DOI:
10.1039/C7RA02063E
(Paper)
RSC Adv., 2017,
7, 19039-19049
Five new 2D and 3D coordination polymers based on two new multifunctional pyridyl–tricarboxylate ligands: hydrothermal syntheses, structural diversity, luminescent and magnetic properties†
Received
19th February 2017
, Accepted 15th March 2017
First published on 30th March 2017
Abstract
Five coordination polymers based on pyridine–tricarboxylate ligands formulated as [Zn(HL1)] (1), [Cd2(L1)(phen)2(OH)]·2H2O (2), [Co(HL1)(H2O)2] (3), [Co3(L1)2(H2O)8]·4(H2O) (4) and [Mn3(L2)2(4,4′-bpy)2(H2O)2] (5) (H3L1 = 4-(2,4-dicarboxylphenyl) picolinic acid, H3L2 = 5-(3′,5′-dicarboxylphenyl) nicotinic acid, phen = 1,10-phenanthroline, 4,4′-bpy = 4,4′-bipyridine) were synthesized under hydrothermal conditions. Compound 1 exhibits a 2D distorted square-grid network based on [Zn(μ-Ocarboxylate)(COO)]n chains. Compound 2 displays a 2D square-grid network based on tetranuclear [Cd4(μ3-OH)2(Ocarboxylate)2(COO)2] units in which Cd(II) ions are bridged by the mixed bridges of (μ3-OH)2(μ-Ocarboxylate)2. Compound 3 exhibits a 2D network based on [Co(COO)]n chains in which Co(II) ions are bridged by a single syn–anti carboxylate bridge (μ-COO). Compound 4 features a 3D framework based on 2D layers which are further interlinked by Co(II) ions, containing [Co(COO)]n chains which Co(II) ions are also bridged by a single syn–anti carboxylate bridge similar with that in 3. Compound 5 shows a 2D network based on trinuclear [Mn3(COO)6(H2O)2] units in which Mn(II) ions are bridged by the mixed bridges of (μ-Ocarboxylate)(COO)2. These compounds have been characterized by IR spectra, TGA and powder XRD pattern. Compounds 1 and 2 exhibit intense luminescence properties in the solid state at room temperature. Magnetic studies for compounds 3–5 demonstrated that the single carboxylate bridge in 3 and 4 transmits weak antiferromagnetic interactions between Co(II) ions while the mixed bridges of (μ-Ocarboxylate)(COO)2 transmit moderate antiferromagnetic interactions between Mn(II) ions in 5.
Introduction
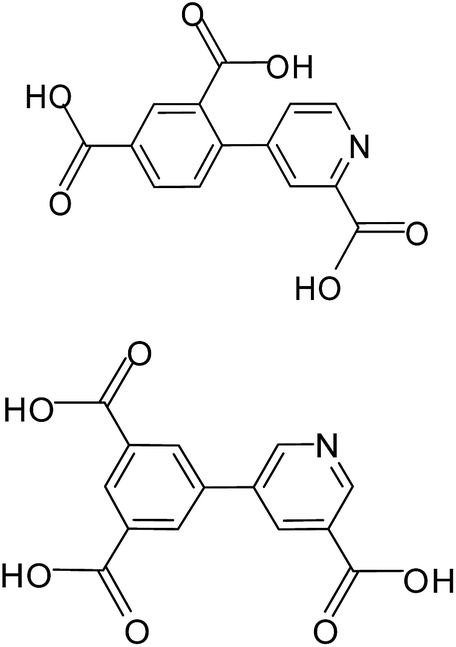
Coordination polymers or metal–organic frameworks (MOFs) have recently attracted great attention due to their intriguing architectures and potential applications in catalysis, gas adsorption, luminescence and magnetism etc.1 However, it is usually difficult to get the accurate control of the target compounds with desired structures and properties in the self-assembling processes, because various and subtle factors can affect the result, such as the coordination geometry of the metal ions, connectivity of the organic ligands, the stoichiometric ratio of reactants, reaction conditions (solvents, temperature, pH values), and the presence of auxiliary ligands.2 An effective synthetic approach to obtaining new compounds with predictable structures and properties to some extent is to chose appropriate organic bridging ligands with multifunctionality and the metal centers with various coordination preferences. In this context, aromatic polycarboxylic acids including N-heterocyclic derivative have been largely explored for the construction of MOFs for their strong and versatile coordination towards metal ions.3–5 Among them, multifunctional pyridyl–polycarboxylate acids, as rigid and versatile bridging ligands, have been studied extensively, leading to a variety of MOFs with diverse topologies and interesting properties reported by us and others.4,5 Here, we are interested in the use of two new π-conjugated pyridyl–tricarboxylate ligands, 4-(2,4-dicarboxylphenyl) picolinic acid (H3L1), 5-(3′,5′-dicarboxylphenyl) nicotinic acid (H3L2) (Scheme 1) which still have not been explored, on the basis of the following considerations: (i) their extreme rigidity is advantageous for the construction of porous structures and the multidentate carboxylate groups can strongly bind metal ions to give extended networks. (ii) The presence of phenyl ring and pyridyl ring that can rotate along the C–C bond to give a twisted conformation, which offers possibilities of creating MOFs with new topologies. Besides, complete or partial deprotonation of such multicarboxylate ligands can also lead to the variation of coordination modes and induce structurally distinct MOFs. Apart from multifunctional carboxylate ligands, 1,10-phenanthroline (phen) and 4,4′-bipyridine (4,4′-bpy) were applied as the simple N-donor auxiliary ligands to tune the structure of the coordination polymers. In this article, we present the syntheses, crystal structures, luminescent and magnetic properties of five coordination polymers, [Zn(HL1)] (1), [Cd2(L1)(phen)2(OH)]·2H2O (2), [Co(HL1)(H2O)2] (3), [Co3(L1)2(H2O)8]·4(H2O) (4) and [Mn3(H3L2)2(4,4′-bpy)2(H2O)2] (5). Compounds 1 and 2 exhibit intense luminescence properties in the solid state at room temperature. Magnetic studies demonstrated that the single carboxylate bridge transmits weak antiferromagnetic (AF) interactions between Co(II) ions in 3 and 4 while the mixed bridges of (μ-Ocarboxylate)(COO)2 transmit moderate AF interactions between Mn(II) ions in 5.
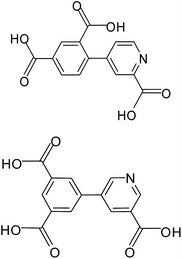 |
| | Scheme 1 4-(2,4-Dicarboxylphenyl) picolinic acid (top, H3L1); 5-(3′,5′-dicarboxylphenyl) nicotinic acid (bottom, H3L2). | |
Experimental section
Materials and physical measurements
The reagents and the organic ligand H3L1 4-(2,4-dicarboxylphenyl) picolinic acid, H3L2 5-(3′,5′-dicarboxylphenyl) nicotinic acid, 1,10-phenanthroline (phen) and 4,4′-bipyridine (4,4′-bpy) were obtained from commercial sources and used without further purification. Elemental analyses were determined on an Elementar Vario ELIII analyzer. The Fourier transform infrared (FTIR) spectra (KBr disk) were recorded in the range 500–4000 cm−1 using KBr pellets on a TENSOR 27 FI-IR spectrophotometer. Powder X-ray diffraction pattern (PXRD) was carried out on a EMPYREAN PANALYTICAL apparatus. The TG curve was recorded on a SDT Q600 Thermal analyzer which was performed under the N2 atmosphere and at a heating rate of 5 °C min−1 over the temperature range of 25–800 °C. The luminescence spectra for the powdered solid samples were measured at room temperature on a Hitachi F-7000 fluorescence spectrophotometer. The UV-vis absorption spectra were measured with an U-3900 spectrophotometer. Magnetic measurements were performed on a Quantum Design MPMS XL7 SQUID magnetometer.
[Zn(HL1)] (1). Zn(NO3)2·6H2O (0.2 mmol, 0.057 g), H3L1 (0.4 mmol, 0.119 g) were dissolved in the solvent of water (10 mL) and transferred to a 23 mL Teflon-lined autoclave. After being stirred in air for 15 min, the mixture was heated at 160 °C for 3 days. After cooling to room temperature at the speed of 5 °C h−1, yellow plate crystals of 1 were collected in a 55% yield based on H3L1. Elem anal. calcd (%) for C14H6ZnNO6: C, 62.37; H, 2.24; N, 5.20. Found: C, 62.52; H, 2.62; N, 5.36%. IR bands (KBr, cm−1): 3432s, 1597s, 1443w, 1388s, 1238w, 1071m, 1016m, 911w, 834m, 709m, 681m.
[Cd2(L1)(phen)2(OH)]·2H2O (2). CdCl2 (0.3 mmol, 0.055 g), H3L1 (0.2 mmol, 0.057 g) and phen (0.3 mmol, 0.059 g) were dissolved in the solvent of water (10 mL) and transferred to a 23 mL Teflon-lined autoclave, thereafter, 600 μL of 1 mol L−1 NaOH aqueous solution was added in the mixture. After being stirred in air for 15 min, the mixture was heated to 160 °C for 3 days. After cooling to room temperature at the speed of 10 °C h−1, colourless crystals of 2 were collected in a 25% yield based on H3L1. Elem anal. calcd (%) for C38H26Cd2N5O9: C, 49.53; H, 2.84; N, 7.60. Found: C, 49.26; H, 2.65; N, 7.35%. IR bands (KBr, cm−1): 3435s, 1638s, 1598s, 1462m, 1431s, 1375s, 1337s, 1097m, 852m, 802m, 767s.
[Co(HL1)(H2O)2] (3). CoCl2·6H2O (0.3 mmol, 0.071 g) and H3L1 (0.2 mmol, 0.057 g) were dissolved in the solvent of water (10 mL) and transferred to a 23 mL Teflon-lined autoclave, thereafter, 200 μL of 1 mol L−1 NaOH aqueous solution was added in the mixture. After being stirred in air for 15 min, the mixture was heated to 160 °C for 3 days. After cooling to room temperature at the speed of 10 °C h−1, light red crystals of 3 were collected in a 42% yield based on H3L1. Elem anal. calcd (%) for C14H11CoNO8: C, 44.23; H, 2.92; N, 3.68. Found: C, 44.50; H, 2.47; N, 3.46%. IR bands (KBr, cm−1): 3394s, 3233s, 1688s, 1624s, 1588s, 1434m, 1364s, 1301m, 1238w, 1132m, 1020m, 943m, 844s, 802s, 760s, 732s.
[Co3(L1)2(H2O)8]·4(H20) (4). CoCl2·6H2O (0.3 mmol, 0.071 g) and H3L1 (0.2 mmol, 0.057 g) were dissolved in the solvent of water (10 mL) and transferred to a 23 mL Teflon-lined autoclave, thereafter, 600 μL of 1 mol L−1 NaOH aqueous solution was added in the mixture. After being stirred in air for 15 min, the mixture was heated to 160 °C for 3 days. After cooling to room temperature at the speed of 10 °C h−1, orange crystals of 4 were collected in a 12% yield based on CoCl2·6H2O. Elem anal. calcd (%) for C28H36Co3N2O24: C, 34.98; H, 3.77; N, 2.91. Found: C, 34.79; H, 3.65; N, 2.65%. IR bands (KBr, cm−1): 3420s, 3290s, 1593s, 1553s, 1473m, 1430s, 1378s, 1362s, 1160w, 1022m, 928m, 915m, 774m, 725m.
[Mn3(H3L2)2(4,4′-bpy)2(H2O)2] (5). MnCl2·4H2O (0.3 mmol, 0.059 g), H3L2 (0.2 mmol, 0.057 g) and 4,4′-bpy (0.3 mmol, 0.047 g) were dissolved in the solvent of water (10 mL) and transferred to a 23 mL Teflon-lined autoclave, thereafter, 600 μL of 1 mol L−1 NaOH aqueous solution was added in the mixture. After being stirred in air for 15 min, the mixture was heated to 160 °C for 3 days. After cooling to room temperature at the speed of 10 °C h−1, light yellow crystals of 5 were collected in a 25% yield based on H3L2. Elem anal. calcd (%) for C48H32Mn3N6O14: C, 53.30; H, 2.98; N, 7.77. Found: C, 53.10; H, 2.78; N, 7.54%. IR bands (KBr, cm−1): 3432s, 1629s, 1569m, 1496m, 1447m, 1416m, 1382s, 1227m, 1120m, 1002w, 901m, 801m, 786m.
Crystal data collection and refinement
Diffraction intensity data were collected at 293 K on a Bruker APEX II diffractometer equipped with a CCD area detector and graphite-monochromated Cu Kα radiation (λ = 1.54184 Å) for compounds 1, 2 and 5; graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) for compounds 3 and 4. Empirical absorption corrections were applied using the SADABS program.6 The structures were solved by the direct method and refined by the full-matrix least-squares method on F2, with all non-hydrogen atoms refined with anisotropic thermal parameters.7 All the hydrogen atoms attached to carbon atoms were placed in calculated positions and refined using the riding model. The hydrogens attached to water molecules were located from the difference Fourier maps and refined isotropically. Crystallographic data for 1–5 have been deposited at the Cambridge Crystallographic Data Center with the deposition numbers of CCDC 1530040–1530044: 1 (1530042), 2 (1530040), 3 (1530043), 4 (1530044) and 5 (1530041).† A summary of the crystallographic data, data collection, and refinement parameters for compounds 1–5 are provided in Table 1. The selected bond lengths and angles are given in Tables S1–S5.†
Table 1 Crystallographic data and structure refinement results for compounds 1–5
| Compound |
1 |
2 |
3 |
4 |
5 |
| Empirical formula |
C14H6ZnNO6 |
C38H26Cd2N5O9 |
C14H11CoNO8 |
C28H36Co3N2O24 |
C48H32Mn3N6O14 |
| Formula weight |
350.58 |
922.45 |
380.17 |
961.38 |
1081.62 |
| Crystal system |
Orthorhombic |
Triclinic |
Monoclinic |
Triclinic |
Triclinic |
| Space group |
Pca2(1) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P2(1)/c |
P |
P |
| a, Å |
15.7203(10) |
11.8521(11) |
18.480(2) |
7.1966(5) |
9.6330(10) |
| b, Å |
13.1516(9) |
12.2200(12) |
9.4446(9) |
10.3116(6) |
10.6019(9) |
| c, Å |
6.1682(4) |
14.6694(15) |
8.0006(7) |
12.2101(8) |
11.2826(10) |
| α, ° |
90 |
68.971(9) |
90 |
102.745(2) |
96.503(7) |
| β, ° |
90 |
68.595(9) |
99.263(3) |
101.788(2) |
104.988(8) |
| γ, ° |
90 |
61.417(10) |
90 |
92.055(2) |
104.111(8) |
| V, Å3 |
1275.26(14) |
1692.3(3) |
1378.2(2) |
862.02(10) |
1060.19(17) |
| Z |
4 |
2 |
4 |
1 |
1 |
| ρcalcd, g cm−3 |
1.826 |
1.810 |
1.832 |
1.852 |
1.694 |
| μ, mm−1 |
2.996 |
1.324 |
1.294 |
1.531 |
7.869 |
| Unique reflections |
1513 |
6091 |
2481 |
4319 |
3784 |
| Rint |
0.0434 |
0.0674 |
0.0240 |
0.0240 |
0.0598 |
| S on F2 |
1.083 |
1.035 |
1.100 |
1.068 |
1.025 |
| R1, wR2[I > 2σ(I)] |
0.0481, 0.1165 |
0.0546, 0.0973 |
0.0240, 0.0620 |
0.0254, 0.0611 |
0.0559, 0.1220 |
| R1, wR2 (all data) |
0.0625, 0.1318 |
0.0891, 0.1248 |
0.0261, 0.0630 |
0.0291, 0.0625 |
0.0862, 0.1405 |
Results and discussion
Synthesis and IR spectral aspects
To explore the H3L1 and H3L2 as ligands toward the self-assembly with metal(II) (Zn, Cd, Mn, Co) to construct coordination polymers, we have tried a lot of hydrothermal reactions by applying different metal(II) salts with H3L1 or H3L2 in the absence or presence of several common auxiliary ligands or potential linkers, such as 1,10-phenanthroline, 4,4′-bipyridine and 4,4′-biphenyldicarboxylic acid and so on. Compound 1 was obtained by hydrothermal reactions applying a mixture of the Zn(NO3)2·6H2O in water with H3L1 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio. Compound 2 was synthesized in a similar way but by using a mixture of CdCl2, with H3L1, phen, and water solution of NaOH as a base in a 3000:2000:3000:6 molar ratio. Compounds 3 and 4 were synthesized by very similar way by using a mixture of CoCl2·6H2O, with H3L1 and water solution of NaOH as a base except for the different numbers of microliter for the same concentration of NaOH water solution (1 M), and CoCl2·6H2O, H3L1 and NaOH are in a 3000:2000:2 and 3000:2000:6 molar ratios, respectively for 3 and 4. Compound 5 was obtained in a similar way by using MnCl2·4H2O, with H3L2, 4,4′-bpy and NaOH water solution in a molar ratio of 3000:2000:3000:6.
:2 molar ratio. Compound 2 was synthesized in a similar way but by using a mixture of CdCl2, with H3L1, phen, and water solution of NaOH as a base in a 3000:2000:3000:6 molar ratio. Compounds 3 and 4 were synthesized by very similar way by using a mixture of CoCl2·6H2O, with H3L1 and water solution of NaOH as a base except for the different numbers of microliter for the same concentration of NaOH water solution (1 M), and CoCl2·6H2O, H3L1 and NaOH are in a 3000:2000:2 and 3000:2000:6 molar ratios, respectively for 3 and 4. Compound 5 was obtained in a similar way by using MnCl2·4H2O, with H3L2, 4,4′-bpy and NaOH water solution in a molar ratio of 3000:2000:3000:6.
The IR spectra of 1–5 show broad absorption bands in the range of 3409 to 3433 cm−1, respectively, attributable to the ν(O–H) vibration of water molecules or hydroxyl (Fig. S1, ESI†). The asymmetric and symmetric stretching vibrations of carboxylate are observed in the range of 1593–1629 and 1362–1388 cm−1, respectively.
Description of the structures
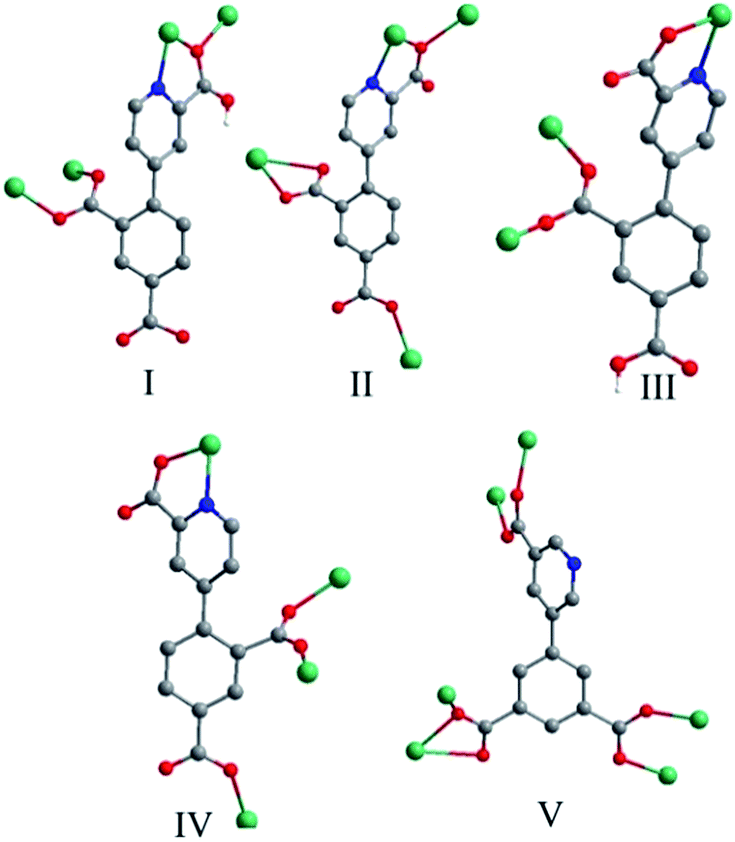
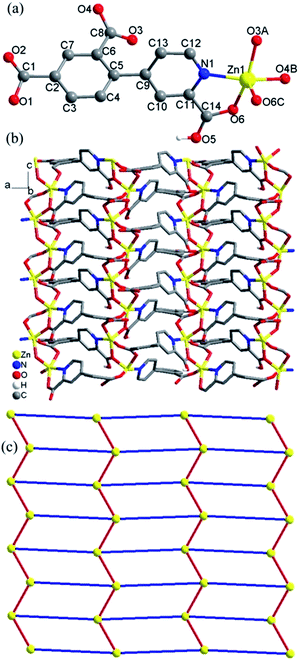
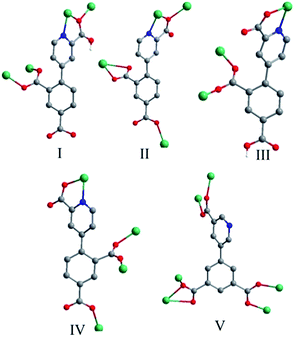
Crystal structure of 1. Compound 1 crystallizes in the orthorhombic space group Pca2(1) and exhibits a 2D framework. The asymmetric unit contains one Zn(II) ion, one HL12− ligand with one protonated carboxyl group and two deprotonated carboxyl groups. The coordination environment of the Zn(II) ion is shown in Fig. 1a. The unique Zn1 ion is five-coordinated in a rectangular pyramid coordination geometry, ligated by one nitrogen atoms and four oxygen atoms from four different HL12− ligands with Zn–N/O distances in the range of 1.992(5)–2.118(6) Å, all the Zn–O and Zn–N distances in 1 are comparable to these in other reported Zn(II) compounds based on the similar pyridine–tricarboxylate ligands.5f–h The HL12− ligand contains a bridging bidentate carboxylate group (μ2-η1:η1-bridging mode), a tridentate picolinic acid which the carboxylate group adopts μ2-η1 bridging mode and the nitrogen atom (N1) is involved in the coordination, and a noncoordinated carboxylate group (mode I, Scheme 2). The dihedral angle between the pyridyl and phenol rings in the HL12− is 67.2°.
 |
| | Fig. 1 (a) Local coordination environment of the Zn2+ center in 1. Symmetry codes: (A) 2 − x, 1 − y, 0.5 + z; (B) 0.5 + x, 1 − y, z; (C) 2.5 − x, y, 0.5 + z. (b) View of the 2D layer of 1. (c) View of the topological net of 1. | |
 |
| | Scheme 2 Coordination modes of H3L1 and H3L2 in compounds 1–5. | |
Adjacent Zn(II) ions are bridged by mixed bridges of (μ-Ocarboxylate)(COO) into 1D zigzag chains along the b direction with Zn⋯Zn distances 3.519(2) Å. Each chains bridged by carboxylate groups are interlinked by HL12− ligands act as μ3 bridge into 2D layers along the ac plane (Fig. 1b). The interchain Zn⋯Zn distances within the layer is in the range of 7.099(1)–10.183(1) Å. Topologically, Zn(II) ions and ligands can be considered as 4-connected nodes and linkers respectively, thus, the whole structure can be simplified as a 2D distorted square-grid layers (Fig. 1c).
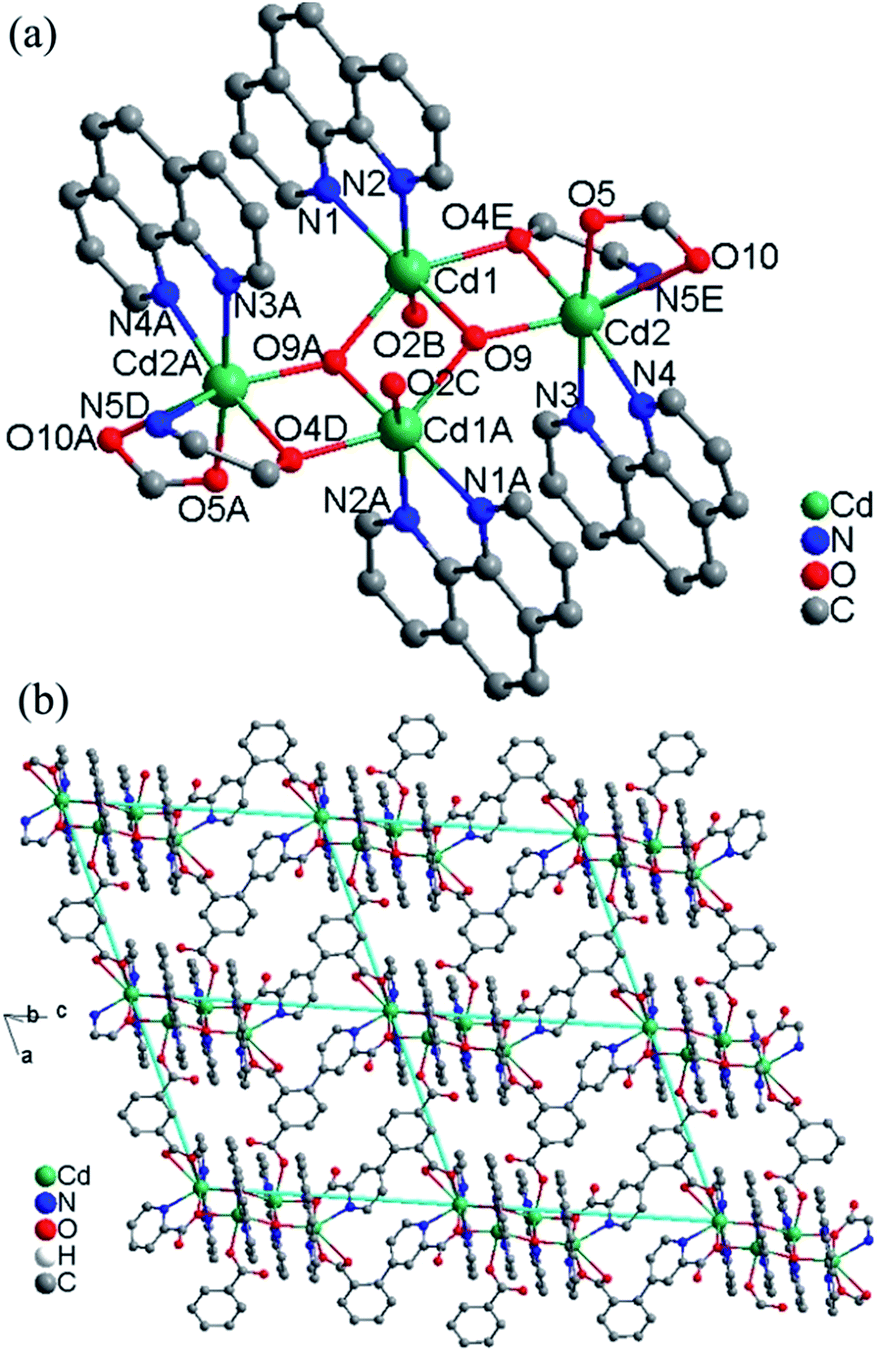
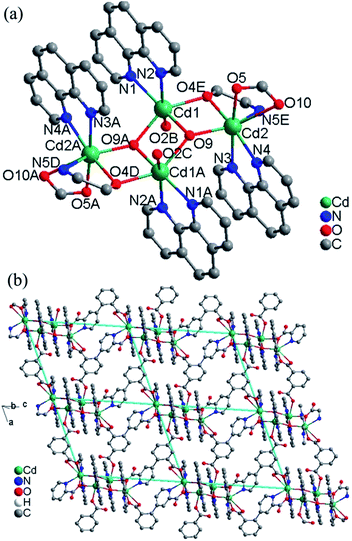
Crystal structure of 2. This compound is also composed of 2D layers, but the layers are based on tetranuclear clusters rather than chains as that in 1. The relevant bond parameters are summarized in Table S2.† As shown in Fig. 2a, the tetranuclear cluster contains a centrosymmetric [Cd4(OH)2]6+ core, in which four Cd(II) atoms are linked by two equivalent μ3-OH bridges to form a planar parallelogram. The geometry can be described as two coplanar and edge-sharing Cd3 triangles that share the edge defined by two centrosymmetry-related and doubly hydroxo-bridged Cd1 atoms, with Cd1⋯Cd1A 3.431(1) Å. The M–O–M angles around the μ3-OH (O9) range from 98.06(2) to 140.83(2)°. The sum of the Cd–O–Cd angles 344.39° around each μ3-OH is in good agreement with the tetrahedral environment of the oxygen atom, and the oxygen atom is placed above and below the Cd4 plane by 0.49 Å. These define a rather flat pyramidal shape for the Cd3O moiety. The tetranuclear [Cd4(μ3-OH)2]6+ core is reinforced by two oxygen atom bridges from two carboxylate groups of two different L13-ligands. There are two different bridging fashions between Cd1 and Cd2 sites. One is the double-bridging motif that contains a μ3-hydroxo (O9) and a carboxylate oxygen atom (O4), with Cd1⋯Cd2 3.572(8) Å; the other is the single-bridging motif of the μ3-hydroxo (O9), with longer Cd2⋯Cd1A distances of 4.253(9) Å. The resulting [Cd4(μ3-OH)2(O)2] cluster is often observed in some complexes with different central metal ions and very similar bridges.8
 |
| | Fig. 2 (a) The tetranuclear cluster in 2. Symmetry codes: (A) 2 − x, 1 − y, 1 − z; (B) 1 + x, y, z; (C) 1 − x, 1 − y, 1 − z; (D) x, y, 1 + z; (E) 2 − x, 1 − y, −z. (b) View of the 2D layer of 2. | |
The asymmetric unit contains two Cd(II) ions, one L13− ligand, two 1,10-phen molecules, one hydroxyl group and two lattice water molecules. The Cd1 ion is ligated by two nitrogen atoms (N1 and N2) from one chelating phen, two carboxylate oxygen atoms (O2 and O4) from two L13− ligands and two hydroxyl oxygen atoms (O9 and O9A) in a distorted octahedral [N2O4] coordination geometry with Cd–N bond lengths ranging from 2.325(6) to 2.405(5) Å, Cd–O bond lengths ranging from 2.251(5) to 2.342(4) Å, all the Cd–O and Cd–N distances in 2 are comparable to these in other reported Cd(II) compounds based on the similar pyridine–tricarboxylate ligands.5f–h The Cd2 ion is ligated by three nitrogen atoms (N3, N4 and N5) from one phen and one L13− ligand, three oxygen atoms (O4, O5 and O10) from two L13− ligands and one hydroxyl oxygen atom (O9) in a distorted mono-capped octahedral [N3O4] coordination geometry with Cd–N bond lengths ranging from 2.354(5) to 2.441(5) Å, Cd–O bond lengths ranging from 2.225(4) to 2.418(4) Å. The L13− ligand acts as a hexadentate bridge ligand (mode II, Scheme 2), using one carboxylate oxygen atom (O4) and one nitrogen atom (N5) from picolinic acid to connect with two Cd(II) ions, one carboxylate group to chelate Cd(II) ion and a monodentate carboxylate oxygen atom to connect with the fourth Cd(II) ion. The dihedral angle between the pyridyl and phenol rings in the L13− is 71.1°.
Each tetranuclear cluster in 2 is linked with four neighbouring tetranuclear clusters by L13− ligands into 2D layer along the ac plane (Fig. 2b). The intertetranuclear clusters Cd⋯Cd distances within the layer is in the range of 8.344(1)–21.462(1) Å. Topologically, Cd(II) ions and ligands can be considered as 4-connected nodes and linkers respectively, thus, the whole structure can be simplified as a 2D 4,4-square-grid layers. The 2D layers are interlinked by the O–H⋯O hydrogen bonds into 3D network (Fig. S2, ESI†).
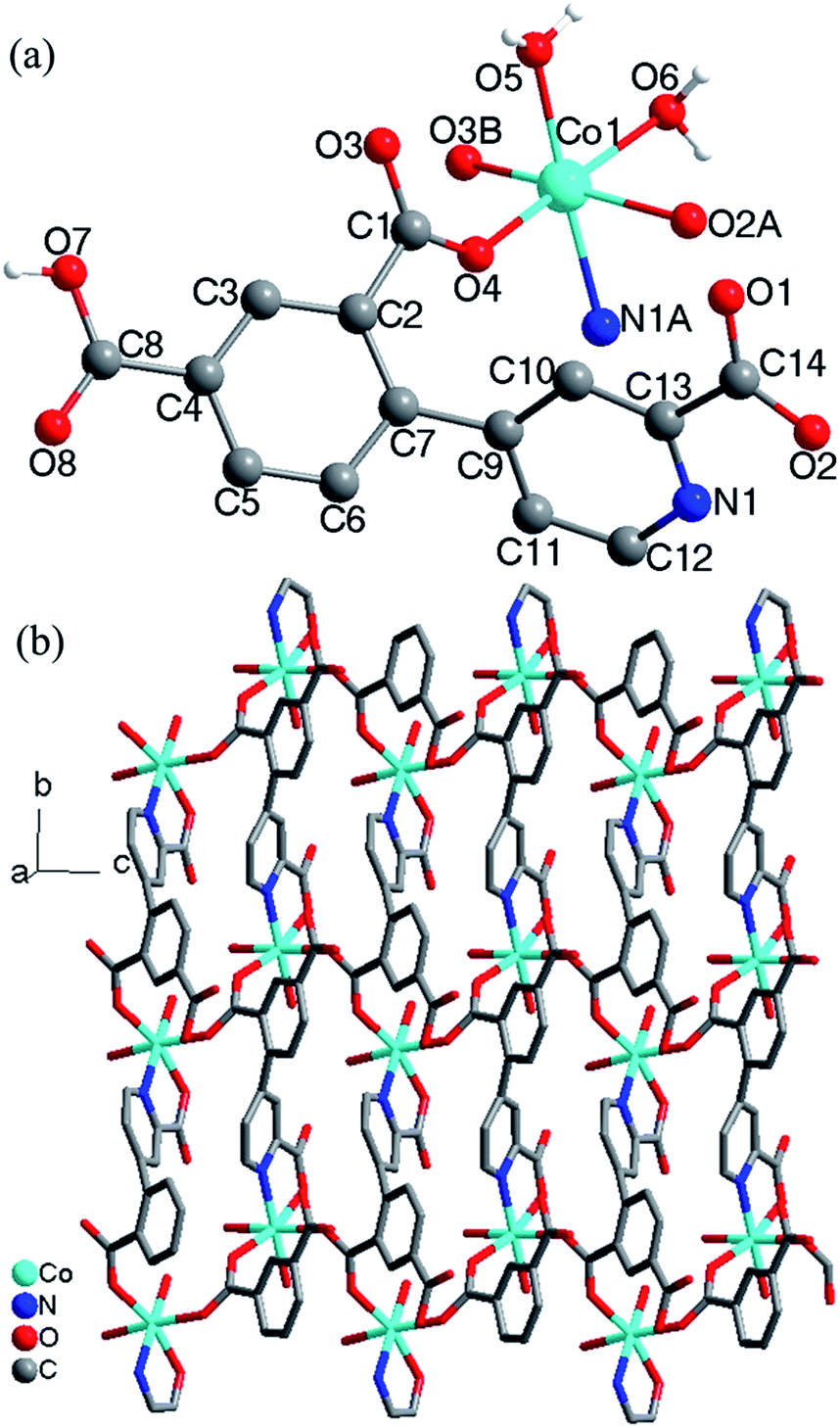
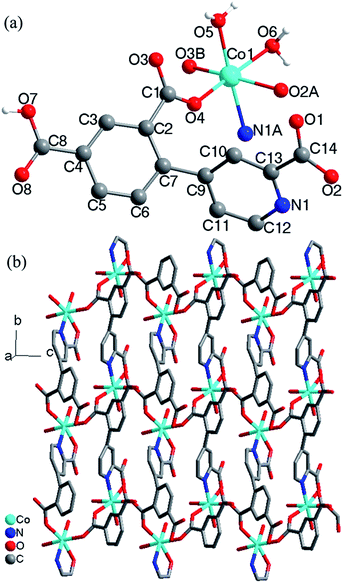
Crystal structure of 3. Compound 3 crystallizes in the monoclinic space group P2(1)/c and exhibits a 2D framework. The asymmetric unit contains one Co(II) ion, one HL12− ligand with one protonated carboxyl group and two deprotonated carboxyl groups and two coordinated water molecules. The coordination environment of the Co(II) ion is shown in Fig. 3a. The unique Co(II) ion is six-coordinated in a octahedral coordination geometry, ligated by one nitrogen atom and four oxygen atoms from three different HL12− ligands and two water molecules with Co–N/O distances in the range of 2.043(2)–2.146(1) Å, all the Co–O and Co–N distances in 3 are comparable to these in other reported Co(II) compounds based on the similar pyridine–tricarboxylate ligands.5f,g Neighbouring Co(II) ions are linked by single carboxylate bridges to generate a 1D [Co(COO)]n chain perpendicular to the a direction (Fig. 3b). The metal ions are arranged in a zigzag mode along the chain. The carboxylate bridge adopts the syn–anti mode, with the Co1–O4–C1–O3 and Co1A–O4–C1–O3 torsion angles being respectively 2.4(3)° and −119.6(2)°. The Co⋯Co distance separated by the carboxylate bridge is 5.178(5) Å. The HL12− ligand contains a bridging bidentate carboxylate group (μ2-η1:η1-bridging mode), a bidentate picolinic acid which the carboxylate group adopts μ1-η1 bridging mode and the nitrogen atom (N1) is involved in the coordination, and a noncoordinated carboxylate group (mode III, Scheme 2). The dihedral angle between the pyridyl and phenol rings in the HL12− is 55.7°.
 |
| | Fig. 3 (a) Local coordination environment of the Co2+ center in 3, symmetry codes: (A) x, 0.5 − y, −0.5 + z; (B) x, 1.5 − y, −0.5 + z. (b) 2D network formed by the HL12− ligands connecting the chains. | |
The [Co(COO)]n chains are interlinked by the HL12− ligands to generate a 2D layers along the a direction (Fig. 3b). The nearest interchain Co⋯Co distances within the layer is 7.342(6) Å. The 2D layers are interlinked by the O–H⋯O hydrogen bonds into 3D network (Fig. S3, ESI†). According to the literature research, Co(II) compounds containing 1D [Co(COO)]n chains which the Co(II) ions are bridged by a single carboxylate bridge are rarely reported.9
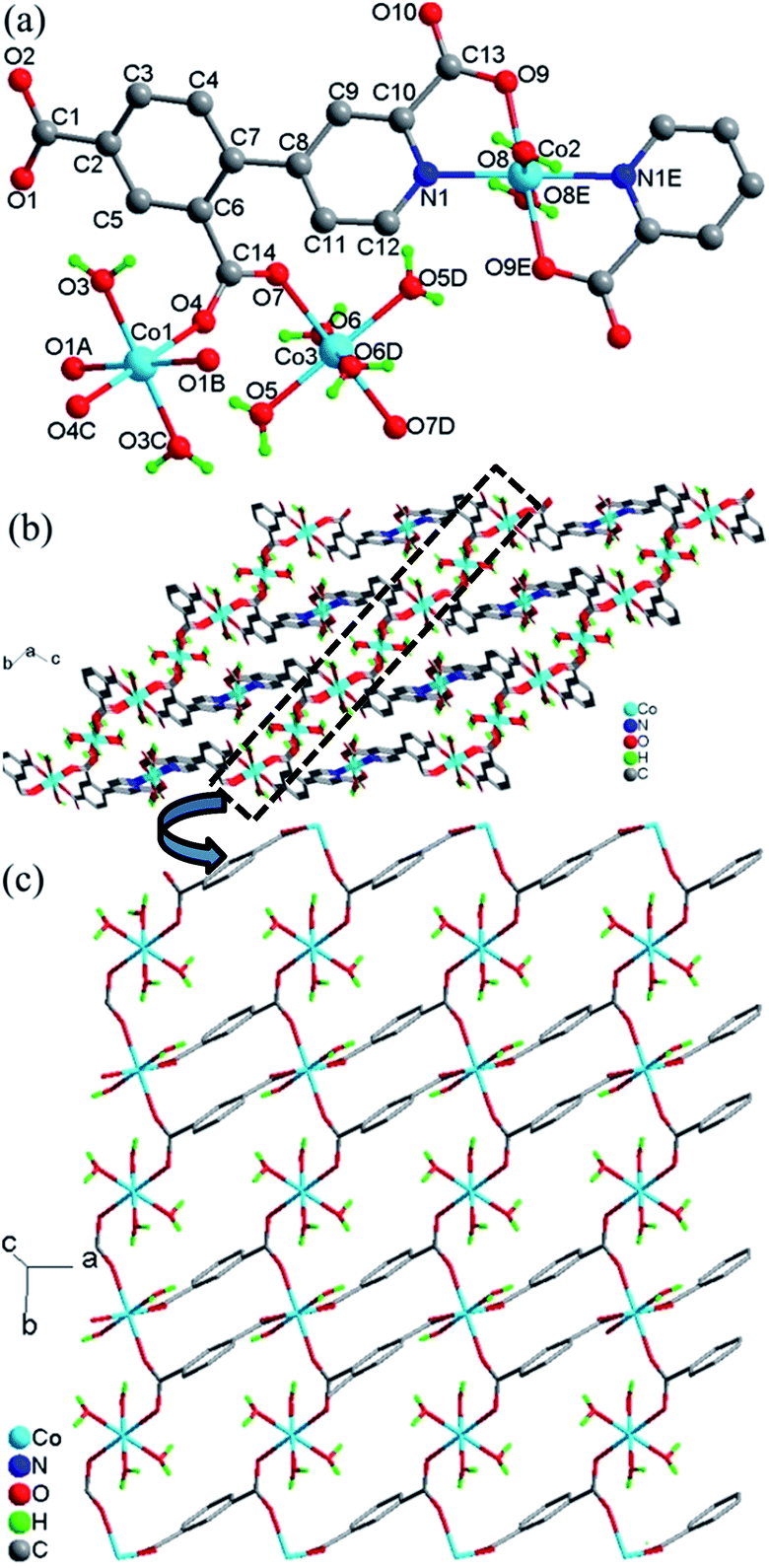
Crystal structure of 4. Compound 4 crystallizes in the triclinic space group P and exhibits a 3D framework. The crystallographically independent unit contains three different Co(II) ions, two L13− ligands, eight coordinated water molecules and four lattice water molecules. As shown in Fig. 4a, all three Co(II) ions are six-coordinated centrosymmetric trans-octahedral geometry. Both Co1 and Co3 ions adopt [O6] geometry which the coordination atoms are from the carboxylate oxygen atoms (O1A, O1B and O4, O4C) and water molecules (O3 and O3C) for Co1; from carboxylate oxygen atoms (O7 and O7D) and water molecules (O5, O5D and O6, O6D) for Co3. Co2 ion adopts [N2O4] geometry which is ligated by the nitrogen atoms (N1 and N1E) and the carboxylate oxygen atoms (O9 and O9E) from the L13− ligands and the water molecules (O8 and O8E). The Co–O/N distances are in the range of 2.067(1)–2.133(1) Å, all the Co–O and Co–N distances in 4 are comparable to these in other reported Co(II) compounds based on the similar pyridine–tricarboxylate ligands.5f,g Co1 and Co3 ions are single-bridged by carboxylate bridges in the syn–anti mode which is similar with that in 3, with Co1–O4–C14–O7 and Co3–O7–C14–O4 torsion angles being respectively 164.3(1)° and −11.5(2)° which are larger than those in 3. The Co⋯Co distance separated by carboxylate bridge is 5.156(3) Å which is slightly smaller than that in 3.
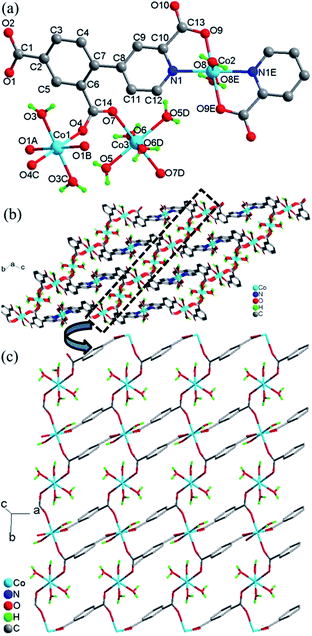 |
| | Fig. 4 (a) Local coordination environment of the Co2+ center in 4, symmetry codes: (A) 1 − x, −y, 2 − z; (B) −1 + x, y, z; (C) −x, −y, 2 − z; (D) −x, 1 − y, 2 − z; (E) −1 − x, 1 − y, 1 − z. (b) View of the whole 3D framework of 4. (c) The unfolded picture of the dotted part of (b) for the structure of 4. | |
The L13− ligand acts as a μ4 bridge to connect metal ions and contains a bridging bidentate carboxylate group (μ2-η1:η1-bridging mode), a bidentate picolinic acid which the carboxylate group adopts μ1-η1 bridging mode and the nitrogen atom (N1) is involved in the coordination, and a mono-coordinated carboxylate group (mode IV, Scheme 2). Along the bc plane, the Co1 and Co3 ions are bridged by (μ-COO) bridges into 1D chains along one direction, and Co1 (Co3) and Co2 ions are also linked by the long L13− ligands into 1D chains (Co1⋯Co2 = 9.916(4) Å, Co3⋯Co2 = 6.422(4) Å) along the other direction, and the angle along the chain of two direction is 26.4(1)° (Fig. 4b). Nevertheless, along the ab plane (Fig. 4c), the chains bridged by the single (μ-COO) bridges between Co1 and Co3 ions are along one direction while the chains linked by the same long L13− ligands between Co1 and Co1 (Co3) ions are along two different directions with Co1⋯Co1 = 7.196(6) Å and Co1⋯Co3 = 9.001(5) Å. Thus, the structure of 3D framework in 4 can be understood in this way: Co1 and Co3 ions are bridged/interlinked by L13− along the ab plane into 2D layers while the Co2 ions interlinked with Co1 and Co3 ions by long L13− ligands are in the plane just perpendicular to the ab plane. This complicated 3D framework can be formed just owing to the rotation of the C–C bond between the phenyl ring and pyridyl ring, and the dihedral angle is 36.7°.
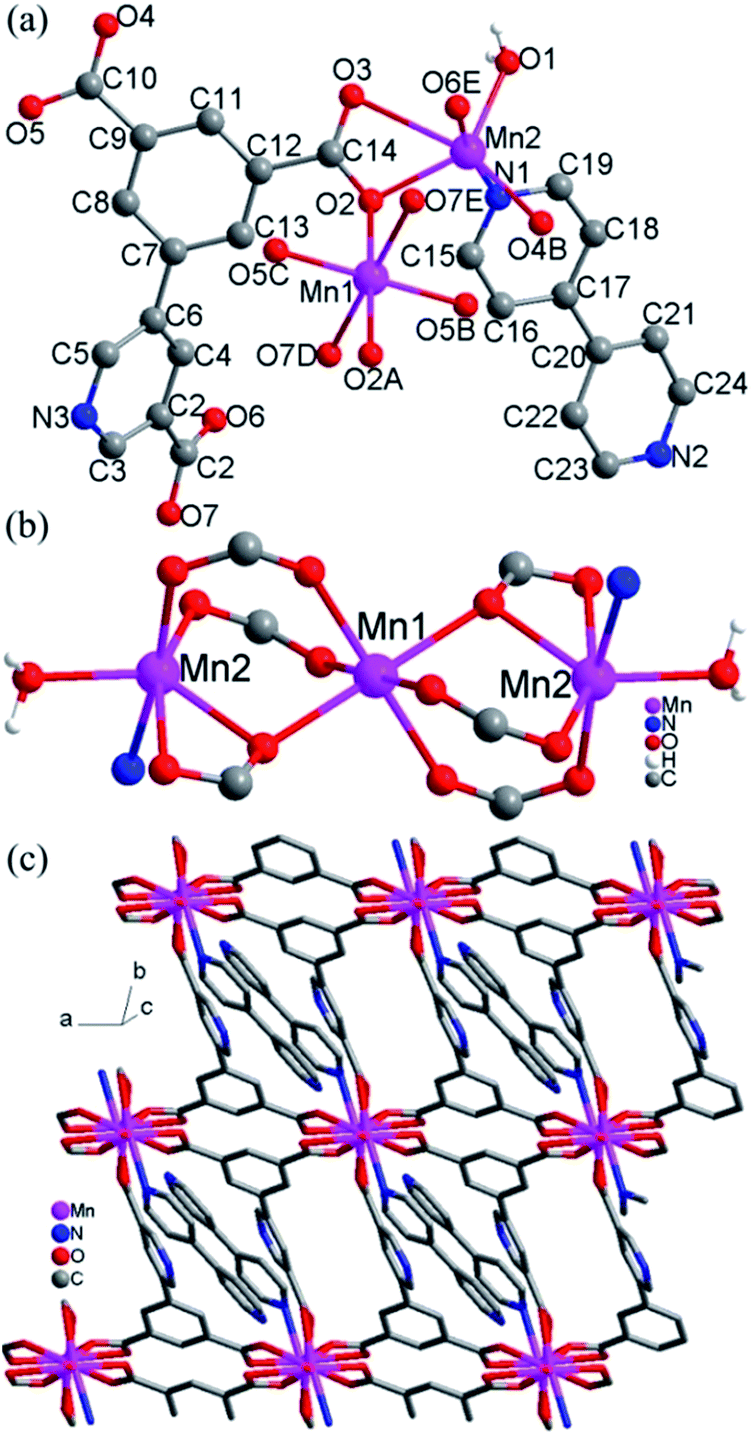
Crystal structure of 5. The structure of 5 consists of 2D layers based on trinuclear [Mn3(COO)6(H2O)2] units. As shown in Fig. 5a, there are two crystallographically independent Mn(II) ions in 5 (Mn1 and Mn2). Mn1 adopts the centrosymmetric trans-octahedral [O6] geometry completed by two axial carboxylate oxygen atoms (O2 and O2A) and four equatorial carboxylate oxygen atoms (O5B, O5C, O7D and O7E). Mn2 assumes a distorted [NO5] geometry defined by four carboxylate oxygen atoms (O2, O3, O4B and O6E), one water molecule (O1) and a nitrogen atom (N1) from 4,4′-bpy. The Mn–N/O bond distances for both Mn1 and Mn2 fall in the range of 2.128(3)–2.654(4) Å while Mn2–O3 can be taken as weak coordination with the longer bond distances 2.654(4) Å. Each Mn1 is connected with two Mn2 ions to give a trinuclear [Mn3(COO)6(H2O)2] unit (Fig. 5b), in which the triple bridges consist of (μ-COO)2(μ2-Ocarboxylate). The Mn⋯Mn distances spanned by the triple bridges are 3.52 Å, with Mn–O–Mn = 105.99(1)°. These angles and distances are similar to those observed for previous Mn(II) compounds with similar bridges.10 The trinuclear Mn(II) compounds with similar bridges have been observed in some discrete molecular species with monocarboxylate,10a–d a few extended frameworks with dicarboxylate ligands10e,11 and a chain compound with a zwitterionic dicarboxylate ligand.10f
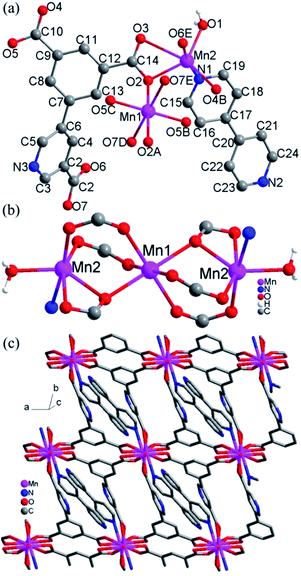 |
| | Fig. 5 (a) Local coordination environments of Mn2+ centers and the ligands in 5, symmetry codes: (A) −x, 1 − y, −z; (B) 1 + x, y, z; (C) −1 − x, 1 − y, −z; (D) −x, 2 − y, −z; (E) x, −1 + y, z. (b) The trinuclear units in 5. (c) 2D layers formed by trinuclear units and ligands L2 in 5. | |
The L23− ligands act as μ6 bridges in which two carboxylate groups adopt μ2-η1:η1-bridging modes, the third carboxylate group adopts μ2-η1:η2-bridging mode and the nitrogen atoms of L23− are not involved in coordination (mode V, Scheme 2). The dihedral angle between the m-phthalic acid plane and the picolinic acid plane of L23− is 120.40(4)°. Neighbouring trinuclear Mn3 units are interlinked by L23− ligands into 2D 4,4-grid layers along the c direction (Fig. 5c). The 4,4′-bpy molecules are mono-coordinated with the Mn(II) ions and locate in the grid formed by Mn3 units and the L23− ligands. The 2D layers are interlinked by the O–H⋯O and O–H⋯N hydrogen bonds into 3D network (Fig. S4, ESI†).
Thermogravimetric analyses and PXRD patterns
Thermogravimetric analyses (TGA) were studied on compounds 1–5 (Fig. S5, ESI†). Compound 1 shows a gradual weight loss of 5% from 25–412 °C, suggesting the gradual decomposition of the framework, and the further sharp weight loss corresponding to the full collapse of the framework of 1. Compound 2 exhibits an initial weight loss of 2% in the temperature range of 25–165 °C, corresponding to the loss of all lattice water molecules (calcd, 3.9%), and then there is continuous weight loss above 165 °C, due to the collapse of the framework of 2. Compound 3 shows no any weight loss in the range of 25–152 °C while shows weight loss of 9.3% in the range of 153–400 °C, corresponding to the loss of all coordinated water molecules (calcd, 10%). The further weight drop above 400 °C suggests the decomposition of the framework. Compound 4 shows a gradual weight loss of 22.2% from 25–400 °C, corresponding to the loss of all lattice and coordinated water molecules (calcd, 22.5%), and the further weight loss corresponding to the decomposition of the framework. Compound 5 exhibits no any weight loss in the range of 25–155 °C while shows weight loss of 3.7% in the relatively narrow range of 155–198 °C, corresponding to the coordinated water molecules (calcd, 4%), and then, there is an initially slow then fast weight loss above 198 °C due to the decomposition of the framework of 5. The observed weight loss values are somewhat less than that expected from the crystallographic data, suggesting the partial loss of water before the measurements.
The PXRD patterns for 1–5 are shown in Fig. S6–S10 in the ESI.† The diffraction peaks of both calculated and observed patterns match well indicative of the purity of the samples. The difference in reflection intensities between the simulated and observed patterns is owing to the different orientation of the crystals in the powder samples.
Photoluminescence
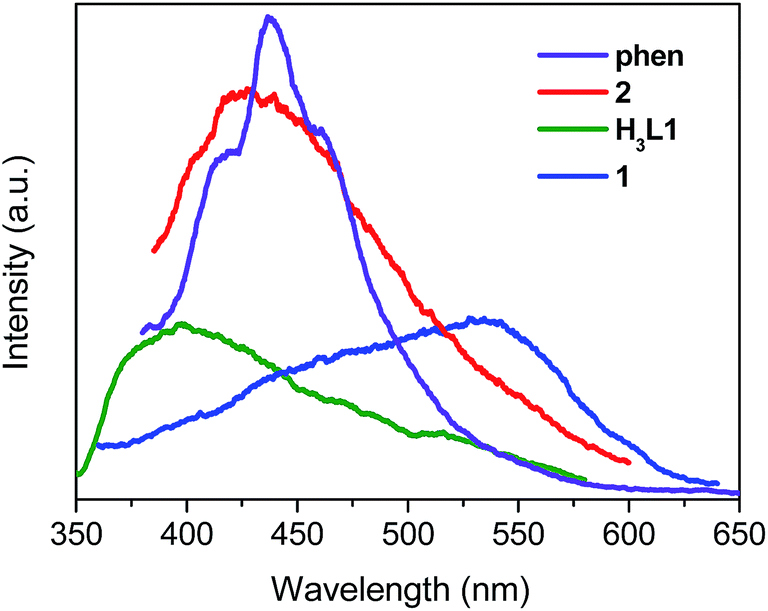
Luminescence properties of Zn(II) and Cd(II) coordination compounds have attracted great attention, due to their potential applications in chemical sensors, photochemistry and electroluminescence displays.12 Here we investigated the solid luminescence properties of compounds 1 and 2 at room temperature (Fig. 6). The free H3L1 ligand exhibits a single broad luminescence emission between 350 nm and 550 nm with a maximum peak at 398 nm (λex = 330 nm). Free phen shows a very intense emission band at 437 nm (λex = 360 nm). The emissions of the organic ligands may be ascribed to the intraligand π* → π or π* → n transitions.13 Upon complexation of the ligands with Zn(II) and Cd(II) ions, the emission peaks occur at 534 nm (λex = 330 nm) for 1, 429 nm (λex = 370 nm) for 2. For compound 1, the emission peak is highly red-shift by 136 nm with respect to the free H3L1 ligand which may be caused by the variations of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels after the organic ligands coordinate to the metal ions. It is difficult to oxidize or to reduce to the d10 configuration of Zn(II) ions, the emission of this compound is neither MLCT nor LMCT in nature.13b,14 Therefore, the emission bands of compound 1 can be assigned to intraligand luminescence emissions. For compound 2, it exhibits emission peak at 429 nm (λex = 370 nm) which are red-shifted with respect to free H3L1 ligands and phen. Thus, the emission of 2 could be attributed to a mixture of characteristics of intraligand and ligand-to-ligand charge transition (LLCT) as reported by others for other d10 metal complexes with N-donor ligands.5h,15 The decay dynamics of the emission bands at 534 nm for 1 and 429 nm for 2 can be well fitted by the double-exponential curve, and their lifetimes are 1.73 and 1.06 ms, respectively (Fig. S11 and S12, ESI†).
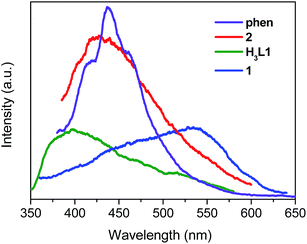 |
| | Fig. 6 The solid-state emission spectra of free H3L1, phen, 1 and 2 at room temperature. | |
Magnetic properties
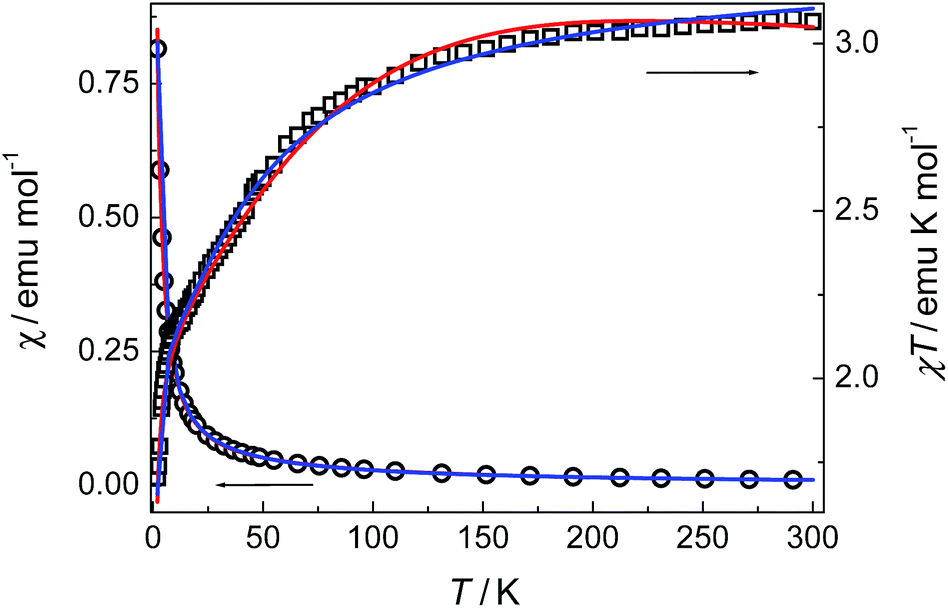
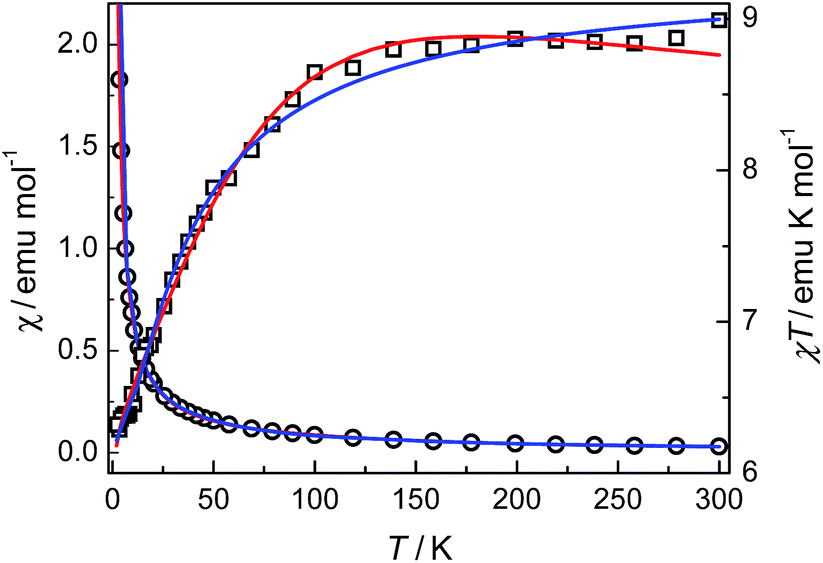
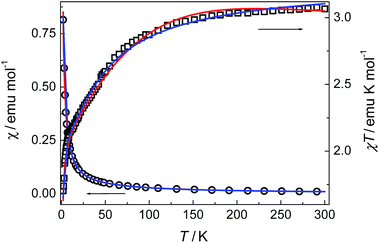
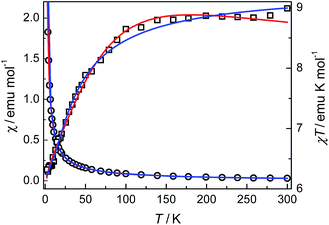
Compounds 3 and 4. The thermal magnetic properties of 3 and 4 were measured under 1000 Oe in the temperature range 2–300 K (Fig. 7 and 8). The two compounds show similar magnetic temperature-dependent behaviours. The χT values per cobalt for 3 and per Co3 unit for 4 at 300 K (3.07 and 8.99 emu K mol−1) are much higher than the spin-only values 1.875 emu K mol−1 for one S = 3/2 ion in 3, and 5.62 emu K mol−1 for three S = 3/2 ions in 4, indicative of the presence of an unquenched orbital moment typical of pseudo-octahedral Co(II). As shown in Fig. 7 and 8, the χT values decrease monotonically upon cooling in the whole temperature range studied, reaching a value of 1.7 emu K mol−1 for 3 and 6.16 emu K mol−1 for 4 at 2 K, while χ increases continuously for 3 and 4. The data above 20 K (3) and 64 K (4) follow the Curie–Weiss law with C = 3.18 emu K mol−1, θ = −10.4 K for 3 and C = 9.12 emu K mol−1, θ = −6.7 K for 4. The relatively small negative θ values indicate weak AF coupling between adjacent Co(II) ions in 3 and 4, which was found in a Co(II) compound with similar bridging mode.9 As far as we know, it was very rarely reported for the examples of Co(II) complexes bridged by single syn–anti carboxylate bridge and their magnetic properties further.9
 |
| | Fig. 7 Plots of χ and χT against T for 3. The solid lines represent the best fits of the data by different equations (see the text): red, eqn (1)–(3); blue, eqn (4). | |
 |
| | Fig. 8 Plots of χ and χT against T for 4. The solid lines represent the best fits of the data by different equations (see the text): red, eqn (1)–(3); blue, eqn (4). | |
Owing to the inherent complication related to the single-ion anisotropy, it is always difficult to exactly evaluate the magnetic parameters for Co(II) systems, and some models at different levels of approximation have been applied.16 Here we have tried two approaches. We firstly employed the effective-spin approach proposed by Lloret et al.17,18
In this approach, the Co(II) ion is treated as Seff = 1/2 spin, which is related to the real spin (S = 3/2) by eqn (1), and the effective spin Hamiltonian for a uniform chain is expressed as eqn (2).
| | |
H = −(25/9)J∑Seff,iSeff,j − G(T,J)βH∑Seff,i
| (2) |
where the fictitious Landé factor
G(
T,
J) is a temperature dependent function including four parameters:
λ (spin–orbital coupling parameter),
α (orbital reduction factor),
Δ (ligand-field distortion factor, assuming an axial distortion), and
J (magnetic exchange parameter). Combining this approximation and the polynomial expression for the susceptibility of AF half-spin chains
18,19 [
eqn (3)]:
| | |
χchain = Nβ2[G(T,J)]2/(kT)[A/B]
| (3) |
where
A = 0.25 + 0.14995
x + 0.30094
x2, and
B = 1 + 1.9862
x + 0.68854
x2 + 6.0626
x3 with
x = 25|
J|/(18
kT). It is noted that the system of
4 can be treated as 1D Co(
II) chains (formed by single carboxylate-bridged Co1 and Co3 ions) plus mononuclear Co(
II) species (Co2) according to the structural data, because Co2 ions are linked with Co1 and Co3 through long H
3L1 ligands and the magnetic interactions through long H
3L
1 ligands can be ignored. The magnetic contribution of the chain (
χchain), in which magnetic coupling (
J) is mediated through the single carboxylate bridge. Then, according to the stoichiometry of the compound, the total susceptibility is
χ =
χchain +
χmono, where
χmono =
Ng2β2S(
S + 1)/3
kT is the contribution from the mononuclear component. The best fits gave
J = −0.26 cm
−1,
λ = −123 cm
−1,
α = 1.44, and
Δ = −967 cm
−1 for
3, and
J = −0.01 cm
−1,
λ = −142 cm
−1,
α = 0.97, and
Δ = −624 cm
−1 for
4. The small negative
J values support weak AF interactions through the single carboxylate bridges in
3 and
4, and the values of the single-ion parameters
λ,
α and
Δ for
3 and
4 fall within the most common ranges expected for octahedral Co(
II) compounds and the
J values suggest the occurrence of weak AF interactions through the single carboxylate bridge in
3 and
4. The carboxylate bridge usually induces AF coupling in the
syn–
syn and
anti–
anti mode, but the
syn–
anti mode may lead to weak antiferro- or ferromagnetic coupling.
18,20 After the literature research, we have found that most of the carboxylate bridged Co(
II) coordination polymers are double carboxylate bridged while the related reports about Co(
II) coordination compounds bridged by only single
syn–
anti carboxylate bridge between Co(
II) ions and the related magnetic properties were very rare.
9 Compounds
3 and
4 are two new examples which Co(
II) ions are bridged only by a single
syn–
anti carboxylate bridge and their magnetic analyses were well done, and our magnetic fit results for
3 and
4 are in accordance with that in literature.
9
Secondly, we adopt the simple phenomenological equation given as eqn (4) proposed by Rueff et al. for low-dimensional Co(II) systems:18,21,22
| | |
χT = Aexp(−E1/kT) + Bexp(−E2/kT)
| (4) |
in which
A +
B is close to the Curie constant
C, and
E1 and
E2 are the “activation energies” associated with single-ion effects and magnetic exchange interactions, respectively.
21 This approach allows one to have rough estimation of the strength of the magnetic exchange interactions.
Very good results have been demonstrated in 1D and 2D Co(II) compounds which E1/k, spin–orbit coupling and site distortion, is on the order of +100 K in almost all of these compounds. As shown in Fig. 7 and 8, the fits using the above expression is quite satisfactory in the whole temperature range. The values obtained are A + B = 3.25 emu K mol−1, E1/k = +34.9 K, and E2/k = +0.43 K for 3; and A + B = 9.31 emu K mol−1, E1/k = +24.4 K, and E2/k = +0.05 K for 4. The A + B values are comparable with the Curie constants obtained reported by the Curie–Weiss fittings (see above). The E1/k values accounting for the effect of spin–ion coupling and site distortion are also consistent with literature values of the order of +100 K. The signs of −E2/k clearly indicate weak AF exchange interactions. The values correspond to J/k = −0.43 K (i.e., J = −0.3 cm−1) for 3 and J/k = −0.05 K (i.e., J = −0.03 cm−1) for 4. It is interesting to note that the J values are comparable with the J = −0.26 cm−1 for 3 and J = −0.01 cm−1 for 4 values obtained from the above firstly used effective-spin approach.
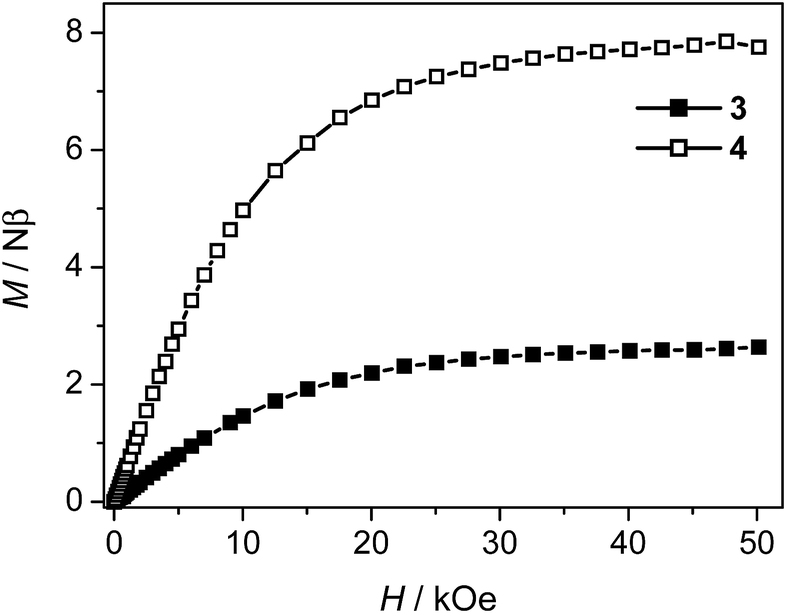
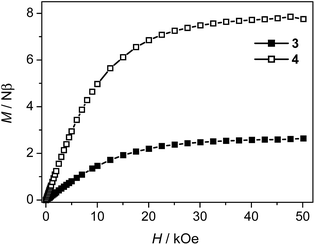
The weak AF interactions in 3 and 4 are confirmed by the isothermal magnetization measured at 2 K (Fig. 9). As the field is increased from 0 to 50 kOe, the magnetization increases slowly in the low field region and is quasi linear in the field range of 0–10 kOe for both 3 and 4. The magnetization values of 3 and 4 at 50 kOe are 2.63 Nβ and 7.75 Nβ respectively, which are in the usual ranges expected for one or three high-spin Co(II) ions with orbital degeneracy.
 |
| | Fig. 9 Magnetization isotherms for 3 and 4 at 2 K. | |
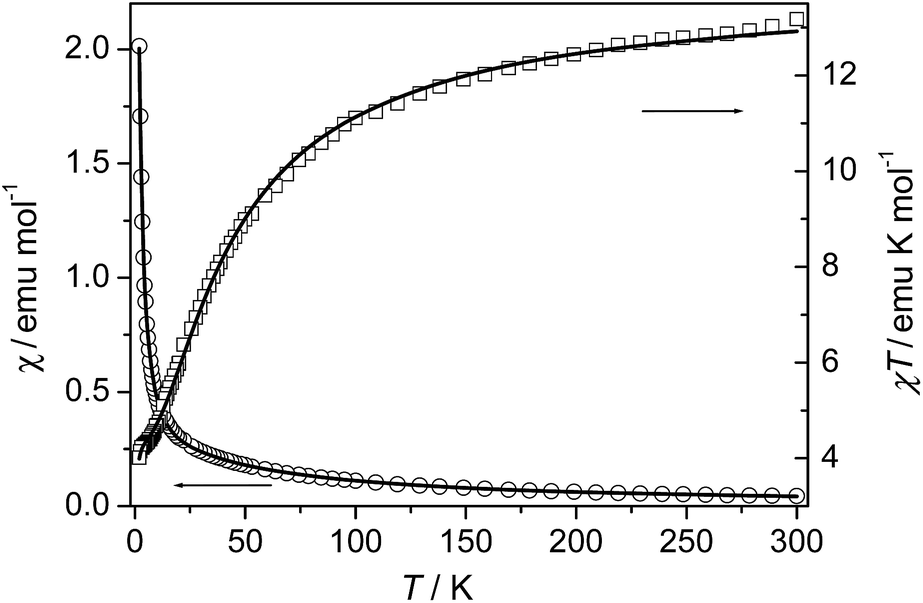
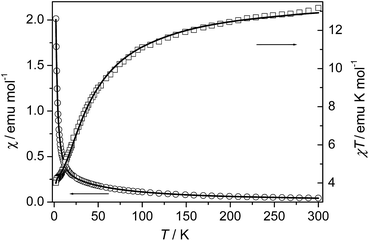
Compound 5. The magnetic susceptibility of compound 5 was measured on a polycrystalline sample under 1 kOe in the range of 2–300 K (Fig. 10). The measured χT value at 300 K is about 13.17 emu K mol−1, almost the same as the spin-only value (13.13 emu K mol−1) for three uncoupled Mn(II) ions with g = 2.00. As the sample is cooled from room temperature, the value of χ increases continuously, while the χT value decreases continuously to a minimum value of about 4.01 emu K mol−1 at 2 K. The data above 35.5 K follow the Curie–Weiss law with C = 14.36 emu K mol−1 and θ = −30.2 K, indicating AF coupling interactions between the Mn(II) centers.
 |
| | Fig. 10 Temperature dependence of χ and χT for 5. The solid lines represent the best fits to the trinuclear model (see the text). | |
The system can magnetically be regarded as an isolated linear trinuclear system. The Heisenberg Hamiltonian for the trinuclear system can be written as H = −J(ScSt1 + ScSt2), where J is the exchange constant between the terminal (spin St1 or St2) and central (Sc) metal ions. By application of van Vleck's equation, an analytical expression of magnetic susceptibility for the trinuclear system can be deduced.10d,19 The best fit parameters are J = −3.75 cm−1 and g = 2.05, suggesting a weak AF interaction through the triple bridge composed of (μ-COO)2(μ2-Ocarboxylate). We note that although some compounds with similar bridges in the previous related reports were characterized structurally and magnetically, no clear correlations was found between the structural parameters and the J values due to the complexity arising from the coexistence of mixed oxygen and carboxylate bridges. Compound 5 shows moderate AF interaction than the previous compounds which may be accounted for the medium Mn–Obridge distance (2.19 Å, 2.22 Å) and the Mn⋯Mn distance compared with the previous reported compounds.10,23
Conclusions
In this work, we have opened up the application of two π-conjugated pyridyl–tricarboxylate ligands, 4-(2,4-dicarboxylphenyl) picolinic acid (H3L1), 5-(3′,5′-dicarboxylphenyl) nicotinic acid (H3L2) as the novel multifunctional tricarboxylate ligands containing a pyridine functionality for the construction of the different 2D or 3D coordination polymers. Thus, five coordination polymers have been successfully generated by hydrothermal self-assembly reactions, using different Zn(II), Cd(II), Co(II) and Mn(II) salts and H3L1 or H3L2 as the unexplored ligands in the absence or presence auxiliary ligands. Compounds 1, 2, 3 and 5 all contain 2D layers based on [Zn(μ-Ocarboxylate)(COO)]n chains, tetranuclear [Cd4(μ3-OH)2(Ocarboxylate)2(COO)2] units, [Co(COO)]n chains and trinuclear [Mn3(COO)6(H2O)2] units respectively. Compound 4 consists of 3D framework which the 2D layers are further interlinked by Co(II) ions. Both compounds 1 and 2 show intense luminescence in the solid state at room temperature. Magnetic studies for 3–5 demonstrated that the single μ-COO bridge transmits weak AF interactions between Co(II) ions in 3 and 4 while the mixed (μ-Ocarboxylate)(COO)2 bridges transmit moderate AF interactions between Mn(II) ions in 5.
Acknowledgements
We are thankful for the financial support from NSFC (21301087, 21361016), the Inner Mongolia autonomous region natural science fund project (2013MS0206), and Programs of Higher-level talents of Inner Mongolia University (SPH-IMU-30105-125135).
Notes and references
-
(a) D. B. Dang, P. Y. Wu, C. He, Z. Xie and C. Y. Duan, J. Am. Chem. Soc., 2010, 132, 14321 CrossRef CAS PubMed;
(b) M. Eddaoudi, D. F. Sava, J. F. Eubank, K. Adil and V. Guillerm, Chem. Soc. Rev., 2015, 44, 228 RSC;
(c) L. J. Murray, M. Dincă and J. R. long, Chem. Soc. Rev., 2009, 38, 1294 RSC;
(d) M. D. Allendor, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC;
(e) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353 RSC;
(f) Y. X. Guo, X. Feng, T. Y. Han, S. Wang, Z. G. Lin, Y. P. Dong and B. Wang, J. Am. Chem. Soc., 2014, 136, 15485 CrossRef CAS PubMed;
(g) D. Zacher, O. Shekhah, C. Wöll and R. A. Fischer, Chem. Soc. Rev., 2009, 38, 1418 RSC;
(h) J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC;
(i) Y. B. He, O. Keefe and B. L. Chen, Chem. Soc. Rev., 2014, 43, 5618 RSC.
-
(a) B. Zheng, H. Dong, J. Bai, Y. Li, S. Li and M. Scheer, J. Am. Chem. Soc., 2008, 130, 7778 CrossRef CAS PubMed;
(b) A. M. Stepenson and D. Ward, Chem. Commun., 2012, 48, 3605 RSC;
(c) L. S. Long, CrystEngComm, 2010, 12, 1354 RSC;
(d) K. H. Li, W. C. Song, Y. W. Li, Y. Q. Chen and X. H. Bu, Cryst. Growth Des., 2012, 12, 1064 CrossRef;
(e) X. M. Chen and M. L. Tong, Acc. Chem. Res., 2007, 40, 162 CrossRef CAS PubMed;
(f) L. N. Zhang, C. Zhang, B. Zhang, C. X. Du and H. W. Hou, CrystEngComm, 2015, 17, 2837 RSC.
-
(a) M. Higuchi, K. Nakamura, S. Horike, Y. Hijikata, N. Yanai, T. Fukushima, J. Kim, K. Kato, M. Takata, D. Watanabe, S. Oshima and S. Kitagawa, Angew. Chem., Int. Ed., 2012, 51, 8369 CrossRef CAS PubMed;
(b) J. C. Yu, Y. J. Cui, C. D. Wu, Y. Yang, B. L. Chen and G. D. Qian, J. Am. Chem. Soc., 2015, 137, 4026 CrossRef CAS PubMed;
(c) X. T. Rao, J. F. Cai, J. C. Yu, Y. B. He, C. D. Wu, W. Zhou, T. Yildirim, B. L. Chen and G. D. Qian, Chem. Commun., 2013, 49, 6719 RSC;
(d) B. Liu, W. P. Wu, L. Hou and Y. Y. Wang, Chem. Commun., 2014, 50, 8731 RSC.
-
(a) Y. Q. Wang, Q. H. Tan, H. T. Liu, W. Sun and Z. L. Liu, RSC Adv., 2015, 5, 86614 RSC;
(b) Q. H. Tan, Y. Q. Wang, X. Y. Guo, H. T. Liu and Z. L. Liu, RSC Adv., 2016, 6, 61725 RSC;
(c) Y. Q. Wang, Q. Yue and E. Q. Gao, Chem.–Eur. J., 2017, 23, 896 CrossRef CAS PubMed;
(d) Y. Q. Wang, A. L. Cheng, P. P. Liu and E. Q. Gao, Chem. Commun., 2013, 49, 6995 RSC;
(e) Y. Q. Wang, Q. H. Tan, X. Y. Guo, H. T. Liu, Z. L. Liu and E. Q. Gao, RSC Adv., 2016, 6, 72326 RSC.
-
(a) N. N. Yang, W. Sun, F. G. Xi, Q. Sui, L. J. Chen and E. Q. Gao, Chem. Commun., 2017, 53, 1747 RSC;
(b) T. Gong, X. Yang, Q. Sui, Y. Qi, F. G. Xi and E. Q. Gao, Inorg. Chem., 2016, 55, 96 CrossRef CAS PubMed;
(c) Q. Yang, J. P. Zhao, B. W. Hu, X. F. Zeng and X. H. Bu, Inorg. Chem., 2010, 49, 3746 CrossRef CAS PubMed;
(d) L. M. Fan, W. L. Fan, B. Lin, X. Z. Liu, X. Zhao and X. T. Zhang, CrystEngComm, 2015, 17, 4669 RSC;
(e) J. Q. Liu, X. F. Li, C. Y. Gu, J. C. S. Silva, A. L. Barros, S. Alves Jr, B. H. Li, F. Ren, S. R. Batten and T. A. Soares, Dalton Trans., 2015, 44, 19370 RSC;
(f) Y. L. Shao, Y. H. Cui, J. Z. Gu, A. M. Kirillov, J. Wu and Y. W. Wang, RSC Adv., 2015, 5, 87484 RSC;
(g) J. Z. Gu, Y. H. Cui, X. X. Liang, J. Wu, D. Y. Lv and A. M. Kirillov, Cryst. Growth Des., 2016, 16, 4658 CrossRef CAS;
(h) J. Z. Gu, Y. H. Cui, J. Wu and A. M. Kirillov, RSC Adv., 2015, 5, 78889 RSC.
- G. M. Sheldrick, Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXTL Version 5.1, Bruker Analytical X-ray Instruments Inc., Madison, Wisconsin, USA, 1998 Search PubMed.
-
(a) X. Y. He, B. C. Noll, A. Beatty, R. E. Mulvey and K. W. Henderson, J. Am. Chem. Soc., 2004, 126, 7444 CrossRef CAS PubMed;
(b) B. Chiari, A. Cinti, O. Crispu, F. Demartin, A. Pasini and O. Piovesana, J. Chem. Soc., Dalton Trans., 2002, 4672 RSC;
(c) N. Srivastav, R. Mutneja, N. Singh, R. Singh, V. Kaur, J. Wagler and E. Kroke, Eur. J. Inorg. Chem., 2016, 1730 CrossRef CAS;
(d) D. M. Kuzyaev, D. L. Vorozhts, N. O. Druzhkov, M. A. Lopatin, E. V. Baranov, A. V. Cherkasov, G. K. Fukin, G. A. Abakumov and M. N. Bochkarev, J. Organomet. Chem., 2012, 698, 35 CrossRef CAS;
(e) C. P. Pradeep, S. Supriya, P. S. Zacharias and S. K. Das, Polyhedron, 2006, 25, 3588 CrossRef CAS.
-
(a) E. Tynan, P. Jensen, N. R. Kelly, P. E. Kruger, A. C. Lees, B. Moubaraki and K. S. Murray, Dalton Trans., 2004, 3440 RSC;
(b) S. J. Retting, R. C. Thompson, J. Trotter and S. Xia, Inorg. Chem., 1999, 38, 1360 CrossRef;
(c) Q. Sun, A. L. Cheng, Y. Q. Wang, Y. Ma and E. Q. Gao, Inorg. Chem., 2011, 50, 8144 CrossRef CAS PubMed.
-
(a) R. L. Rardin, P. Poganiuch, A. Bino, D. P. Goldberg, W. B. Tolman, S. C. Liu and S. J. Lippard, J. Am. Chem. Soc., 1992, 114, 5240 CrossRef CAS;
(b) R. A. Reynolds III, W. R. Dunham and D. Coucouvanis, Inorg. Chem., 1998, 37, 1232 CrossRef PubMed;
(c) G. Fernndez, M. Corbella, J. Mahia and M. A. Maestro, Eur. J. Inorg. Chem., 2002, 2502 CrossRef;
(d) S. Menage, S. E. Vitols, P. Bergerat, E. Codjovi, O. Kahn, J. J. Girerd, M. Guillot, X. Solans and T. Calvet, Inorg. Chem., 1991, 30, 2666 CrossRef CAS;
(e) H. Tian, Q. X. Jia, J. Y. Zhang and E. Q. Gao, Inorg. Chim. Acta, 2010, 363, 2481 CrossRef CAS;
(f) Q. H. Tan, Y. Q. Wang, H. T. Liu and Z. L. Liu, Inorg. Chem. Commun., 2015, 58, 67 CrossRef CAS.
-
(a) B. Liu, R. Q. Zou, R. Q. Zhong, S. Han, H. Shioyama, T. Yamada, G. Maruta, S. Takeda and Q. Xu, Microporous Mesoporous Mater., 2008, 111, 470 CrossRef CAS.
-
(a) M. Dai, X.-R. Su, X. Wang, B. Wu, Z.-G. Ren, X. Zhou and J.-P. Lang, Cryst. Growth Des., 2014, 14, 240 CrossRef CAS;
(b) Y. Shen, C. C. Fan, Y. Z. Wei, J. Hu, H. B. Zhu and Y. Zhao, Cryst. Growth Des., 2016, 16, 5859 CrossRef CAS;
(c) L. Liu, C. Huang, Z. Wang, D. Wu, H. Hou and Y. Fan, CrystEngComm, 2013, 15, 7095 RSC;
(d) X.-W. Wang, J.-Z. Chen and J.-H. Liu, Cryst. Growth Des., 2007, 7, 1227 CrossRef CAS;
(e) X. He, C.-Z. Lu and D.-Q. Yuan, Inorg. Chem., 2006, 45, 5760 CrossRef CAS PubMed;
(f) Y.-Z. Tang, G.-X. Wang, Q. Ye, R.-G. Xiong and R.-X. Yuan, Cryst. Growth Des., 2007, 7, 2382 CrossRef CAS;
(g) K. C. Wang, T. L. Liu, Y. J. Liu, X. Tian, J. Sun and Q. H. Zhang, CrystEngComm, 2016, 18, 8301 RSC.
-
(a) A. Niu, J. Yang, J. Guo, W. Q. Kan, S. Y. Song, P. Du and J. F. Ma, Cryst. Growth Des., 2012, 12, 2397 CrossRef;
(b) L. Wen, Z. Lu, J. Lin, Z. Tian, H. Zhu and Q. Meng, Cryst. Growth Des., 2007, 7, 93 CrossRef CAS;
(c) X. X. Wang, X. Q. Wang, X. Y. Niu and T. P. Hu, CrystEngComm, 2016, 18, 7471 RSC.
-
(a) L. Wen, Y. Li, Z. Lu, J. Lin, C. Duan and Q. Meng, Cryst. Growth Des., 2006, 6, 530 CrossRef CAS;
(b) L. P. Zhang, J. F. Ma, J. Yang, Y. Y. Pang and J. C. Ma, Inorg. Chem., 2010, 49, 1535 CrossRef CAS PubMed.
-
(a) D. M. Chen, X. Z. Ma, W. Shi and P. Cheng, Cryst. Growth Des., 2015, 15, 3999 CrossRef CAS;
(b) S. L. Wang, F. L. Hu, J. Y. Zhou, Y. Zhou, Q. Huang and J. P. Lang, Cryst. Growth Des., 2015, 15, 4087 CrossRef CAS.
-
(a) R. L. Carlin, Magnetochemistry, Springer-Verlag, Berlin, Heidelberg, 1986 Search PubMed;
(b) O. Kahn, MolecularMagnetism, VCH, New York, 1993 Search PubMed;
(c) F. Lloret, M. Julve, J. Cano, R. Ruiz-García and E. Pardo, Inorg. Chim. Acta, 2008, 361, 3432 CrossRef CAS and references therein.
- F. Lloret, M. Julve, J. Cano, R. Ruiz-Garcia and E. Pardo, Inorg. Chim. Acta, 2008, 361, 3432 CrossRef CAS.
- H. Tian, A. L. Cheng and E. Q. Gao, Dalton Trans., 2016, 45, 18696 RSC.
- O. Kahn, Molecular Magnetism, Wiley-VCH, Weinheim, 1993 Search PubMed.
-
(a) A. Rodríguez-Fortea, P. Alemany, S. Alvarez and E. Ruiz, Chem.–Eur. J., 2001, 7, 627 CrossRef;
(b) J. Y. Zhang, Y. Ma, A. L. Cheng, Q. Yue, Q. Sun and E. Q. Gao, Dalton Trans., 2011, 40, 7219 RSC.
-
(a) J. M. Rueff, N. Masciocchi, P. Rabu, A. Sironi and A. Skoulios, Chem.–Eur. J., 2002, 8, 1813 CrossRef CAS PubMed;
(b) J. M. Rueff, N. Masciocchi, P. Rabu, A. Sironi and A. Skoulios, Eur. J. Inorg. Chem., 2001, 11, 2843 CrossRef.
- Y. Q. Wang, K. Wang, Q. Sun, H. Tian, E. Q. Gao and Y. Song, Dalton Trans., 2009, 9854 RSC.
-
(a) C. J. Milios, T. C. Stamatatos, P. Kyritsis, A. Terzis, C. P. Raptopoulou, R. Vicente, A. Escuer and S. P. Perlepes, Eur. J. Inorg. Chem., 2004, 2885 CrossRef CAS;
(b) A. Escuer, B. Cordero, X. Solans, M. Font-Bardia and T. Calvet, Eur. J. Inorg. Chem., 2008, 5082 CrossRef CAS;
(c) V. Gomez and M. Corbella, Eur. J. Inorg. Chem., 2009, 4471 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1530040–1530044. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra02063e |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Zhi-Liang Liu
*a,
Zhi-Liang Liu