.99 (t, J = 10.4 Hz, 1H), 7.94 (d, J = 1.7 Hz, 1H), 7.89–7.78 (m, 2H), 7.17–7.10 (m, 1H), 7.05 (dd, J = 9.9, 1.9 Hz, 1H), 5.61–5.38 (m, 1H), 3.03–2.80 (m, 1H), 2.26–1.98 (m, 4H), 1.87–1.64 (m, 5H), 1.17–1.00 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 180.12, 173.49, 160.36 (d, J = 251.3 Hz), 137.10 (d, J = 10.0 Hz), 136.95, 135.25, 133.32 (q, J = 33.8. Hz), 132.31, 130.06 (d, J = 4.6 Hz), 127.22 (q, J = 3.2 Hz), 127.98 (d, J = 3.1 Hz), 123.86 (q, J = 280.1 Hz), 123.52 (d, J = 14.0 Hz), 122.36 (q, J = 283.3 Hz), 118.26 (d, J = 23.3 Hz), 114.80, 110.19, 67.73, 65.97 (q, J = 61.4 Hz), 32.82, 23.86, 20.90. 19F NMR (376 MHz, CDCl3) δ −62.04, −78.37, −113.90. HRMS (EI) calcd for C24H18F7N3O2S [M + H]+: 546.1081, found: 546.1086.
4-(4-Oxo-2-thioxo-1-(4-(2,2,2-trifluoro-1-hydroxyethyl)phenyl)-1,3-diazaspiro[4.4]nonan-3-yl)-2-(trifluoromethyl)benzonitrile (13h). 1H NMR (400 MHz, CDCl3) δ 7.98 (dd, J = 4.7, 3.2 Hz, 2H), 7.90–7.83 (m, 1H), 7.69 (d, J = 8.2 Hz, 2H), 7.39 (d, J = 8.3 Hz, 2H), 5.25–5.05 (m, 1H), 3.07–2.49 (m, 1H), 2.34 (dt, J = 13.2, 6.2 Hz, 2H), 2.23–2.11 (m, 2H), 1.94–1.82 (m, 2H), 1.66–1.44 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 180.19, 176.05, 137.19, 136.74, 135.58, 135.20, 133.61 (q, J = 37.0 Hz), 132.19, 130.21, 129.09, 127.12 (q, J = 4.8 Hz), 123.73 (q, J = 279.6 Hz), 121.84 (q, J = 270.7 Hz), 114.83, 110.17, 75.27, 72.10 (q, J = 32.3 Hz), 36.16, 25.12. 19F NMR (376 MHz, CDCl3) δ −61.96, −78.19. HRMS (EI) calcd for C23H17F6N3O2S [M + H]+: 514.1018, found: 514.1023.
4-(4-Oxo-2-thioxo-1-(4-(2,2,2-trifluoro-1-hydroxyethyl)phenyl)-1,3-diazaspiro[4.5]decan-3-yl)-2-(trifluoromethyl)benzonitr-ile (13i). 1H NMR (400 MHz, CDCl3) δ 8.05–7.90 (m, 2H), 7.83 (dd, J = 8.2, 1.7 Hz, 1H), 7.67 (d, J = 8.2 Hz, 2H), 7.31 (t, J = 9.3 Hz, 2H), 5.12 (q, J = 6.6 Hz, 1H), 3.28–2.83 (m, 1H), 2.19–2.00 (m, 4H), 1.70 (dd, J = 17.4, 7.1 Hz, 5H), 1.17–1.00 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 180.07, 173.81, 137.12, 135.76, 135.21, 133.57 (q, J = 34.0 Hz), 132.36, 130.69, 128.99, 127.32 (q, J = 5.0 Hz), 124.05 (q, J = 278.0 Hz), 121.90 (q, J = 273.0 Hz), 114.86, 110.08, 72.05 (q, J = 32.0 Hz), 67.62, 32.75, 23.88, 20.69. 19F NMR (376 MHz, CDCl3) δ −61.94, −78.07. HRMS (EI) calcd for C24H19F6N3O2S [M + H]+: 528.1175, found: 528.1183.
4-(7,7-Dimethyl-4-oxo-2-thioxo-1-(4-(2,2,2-trifluoro-1-hydro-xyeth-yl)phenyl)-1,3-diazaspiro[4.5]decan-3-yl)-2-(trifluoromethyl)benz-onitrile (13j). 1H NMR (400 MHz, CDCl3) δ 8.04–7.89 (m, 2H), 7.90–7.77 (m, 1H), 7.75–7.64 (m, 2H), 7.28 (t, J = 6.1 Hz, 2H), 5.27–5.01 (m, 1H), 3.17–2.69 (m, 1H), 2.36–2.20 (m, 1H), 2.21–2.10 (m, 1H), 1.98–1.86 (m, 1H), 1.73–1.60 (m, 3H), 1.57–1.46 (m, 2H), 1.22 (d, J = 14.6 Hz, 3H), 0.98–0.90 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 180.04, 174.36, 137.23, 135.66, 135.17, 133.57 (q, J = 34.0 Hz), 132.40, 131.28, 128.98, 127.32 (q, J = 5.0 Hz), 124.05 (q, J = 281.2 Hz), 121.89 (q, J = 272.5 Hz), 114.84, 110.11, 72.09 (q, J = 32.5 Hz), 68.81, 43.20, 37.41, 35.03, 32.48, 32.19, 26.36, 17.95. 19F NMR (376 MHz, CDCl3) δ −61.96, −78.04. HRMS (EI) calcd for C26H23F6N3O2S [M + H]+: 556.1488, found: 556.1494.
Compounds 14b–d and 15b–e were synthesized by a similar procedure as described for compound 14a.
4-(3-(3-Fluoro-4-(2,2,2-trifluoro-1-(methylamino)ethyl)phenyl)-4,4-dimethyl-5-oxo-2-thioxoimidazolidin-1-yl)-2-(trifluoromethyl)ben-zonitrile (14a). 1H NMR (400 MHz, CDCl3) δ 8.04–7.91 (m, 2H), 7.89–7.78 (m, 1H), 7.68 (t, J = 7.9 Hz, 1H), 7.22–7.16 (m, 1H), 7.16–7.05 (m, 1H), 4.75–4.38 (m, 1H), 2.49 (s, 3H), 1.70 (ddd, J = 11.6, 10.6, 7.5 Hz, 1H), 1.62 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 179.78, 174.59, 162.34 (d, J = 251.6 Hz), 136.89, 136.80 (d, J = 9.8 Hz), 135.29, 133.66 (q, J = 33.2 Hz), 132.13, 130.13 (d, J = 4.0 Hz), 127.08 (q, J = 4.1 Hz), 126.02 (d, J = 3.8 Hz), 125.07 (q, J = 277.2 Hz), 123.59 (q, J = 14.0 Hz) 121.84 (q, J = 273.1 Hz), 117.49 (q, J = 23.9 Hz), 114.76, 110.36, 66.59, 58.65 (q, J = 27.5 Hz), 34.84, 23.81. 19F NMR (376 MHz, CDCl3) δ −61.88, −78.43, −78.45, −113.50, −113.52. HRMS (EI) calcd for C22H17F7N4OS [M + H]+: 519.1084, found: 519.1093.
4-(3-(3-Fluoro-4-(2,2,2-trifluoro-1-((2-hydroxyethyl)amino)ethyl) phenyl)-4,4-dimethyl-5-oxo-2-thioxoimidazolidin-1-yl)-2-(trifluoro-methyl)benzonitrile (14b). 1H NMR (400 MHz, CDCl3) δ 8.04–7.91 (m, 2H), 7.85 (t, J = 10.9 Hz, 1H), 7.68 (t, J = 7.9 Hz, 1H), 7.18 (t, J = 9.0 Hz, 1H), 7.12 (d, J = 9.9 Hz, 1H), 4.67 (q, J = 7.0 Hz, 1H), 3.91–3.51 (m, 2H), 3.13–2.51 (m, 2H), 2.28–1.90 (m, 2H), 1.62 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 179.81, 174.58, 161.20 (d, J = 251.0 Hz), 136.92, 136.91 (d, J = 9.9 Hz), 135.31, 133.69 (q, J = 33.1 Hz), 132.18, 130.05 (d, J = 4.0 Hz), 127.10 (q, J = 4.8 Hz), 126.08 (d, J = 3.0 Hz), 124.93 (q, J = 281.1 Hz), 123.84 (d, J = 13.8 Hz), 121.85 (q, J = 273.1 Hz), 117.58 (d, J = 24.3 Hz), 114.76, 110.36, 66.62, 61.26, 56.93 (q, J = 24.3 Hz), 49.51, 23.81. 19F NMR (376 MHz, CDCl3) δ −61.94, −73.19, −113.75. HRMS (EI) calcd for C23H19F7N4O2S [M + H]+: 549.1190, found: 549.1191.
4-(3-(3-Fluoro-4-(2,2,2-trifluoro-1-((1-hydroxy-2-methylpro-pan-2-yl)amino)ethyl)phenyl)-4,4-dimethyl-5-oxo-2-thioxoi-midazolidin-1-yl)-2-(trifluoromethyl)benzonitrile (14c). 1H NMR (400 MHz, CDCl3) δ 7.91 (d, J = 8.6 Hz, 2H), 7.82–7.74 (m, 1H), 7.59–7.45 (m, 2H), 7.23 (dd, J = 18.1, 4.6 Hz, 2H), 4.32–4.11 (m, 1H), 3.41–3.24 (m, 1H), 3.22–3.07 (m, 1H), 2.14–1.85 (m, 1H), 1.69–1.61 (m, 1H), 1.53 (s, 6H), 0.95 (d, J = 11.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 179.80, 174.58, 161.35 (d, J = 257.0 Hz), 136.92, 136.69 (d, J = 10.0 Hz), 135.30, 133.65 (q, J = 33.0 Hz), 132.15, 130.05 (d, J = 4.0 Hz), 127.09 (q, J = 5.0 Hz), 126.79 (d, J = 14.1 Hz), 126.13 (d, J = 4.0 Hz), 124.81 (q, J = 280.3 Hz), 121.85 (q, J = 273.1 Hz), 117.53 (d, J = 24.0 Hz), 114.76, 110.33, 69.35, 66.54, 54.91, 51.21 (q, J = 29.3 Hz), 24.81, 23.93, 23.82. 19F NMR (376 MHz, CDCl3) δ −61.95, −74.66, −114.24. HRMS (EI) calcd for C25H23F7N4O2S [M + H]+: 577.1503, found: 577.1504.
4-(3-(4-(1-((2,3-Dihydroxypropyl)amino)-2,2,2-trifluoroethyl)-3-fluorophenyl)-4,4-dimethyl-5-oxo-2-thioxoimidazolidin-1-yl)-2-(trifluoromethyl)benzonitrile (14d). 1H NMR (400 MHz, CDCl3) δ 8.10–7.91 (m, 2H), 7.90–7.74 (m, 1H), 7.74–7.58 (m, 1H), 7.18 (t, J = 9.6 Hz, 1H), 7.13 (d, J = 10.0 Hz, 1H), 4.73–4.51 (m, 1H), 3.92–3.69 (m, 2H), 3.67–3.52 (m, 1H), 2.92–2.83 (m, 1H), 2.82–2.70 (m, 1H), 2.49–2.05 (m, 2H), 1.62 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 179.82, 174.55, 161.20 (d, J = 261.1 Hz), 137.03 (d, J = 8.5 Hz), 136.99, 136.89, 135.31, 133.69 (q, J = 33.4 Hz), 132.17, 129.94 (d, J = 5.1 Hz), 127.11 (q, J = 5.1 Hz), 126.14 (d, J = 4.1 Hz), 124.84 (q, J = 277.3 Hz), 119.15 (q, J = 270.6 Hz), 117.68 (d, J = 24.1 Hz), 114.74, 110.41, 70.15 (d, J = 30.2 Hz), 66.62, 65.02 (d, J = 12.3 Hz), 57.20 (q, J = 30.3 Hz), 50.21, 23.83. 19F NMR (377 MHz, CDCl3) δ −61.99, −73.69, −73.71, −73.72, −113.60, −113.61, −113.66, −113.67. HRMS (EI) calcd for C24H21F7N4O3S [M + H]+: 579.1295, found: 579.1299.
4-(3-(4-(1-Amino-2,2,2-trifluoroethyl)phenyl)-4,4-dimethyl-5-oxo-2-thioxoimidazolidin-1-yl)-2-(trifluoromethyl)benzonitrile (15a). 1H NMR (400 MHz, CDCl3) δ 8.12–7.93 (m, 2H), 7.91–7.76 (m, 1H), 7.74–7.55 (m, 2H), 7.35 (d, J = 8.4 Hz, 2H), 4.66–4.40 (m, 1H), 1.93–1.77 (m, 2H), 1.60 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 179.86, 174.90, 137.15, 137.09, 135.66, 135.27, 133.59 (q, J = 33.1 Hz), 132.24, 129.84, 129.53, 127.14 (q, J = 4.3 Hz), 125.37 (q, J = 280.6 Hz), 121.89 (q, J = 272.7 Hz), 114.83, 110.22, 66.53, 57.51 (q, J = 31.2 Hz), 23.75. 19F NMR (376 MHz, CDCl3) δ −61.95, −76.41. HRMS (EI) calcd for C21H16F6N4OS [M + H]+: 487.1022, found: 487.1025.
4-(4,4-Dimethyl-5-oxo-2-thioxo-3-(4-(2,2,2-trifluoro-1-(methyl-amino)ethyl)phenyl)imidazolidin-1-yl)-2-(trifluoromethyl)ben-zonitrile (15b). 1H NMR (400 MHz, CDCl3) δ 8.05–7.95 (m, 2H), 7.89–7.82 (m, 1H), 7.61 (d, J = 8.3 Hz, 2H), 7.40–7.32 (m, 2H), 4.23–4.07 (m, 1H), 2.47 (s, 3H), 1.61 (d, J = 2.0 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 179.82, 174.92, 137.11, 135.99, 135.71, 135.26, 133.57 (q, J = 33.8 Hz), 132.24, 130.06, 129.85, 127.12 (q, J = 5.1 Hz), 125.14 (q, J = 280.2 Hz), 121.89 (q, J = 272.5 Hz), 114.83, 110.21, 66.53, 66.02 (q, J = 32.1 Hz), 34.85, 23.76. 19F NMR (377 MHz, CDCl3) δ −61.98, −73.81. HRMS (EI) calcd for C22H18F6N4OS [M + H]+: 501.1178, found: 501.1188.
4-(4,4-Dimethyl-5-oxo-2-thioxo-3-(4-(2,2,2-trifluoro-1-((2-hydroxy-ethyl)amino)ethyl)phenyl)imidazolidin-1-yl)-2-(trifluoromethyl) benzonitrile (15c). 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 8.4 Hz, 2H), 7.92–7.77 (m, 1H), 7.62 (t, J = 13.0 Hz, 2H), 7.42–7.27 (m, 2H), 4.28 (q, J = 7.2 Hz, 1H), 3.85–3.51 (m, 2H), 2.97–2.65 (m, 2H), 2.01 (s, 2H), 1.61 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 179.83, 174.89, 137.07, 136.11, 135.77, 135.26, 133.60 (q, J = 33.2 Hz), 132.23, 129.97, 129.93, 127.14 (q, J = 4.8 Hz), 125.13 (q, J = 281.2 Hz), 121.88 (q, J = 273.2 Hz), 114.82, 110.23, 66.54, 64.06 (q, J = 28.6 Hz), 61.37, 49.41, 23.77. 19F NMR (376 MHz, CDCl3) δ −61.96, −73.78. HRMS (EI) calcd for C23H20F6N4O2S [M + H]+: 531.1284, found: 531.1288.
4-(4,4-Dimethyl-5-oxo-2-thioxo-3-(4-(2,2,2-trifluoro-1-((1-hydroxy-2-methylpropan-2-yl)amino)ethyl)phenyl)imida-zolidin-1-yl)-2-(trifluoromethyl)benzonitrile (15d). 1H NMR (400 MHz, CDCl3) δ 7.91 (d, J = 8.6 Hz, 2H), 7.82–7.74 (m, 1H), 7.51 (d, J = 8.1 Hz, 2H), 7.26 (d, J = 8.2 Hz, 2H), 4.40–4.15 (m, 1H), 3.39–3.23 (m, 1H), 3.24–3.09 (m, 1H), 2.15–1.87 (m, 1H), 1.79–1.64 (m, 2H), 1.53 (s, 6H), 0.95 (d, J = 11.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 179.80, 174.88, 139.13, 137.06, 135.48, 135.25, 133.60 (q, J = 33.1 Hz), 132.18, 129.96, 129.51, 127.13 (q, J = 4.9 Hz), 125.26 (q, J = 279.3 Hz), 121.88 (q, J = 273.4 Hz), 114.81, 110.24, 69.46, 66.47, 58.32 (q, J = 29.6 Hz), 55.04, 25.05, 24.27, 23.80. 19F NMR (376 MHz, CDCl3) δ −61.97, −74.32. HRMS (EI) calcd for C25H24F6N4O2S [M + H]+: 559.1597, found: 559.1600.
4-(3-(4-(1-((2,3-Dihydroxypropyl)amino)-2,2,2-trifluoroethyl)phen-yl)-4,4-dimethyl-5-oxo-2-thioxoimidazolidin-1-yl)-2-(trifluorometh-yl)benzonitrile (15e). 1H NMR (400 MHz, CDCl3) δ 8.06–7.94 (m, 2H), 7.88–7.80 (m, 1H), 7.66–7.55 (m, 2H), 7.41–7.32 (m, 2H), 4.25 (qd, J = 7.1, 2.3 Hz, 1H), 3.87–3.76 (m, 1H), 3.73 (dt, J = 11.3, 3.3 Hz, 1H), 3.60 (ddd, J = 11.5, 6.3, 5.4 Hz, 1H), 2.84 (dt, J = 12.0, 3.6 Hz, 1H), 2.77–2.67 (m, 1H), 2.20 (s, 1H), 1.75–1.64 (m, 2H), 1.61 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 179.84, 174.93, 137.03, 135.90, 135.77, 135.26, 133.63 (q, J = 33.3 Hz), 132.19, 130.02, 129.88, 127.13 (q, J = 4.8 Hz), 124.83 (q, J = 281.3 Hz), 118.27 (q, J = 278.3 Hz), 114.79, 110.29, 70.21 (d, J = 36.2 Hz), 66.54, 65.12 (d, J = 8.1 Hz), 64.33 (q, J = 28.2 Hz), 50.37 (d, J = 45.3 Hz), 23.79. 19F NMR (376 MHz, CDCl3) δ −61.97, −73.75, −73.78. HRMS (EI) calcd for C24H22F6N4O3S [M + H]+: 561.1390, found: 561.1390.
4-(4,4-Dimethyl-5-oxo-2-thioxo-3-(4-(2,2,2-trifluoro-1-hydroxyeth-yl)phenyl)imidazolidin-1-yl)-2-(trifluoromethyl)benzoic acid (15f). In a 20 mL tube sealing, 13b (49 mg, 0.1 mmol), concentrated H2SO4 (5 mL), were added. The mixture was sealed and stirred for 24 h at 100 °C. After the reaction, the mixture was poured into 100 mL ice water and saturated sodium bicarbonate was added to adjust pH to 4. The mixture was washed with EA (30 mL × 3) and the organic layer was separated and dried over Na2SO4. After filtration and evaporation, the resulting crude product was purified by column chromatography with petrol ether as eluent to afford 15f (43 mg, 84% yield).
1H NMR (400 MHz, DMSO) δ 8.06 (d, J = 1.7 Hz, 1H), 8.02–7.92 (m, 1H), 7.88 (dd, J = 8.2, 1.7 Hz, 1H), 7.68 (t, J = 9.4 Hz, 2H), 7.48 (t, J = 10.3 Hz, 2H), 5.52–5.13 (m, 1H), 1.52 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 185.74, 180.49, 172.60, 142.00, 141.17, 140.70, 138.48, 135.54, 134.91, 133.94, 132.82 (q, J = 2.9 Hz), 132.08 (q, J = 32.1 Hz), 130.17 (q, J = 282.7 Hz), 128.37, (q, J = 273.0 Hz), 75.18 (q, J = 29.8 Hz), 71.42, 28.23. 19F NMR (376 MHz, CDCl3) δ −53.41, −71.81. HRMS (EI) calcd for C21H16F6N2O4S [M + H]+: 507.0808, found: 507.0817.
4-(4,4-Dimethyl-5-oxo-2-thioxo-3-(4-(2,2,2-trifluoro-1-hydroxyeth-yl)phenyl)imidazolidin-1-yl)-2-(trifluoromethyl)benza-mide (15g). To a mixture of 13b (49 mg, 0.1 mmol) and sodium hydroxide (4.0 M in water, 0.25 mL, 1.0 mmol) in DMSO (2.5 mL) was added hydrogen peroxide (30% aqueous solution, 0.05 mL, 0.49 mmol). The reaction mixture was heated to 35 °C. After 40 minutes, 20 mL H2O was added, the mixtures was extracted with ethyl acetate. The organic layer was separated and dried over Na2SO4. After filtration and evaporation, the resulting crude product was purified by column chromatography with petrol ether as eluent to afford 15g (39 mg, 77% yield).
1H NMR (400 MHz, DMSO) δ 8.19–8.10 (m, 1H), 8.04–7.91 (m, 1H), 7.87–7.78 (m, 1H), 7.70 (dd, J = 16.7, 7.3 Hz, 4H), 7.46 (d, J = 8.2 Hz, 2H), 7.09–6.91 (m, 1H), 5.37–5.24 (m, 1H), 1.51 (s, 6H). 13C NMR (101 MHz, DMSO) δ 181.07, 175.85, 168.88, 137.55, 137.21, 136.47, 134.84, 133.55, 130.18, 129.51, 129.17, 127.84 (q, J = 5.1 Hz), 126.53, 125.43 (q, J = 283.1 Hz), 123.81 (q, J = 273.3 Hz), 70.47 (q, J = 21.1 Hz), 66.62, 23.51. 19F NMR (376 MHz, DMSO) δ −57.99, −76.56. HRMS (EI) calcd for C21H17F6N3O3S [M + H]+: 506.0968, found: 506.0975.
4-(4,4-Dimethyl-5-oxo-2-thioxo-3-(4-(2,2,2-trifluoro-1-methoxyeth-yl)phenyl)imidazolidin-1-yl)-2-(trifluoromethyl)benzo-nitrile (15h). In a 25 mL round-bottomed flask, 13b (49 mg, 0.1 mmol), DMF (5 mL), and NaH 60% (12 mg, 0.3 mmol) were added. The mixture was stirred and simultaneously CH3I (12.3 mg 0.3 mmol) was added then, the reaction was stirred for 2 h at room temperature. After the reaction, 20 mL saturated NH4Cl was added, the mixtures was extracted with ethyl acetate. The organic layer was separated and dried over Na2SO4. After filtration and evaporation, the resulting crude product was purified by column chromatography with petrol ether as eluent to afford 15h (40 mg, 77% yield).
1H NMR (400 MHz, CDCl3) δ 8.22–7.93 (m, 2H), 7.86–7.84 (m, 1H), 7.63–7.57 (m, 2H), 7.39–7.34 (m, 2H), 4.61 (dd, J = 11.0, 4.6 Hz, 1H), 3.52–3.48 (m, 3H), 1.61 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 179.83, 174.88, 137.11, 136.19, 135.26, 133.52 (q, J = 33.0 Hz), 132.26, 129.82, 129.57, 127.15 (q, J = 4.8 Hz), 123.64 (q, J = 281.0 Hz), 121.85 (q, J = 276.3 Hz), 114.84, 110.20, 80.79 (q, J = 31.3 Hz), 66.56, 58.99, 23.75. 19F NMR (376 MHz, CDCl3) δ −61.96, −62.02, −76.36, −76.47. HRMS (EI) calcd for C22H17F6N3O2S [M + H]+: 502.1018, found: 502.1020.
4-(4,4-Dimethyl-5-oxo-2-thioxo-3-(4-(2,2,2-trifluoroacetyl)phenyl) imidazolidin-1-yl)-2-(trifluoromethyl)benzonitrile (15i). In a 25 mL round-bottomed flask, 13b (49 mg, 0.1 mmol), DMSO (5 mL), and IBX (84 mg, 0.3 mmol) were added. The mixture was stirred for 12 h at room temperature. After the reaction, the mixture was poured over and filtered through a silica gel pad under vacuum the filtrate was added with EA (15 mL) and washed with water (20 mL × 3) and saturated brine (15 mL × 3). The organic layer was separated and dried over Na2SO4. After filtration and evaporation, the resulting crude product was purified by column chromatography with petrol ether as eluent to afford 15i (45 mg, 90% yield).
1H NMR (400 MHz, CDCl3) δ 8.24 (dd, J = 22.9, 8.3 Hz, 2H), 7.99 (dd, J = 9.2, 4.7 Hz, 2H), 7.92–7.77 (m, 1H), 7.58 (dd, J = 22.9, 8.6 Hz, 2H), 1.79–1.52 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 179.73, 179.49 (q, J = 35.1 Hz), 174.43, 141.68, 136.84, 135.35, 133.69 (q, J = 34.9 Hz), 132.18, 131.66, 130.62, 130.55, 127.07 (q, J = 5.1 Hz), 121.86 (q, J = 273.4 Hz), 116.46 (q, J = 290.1 Hz), 114.76, 110.42, 66.76, 23.95. 19F NMR (376 MHz, CDCl3) δ −61.97, −71.55. HRMS (EI) calcd for C21H13F6N3O2S [M + H]+: 486.0705, found: 486.0708.
4.3 Cell proliferation inhibition assay
Cell viability was determined using the CCK8 assay method. The cells were seeded in a 96-well plate at 3 × 103 cells per well for 24 hours (37 °C, 5% CO2), and an equal volume of medium containing increasing concentration of inhibitors was added to each well. After a 5 days incubation, CCK8 reagent was added for 1.5 hours. The light absorption (OD) of the 96-well plate was measured at 490 nm using a SpectraMAX M5 microplate spectrophotometer (Molecular Devices). All experiments were performed in triplicate. The percentage of viable cells was calculated and compared with that of the control cells, the half maximal inhibitory concentration (IC50) was calculated by GraphPad Prism5 software.4.4 Western blot analysis
Cells were treated with a series of concentrations of 13b and ENT for 5 days at 37 °C, then the cells were harvested, washed in ice-cold PBS, analyzed with RIPA buffer (10 mM Tris–HCl (pH 7.8), 1% NP40, 0.15 M NaCl, 1 mM EDTA, 10 μM aprotinin, 1 mM NaF and 1 mM Na3VO4), protease inhibitors, phosphatase cocktails A and B, and PMSF (1 mM). Protein concentration was determined by the BCA Protein Assay Kit (beyotime#p0012s), the sample proteins were separated by 10% SDS-PAGE gel and transferred onto 0.2 um polyvinylidene difluoride membranes (millpore#ISEQ00010), then incubated overnight at 4 °C with the AR or PSA antibody overnight at the indicated concentrations in 5% BSA/TBST buffer with gently shaking, then washing with 1 × TBS/T 3 times and followed by incubation for 1.5 hour with a 1/5000 dilution of secondary HRP antibody in 5% nonfat milk/TBST. The target blots were detected with a chemiluminescence system.4.5 In vivo model
The animal studies were carried out under protocols approved by the animal protection law of the People's Republic of China Care and Use Committees. All rodent studies were also carried out in accordance with the guidelines of Institutional Animal Care and Treatment Committee of Sichuan University in China. The male CB17 SCID mice were purchased (Beijing HFK Bioscience Co. ltd., Beijing, China). LNCap-AR cells were harvested during the exponential-growth phase, washed twice with serum-free medium, and re-suspended at a concentration of 1 × 107 mL−1. One hundred microliters of the cell suspension was injected subcutaneously into the hind flank of each male CB17 SCID mouse (6–7 weeks old) after castrated 2 days. The tumors were allowed to grow to 100–150 mm3, at which point the mice were randomized into 5 groups (8 mice for each group). The mice were dosed orally with 13b (15 mg per kg q.d, 15 mg per kg q.3d, or 5 mg per kg q.3d), vehicle, and the reference compound ENT (15 mg per kg q.d). Tumor growth and body weight were measured every 3 days using vernier calipers for the duration of the treatment. The volume was calculated as follows: tumor volume = a × b2/2 (a, long diameter; b, short diameter).Acknowledgements
This work is financially supported by Hinova Pharmaceuticals Inc. and also partially supported by National Natural Science Foundation of China (No. 81472418, 81672951). We also thank the State Key Laboratory of Biotherapy and Cancer Center of Sichuan University for providing analytical service.References and notes
- Y. N. Wong, R. Ferraldeschi, G. Attard and J. de Bono, Nat. Rev. Clin. Oncol., 2014, 11, 365 CrossRef CAS PubMed.
- J. K. Rane, D. Pellacani and N. J. Maitland, Nat. Rev. Urol., 2012, 9, 595 CrossRef CAS PubMed.
- J. K. Myung, C. A. Banuelos, J. G. Fernandez, N. R. Mawji, J. Wang, A. H. Tien, Y. C. Yang, I. Tavakoli, S. Haile, K. Watt, I. J. McEwan, S. Plymate, R. J. Andersen and M. D. Sadar, J. Clin. Invest., 2013, 123, 2948 CAS.
- L. Yin, Q. Hu and R. Hartmann, Int. J. Mol. Sci., 2013, 14, 13958 CrossRef PubMed.
- T. M. Beer, A. J. Armstrong, D. E. Rathkopf, Y. Loriot, C. N. Sternberg, C. S. Higano, P. Iversen, S. Bhattacharya, J. Carles, S. Chowdhury, I. D. Davis, J. S. de Bono, C. P. Evans, K. Fizazi, A. M. Joshua, C. S. Kim, G. Kimura, P. Mainwaring, H. Mansbach, K. Miller, S. B. Noonberg, F. Perabo, D. Phung, F. Saad, H. I. Scher, M. E. Taplin, P. M. Venner, B. Tombal and P. Investigators, N. Engl. J. Med., 2014, 371, 424 CrossRef PubMed.
- A. Parray, H. R. Siddique, S. Nanda, B. R. Konety and M. Saleem, Biol.: Targets Ther., 2012, 6, 267 CAS.
- C. Helsen, T. Van den Broeck, A. Voet, S. Prekovic, H. Van Poppel, S. Joniau and F. Claessens, Endocr.-Relat. Cancer, 2014, 21, T105 CrossRef CAS PubMed.
- A. M. Moilanen, R. Riikonen, R. Oksala, L. Ravanti, E. Aho, G. Wohlfahrt, P. S. Nykanen, O. P. Tormakangas, J. J. Palvimo and P. J. Kallio, Sci. Rep., 2015, 5, 12007 CrossRef PubMed.
- C. Tran, S. Ouk, N. J. Clegg, Y. Chen, P. A. Watson, V. Arora, J. Wongvipat, P. M. Smith-Jones, D. Yoo, A. Kwon, T. Wasielewska, D. Welsbie, C. D. Chen, C. S. Higano, T. M. Beer, D. T. Hung, H. I. Scher, M. E. Jung and C. L. Sawyers, Science, 2009, 324, 787 CrossRef CAS PubMed.
- Y. M. Ning, W. Pierce, V. E. Maher, S. Karuri, S. H. Tang, H. J. Chiu, T. Palmby, J. F. Zirkelbach, D. Marathe, N. Mehrotra, Q. Liu, D. Ghosh, C. L. Cottrell, J. Leighton, R. Sridhara, A. Ibrahim, R. Justice and R. Pazdur, Clin. Cancer Res., 2013, 19, 6067 CrossRef CAS PubMed.
- H. I. Scher, K. Fizazi, F. Saad, M. E. Taplin, C. N. Sternberg, K. Miller, R. de Wit, P. Mulders, K. N. Chi, N. D. Shore, A. J. Armstrong, T. W. Flaig, A. Flechon, P. Mainwaring, M. Fleming, J. D. Hainsworth, M. Hirmand, B. Selby, L. Seely, J. S. de Bono and AInvestigators, N. Engl. J. Med., 2012, 367, 1187 CrossRef CAS PubMed.
- M. Korpal, J. M. Korn, X. Gao, D. P. Rakiec, D. A. Ruddy, S. Doshi, J. Yuan, S. G. Kovats, S. Kim, V. G. Cooke, J. E. Monahan, F. Stegmeier, T. M. Roberts, W. R. Sellers, W. Zhou and P. Zhu, Cancer Discovery, 2013, 3, 1030 CrossRef CAS PubMed.
- W. C. Black, C. I. Bayly, D. E. Davis, S. Desmarais, J. P. Falgueyret, S. Leger, C. S. Li, F. Masse, D. J. McKay, J. T. Palmer, M. D. Percival, J. Robichaud, N. Tsou and R. Zamboni, Bioorg. Med. Chem. Lett., 2005, 15, 4741 CrossRef CAS PubMed.
- J. A. Gibbons, T. Ouatas, W. Krauwinkel, Y. Ohtsu, J. S. van der Walt, V. Beddo, M. de Vries and J. Mordenti, Clin. Pharmacokinet., 2015, 54, 1043 CrossRef CAS PubMed.
- J. Jiang, X. Pang, L. Li, X. Dai, X. Diao, X. Chen, D. Zhong, Y. Wang and Y. Chen, Drug Des., Dev. Ther., 2016, 10, 2181 CrossRef PubMed.
- N. J. Clegg, J. Wongvipat, J. D. Joseph, C. Tran, S. Ouk, A. Dilhas, Y. Chen, K. Grillot, E. D. Bischoff, L. Cai, A. Aparicio, S. Dorow, V. Arora, G. Shao, J. Qian, H. Zhao, G. Yang, C. Cao, J. Sensintaffar, T. Wasielewska, M. R. Herbert, C. Bonnefous, B. Darimont, H. I. Scher, P. Smith-Jones, M. Klang, N. D. Smith, E. De Stanchina, N. Wu, O. Ouerfelli, P. J. Rix, R. A. Heyman, M. E. Jung, C. L. Sawyers and J. H. Hager, Cancer Res., 2012, 72, 1494 CrossRef CAS PubMed.
- H. Cheng, Y. Pei, F. Leng, J. Li, A. Liang, D. Zou, Y. Wu and Y. Wu, Tetrahedron Lett., 2013, 54, 4483 CrossRef CAS.
- N. D. Smith, C. Bonnefous and J. D. Julien, WO 2011103202A2, 2011.
- M. D. Balabas, M. J. Evans, C. L. Sawyers, S. Yang, D. Hosfield and G. L. Greene, WO 2014066799A2, 2014.
- Y. Chen and Y. Gong, WO 2014190895A1, 2014.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02142a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |