
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCytotoxic isovaleryl sucrose esters from Ainsliaea yunnanensis: reduction of mitochondrial membrane potential and increase of reactive oxygen species levels in A549 cells†
Xin Fangab,
Zhi-Guo Zhuoa,
Xi-Ke Xua,
Ji Yea,
Hui-Liang Lia,
Yun-Heng Shen *a and
Wei-Dong Zhang*abc
*a and
Wei-Dong Zhang*abc
aDepartment of Phytochemistry, School of Pharmacy, Second Military Medical University, Shanghai 200433, P. R. China. E-mail: shenyunheng@hotmail.com; wdzhangy@hotmail.com
bShanghai University of Traditional Chinese Medicine, Shanghai 201203, P. R. China
cShanghai Institute of Pharmaceutical Industry, Shanghai 200400, P. R. China
First published on 11th April 2017
Abstract
Eight isovaleryl sucrose esters, named ainslosides A–H (1–8), were isolated from Ainsliaea yunnanensis Franch. Their structures, including the absolute configurations of the sugar residues, were elucidated by extensive analysis of NMR spectra and acid hydrolysis. All compounds were tested in vitro for cytotoxicity against four human tumour cell lines, A549, HCT116, MDA-MB-231, and BEL7404. Among the compounds tested, ainsloside B (2) showed potent cytotoxicity against the A549 cell line with an IC50 value of 3.3 μM. Flow cytometry analysis showed that compound 2 can arrest the cell cycle at the G0/G1 phase and induce cell apoptosis in A549 cells. Further studies indicated that the apoptosis-inducing effect of compound 2 may be involved in the reduction of mitochondrial membrane potential (MMP) and increase of reactive oxygen species (ROS) level in A549 cells.
Introduction
The genus Ainsliaea of Asteraceae comprises about 70 species, which is mainly distributed in the southeast of Asia. Many Ainsliaea species have been long used for the treatment of various diseases, such as rheumatism, traumatic injury, enteritis dysentery, sore throat, and urological and gynecological diseases.1 The chemical constituents and crude extracts from Ainsliaea species have been reported to have various biological activities, such as anti-microbial,2 cytotoxic,3 antiviral,4 antioxidant,5 and anti-inflammation6–8 activities. Our previous investigations on Ainsliaea species have reported the isolation of a series of sesquiterpenoids, triterpenoids, sesquiterpenoid lactone dimers and trimers, of which the first two guaianolide trimers, ainsliatrimers A and B from A. fulvioides,9 and a new guaianolide dimer with an unusual carbon skeleton, ainsliadimer A from A. macrocephala,10 showed potent cytotoxicity and anti-inflammatory activities. Ainsliaea yunnanensis Franch., a kind of perennial plant of the genus Ainsliaea, is distributed exclusively in China. In Chinese folk medicine, the whole plants of A. yunnanensis have been used for the treatment of traumatic injury and rheumatism pain.11 However, few studies of the chemical constituents and bioactivities of A. yunnanensis were reported except for the isolation of several sesquiterpenoids, triterpenoids, and some phenoloids.12,13 As part of our continuing efforts to discover structurally interesting bioactive compounds from Ainsliaea species, eight isovaleryl sucrose esters, ainslosides A–H (1–8), were isolated from the whole plants of A. yunnanensis. Compounds 1–8 were in vitro tested for cytotoxicity against four human tumour cell lines A549, HCT116, MDA-MB-231, and BEL7404, and cell cycle arresting and apoptosis induction of ainsloside B (2) in A549 cell line. Herein, we described the isolation, structural elucidation, and in vitro antitumor evaluation (Fig. 1).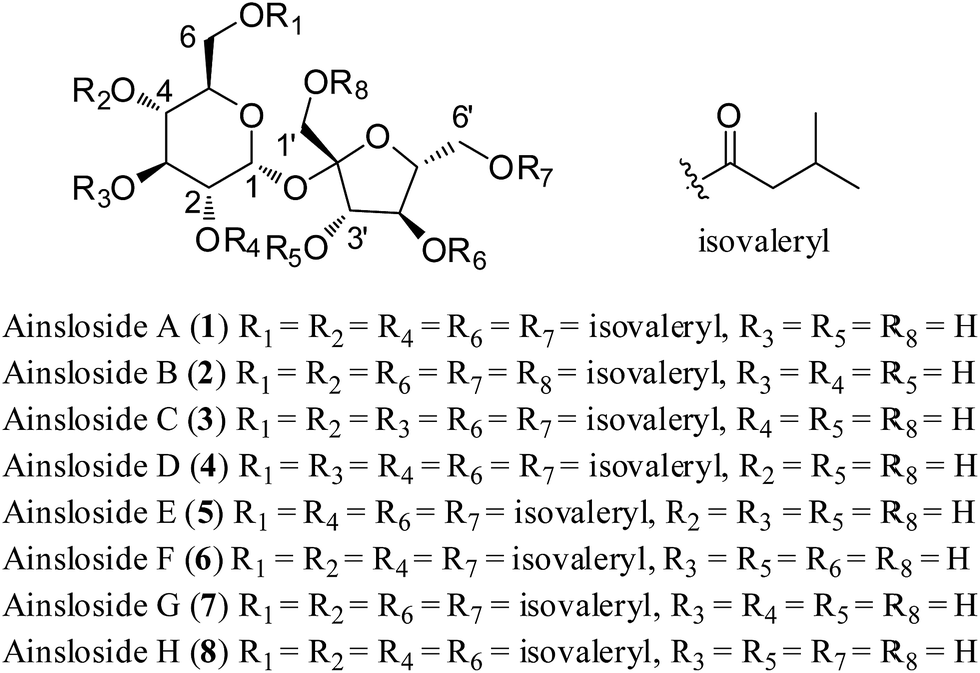 | ||
| Fig. 1 Structures of compounds 1–8. | ||
Results and discussion
Ainsloside A (1) was obtained as colourless oil. Its molecular formula was determined to be C37H62O16 as deduced from positive HR-ESI-MS (m/z 785.3942 [M + Na]+, calcd 785.3936), indicating seven degrees of unsaturation. The IR spectrum of 1 showed characteristic absorption bands of hydroxyl group (3471 cm−1) and carbonyl group (1743 cm−1). The 13C and DEPT NMR spectra (Table 1) displayed 37 carbon signals, in which 12 oxygenated carbon resonances were observed in the region 60 < δ < 105 ppm (δC 105.5, 90.1, 78.1, 77.5, 77.0, 72.4, 70.6, 70.0, 68.8, 64.0, 63.6, 62.1), implying the presence of a disaccharide residue. Characteristic signals from the NMR spectra, including an anomeric proton at δH 5.64 (d, J = 3.5 Hz) and the corresponding anomeric carbon at δC 90.1, a hydroxymethyl at δC 62.1, an hemiketal anomeric carbon at δC 105.5, and two hydroxymethyls at δC 63.6 and 64.0, exhibited quite similarity with those of sucrose previously reported in literatures,14 indicating that the disaccharide moiety should be a sucrose residue. The 1H–1H COSY correlations of H-1/H-2/H-3/H-4/H-5/H-6 and H-3′/H-4′/H-5′/H-6′, in combination with the HMBC correlation from the anomeric proton (δH 5.64) of glucopyranosyl unit to the hemiketal anomeric carbon at δC 105.5, supported the above inference. In addition, the relative configuration of the anomeric proton of glucopyranosyl residue was determined to be α-oriented based on the small coupling constant (J = 3.5 Hz) between H-1 and H-2,15 while the relative configuration of fructose was assigned to be β-orientation through the NOESY correlations of H-3′ with H-1′ and H-5′ (Fig. 2). This conclusion also can be confirmed by comparing their 13C NMR data with those of α-glucose and β-fructose previously reported in literatures.16 Moreover, compound 1 was submitted alkaline hydrolysis, and the hydrolysate was extracted with CHCl3 to afford isovaleric acid. The subsequent acid hydrolysis yielded two sugar units, and the sugar units were identified as D-glucose and D-fructose through the HPLC analysis and comparing their corresponding specific optical rotations with those of authentic samples (glucose: [α]23D +53; fructose: [α]23D −149). In the high field region of the 1H NMR spectrum, three groups of overlapping signals, corresponding to methylene, methine, and methyl protons, were observed occurring at δH 2.22–2.28 (10H, m), 2.06–2.14 (5H, m), and 0.93–0.98 (30H, m), respectively (Table 1). Combining with the 13C and DEPT NMR, empirical calculations indicated the presence of a total of ten methyls, five methines and five methylenes. Also, the 13C NMR spectrum of 1 exhibited five ester carbonyl resonances at δC 173.0, 172.9, 172.6, 172.5 and 172.4. Therefore, it may be proposed that the structure of 1 composed of a sucrose moiety and five isovaleryl moieties. Compared with sucrose,14 the 13C chemical shifts of 1 at C-2, C-4, C-6, C-4′, and C-6′, were shifted to downfield, while their adjacent carbons were shifted to upfield, implying that the five isovaleryl moieties may be substituted at C-2, C-4, C-6, C-4′, and C-6′, respectively. This deduction was further evidenced on the basis of analysis of the HMBC spectrum which showed cross-peaks between H-4′ (δH 5.17), H-4 (δH 4.93), H-6 (δH 4.20, 4.15), H-6′ (δH 4.30), H-2 (δH 4.90) and the corresponding ester carbonyls at δC 173.0, 172.9, 172.6, 172.5, and 172.4, respectively (Fig. 2). Thus, compound 1 was deduced as 2,4,4′,6,6′-pentakis-isovaleryl-β-D-fructofuranosyl-α-D-glucopyranoside, and given a trivial name ainsloside A.| No. | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | δH | δC | |
| 1 | 5.64 d (3.5) | 90.1 | 5.55 d (3.5) | 92.2 | 5.52 d (3.5) | 91.9 | 5.57 d (3.5) | 89.7 |
| 2 | 4.90 dd (10.0, 3.5) | 72.4 | 3.66 m | 72.3 | 3.77 m | 70.9 | 4.89 dd (10.0, 3.5) | 69.8 |
| 3 | 4.05 m | 70.0 | 3.88 t (9.5) | 72.2 | 5.25 t (9.5) | 73.4 | 5.40 t (10.0) | 72.2 |
| 4 | 4.93 t (10.0) | 70.6 | 4.87 t (9.5) | 70.2 | 5.07 t (10.0) | 67.3 | 3.49 t (9.5) | 69.4 |
| 5 | 4.26 m | 68.8 | 4.19 m | 68.9 | 4.29 m | 68.8 | 4.22 m | 70.9 |
| 6 | 4.20 dd (12.0, 2.0) | 62.1 | 4.15 m | 62.1 | 4.18 dd (12.5, 2.0) | 61.8 | 4.36 m | 63.0 |
| 4.15 dd (12.0, 5.5) | 4.15 m | |||||||
| 1′ | 3.66 m | 63.6 | 4.32 m | 63.0 | 3.76 m | 64.1 | 3.58 q (14.5) | 63.7 |
| 3.68 m | ||||||||
| 2′ | — | 105.5 | — | 104.3 | — | 105.0 | — | 105.3 |
| 3′ | 4.34 t (7.0) | 77.0 | 4.25 d (7.0) | 77.2 | 4.31 m | 78.0 | 4.34 m | 76.8 |
| 4′ | 5.17 t (7.0) | 77.5 | 5.16 d (7.0) | 77.4 | 5.20 t (6.5) | 77.5 | 5.16 t (7.0) | 77.3 |
| 5′ | 4.12 m | 78.1 | 4.08 m | 78.0 | 4.12 m | 78.6 | 4.08 m | 78.4 |
| 6′ | 4.30 m | 64.0 | 4.31 m | 64.1 | 4.34 m | 64.3 | 4.43 m | 64.2 |
| 4.27 dd (12.0, 3.5) | ||||||||
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
— | 173.0 | — | 173.0 | — | 174.1 | — | 174.1 |
| 172.9 | 172.9 | 172.6 | 173.5 | |||||
| 172.6 | 172.7 | 172.6 | 172.8 | |||||
| 172.5 | 172.6 | 172.5 | 172.7 | |||||
| 172.4 | 172.2 | 171.5 | 172.0 | |||||
| CH2 | 2.22–2.28 m | 43.2 | 2.21–2.26 m | 43.2 | 2.15–2.26 m | 43.3 | 2.16–2.26 m | 43.4 |
| 43.0 | 43.1 | 43.0 | 43.0 | |||||
| 43.0 | 43.1 | 43.0 | 43.0 | |||||
| 42.9 | 43.0 | 43.0 | 42.8 | |||||
| 42.8 | 42.8 | 42.8 | 42.7 | |||||
| CH | 2.06–2.14 m | 25.7 | 2.06–2.14 m | 25.7 | 2.02–2.13 m | 25.7 | 2.01–2.12 m | 25.6 |
| 25.7 | 25.6 | 25.6 | 25.6 | |||||
| 25.6 | 25.6 | 25.6 | 25.5 | |||||
| 25.5 | 25.6 | 25.4 | 25.5 | |||||
| 25.4 | 25.4 | 25.3 | 25.2 | |||||
| CH3 | 0.93–0.98 m | 22.3 × 10 | 0.92–0.98 m | 22.3 × 10 | 0.92–0.98 m | 22.3 × 10 | 0.90–0.99 m | 22.3 × 10 |
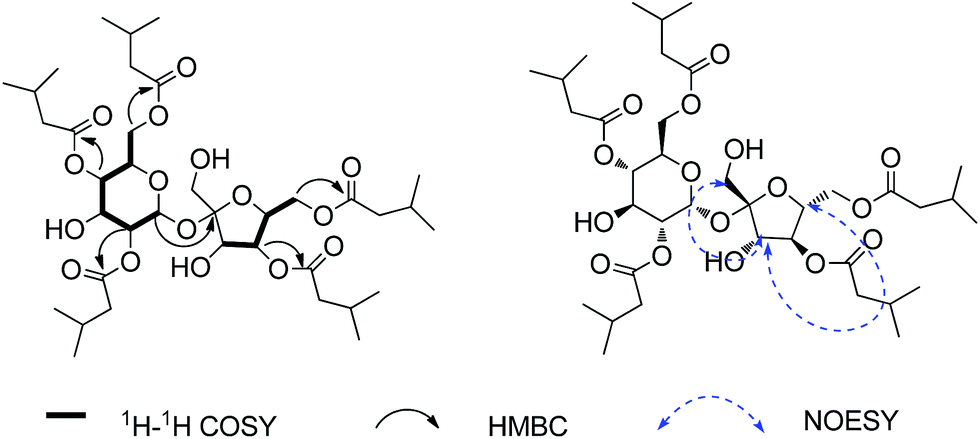 | ||
| Fig. 2 Key 1H–1H COSY, HMBC, and NOESY correlations for compound 1. | ||
Ainslosides B–D (2–4) shared the same molecular formula C37H62O16 as ainsloside A (1) by analysis of HR-ESI-MS and NMR spectroscopy, and their IR spectra suggested the presence of hydroxyl group and ester carbonyl group based on characteristic absorption bands. The 13C and DEPT NMR spectra (Table 1) of all the compounds also showed twelve carbon signals in the region 60 < δ < 105 ppm which revealed the same sucrose moiety as compound 1. Deducting the sucrose moiety, in combination with the 1H NMR spectrum (Table 1), the left 25 carbons were assigned as ten methyls, five methylenes, five methines, and five ester carbonyls, which belonged to five isovaleryls. Comprehensive analyses of 1D and 2D NMR spectra of ainslosides B–D (2–4) suggested that these compounds were isovaleryl substituted derivatives of sucrose similar to compound 1 with different substitution pattern. The linkage positions of the isovaleryls were assigned on the basis of interpretation of the HMBC spectrum. In the HMBC spectrum of compound 2, it could be observed the cross-peaks of H-4′ (δH 5.16), H-4 (δH 4.87), H-6 (δH 4.15), H-6′ (δH 4.31), H-1′ (δH 4.32) with the corresponding ester carbonyls of five isovaleryls at δC 173.0, 172.9, 172.7, 172.6, and 172.2, respectively. So, compound 2 was deduced as 1′,4,4′,6,6′-pentakis-isovaleryl-β-D-fructofuranosyl-α-D-glucopyranoside, and named ainsloside B. In the same way, compound 3 was determined as 3,3′,4,4′,6-pentakis-isovaleryl-β-D-fructofuranosyl-α-D-glucopyranoside by the long-range HMBC correlations between H-3 (δH 5.25), H-3′ (δH 4.31), H-6 (δH 4.18), H-4′ (δH 5.20), H-4 (δH 5.07) and the corresponding ester carbonyls of five isovaleryls at δC 174.1, 172.6, 172.6, 172.5, and 171.5, respectively, and named ainsloside C. Meanwhile, by the key HMBC correlations of H-3 (δH 5.40), H-6 (δH 4.36), H-6′ (δH 4.27), H-4′ (δH 5.16), H-2 (δH 4.89) with the corresponding ester carbonyls of five isovaleryls at δC 174.1, 173.5, 172.8, 172.7, and 172.0, respectively, the structure of compound 4 was deduced as 2,3,4′,6,6′-pentakis-isovaleryl-β-D-fructofuranosyl-α-D-glucopyranoside, and named ainsloside D (Fig. 3).
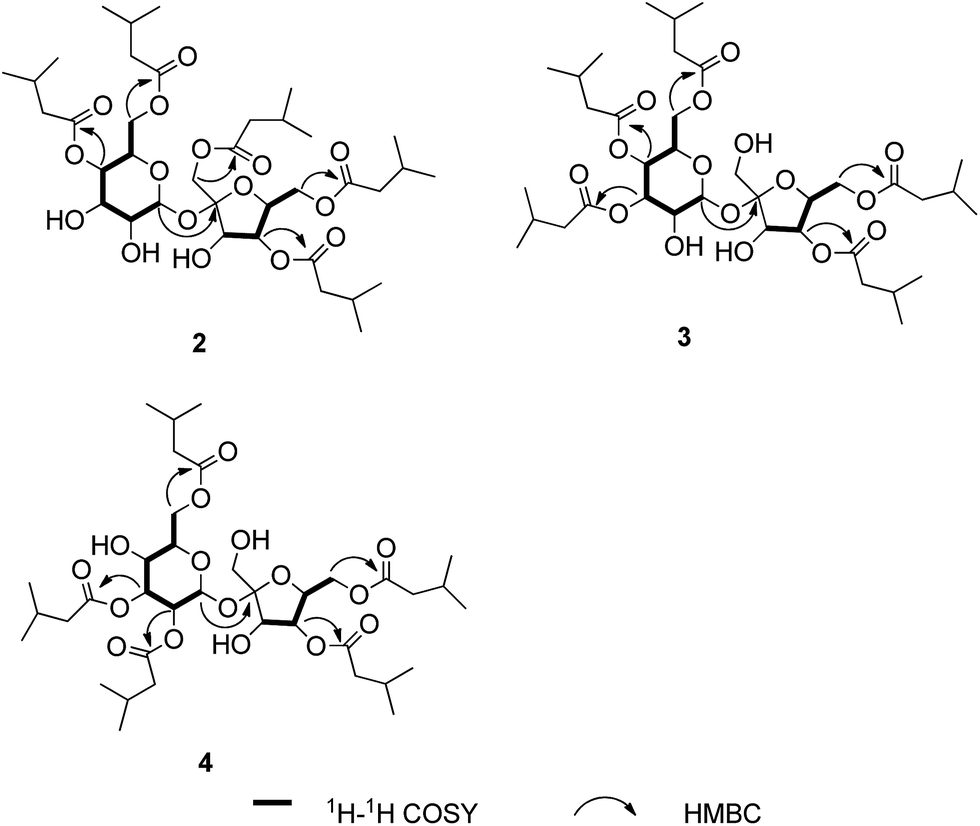 | ||
| Fig. 3 Key 1H–1H COSY, and HMBC correlations for compounds 2–4. | ||
Ainsloside E (5) was obtained as colourless oil. The molecular formula of 5 was inferred as C32H54O15 based on the HR-ESI-MS (m/z 701.3365 [M + Na]+, calcd 701.3360), implying the existence of six degrees of unsaturation. The IR spectrum of 5 showed characteristic absorption bands of hydroxyl group (3467 cm−1), and ester carbonyl group (1739 cm−1). In the 13C and DEPT NMR spectra of 5 (Table 2), 12 carbon resonances in the region 60 < δ < 105 ppm showed characteristic signals of sucrose (δC 104.9, 90.1, 78.0, 77.3, 76.8, 72.0, 70.7, 70.7, 70.6, 64.4, 63.4, 63.2), in agreement with those of previously reported for sucrose in literature,14 revealing the presence of sucrose moiety. The 1H–1H COSY correlations of H-1/H-2/H-3/H-4/H-5/H-6 and H-3′/H-4′/H-5′/H-6′ further supported the above deduction. Similar to compounds 1–4, in the high field region of the 1H NMR spectrum of 5 (Table 2), three groups of overlapping proton signals for methylene, methine, and methyl signals were observed occurring in the region δH 2.20–2.29 (8H, m), 2.06–2.12 (4H, m) and 0.93–0.97 (24H, m), respectively. Meanwhile, the 13C and DEPT NMR spectra of 5 only exhibited four ester carbonyls at δC 173.9, 173.0, 172.8, and 172.6, eight methyls, four methylenes, and four methines. The information implied that there were only four isovaleryls in the structure of 5. Therefore, it could be concluded that compound 5 was a sucrose derivative with four isovaleryl groups. The downfield shifted C-2, C-6, C-4′, and C-6′ and their upfield shifted adjacent carbons, disclosed that the four isovaleryl groups were attached to C-2, C-6, C-4′, and C-6′ positions, respectively. In the HMBC spectrum, the proton signals of the sucrose residue were observed the correlations from H-6 (δH 4.36) to the ester carbonyl at δC 173.9, from H-6′ (δH 4.31) to the ester carbonyl at δC 173.0, from H-4′ (δH 5.16) to the ester carbonyl at δC 172.8, and from H-2 (δH 4.78) to ester carbonyl at δC 172.6, revealing that four isovaleroxyls were substituted at C-2, C-4′, C-6, and C-6′ positions of sucrose residue, respectively (Fig. 4). Thus, compound 5 were deduced as 2,4′,6,6′-tetrakis-isovaleryl-β-D-fructofuranosyl-α-D-glucopyranoside, and named ainsloside E.
| No. | 5 | 6 | 7 | 8 | ||||
|---|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | δH | δC | |
| 1 | 5.53 d (3.5) | 90.1 | 5.54 d (3.5) | 89.5 | 5.44 d (3.5) | 91.7 | 5.67 d (3.5) | 90.1 |
| 2 | 4.78 dd (10.0, 3.5) | 72.0 | 4.87 dd (10.0, 4.0) | 72.4 | 3.69 dd (9.5, 3.5) | 72.2 | 4.87 dd (10.5, 4.0) | 72.6 |
| 3 | 4.02 t (9.5) | 70.6 | 4.04 dd (10.0, 4.0) | 70.0 | 3.94 t (9.5) | 71.8 | 4.05 t (10.0) | 69.8 |
| 4 | 3.40 t (9.5) | 70.7 | 4.94 t (9.5) | 70.7 | 4.84 t (10.0) | 70.1 | 4.95 t (10.0) | 70.4 |
| 5 | 4.08 m | 70.7 | 4.21 m | 68.7 | 4.20 m | 69.2 | 4.26 m | 68.9 |
| 6 | 4.36 m | 63.2 | 4.21 m | 62.1 | 4.16 m | 62.2 | 4.25 m | 61.7 |
| 4.13 dd (13.0, 6.0) | 4.14 dd (12.5, 5.0) | |||||||
| 1′ | 3.58 q (12.0) | 63.4 | 3.62 q (7.0) | 64.2 | 3.76 d (12.5) | 63.9 | 3.61 q (12.5) | 64.0 |
| 3.61 d (13.0) | ||||||||
| 2′ | — | 104.9 | — | 104.5 | — | 104.7 | — | 105.5 |
| 3′ | 4.08 m | 78.0 | 4.18 m | 78.3 | 4.25 d (7.0) | 78.7 | 4.36 d (7.0) | 76.9 |
| 4′ | 5.16 t (7.0) | 77.3 | 4.02 m | 75.8 | 5.17 t (7.0) | 78.0 | 5.26 t (6.5) | 76.7 |
| 5′ | 4.31 m | 76.8 | 3.94 m | 79.6 | 4.12 m | 78.5 | 3.97 m | 81.5 |
| 6′ | 4.31 m | 64.4 | 4.31 m | 64.1 | 4.34 dd (12.0, 5.0) | 64.7 | 3.84 dd (13.0, 3.0) | 61.4 |
| 3.67 dd (13.0, 4.0) | ||||||||
| CO |
— | 173.9 | — | 173.3 | — | 173.1 | — | 173.1 |
| 173.0 | 172.9 | 172.8 | 172.9 | |||||
| 172.8 | 172.8 | 172.7 | 172.8 | |||||
| 172.6 | 172.4 | 172.6 | 172.7 | |||||
| CH2 × 4 | 2.20–2.29 m | 43.1 | 2.23–2.29 m | 43.2 | 2.18–2.27 m | 43.2 | 2.23–2.30 m | 43.2 |
| 43.0 | 43.0 | 43.1 | 43.1 | |||||
| 42.9 | 43.0 | 43.0 | 42.9 | |||||
| 42.9 | 42.9 | 42.8 | 42.8 | |||||
| CH × 4 | 2.06–2.12 m | 25.7 | 2.06–2.14 m | 25.7 | 2.03–2.14 m | 25.7 | 2.06–2.13 m | 25.7 |
| 25.7 | 25.7 | 25.7 | 25.7 | |||||
| 25.6 | 25.6 | 25.6 | 25.7 | |||||
| 25.4 | 25.5 | 25.4 | 25.4 | |||||
| CH3 × 8 | 0.93–0.97 m | 22.3 × 8 | 0.94–0.98m | 22.3 × 8 | 0.92–0.98 m | 22.3 × 8 | 0.93–0.97 m | 22.3 × 8 |
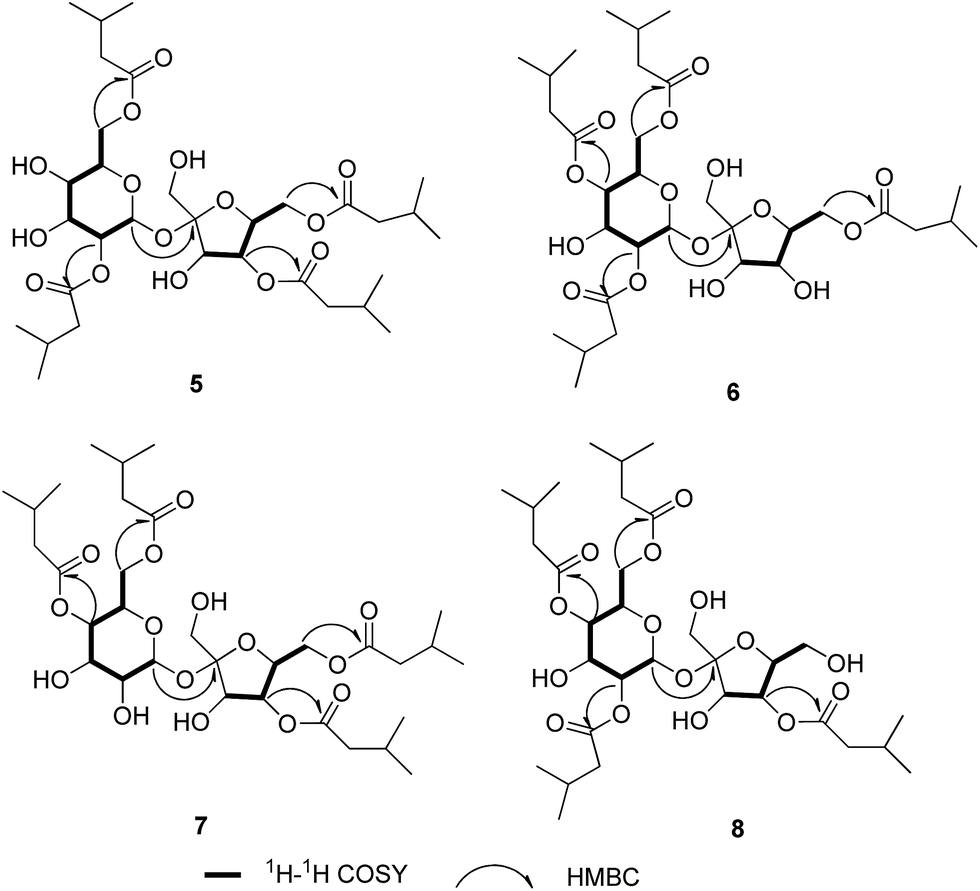 | ||
| Fig. 4 Key 1H–1H COSY, HMBC, and NOESY correlations for compound 5–8. | ||
Ainslosides F–H (6–8) shared the same molecular formula C32H54O15 as that of compound 5 due to analysis of their HR-ESI-MS spectra. The IR spectra indicated the presence of hydroxyl and ester carbonyl based on their characteristic absorption bands. The 1H and 13C NMR spectra (Table 2) of 6–8 exhibited the same functionalities as ainsloside E (5), including 12 carbon resonances for a sucrose in the region 60 < δ < 105 ppm and four ester carbonyls, eight methyls, four methylenes, and four methines in 13C NMR and DEPT spectra, together with three groups of overlapping proton signals for methylene, methine, and methyl in the 1H NMR spectrum (Table 1). The above evidences indicated that compounds 6–8 were four isovaleryls substituted sucrose ester similar to compound 5 with different substitution pattern.
The linkage positions of four isovaleryls were determined by HMBC experiment. In the HMBC spectrum of 6, the sucrose protons at δH 4.94 (H-4), 4.31 (H-6′), 4.21 (H-6), 4.87 (H-2) were correlated with the corresponding ester carbonyls of four isovaleryls at δC 173.3, 172.9, 172.8, and 172.4, respectively, implying that four isovaleroyoxyls were linked to C-4, C-6′, C-6, and C-2 positions of sucrose moiety, respectively. Therefore, the structure of 6 was deduced as 2,4,6,6′-tetrakis-isovaleryl-β-D-fructofuranosyl-α-D-glucopyranoside, and named ainsloside F. Similarly, the HMBC correlations from H-4 (δH 4.84) to the ester carbonyl at δC 173.1, from H-6 (δH 4.16) to the ester carbonyl at δC 172.8, from H-4′ (δH 5.17) to the ester carbonyl at δC 172.7, and from H-6′ (δH 4.34) to the ester carbonyl at δC 172.6, proposed the structure of 7 as 4,4′,6,6′-tetrakis-isovaleryl-β-D-fructofuranosyl-α-D-glucopyranoside, given a trivial name ainsloside G. The structure of 8 was identified as 2,4,4′,6-tetrakis-isovaleryl-β-D-fructofuranosyl-α-D-glucopyranoside on the basis of the HMBC correlations between H-4′ (δH 5.26) with the ester carbonyl at δC 173.1, between H-2 (δH 4.87) with the ester carbonyl at δC 172.9, between H-4 (δH4.95) with the ester carbonyl at δC 172.8, and between H-6 (δH 4.25) with the ester carbonyl at 172.7 (Fig. 4).
Sucrose esters are of interest in such applications as fat substitutes, bleaching boosters, and emulsifiers in the food and cosmetic industries.17,18 Meanwhile, they also possessed bioactivities such as antimicrobial, antitumor, and insecticidal activities.19 Their properties depend strongly on their compositions in terms of degree of substitution and regiochemistry.20 Previous studies showed the hydroxyl group at OH-2 is very reactive in water just as it is in organic solvents,21 while the ester at OH-6 is much more stable in synthesis.22 In our studies, the content of ainsloside A is much more than others', which suggests that the esters at OH-2, OH-4, OH-4′, OH-6 and OH-6′ might be more stable or important in plant, at least in this species.
All isolates were evaluated for in vitro cytotoxic activity against four human tumour cell lines (A549, HCT116, MDA-MB-231, and BEL7404), using MTT assay with doxorubicin as positive control (Table 3). Among these isolates, compounds 1, 2, and 5 showed strong inhibitory activity against A549 cell line with IC50 values of 9.74, 3.30, and 10.75 μM, compounds 1, 2, 4, and 5 exhibited weak inhibitory activity against HCT116 cell line with IC50 values of 10.77, 10.25, 16.56, and 16.10 μM, In addition, compounds 1, 2, 4, and 5 also showed weak inhibitory activity against BEL7404 cell line with IC50 values of 11.45, 11.23, 15.95, and 12.77 μM. Of all the isolates tested, compound 2 displayed the strongest inhibitory activity against the proliferation of A549 cell line in a dose and time dependent manner (Fig. S84, see ESI†).
| No. | IC50 (μM) | |||
|---|---|---|---|---|
| A549 | HCT116 | MDA-MB-231 | BEL7404 | |
| 1 | 9.74 ± 0.71 | 10.77 ± 0.87 | 39.10 ± 1.87 | 11.45 ± 0.57 |
| 2 | 3.30 ± 0.22 | 10.25 ± 0.53 | 19.59 ± 0.69 | 11.23 ± 0.61 |
| 3 | >100 | 68.51 ± 0.86 | >100 | 95.48 ± 4.47 |
| 4 | 19.93 ± 0.93 | 16.56 ± 0.67 | 38.23 ± 1.23 | 15.95 ± 1.31 |
| 5 | 10.75 ± 1.07 | 16.10 ± 1.12 | 25.54 ± 2.13 | 12.77 ± 0.78 |
| 6 | 60.06 ± 2.71 | 23.11 ± 1.73 | 95.19 ± 3.57 | 29.86 ± 1.75 |
| 7 | 46.19 ± 3.21 | 23.65 ± 2.01 | >100 | 53.61 ± 1.31 |
| 8 | 66.00 ± 1.17 | 54.82 ± 3.15 | >100 | 80.03 ± 2.16 |
| Doxorubicin | 0.034 ± 0.002 | 0.045 ± 0.003 | 0.11 ± 0.01 | 0.12 ± 0.01 |
Cell cycle dysregulation contributes to the aberrant cell proliferation and development of cancer.23,24 Moreover, targeting apoptosis is considered as one of the major strategies for developing anticancer drugs and most clinically used anticancer medicines can induce cell apoptosis.25 Considering the strong inhibition of compound 2 against the proliferation of A549 cells, compound 2 were further studied for its cell cycle arresting and apoptosis induction in A549 cells.
To determine if compound 2 can arrest cell-cycle progression, A549 cells were exposed to various concentrations of compound 2 for 24 hours, and the distribution of cells in the cycle was determined by flow-cytometric analysis. As shown in Fig. 5, A549 cells treated with compound 2 exhibited a dose dependent increase in the proportion of cells in the G0/G1 phase and a decrease of cells in the G2/M phase compared to the untreated control. This indicated that the growth inhibition induced by 2 in a dose-dependent manner occurs through the arrest of A549 cells in G0/G1 phase.
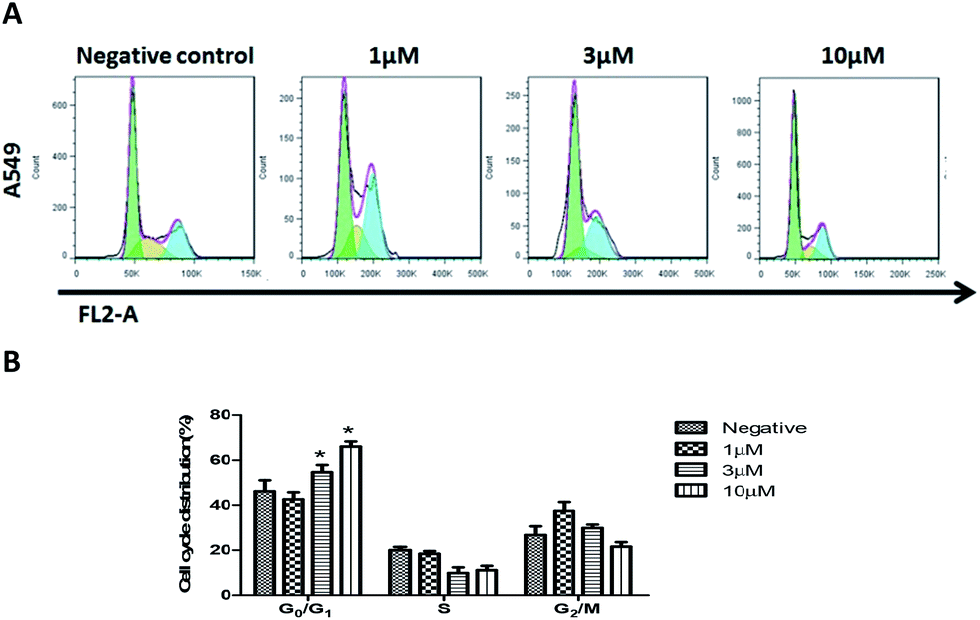 | ||
| Fig. 5 Cell cycle assay of A549 cells exposed to different concentrations of compound 2 were examined by flow cytometry (A). The effect of compound 2 on the cell cycle and the expression of cell cycle regulators in A549 cells (B). The results are represented as the mean ± SEM for three independent experiments with similar results. *P < 0.05 vs. the control. | ||
In addition, we examined compound 2-induced apoptosis in A549 cells by using annexin V-FITC/PI double staining method. The X- and Y-axis represented annexin V-FITC and PI staining, respectively. The right upper and lower right quadrant of the figure represented late and early stages of cell apoptosis, respectively. As illustrated in Fig. 6, compound 2 induced a dose-dependent increase in A549 apoptosis when treated with 10 μM of 2 for 24 h. These results confirmed that the cytotoxicity of compound 2 to A549 cells is associated with arrest of cell-cycle progression and induction of apoptosis.
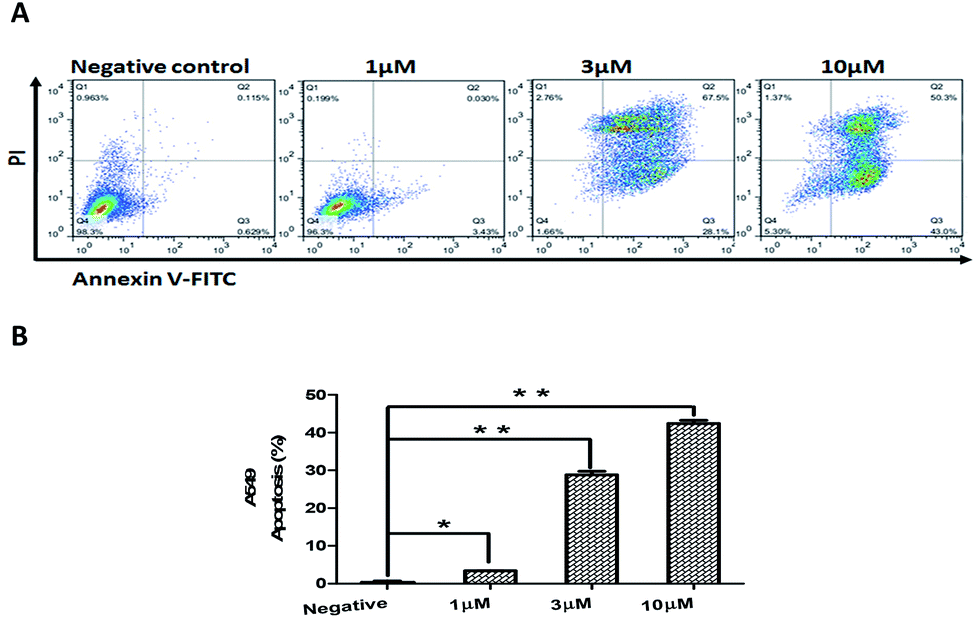 | ||
| Fig. 6 Compound 2-induced apoptosis in A549 cells. (A) Apoptosis was evaluated using an annexin V-FITC apoptosis detection kit and flow cytometry. The representative pictures are from A549 cells incubated with different concentrations. (B) Compound 2 induced apoptosis in A549 cells in a dose dependent manner. The data are expressed as the means ± SEM of three independent experiments with the similar results. *P < 0.05; **P < 0.01 vs. the control. | ||
Depolarization of mitochondrial membrane potential (MMP) is one of the characteristic event of apoptosis.26,27 To investigate whether compound 2-induced apoptosis was associated with mitochondrial dysfunction, we analyzed MMP changes of the A549 cells by staining with mitochondria sensitive Rhodamine 123, and the results showed the MMP of the A549 cells was decreased dose-dependently after treatment with different concentration of compound 2 for 24 h. The experiment indicated that compound 2-induced A549 cells apoptosis may be associated with the disruption of MMP (Fig. 7).
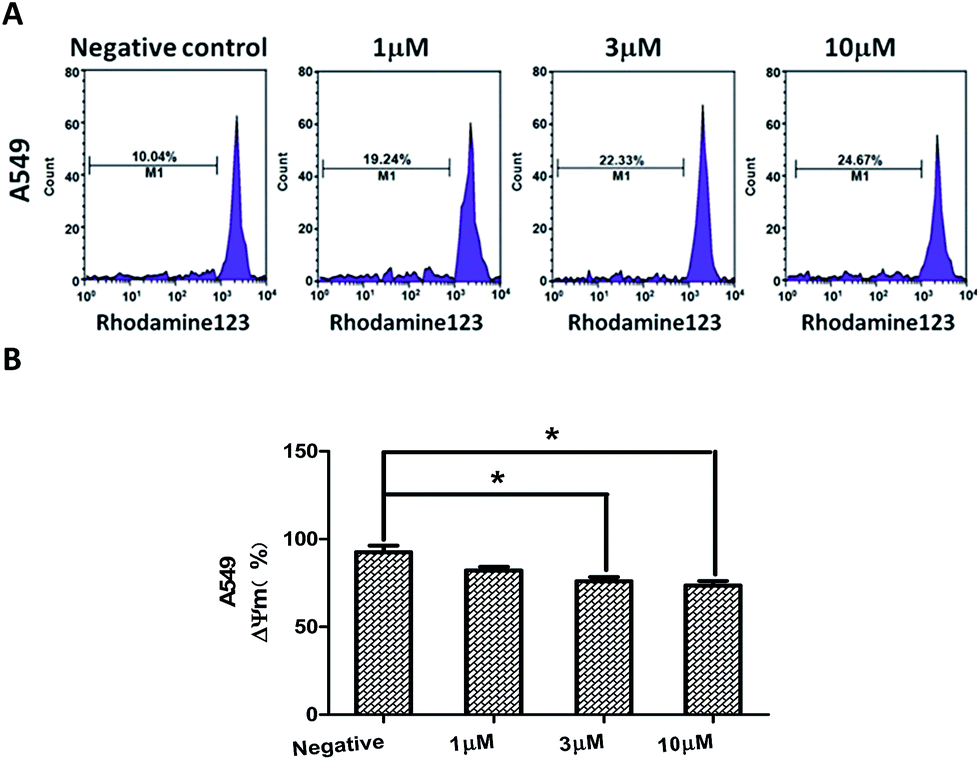 | ||
| Fig. 7 The MMP of A549 cells treated with compound 2 (A). The loss of the MMP in A549 cells following treatment of compound 2 (B). The data are expressed as the means ± SEM for three independent experiments with the similar results. *P < 0.05 vs. the control. | ||
Previous studies showed that ROS (reactive oxygen species) can induce apoptotic cell death in various types of cancer cell after treatment with anticancer drugs.28–30 So, the association between compound 2-induced apoptosis with elevated levels of ROS in A549 cells was studied. When A549 cells were treated with compound 2 for 24 h, the DCFH-DA fluorescence was shifted to a higher intensity, indicating an increase in ROS levels compared to the untreated control (Fig. 8). These data suggested that the level of intracellular ROS may be involved in compound 2-induced apoptosis in A549 cells.
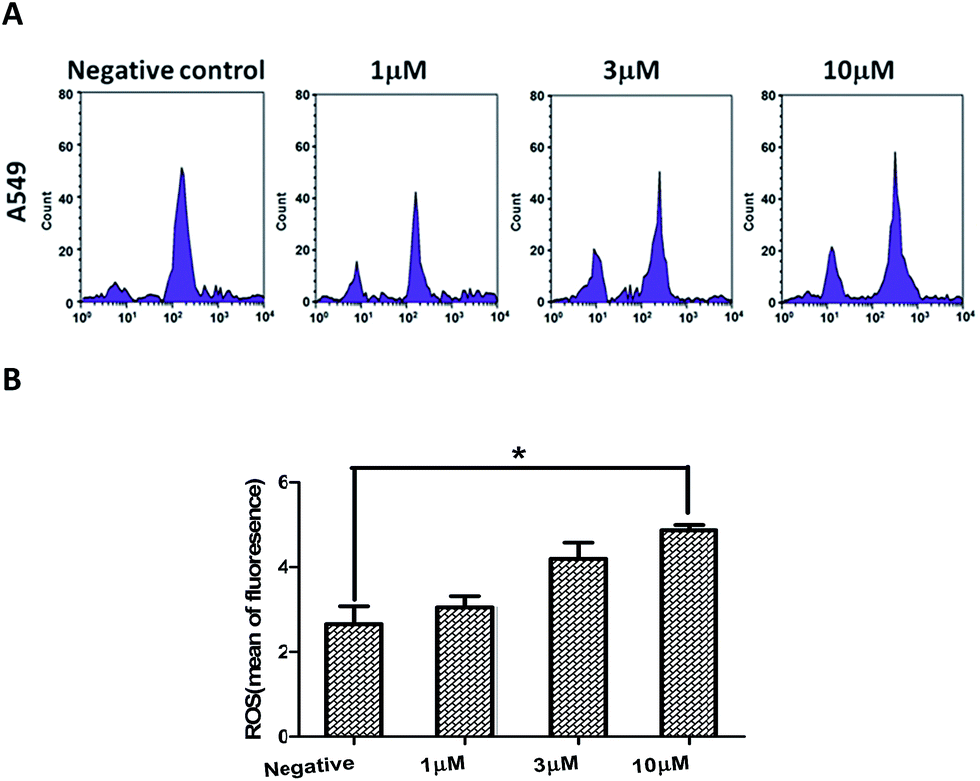 | ||
| Fig. 8 Effects of compound 2 on intracellular ROS accumulation in A549 cells (A). The results are presented as the percentage distributions for specific phases (B). The data are expressed as the means ± SEM for three independent experiments with the similar results. *P < 0.05 vs. the control. | ||
Experimental
General experimental procedures
1D and 2D NMR spectra were determined with a Bruker Avance-500 spectrometer in CDCl3 with TMS as internal standard. ESI-MS were acquired on an Agilent LC/MSD Trap XCT mass spectrometer, whereas HR-ESI-MS were measured using a Waters Q-TOF micro mass spectrometer. Optical rotations were obtained with a JASCO P-2000 polarimeter. UV spectra were obtained on a Shimadzu UV-2550 spectrometer. IR spectra were recorded on a Bruker FTIR Vector 22 spectrometer using KBr pellets. Column chromatography (CC) was performed on silica gel (100–200, 200–300 mesh, Yantai, China), Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Sweden) and YMC-Gel ODS-A (50 μm; YMC, Milford, MA). Preparative TLC (0.4–0.5 mm) was conducted with glass plates pre-coated with silica gel GF254 (Yantai, China). A semi-preparative column (Agilent ZORBAX SB-C18, 5 μm, 9.4 × 250 mm) was used for HPLC (Shimadzu LC-2010A HT), and an analytical column (YMC-Pack NH2, 5 μm, 12 nm, 4.6 × 250 mm) was used for analysis of sugar units on a HPLC (Agilent 1100) instrument with an ELSD detector (SEDEX 85).HPLC grade acetonitrile was purchased from Merck (Darmstadt, Germany). Water was purified by a Milli-Q system from Millipore (Bedford, MA, USA). All other reagents were of analytical grade and obtained from conventional commercial sources.
Human cell lines A549 (lung adenocarcinoma cells), HCT116 (colon carcinoma cells), MDA-MB-231 (breast cancer cells) and BEL7404 (hepatoma cancer cells) were obtained from the Cell Bank of Shanghai Institute of Biochemistry & Cell Biology, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences.
Plant material
The whole plants of Ainsliaea yunnanensis were collected in Baoshan city, Yunnan province, in August 2013 and authenticated by Prof. Bao-Kang Huang in the Department of Pharmacognosy, Second Military Medical University. A voucher specimen (20130813001) was deposited at the Herbarium of School of Pharmacy, Second Military Medical University, Shanghai, the People's Republic of China.Extraction and isolation
The air-dried whole plants of A. yunnanensis (15.0 kg) were refluxed with 95% ethanol and 80% ethanol for each twice. The combined extract was chromatographed by a silica gel (100–200 mesh) column eluted successively with petroleum ether (PE), CHCl3, EtOAc, and n-BuOH, respectively. The EtOAc extract (100 g) was subjected to silica gel column (100–200 mesh, 200 g) chromatography (CC) eluted with gradient CHCl3/MeOH (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1) to give 9 fractions (Fr.1–Fr.9) based on TLC analysis. The Fr.1 (16.6 g) was subjected to column chromatography on reverse-phase silica gel (ODS) eluted successively with gradient MeOH–H2O to afford 8 fractions (Fr.1-1 to Fr.1-8). Subfraction Fr.1-2 (0.4 g) was subjected to semi-preparative HPLC (MeOH/H2O, 70:30) to give ainsloside B (2, 26 mg) and ainsloside C (3, 52 mg). Fr.1-3 (3.5 g) was subjected to a silica gel CC (ϕ 2.5 × 30 cm; 200–300 mesh, 50 g) eluted with gradient PE/EtOAc (20:1 to 1:1) to give ainsloside A (1, 610 mg). From subfraction Fr.1-3-4 (0.9 g), ainsloside D (4, 28 mg) and ainsloside E (5, 21 mg) were isolated after CC over Sephadex LH-20 (ϕ 2.5 × 150 cm; MeOH) followed by semi-preparative HPLC (MeOH/H2O, 65:35). Fr.1-5 (3.3 g) was chromatographed on silica gel (ϕ 2.5 × 30 cm; 50 g, 200–300 mesh) eluted with gradient PE/EtOAc (20:1 to 1:1) followed by semi-preparative HPLC (MeOH/H2O, 65:35) to give ainsloside F (6, 20 mg). Fr.1-7 (3.0 g) was chromatographed on silica gel column (ϕ 2.5 × 30 cm; 40 g, 200–300 mesh) eluted with gradient PE/EtOAc (20:1 to 1:1) to give four subfractions (Fr.1-7a–Fr.1-7d). Fr.1-7b (0.2 g) was subjected to semi-preparative HPLC (MeOH/H2O, 60:40) to give ainsloside G (7, 23.0 mg). Fr.1-7d (0.3 g) was subjected to semi-preparative HPLC (MeOH/H2O, 60:40) to give ainsloside H (8, 25 mg).
:1 to 1:1) to give 9 fractions (Fr.1–Fr.9) based on TLC analysis. The Fr.1 (16.6 g) was subjected to column chromatography on reverse-phase silica gel (ODS) eluted successively with gradient MeOH–H2O to afford 8 fractions (Fr.1-1 to Fr.1-8). Subfraction Fr.1-2 (0.4 g) was subjected to semi-preparative HPLC (MeOH/H2O, 70:30) to give ainsloside B (2, 26 mg) and ainsloside C (3, 52 mg). Fr.1-3 (3.5 g) was subjected to a silica gel CC (ϕ 2.5 × 30 cm; 200–300 mesh, 50 g) eluted with gradient PE/EtOAc (20:1 to 1:1) to give ainsloside A (1, 610 mg). From subfraction Fr.1-3-4 (0.9 g), ainsloside D (4, 28 mg) and ainsloside E (5, 21 mg) were isolated after CC over Sephadex LH-20 (ϕ 2.5 × 150 cm; MeOH) followed by semi-preparative HPLC (MeOH/H2O, 65:35). Fr.1-5 (3.3 g) was chromatographed on silica gel (ϕ 2.5 × 30 cm; 50 g, 200–300 mesh) eluted with gradient PE/EtOAc (20:1 to 1:1) followed by semi-preparative HPLC (MeOH/H2O, 65:35) to give ainsloside F (6, 20 mg). Fr.1-7 (3.0 g) was chromatographed on silica gel column (ϕ 2.5 × 30 cm; 40 g, 200–300 mesh) eluted with gradient PE/EtOAc (20:1 to 1:1) to give four subfractions (Fr.1-7a–Fr.1-7d). Fr.1-7b (0.2 g) was subjected to semi-preparative HPLC (MeOH/H2O, 60:40) to give ainsloside G (7, 23.0 mg). Fr.1-7d (0.3 g) was subjected to semi-preparative HPLC (MeOH/H2O, 60:40) to give ainsloside H (8, 25 mg).
Sugar analysis
A solution of 1 (35 mg) in 5% KOH (5 mL) was heated at 95 °C for 3 hours. The reaction mixture was acidified to PH 4 with 1 M HCl, then extracted with CHCl3 (3 × 10 mL) and n-BuOH(3 × 10 mL) each three times. The CHCl3 layer was concentrated to afford isovaleric acid, which was elucidated by extensive analysis of 1D NMR spectra. The n-BuOH layer was hydrolyzed in 2 M HCl–MeOH (1:1, v/v, 10 mL) at 90 °C for 2 h by heating in water bath. The aqueous phase was extracted with n-BuOH (3 × 10 mL) after acid hydrolysis and concentrated to yield a colourless solid. The residue was dissolved in H2O and directly analyzed by HPLC with an ELSD detector with reference standards (MeCN–H2O, 75/25): D-glucose eluted at 16.308 min, and D-fructose at 13.116 min. Each of these eluates was individually collected, concentrated, and dissolved in H2O. The obtained sugar residues were identified as D-glucose, [α]23D +53 (c 0.113, H2O), and D-fructose, [α]23D −149 (c 0.118, H2O), through comparisons of their specific rotations with those of the corresponding authentic samples.
Isovaleric acid
1H NMR (in CDCl3, 500 MHz) δ 2.23 (2H, d, J = 7.5 Hz), 2.12 (1H, m), 0.99 (6H, d, J = 7.0 Hz); 13C NMR δ 178.8 (–COOH), 43.0 (CH2), 25.5 (CH), 23.3 (CH3), 22.3 (CH3).Cytotoxic activity assays
The viability of cells was determined by MTT assay to detect functional mitochondria in living cells (Mosmann, 1983) using doxorubicin as positive control. Human cell lines A549 (lung adenocarcinoma cells), HCT116 (colon carcinoma cells), MDA-MB-231 (breast cancer cells) and BEL7404 (hepatoma cancer cells) were incubated at 37 °C in a Dulbecco's minimum essential medium (DMEM) containing 10% FBS and penicillin–streptomycin solution. About 1 × 104 cells per wells were seeded into 96-well microtiter plates. After twenty-four hours post-seeding, cells were treated with vehicle control or various concentrations of samples for 48 h. 20 μL of MTT solution (5 mg mL−1, Sigma Aldrich, St. Louis, MO, USA) was added to each well and the tumor cells were incubated at 37 °C in a humidified atmosphere of 5% CO2 air for 4 h. Upon removal of MTT/medium, 150 μL of DMSO was added to each well and the plate was agitated at oscillator for 5 min to dissolve the MTT-formazan. The assay plate was read at a wavelength of 570 nm using a microplate reader (Bio-Tek, Winooski, Vermont, USA).Cell cycle arresting assay
A549 cells were incubated with compound 2 for 24 h, and then trypsinized, washed with PBS, and fixed with 70% of cold absolute ethanol at 4 °C overnight. After washed twice with PBS, the cells were incubated with 100 μL of RNase A for 30 min and then stained with 1 mL PI staining solution (3.8 mM sodium citrate, 50 μg mL−1 PI in PBS, Beyotime) for 10 min at room temperature in the dark. The DNA content of cells and cell cycle distribution were analyzed by flow cytometry (BD FACS Calibur).Cell apoptosis assay
The apoptotic cells were quantified by using the annexin V-FITC and PI double staining kit (DOJINDO, Japan). Briefly, A549 cells were incubated with compound 2 for 24 h. Then the cells were centrifuged, and washed with ice-cold PBS. The cultures were suspended in 400 μL of binding buffer containing 5 μL annexin V-FITC (10 μg mL−1) for 15 min in the dark, and then incubated with 10 μL of PI (20 μg mL−1) for 5 min. The cells were immediately analyzed by flow cytometry (BD FACSC Calibur).Mitochondrial membrane potential measurement
The mitochondrial membrane potential (MMP) was measured by flow cytometry using Rhodamine 123 (Rh-123) as fluorochrome. A549 cells were treated with compound 2 for 24 h, and then incubated with 5 μg mL−1 of Rh-123 for 30 min at room temperature in the dark. After centrifuged and washed twice with PBS, the cells were resuspended in 1000 μL of PBS and analyzed using flow cytometry with excitation and emission wavelength of 488 and 530 nm, respectively.Intracellular ROS production measurement
ROS levels were detected using a flow cytometer and a microplate spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). After treatment, A549 cells were harvested and washed with PBS and suspended in DMEM containing 10 μM 5(6)-carboxy-2′, 7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA; Invitrogen) at 37 °C for 20 min. The cells were washed again, and flow cytometry analysis was performed.Statistical analysis
Data were analyzed using GraphPad Prism software (GraphPad software Inc., San Diego, CA, USA). The comparison between two groups was analyzed by unpaired Student t-test, and multiple comparisons were compared by one-way ANOVA analysis of variance followed by Tukey post hoc test. Statistical significance was determined as P < 0.05.Conclusions
Isovaleryl sucrose esters are a kind of rare second metabolites, which have been sporadically isolated from some plants.31–33 In current study, eight isovaleryl sucrose esters were obtained from the whole plants of A. yunnanensis. It is the first report of poly isovaleryl sucrose derivatives from Ainsliaea species. Interestingly, ainsloside B (2), a poly isovaleryl sucrose derivative with 1′,4,4′,6,6′-pentakis-isovaleryl substitution, exhibited potent inhibition against the proliferation of A549 with IC50 value of 3.3 μM, while other compounds showed weak inhibition or being inactive. It could be speculated that the quantity and substitution pattern of isovaleryl may affect cytotoxicities of these compounds. Meanwhile, ainsloside B (2) can arrest cell cycle of A549 at G0/G1 phase and induce A549 cell apoptosis in dose-dependent manner. Further investigation indicated that the apoptosis-inducing effect of compound 2 may be involved in reduction of mitochondrial membrane potential (MMP) and increase of reactive oxygen species (ROS) levels in A549 cells. Therefore, polyisovaleryl sucrose esters could be a type of important bioactive constituents of Ainsliaea plants.Acknowledgements
The work was supported by Professor of Chang Jiang Scholars Program, NSFC (81573318, 81230090, 81520108030, 81373301, 1302658), Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products (10DZ2251300), the Scientific Foundation of Shanghai China (12401900801, 13401900101), National Major Project of China (2011ZX09307-002-03) and the National Key Technology R&D Program of China (2012BAI29B06).Notes and references
- R. Wang, Y. X. Tang and X. Y. Shang, J. Chin. Med. Mater., 2012, 35, 1171–1175 CAS.
- R. Y. Qiu, J. Xu, W. Xu, J. Xiong, Y. H. Liu and B. Yuan, J. Chin. Pharm. Sci., 2009, 18, 13–14 Search PubMed.
- S. Z. Choi, M. C. Yang, S. U. Choi and K. R. Lee, Arch. Pharmacal Res., 2006, 29, 203–208 CrossRef CAS.
- M. M. Fan, S. L. Yang, Y. S. Wang, Y. Huang, H. Jian, Y. Rao, Q. P. Wu and X. Qian, IPC A61K36/28(2006.01)I, Patent 101293003, 2008.
- H. I. Moon, O. P. Zee and M. S. Shin, Han'guk Nonghwa Hakhoechi, 1999, 42, 162–165 CAS.
- Z. J. Wu, X. K. Xu, H. W. Zeng, Y. H. Shen, J. M. Tian, J. Su, H. L. Li, L. Shan, R. H. Liu and W. D. Zhang, Planta Med., 2011, 77, 5517–5520 Search PubMed.
- H. Wang, T. Wu, M. Yan, G. Liu, P. Li, X. Q. Zhang, W. C. Ye and L. Y. Zhang, Chem. Pharm. Bull., 2009, 57, 597–599 CrossRef CAS PubMed.
- X. Chen, J. S. Miao, H. Wang, F. Zhao, J. Hu, P. Gao, Y. Wang, L. Y. Zhang and M. Yan, J. Ethnopharmacol., 2015, 170, 72–80 CrossRef CAS PubMed.
- Y. Wang, Y. H. Shen, H. Z. Jin, J. J. Fu, X. J. Hu, J. J. Qin, J. H. Liu, M. Chen, S. K. Yan and W. D. Zhang, Org. Lett., 2008, 10, 5517–5520 CrossRef CAS PubMed.
- Z. J. Wu, X. K. Xu, Y. H. Shen, J. Su, J. M. Tian, S. Liang, H. L. Li, R. H. Liu and W. D. Zhang, Org. Lett., 2008, 10, 2397–2400 CrossRef CAS PubMed.
- R. Wang, Z. Sun, E. L. Wang, Z. Z. Yuan, J. J. Li and X. Y. Shang, J. Chin. Med. Mater., 2013, 36, 61–64 CAS.
- J. J. Li, E. L. Wang, Z. Z. Yuan, C. Y. Wu, L. H. Yang and X. Y. Shang, China J. Chin. Mater. Med., 2013, 38, 3918–3922 CAS.
- J. Yang, H. Y. Wu, Q. H. Li, P. Yi, Y. Min, W. Liu, X. D. Yang and L. Li, Adv. Mater. Res., 2012, 554–556, 1845–1848 CrossRef CAS.
- W. Li, Y. N. Sun, X. T. Yan, S. Y. Yang, S. B. Song, Y. M. Lee and Y. H. Kim, J. Agric. Food Chem., 2013, 61, 7081–7088 CrossRef CAS PubMed.
- J. Duus, C. H. Gotfredsen and K. Bock, Chem. Rev., 2000, 100, 4589–4614 CrossRef CAS PubMed.
- K. Bock and C. Pedersen, Adv. Carbohydr. Chem. Biochem., 1983, 41, 27–66 CrossRef CAS.
- C. C. Akoh, Food Sci. Technol., 2002, 117, 695–727 CAS.
- I. Janicot, A. Bouchu, G. Descotes and E. Wong, Tenside, Surfactants, Deterg., 1996, 33, 290–296 CAS.
- M. Ferrera, J. Soliverib, F. J. Ploua, N. Lopez-Cortesa, D. Reyes-Duartea, M. Christensenc, J. L. Copa-Patinob and A. Ballesterosa, Enzyme Microb. Technol., 2005, 36, 391–398 CrossRef.
- Y. Queneau, S. Jarosz, B. Lewandowski and J. Fitremann, Adv. Carbohydr. Chem. Biochem., 2007, 61, 218–292 CrossRef.
- S. Thevenet, A. Wernicke, S. Belniak, G. Descotes, A. Bouchu and Y. Queneau, Carbohydr. Res., 1999, 318, 52–66 CrossRef CAS.
- V. Molinier, K. Wisniewski, A. Bouchu, J. Fitremann and Y. Queneau, J. Carbohydr. Chem., 2003, 22, 657–669 CrossRef CAS.
- D. F. Amanatullah, A. T. Reutens, B. T. Zafonte, M. Fu, S. Mani and R. G. Pestell, Front. Biosci., 2000, 5, 372–390 CrossRef.
- Z. A. Stewart, M. D. Westfall and J. A. Pietenpol, Trends Pharmacol. Sci., 2003, 24, 139–145 CrossRef CAS PubMed.
- I. M. Ghobrial, T. E. Witzig and A. A. Adjei, Ca-Cancer J. Clin., 2005, 55, 178–194 CrossRef PubMed.
- M. J. Wu, H. Zhang, J. H. Hu, Z. Y. Weng, C. Y. Li, H. Li, Y. Zhao, X. F. Mei, F. Ren and L. H. Li, PLoS One, 2013, 8, 1–8 Search PubMed.
- G. W. Wang, C. Lv, Z. R. Shi, R. T. Zeng, X. Y. Dong, W. D. Zhang, R. H. Liu, L. Shan and Y. H. Shen, PLoS One, 2014, 9, 1–19 Search PubMed.
- K. Gong and W. Li, Free Radical Biol. Med., 2011, 51, 2259–2271 CrossRef CAS PubMed.
- W. P. Tsang, S. P. Chau, S. K. Kong, K. P. Fung and T. T. Kwok, Life Sci., 2003, 73, 2047–2058 CrossRef CAS PubMed.
- Y. Tang, R. Chen, Y. Huang, G. Li, Y. Huang, J. Chen, L. Duan, B. T. Zhu, J. B. Thrasher, X. Zhang and B. Li, Mol. Cancer Ther., 2014, 13, 1526–1536 CrossRef CAS PubMed.
- A. T. Tchinda, P. Tane, J. F. Ayafor and J. D. Conolly, Phytochemistry, 2003, 63, 841–846 CrossRef CAS PubMed.
- A. P. Castorena, M. Luna, M. Martinez and E. Maldonado, Carbohydr. Res., 2012, 352, 211–214 CrossRef PubMed.
- N. J. Toyang, M. A. Krause, R. M. Fairhurst, P. Tane, J. Bryant and R. Verpoorte, J. Ethnopharmacol., 2013, 147, 618–621 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01986f |
| This journal is © The Royal Society of Chemistry 2017 |