
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An ultrasensitive and label-free electrochemical DNA biosensor for detection of DNase I activity†
Chen Liab,
Xuejuan Chenac,
Nan Wangac and
Bailin Zhang *a
*a
aState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: blzhang@ciac.ac.cn
bUniversity of Science and Technology of China, Hefei 230026, P. R. China
cUniversity of Chinese Academy of Sciences, Beijing, 100049, P. R. China
First published on 18th April 2017
Abstract
An ultrasensitive and label-free DNA biosensor for the detection of deoxyribonuclease I (DNase I) activity was comprehensively studied based on electrochemical methods by cyclic voltammetry (CV) and square wave voltammetry (SWV). For the preparation of the DNA biosensor, the arbitrary 76-bp single-stranded DNA reporter probes and electrochemical indicator methylene blue (MB) were modified on the Au electrode. Initially, the sensor generated a strong signal because of the redox indicator MB. In the presence of DNase I, the enzyme cleaved single-stranded DNA at their phosphate backbone, and then the MB was removed from the electrode, resulting in the reduction of current signals. The differential current signals were directly proportional to the logarithm of the concentration of DNase I in the range of 10−5 units per mL to 0.5 units per mL with a detection limit of 10−6 units per mL (S/N = 3). The sensor exhibits a wide linear range and superior sensitivity and can be applied in more bioanalytical systems.
1 Introduction
Deoxyribonuclease I is a glycoprotein that acts as an important enzyme on either double-stranded or single-stranded DNA. On the phosphate backbone, it nonspecifically cleaves the DNA, generating a series of di-, tri-, and oligonucleotides which have hydroxylated 3′ terminal and phosphorylated 5′ terminal.1,2 DNase I, a useful diagnostic biomarker,3–11 is responsible for the fragmentation of genomic content during apoptosis.12,13 Recently, some studies have shown that several diseases are relevant to the DNase I activity. Patients who have prostate cancer4,14 and systemic lupus erythematosus (SLE) exhibited reduced DNase I activity,6,7,11 while those who were suffering from acute myocardial infarction (AMI) showed increased DNase I activity.3,5 Thus, the measurement of DNase I activity might be regarded as an innovative diagnostic marker for the early diagnosis of SLE or AMI.The activity of DNase I relies on Ca2+, Mg2+, or Mn2+ ions and requires an environment close to neutral. Considering the complexity of determination of DNase I in clinical samples, several assays depended on the enzymatic activity have been reported,15 such as immunochemical assays and single radial enzyme diffusion (SRED) with high sensitivity and convenience.16 However, to yield results, immunochemical assays require multiple washing steps and SRED method is time-consuming. At the meantime, the presence of endogenous inhibitor, like G-actin,17 can potentially hinder reliability of the measurement. Recently, Lee et al. reported an assay method utilizing the preferential interaction of graphene oxide surface with ssDNA.18 Islam et al. demonstrated a method of electrochemical assay for the ENases activity with graphene monolayer as an electrode and substrate for immobilization.19 Their strategy was based on the π–π staking interaction between aromatic ring structure of exposed nucleobases and the hexagonal structure of graphene. Their approach suggested several advantages over conventional methods, but still had typical limitations in fluorescent detection. Hence, it is of great importance to explore a rapid credible assay to measure DNase I in a clinical testing.
The electrochemical sensor becomes one of the most promising tools due to its sensitivity, convenience, rapidity, and economy in real operations. This sensor can fall into single signaling electrochemical type by target-induced conformational changes or ratiometric one by coupled “signal-on/off” dual-signaling.20 Up to now all kinds of DNA structures have been applied to electrochemical experimental study, such as single-strand,21 double helix,22 hand-in-hand,23 sandwich,24 super sandwich,25 hairpin,26 and dual-hairpin27 structures. Hence electrochemical DNA biosensor has been one of the most promising detection systems for nucleic acid detection due to their sensitive and rapid response, convenient and simple operation, low cost, and suitability for mass production.28,29
In this paper, we proposed an ultrasensitive and label-free electrochemical DNA biosensor for the detection of DNase I activity on the bare gold electrode without any other structures. Firstly, the Au electrode, easily fabricated by arbitrary 76-bp single-stranded DNA reporter probes and blocked the nonspecific binding sites by 6-mercapto-1-hexanol (MCH), was characterized using electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV). Then electrochemical indicator methylene blue (MB) was added and interacted with the DNA probes, generating obvious electrochemical signals screened by square wave voltammetry (SWV). When the DNase I existed, the part of DNA was cleaved and SWV signals of MB decreased as described in Scheme 1. The differential current signals were directly proportional to the logarithm of concentrations of DNase I, which is related to the DNase I activity. In addition, this electrochemical detection platform can be applied in many other bioanalytical systems.
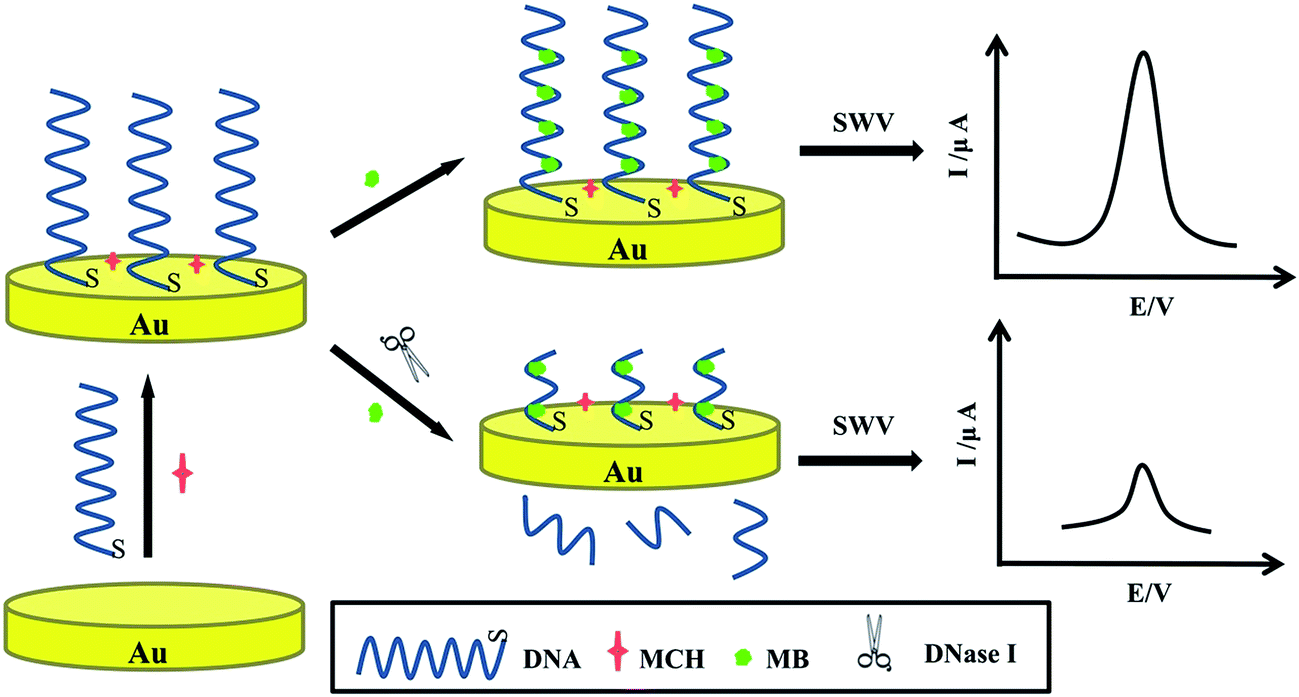 | ||
| Scheme 1 Schematic diagrams of the detection of DNase I activity based on electrochemical method by single-stranded DNA reporter probes. | ||
2 Experimental sections
2.1 Fabrication of DNA biosensor
The Au electrode (Φ 2.0 mm) was firstly pretreated in piranha solution which was freshly prepared (the volume ratio of 98% H2SO4 to 30% H2O2 equals 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) for 5 min (Caution! Piranha solution has strong corrosion of organic matter!), and polished with 1.0 μm and 0.05 μm Al2O3 polishing powder carefully until a mirror surface achieved, and then cleaned ultrasonically with ultrapure water and ethanol at least three times. Thereafter, the above electrode was electrochemically polished by voltammetric scanning in a 0.5 M H2SO4 solution. In the end, the electrode was rinsed with ultrapure water and dried in the nitrogen.
:3) for 5 min (Caution! Piranha solution has strong corrosion of organic matter!), and polished with 1.0 μm and 0.05 μm Al2O3 polishing powder carefully until a mirror surface achieved, and then cleaned ultrasonically with ultrapure water and ethanol at least three times. Thereafter, the above electrode was electrochemically polished by voltammetric scanning in a 0.5 M H2SO4 solution. In the end, the electrode was rinsed with ultrapure water and dried in the nitrogen.
The thiol-labeled DNA reporter probe solution (20 μL, 1.0 μM) was carefully dripped to the surface of the pure electrode for 6 h to form monolayer and rinsed with washing solution to remove the weak adsorbates. Subsequently, to block the nonspecific binding sites, the above modified surface was immersed in MCH (2.0 mM) for 1 h on surface and rinsed with washing solution thoroughly. After that, the electrode was immersed in the stirring MB solution (20 μM) for 5 min to accumulate MB onto the surface-hybridized DNA probes. Finally, the DNA/MCH/Au electrode was incubated in proportional concentrations of DNase I for 1 h, along with extensively rinsed by the washing solution and characterized by SWV in 20 mM Tris–HCl electrolyte buffer (pH 7.4).
3 Results and discussion
3.1 Mechanism of the DNA biosensor
Scheme 1 illuminated the working mechanism of label-free electrochemical DNA biosensor to detect DNase I activity. Initially, DNA reporter probes were self-assembled steadily forming a uniform monomolecular layer by Au–S bond on the gold electrode that blocked nonspecific binding sites by MCH. Then the electrode was immersed in MB solution. MB, a frequently used electroactive marker, could readily bind to the guanine bases on the DNA via electrostatic attraction or intercalation30,31 and could be electronically well bonded to the modified electrode surface. Yang et al.32 have been demonstrated the relationship between the peak currents and the number of guanine bases in the oligonucleotide was linear. In the absence of DNase I, the DNA guanine was affected as a fixator to attract the redox-active MB to approach to the electrode surface, causing strong electrochemical current signals. After incubated in the DNase I solution, the strands of DNA with MB were cut off and entered into the solution. Due to the cleavage effect, the reduced electrochemical signals were associated with various concentrations of DNase I, which could be used for the quantitative determination of DNase I. Then a sensitive DNA biosensor to detect DNase I has been fabricated.3.2 Characterization of the DNA biosensor
CVs and EIS were investigated in Fig. 1 to monitor the each fabrication process of the electrochemical DNA biosensor. In Fig. 1A, compared with bare Au electrode (curve a), there was an obvious decrease in peak current of the DNA probes modified electrode (curve b) and the redox peak potential difference increased owing to the repulsive interaction between the negatively charged backbones of DNA and [Fe(CN)6]3−/4− anions. As the DNA/Au electrode was ulterior blocked by MCH, which impeded the interfacial electron transfer kinetics of the probes, the peak currents decreased and the redox peak potential difference increased correspondingly (curve c). However, after incubation in DNase I solution, the peak current increased and the redox peak potential difference decreased (curve d), indicating the cleavage of DNA by DNase I.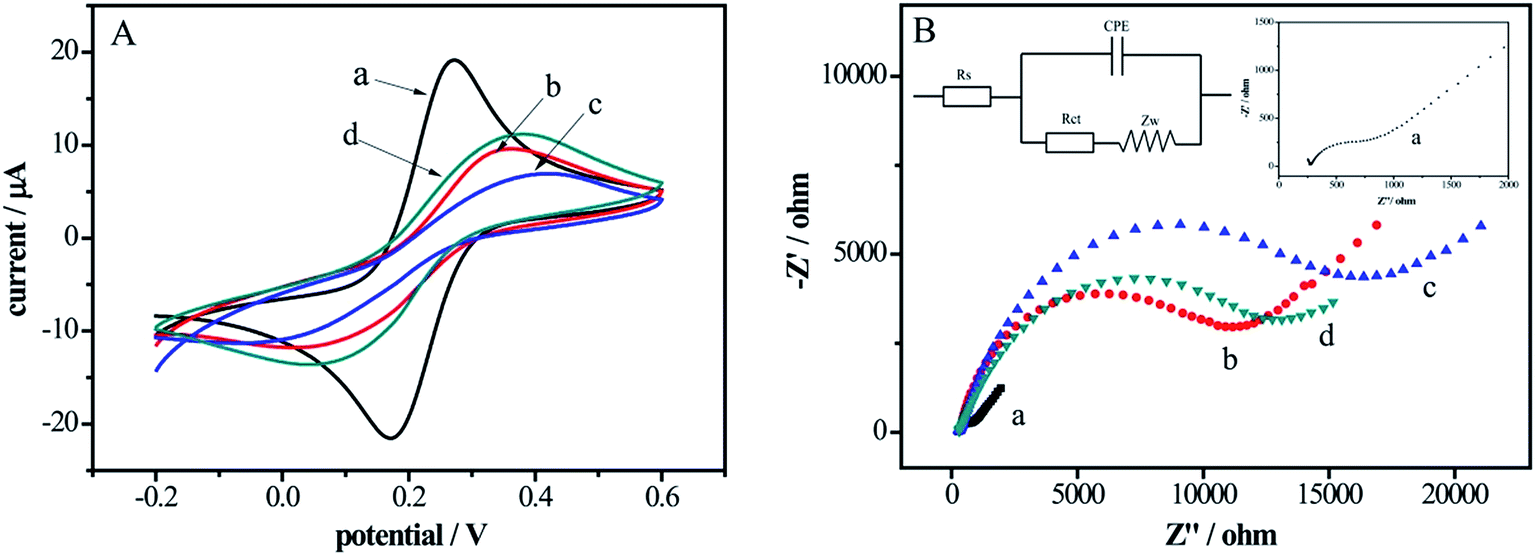 | ||
| Fig. 1 (A) Cyclic voltagramms at a potential scan rate of 50 mV s−1 (vs. SCE); (B) Nyquist plots of electrochemical impendence spectra under excitation of a sinusoidal perturbation signal of 5 mV amplitude in the frequency range from 0.1 Hz to 100 kHz at a potential of 0.2 V of bare Au electrode (a), DNA/Au electrode (b), DNA/MCH/Au electrode (c), DNA/MCH/Au electrode incubation with DNase I solution for 1 h (d) in 5 mM [Fe(CN)6]3−/4−containing 0.1 M KCl; insets: equivalent circuit model for electrochemical impedance measurement system (the upper left corner) and the enlarged curve a in (B) (the upper right corner). | ||
Fig. 1B showed the Nyquist plots of EIS at different functional steps of Au electrodes. In the Nyquist plots, the interfacial electron transfer resistance (Rct) was expressed by the diameter of the semicircle portion and the increase of the diameter reflected that of the Rct.33 The EIS data was fitted in the equivalent circuit in the upper left inset of Fig. 1B. As shown in curve a (enlarged in the upper right inset, Fig. 1B), the Rct of bare Au electrode was a tiny semicircle domain, which demonstrated a quick electron transfer of [Fe(CN)6]3−/4−. The Rct increased significantly along with the assembling of DNA probes (curve b) on the electrode surface thanks to the negatively charged DNA hindered the interfacial electron transfer. With MCH blocking the nonspecific sites, the Rct further increased (curve c). However, after the electrode was reacted with the DNase I solution (curve d), the Rct decreased due to interfacial electron transferring easily caused by the short DNA strands. The EIS results are in keeping with the measurements of the CVs (Fig. 1A), which proves that the DNA biosensor has been successfully established.
3.3 Feasibility of the DNA biosensor
To verify the feasibility of the DNA biosensor, the SWV signals of the proposed biosensor in the different modified electrodes were compared in supporting electrolyte solution. As shown in Fig. 2, without electrochemical indicator MB modification on the electrode surface, there were no electrochemical signals on bare Au electrode (curve a), DNA/Au electrode (curve b) and electrode (curve c). With the modification of the MB on the DNA/MCH/Au electrode, the signals increased obviously (curve d) owing to the specific intercalation between the guanine bases of DNA and MB. While after incubated in 0.01 units per mL DNase I, the peak current of MB decreased (curve e) due to the removal of MB on the DNA strands from electrode surface as a result of cleavage of enzyme. These results perfectly agree with the CVs and EIS given in Fig. 1, indicating that the biosensor can be utterly used for the detection of DNase I.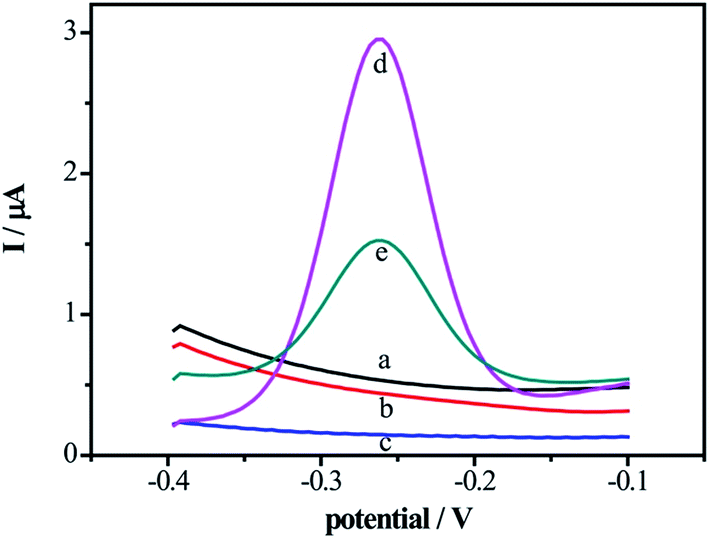 | ||
| Fig. 2 The square wave voltammetric curves of MB on the bare Au electrode (a), DNA/Au electrode (b), DNA/MCH/Au electrode (c), DNA/MCH/Au electrode modified by the MB before (d) and after (e) incubation with DNase I solution for 1 hour in 20 mM Tris–HCl buffer containing 20 mM NaCl at frequency of 15 Hz, amplitude of 25 mV, potential increment of 4 mV. | ||
3.4 Sensitivity of the DNA biosensor
Under the optimical experiment conditions in ESI (Fig. S1†), the sensitivity of electrochemical DNA biosensor was measured with a series different concentrations of DNase I. The SWV signals of the DNA/MCH/Au electrode incubacted with DNase I at various concentrations were elucidated in Fig. 3A, from which the MB currents decreased with the increasing concentrations of the DNase I (curve a–h). The correlation between the concentrations of DNase I and the peak currents of MB was analyzed and a linear relationship (ΔI/μA = 2.0525 + 0.36032log[C]/units per mL) was obtained between the logarithm of DNase I concentration and the peak current of MB. At the meantime, the linear range was 10−5 units per mL to 0.5 units per mL and the detection of limit was 10−6 units per mL (S/N = 3) as shown in Fig. 3B. The sensitivity of the proposed approach shows pretty performance in the matter of analytical range and detection limit. Compared with the other reported methods to detect DNase I (Table S1 in ESI†), the sensitivity of this approach is several orders of magnitude higher than those of others.
 | ||
| Fig. 3 (A) The square wave voltammetric curves of biosensor in 20 mM Tris–HCl buffer containing 20 mM NaCl after incubation with different concentrations of DNase I (from a to h: 0.5, 0.1, 0.05, 0.01, 0.001, 0.0001, 0.00001, 0 units per mL) (B) the calibration curve for the DNase I detection between differential SWV peak currents and DNase I concentrations. The error bars represent the standard deviations of three repetitive experiments. | ||
3.5 Specificity of the DNA biosensor
The specificity of new assay method is a critical index in the analytical chemistry. In order to evaluate this index, RNase A, Hind III, trypsin and ATP, were tested under the same experimental conditions by monitored the current signal differences of the system. Their abilities to cleave the DNA were shown in Fig. 4. Only when the DNase I was applied in the system, was the remarkable current signal changes observed. Even though the concentrations of these other samples are dozens of times higher than that of DNase I, the current signal changes of them were observed much weaker. These results obviously confirmed that the specificity of our DNA biosensor for DNase I detection was very well through the DNA cleavage reaction.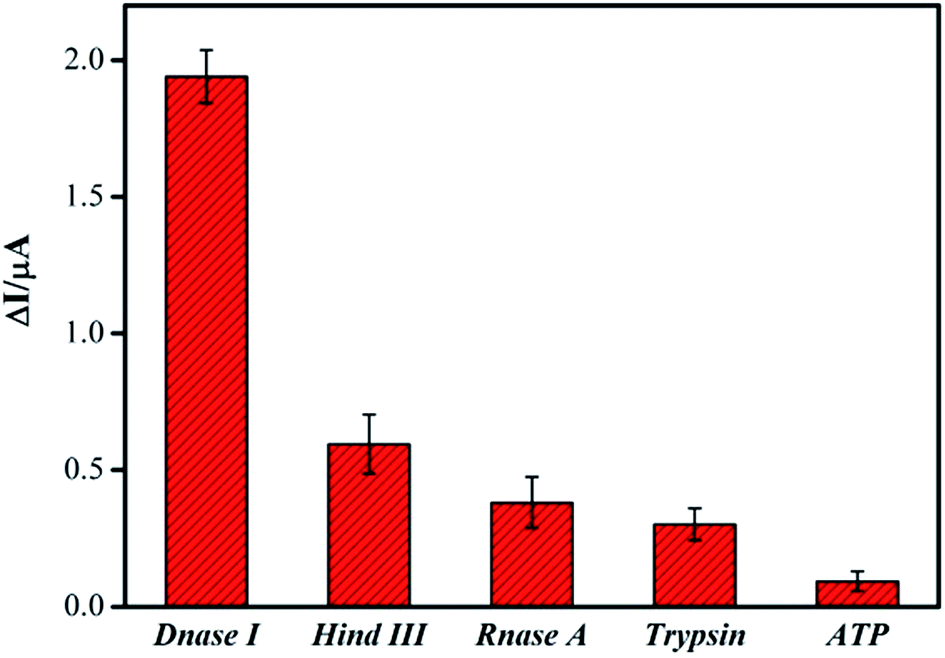 | ||
| Fig. 4 The selectivity of the DNA biosensor for detection of DNase I in 20 mM Tris–HCl buffer containing 20 mM NaCl after incubation 1 h. The concentration of DNase I is 0.5 units per mL, while the concentrations of Hind III, RNase A, Trypsin 1:300, ATP was 50 units per mL, 50 units per mL, 10 mg mL−1, 10 mg mL−1, respectively. The error bars represent the standard deviations of three repetitive measurements. | ||
3.6 Practical analysis in real samples
To evaluate the application of the proposed sensor in real sample analysis, a fetal bovine serum samples for determining the DNase I activity was demonstrated by the DNA biosensor system. In the recovery tests, we preformed target DNase I in three different concentrations in 20% fetal bovine serum. As illustrated in Table 1, the recoveries were in the range of 96.6–107.9% and the relative standard deviation (RSD) was less than 7%. According to these results, it is high-efficient and accurate by the proposed electrochemical DNA biosensor to measure DNase I activity in serum samples.| Sample | Added DNase I (units per mL) | Measured DNase I (units per mL) | Recovery (%) | RSD (%) |
|---|---|---|---|---|
| 1 | 0.0050 | 0.0053 | 105.7 | 5.17 |
| 2 | 0.050 | 0.054 | 107.9 | 6.95 |
| 3 | 0.40 | 0.39 | 96.60 | 0.30 |
4 Conclusions
In brief, a sensitive and unlabeled DNA electrochemical biosensor for DNase I activity detection was developed. In the presence of DNase I, the arbitrary 76-bp DNA strands immobilized on gold electrode were cleaved and removed, giving rise to the decreased SWV signals. These signals were linearly related to the different concentrations of DNase I. This biosensor can be easily fabricated with high sensitivity (the LOD is 10−6 units per mL) and a linear working range is 10−5 units per mL to 0.5 units per mL (S/N = 3). What's more, this sensing method exhibited good applicability in the fetal bovine serum samples by making use of immobilization-free technology and could be acted as a facile and high-efficiency platform for the detection of other biomolecules.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21375122).References
- M. Kunitz, J. Gen. Physiol., 1950, 33, 349–362 CrossRef CAS PubMed.
- S. Vanecko and M. Laskowski, J. Biol. Chem., 1961, 236, 3312–3316 CAS.
- K. Arakawa, Y. Kawai, T. Kumamoto, N. Morikawa, M. Yoshida, H. Tada, R. Kawaguchi, K. Taniguchi, I. Miyamori and Y. Kominato, Eur. Heart J., 2005, 26, 2375–2380 CrossRef CAS PubMed.
- A. V. Cherepanova, S. N. Tamkovich, O. E. Bryzgunova, V. V. Vlassov and P. P. Laktionov, Ann. N. Y. Acad. Sci., 2008, 1137, 218–221 CrossRef CAS PubMed.
- Y. Kawai, M. Yoshida, K. Arakawa, T. Kumamoto, N. Morikawa, K. Masamura, H. Tada, S. Ito, H. Hoshizaki and S. Oshima, Circulation, 2004, 109, 2398–2400 CrossRef CAS PubMed.
- M. Macanovic and P. J. Lachmann, Clin. Exp. Immunol., 1997, 108, 220–226 CAS.
- M. Macanovic, D. Sinicropi, S. Baughman, S. Thiru and P. Lachmann, Clin. Exp. Immunol., 1996, 106, 243 CAS.
- K. Malícková, D. Duricová, M. Bortlík, Z. Hrušková, B. Svobodová, N. Machková, V. Komárek, T. Fucíková, I. Janatková and T. Zima, Autoimmune Dis., 2011, 2011, 945861 Search PubMed.
- N. Morikawa, Y. Kawai, K. Arakawa, T. Kumamoto, I. Miyamori, H. Akao, M. Kitayama, K. Kajinami, J.-D. Lee and H. Takeshita, Eur. Heart J., 2007, 28, 2992–2997 CrossRef CAS PubMed.
- B. Zhu, Y. Gong, P. Chen, H. Zhang, T. Zhao and P. Li, Mol. Cell. Biochem., 2014, 393, 23–32 CrossRef CAS PubMed.
- F. M. Valle, E. Balada, J. Ordi-Ros and M. Vilardell-Tarres, Autoimmun. Rev., 2008, 7, 359–363 CrossRef CAS PubMed.
- N. C. Mishra, Nucleases: Molecular Biology and Applications, Wiley-Interscience, Hoboken, NJ, USA, 1st edn, 2002 Search PubMed.
- S. Moore, in The Enzymes, ed. D. B. Paul, Academic Press, USA, 3rd edn, 1981, ch. 4, vol. 14, pp. 281–296 Search PubMed.
- S. N. Tamkovich, A. V. Cherepanova, E. V. Kolesnikova, E. Y. Rykova, D. V. Pyshnyi, V. V. Vlassov and P. P. Laktionov, Ann. N. Y. Acad. Sci., 2006, 1075, 191–196 CrossRef CAS PubMed.
- D. Sinicropi, D. L. Baker, W. S. Prince, K. Shiffer and S. Shak, Anal. Biochem., 1994, 222, 351–358 CrossRef CAS PubMed.
- D. Nadano, T. Yasuda and K. Kishi, Clin. Chem., 1993, 39, 448–452 CAS.
- J. S. Ulmer, A. Herzka, K. J. Toy, D. L. Baker, A. H. Dodge, D. Sinicropi, S. Shak and R. A. Lazarus, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8225–8229 CrossRef CAS.
- J. Lee, Y. K. Kim and D. H. Min, Anal. Chem., 2011, 83, 8906–8912 CrossRef CAS PubMed.
- K. Islam, R. Chand, D. Han, I.-S. Shin and Y.-S. Kim, J. Electrochem. Soc., 2014, 161, B261–B264 CrossRef.
- X. Cao, J. Xia, H. Liu, F. Zhang, Z. Wang and L. Lu, Sens. Actuators, B, 2017, 239, 166–171 CrossRef CAS.
- M. N. Stojanovic, P. De Prada and D. W. Landry, J. Am. Chem. Soc., 2000, 122, 11547–11548 CrossRef CAS.
- Y.-H. Liu, H.-N. Li, W. Chen, A.-L. Liu, X.-H. Lin and Y.-Z. Chen, Anal. Chem., 2012, 85, 273–277 CrossRef PubMed.
- C. Li, Z. Wang, T. Gao, A. Duan and G. Li, Chem. Commun., 2013, 49, 3760–3762 RSC.
- K. Ikebukuro, C. Kiyohara and K. Sode, Anal. Lett., 2004, 37, 2901–2909 CrossRef CAS.
- Y. Jiang, N. Liu, W. Guo, F. Xia and L. Jiang, J. Am. Chem. Soc., 2012, 134, 15395–15401 CrossRef CAS PubMed.
- X. Wei, W. Lin, N. Ma, F. Luo, Z. Lin, L. Guo, B. Qiu and G. Chen, Chem. Commun., 2012, 48, 6184–6186 RSC.
- X. He, G. Wang, G. Xu, Y. Zhu, L. Chen and X. Zhang, Langmuir, 2013, 29, 14328–14334 CrossRef CAS PubMed.
- Y. Hu, X. Xu, Q. Liu, L. Wang, Z. Lin and G. Chen, Anal. Chem., 2014, 86, 8785–8790 CrossRef CAS PubMed.
- Z. Liu, W. Zhang, S. Zhu, L. Zhang, L. Hu, S. Parveen and G. Xu, Biosens. Bioelectron., 2011, 29, 215–218 CrossRef CAS PubMed.
- M. Jarczewska, Ł. Górski and E. Malinowska, Sens. Actuators, B, 2016, 226, 37–43 CrossRef CAS.
- Y. Jin, X. Yao, Q. Liu and J. Li, Biosens. Bioelectron., 2007, 22, 1126–1130 CrossRef CAS PubMed.
- W. Yang, M. Ozsoz, D. B. Hibbert and J. J. Gooding, Electroanalysis, 2002, 14, 1299–1302 CrossRef CAS.
- I. O. K'owino and O. A. Sadik, Electroanalysis, 2005, 17, 2101–2113 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01995e |
| This journal is © The Royal Society of Chemistry 2017 |