
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification and functional application of a new malonyltransferase NbMaT1 towards diverse aromatic glycosides from Nicotiana benthamiana†
Yuyu Liu,
Xiaohui Wang,
Ting Mo,
Yaru Yan,
Yuelin Song,
Yunfang Zhao,
Jun Li,
Shepo Shi,
Xiao Liu * and
Pengfei Tu*
* and
Pengfei Tu*
Modern Research Center for Traditional Chinese Medicine, School of Chinese Materia Medica, Beijing University of Chinese Medicine, Beijing 100029, People's Republic of China. E-mail: fcliuxiao@163.com; pengfeitu@163.com; Fax: +86-10-82802750; Fax: +86-10-64286350; Tel: +86-10-82802750 Tel: +86-10-64286350
First published on 12th April 2017
Abstract
Recently, owing to the important pharmaceutical properties of malonylated glycosides, their chemoenzymatic synthesis using malonyltransferase has received significant attention. In the present study, a new malonyltransferase, NbMaT1, was identified from Nicotiana benthamiana. Extensive enzymatic assays revealed its significant substrate tolerance based on HPLC-UV and HR-MS analyses. Moreover, 16 of the tested glycosides including flavone glycosides, flavonol glycosides, dihydroflavone glycosides, isoflavone glycosides, coumarin glycosides, and phenylethyl chromone glycosides with various sugar moieties (such as glucose, xylose, and galactose) substituted at different positions of their skeleton could be accepted by NbMaT1 to conduct the corresponding malonylation. Among these, enzymatic malonylation of phenylethyl chromone glycosides as well as xylosides and galactosides has rarely been reported earlier. Furthermore, one-pot synthesis using the known malonyl-CoA synthetase MatB and NbMaT1 as well as an unnatural fusion protein MatB–NbMaT1 was designed, which allowed malonic acid to be directly used in the malonylation reaction without the addition of expensive malonyl-CoA. Moreover, a remarkably improved conversion rate was observed for all the tested substrates, with both commercial and industrial application values. The malonylated product of the bioactive flavonoid diglycoside icariin was prepared and NMR spectroscopy revealed that the malonyl group was specifically transferred onto the 6-OH group of the glucose moiety. NbMaT1 was expected to be a universal and effective tool for chemoenzymatic synthesis of diverse bioactive-malonylated glycoside derivatives for drug discovery.
1. Introduction
The glycosides of various classes are widely distributed in nature and they possess diverse pharmacological activities.1,2 As one of the major secondary metabolites in plants, these compounds are often present acylated with aliphatic or aromatic acids at specific hydroxyl groups of their sugar moieties.3 For example, in acylated flavonoid glycosides, acyl-free precursor flavonoid glycosides have been reported to exhibit various biological activities including anti-oxidant, anti-carcinogenic, anti-microbial, and cardio-protective.4–6 However, their relatively weak solubility and stability in lipophilic and/or hydrophobic media have been a critical drawback, which restricts their further applications.7,8 Acylation modification has become an optional way to solve these limitations, and many investigations have reported that the acylated products obviously showed improved stabilities, increased lipophilicity, increased solubilities, altered physical properties as well as enhanced intra- and intermolecular hydrophobic interactions, which were likely a result of the presence of the bulky acyl moiety.9–12 These remarkable characteristics have gradually made the acylated glycosides a hot research topic and they are used as desirable candidates in pharmaceutical, food, and cosmetic preparations.13–17 Thus, exploring the rich pool of bioactive acylated glycosides for the design of new candidate drugs is a compelling target for pharmacological applications.However, on the one hand, many potentially useful acylated glycosides occur only in trace amounts in natural sources. On the other hand, the one-step selective chemical acylation of glycosides is hindered because of the rigorous conditions, uncontrolled process, poor specificity and unpredictability of the products since it is hard to discriminate the various hydroxyl groups among the diverse sugar units (either the multiple sugar units or a single carbohydrate moiety).3,4
Acyltransferases, which transfer acyl molecules onto sugar moieties, are involved in the biosynthesis of diverse bioactive natural acylated glycosides. Considering its high catalytic efficiency, mild reaction condition, controllable process, and environmentally friendly advantages, the manipulation of acylation reactions catalysed by acyltransferases may represent an advantageous shortcut for the synthesis of acylated glycosides.8–10,13
Recently, biochemical investigations of secondary metabolic pathways have led to the discovery of a large family of acyltransferases named BAHD that catalyse the acyl moiety (acetyl-, malonyl-, tigloyl-, benzoyl-, and hydroxycinnamoyl coenzyme A (CoA) thioesters) transfer reactions to form a diverse group of plant metabolites including small volatile esters, modified anthocyanins, as well as constitutive defense compounds and phytoalexins.18,19 Structural research has revealed that two conserved motifs belonging to this family include an HXXXDG domain located near the center portion and a DFGWG motif located near the carboxyl terminus, which play important roles in the catalytic processes.20,21
In this study, a new BAHD acyltransferase gene, NbMaT1, was cloned from Nicotiana benthamiana. Sequence alignment indicated that NbMaT1 may be an ortholog of the previously reported malonyltransferase NtMaT1 obtained from N. tabacum.22–24 Extensive enzymatic assays have revealed the significant substrate promiscuities of NbMaT1 in which 16 of the tested glycosides belonging to various structural types including flavonoid glycosides, coumarin glycosides, and phenylethyl chromone glycosides could be accepted as an acyl acceptor, with malonyl-CoA as the acyl donor to conduct the corresponding malonylation reaction. Unlike NtMaT1 that mainly conducts malonylation of flavonoid 7-O-glucosides and flavonoid 3-O-glucosides, the sugar moieties malonylated by NbMaT1 were substituted at different positions of the flavonoid. However, in addition to the usual glucosyl moiety, flavonoid glycosides substituted with the xylosyl and/or galactosyl group were also accepted as positive substrates by NbMaT1. The enzymatic malonylation of phenylethyl chromone glycosides as well as xylosides and galactosides have rarely been reported earlier. Moreover, considering the high price and instability of malonyl-CoA, an efficient malonyl-CoA production system catalysed by a known malonyl-CoA synthetase, MatB, from Arabidopsis thaliana, was constructed.25–30 Furthermore, a one-pot synthesis system using MatB and NbMaT1 as well as an unnatural fusion protein MatB–NbMaT1 was constructed, which allowed malonic acid to be directly used in the subsequent malonylation reaction without the addition of malonyl-CoA and remarkably improved the conversion rate for all the tested substrates. The bioactive flavonoid diglycoside icariin (9) was subjected to this one-pot reaction system for the preparative scale enzymatic synthesis, and a new compound 9a with the malonyl group substituted at the 6-OH group of the glucose moiety was obtained. The unprecedentedly remarkable promiscuity of NbMaT1 towards diverse glycosides acceptors as well as the effective one-pot synthesis system makes NbMaT1 a versatile tool for the chemo-enzymatic synthesis of diverse malonylated glycosides derivatives and also a potentially general functional part for secondary metabolism pathway, design, and construction of bioactive natural and unnatural malonylated glycosides in synthetic biology for lead compounds discovery in drug R&D.
2. Materials and methods
2.1. Chemicals
The reference standards malonyl-CoA and CoA were purchased from Sigma-Aldrich (St. Louis, USA), ATP was purchased from Biodee Biotechnology Co., Ltd. (Beijing, China), malonate was purchased from Aladdin Industrial Co., Ltd. (Shanghai, China), and various types of tested substrates for the enzymatic reaction were purchased from Chengdu Push Biotechnology Co., Ltd. (Chengdu, China) and Chengdu Biopurify Phytochemicals Ltd. (Chengdu, China).2.2. Gene cloning and the heterogeneous expression of NbMaT1, MatB, and fusion gene MatB–NbMaT1
Total RNA samples were prepared from 100 mg of young fresh leaves of N. benthamiana and A. thaliana using a Plant RNA kit (OMEQA Bio-Tek, Inc. GA, USA), and their cDNAs were obtained using M-MLV reverse transcriptase (Promega, USA). All PCR were processed using high fidelity KOD-Plus-Neo DNA polymerase (TOYOBO, Osaka, Japan). The NbMaT1 gene and MatB gene were amplified using cDNA of N. benthamiana and A. thaliana as the template, respectively, using RT-PCR with primer pairs as follows: NbMaT1![[B with combining low line]](https://www.rsc.org/images/entities/i_char_0042_0332.gif)
![[a with combining low line]](https://www.rsc.org/images/entities/i_char_0061_0332.gif)
![[m with combining low line]](https://www.rsc.org/images/entities/i_char_006d_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[I with combining low line]](https://www.rsc.org/images/entities/char_0049_0332.gif) -F (GGA TCC ATG GCA TCT GTG ATT GAG CAA TGT C), NbMaT1–
-F (GGA TCC ATG GCA TCT GTG ATT GAG CAA TGT C), NbMaT1–![[S with combining low line]](https://www.rsc.org/images/entities/i_char_0053_0332.gif)
![[l with combining low line]](https://www.rsc.org/images/entities/i_char_006c_0332.gif) -R (GTC GAC CTA CAG AAA GCT AAT CCC GTG GGT G); MatB–-F (5′-CGC GGA TCC ATG GAA GTG TTT AAA GCA GC-3′), MatB–-R (5′-GTC GAC TTA TTC TTG ATT TTC CAG AGA TTT C-3′). To construct the MatB–NbMaT1 fusion gene, the stop codon of MatB was removed and a three amino acid linker (Gly-Ser-Gly) was introduced between the open reading frames of MatB and NbMaT1 through two rounds of PCR with primers MatB-linker-NbMaT1-F (5′-CTC TGG AAA ATC AAG AAG GCA GCG GAA TGG CAT CTG TGA TTG AGC-3′) and MatB-linker-NbMaT1-R (5′-GCT CAA TCA CAG ATG CCA TTC CGC TGC CTT CTT GAT TTT CCA GAG-3′). Underlining indicates the restriction sites. The amplified DNA fragments were digested with the corresponding restriction enzymes (BamHI and SalI, Takara, Dalian, China) and inserted into the counterpart sites in pET-28a vector (Novagen). The fragments of the vector and genes were recovered using a Nucleic Acid Purification Kit (Axygen, Union City, USA) and then ligated with T4 DNA ligase (BioLabs, Beijing, China) at 25 °C for 30 minutes. The recombinant plasmid was transformed into Escherichia coli Transetta DE3 (TransGen, Beijing, China) before analysis. The sequence similarities were calculated using the NCBI BLAST tool (http://www.blast.ncbi.nlm.nih.gov/). Gene-specific primers were designed using a primer design software. Sequence alignment and analysis were performed via a DNAMAN software.
-R (GTC GAC CTA CAG AAA GCT AAT CCC GTG GGT G); MatB–-F (5′-CGC GGA TCC ATG GAA GTG TTT AAA GCA GC-3′), MatB–-R (5′-GTC GAC TTA TTC TTG ATT TTC CAG AGA TTT C-3′). To construct the MatB–NbMaT1 fusion gene, the stop codon of MatB was removed and a three amino acid linker (Gly-Ser-Gly) was introduced between the open reading frames of MatB and NbMaT1 through two rounds of PCR with primers MatB-linker-NbMaT1-F (5′-CTC TGG AAA ATC AAG AAG GCA GCG GAA TGG CAT CTG TGA TTG AGC-3′) and MatB-linker-NbMaT1-R (5′-GCT CAA TCA CAG ATG CCA TTC CGC TGC CTT CTT GAT TTT CCA GAG-3′). Underlining indicates the restriction sites. The amplified DNA fragments were digested with the corresponding restriction enzymes (BamHI and SalI, Takara, Dalian, China) and inserted into the counterpart sites in pET-28a vector (Novagen). The fragments of the vector and genes were recovered using a Nucleic Acid Purification Kit (Axygen, Union City, USA) and then ligated with T4 DNA ligase (BioLabs, Beijing, China) at 25 °C for 30 minutes. The recombinant plasmid was transformed into Escherichia coli Transetta DE3 (TransGen, Beijing, China) before analysis. The sequence similarities were calculated using the NCBI BLAST tool (http://www.blast.ncbi.nlm.nih.gov/). Gene-specific primers were designed using a primer design software. Sequence alignment and analysis were performed via a DNAMAN software.
2.3. The expression and purification of recombinant NbMaT1, MatB, and fusion MatB–NbMaT1
Cells harboring the corresponding plasmid were cultured to the optical density of 0.6 at 600 nm in an LB medium containing 40 μg mL−1 kanamycin and 37 μg mL−1 chloramphenicol at 37 °C, 200 rpm and were induced by 0.5 mM isopropyl β-D-thiogalactopyranoside (IPTG) at 23 °C for 16 h. The cells were obtained by centrifugation at 7000 × g for 20 min and resuspended in lysis buffer (20 mM imidazole, 500 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol (DTT), and 3% glycerol in 20 mM phosphate buffer, pH 7.4) before being lysed in an ice bath using an Ultrasonic Processor (VERNON HILLS, Illinois, USA). Supernatants containing soluble proteins were filtered through 0.45 μm Millex Syringe-driven Filter Units (Merck KGaA, Darmstadt, Germany) after centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 40 min at 4 °C. The obtained supernatants were loaded onto a column comprising Ni-NTA His-Bind Resin (GE Healthcare, Uppsala, Sweden), which was equilibrated in a binding buffer (20 mM phosphate buffer, 20 mM imidazole, 500 mM NaCl, 3% glycerol, pH 7.4). Subsequently, the column was washed with the binding buffer to remove the unbound contaminant proteins. The His-tagged protein was eluted with an elution buffer (20 mM phosphate buffer, linear gradient from 20 to 500 mM imidazole, 500 mM NaCl, 3% glycerol, pH 7.4). Further purification of the targeted proteins was performed with a buffer (50 mM Tris–HCl, 100 mM NaCl, 1 mM DTT, 3% glycerol, pH 7.4) using a PD-10 column (GE Healthcare, Uppsala, Sweden). The protein concentration was determined by the Bradford method using BSA as the standard.
000 × g for 40 min at 4 °C. The obtained supernatants were loaded onto a column comprising Ni-NTA His-Bind Resin (GE Healthcare, Uppsala, Sweden), which was equilibrated in a binding buffer (20 mM phosphate buffer, 20 mM imidazole, 500 mM NaCl, 3% glycerol, pH 7.4). Subsequently, the column was washed with the binding buffer to remove the unbound contaminant proteins. The His-tagged protein was eluted with an elution buffer (20 mM phosphate buffer, linear gradient from 20 to 500 mM imidazole, 500 mM NaCl, 3% glycerol, pH 7.4). Further purification of the targeted proteins was performed with a buffer (50 mM Tris–HCl, 100 mM NaCl, 1 mM DTT, 3% glycerol, pH 7.4) using a PD-10 column (GE Healthcare, Uppsala, Sweden). The protein concentration was determined by the Bradford method using BSA as the standard.
2.4. The enzymatic assays of NbMaT1
The reaction mixture routinely consisted of 0.5 mM malonyl-CoA, 0.4 mM malonyl acceptor, and 50 μg of purified NbMaT1 protein in 150 μL of potassium phosphate reaction buffer (100 mM, pH 7.5). After incubation at 30 °C for 12 hours, the reaction was terminated by adding a double volume of methanol. After being vortexed and centrifuged at 15000 rpm for 1 hour, the supernatant was analysed using an Agilent 1260 Series HPLC system with a SHESHIDO C18 Column (4.6 mm I.D. × 250 mm, 5 μm). The mobile phase comprised a solvent A (0.1% formic acid) and solvent B (acetonitrile) with a gradient elution procedure as follows: A:B (v/v): 0 min (98:2), ∼15 min (85:15), ∼25 min (60:40), ∼30 min (40:60), and ∼35 min (0:100) at 1.0 mL min−1. HR-ESI-MS data were obtained using an LCMS-IT-TOF system equipped with a Prominence UFLC system and an ESI interface (Shimadzu, Kyoto, Japan) with ultra-high purity He as the collision gas and N2 as the nebulizing gas. The optimized ESI source parameters were as follows: sheath gas flow rate, 1.5 mL min−1; auxiliary gas flow rate, 1.5 mL min−1; spray voltage, 4.5 kV; and capillary temperature, 200 °C. The spectra were obtained in the 100–1000 m/z range for a full scan MS analysis.
2.5. The enzymatic synthesis and analysis of malonyl-CoA
Malonyl-CoA was enzymatically synthesized by MatB using malonic acid as the substrate. The reaction mixture contained 3 mM CoA, 6 mM malonic acid, 2 mM DTT, 6 mM ATP, and 10 mM MgCl2 in 100 mM potassium phosphate (pH 7.5). The reaction was initiated by adding 0.5 μg μL−1 of purified MatB protein. After incubation at 30 °C for 12 h, the reaction was terminated by the addition of 4% ammonium acetate. The reaction mixture was filtered and the supernatant was then analysed using the TOSOH TSKgel ODS Column (4.6 mm I.D. × 250 mm, 5 μm) HPLC system as described above. The mobile phase comprised solvent A (0.1% trifluoroacetic acid) and solvent B (acetonitrile) with the gradient elution procedure as follows: A:B (v/v): 0 min (100:0), ∼5 min (100:0), ∼35 min (40:60), ∼38 min (40:60), and ∼43 min (100:0). The flow rate was 1.0 mL min−1. The peaks were measured at 254 nm.
2.6. Enzymatic assays using the one-pot reaction system and the fusion protein MatB–NbMaT1
The standardized enzymatic assays of the one-pot reaction system comprised 0.4 mM malonyl acceptors, 6 mM ATP, 3 mM CoA, 6 mM malonic acid, 2 mM DTT, and 10 mM MgCl2 in 150 μL of potassium phosphate buffer (100 mM, pH 7.5) with the purified MatB (75 μg) and NbMaT1 (50 μg), which were both added respectively. The reaction system using the fusion protein was with the same composition except for the catalyst used was 125 μg of the purified fusion enzyme MatB–NbMaT1. After incubation at 30 °C for 12 hours, the reactions were terminated by adding 300 μL of methanol. Subsequent post-processing and analysis methods of the reaction samples were same as described above.2.7. Preparation of the enzymatic product using NbMaT1
The preparative scale reaction comprised 0.012 mmol of the malonyl acceptor icariin (9), 0.18 mmol of ATP, 0.09 mmol of CoA, 2 mM DTT, 10 mM MgCl2, 0.18 mmol of malonic acid, 15 mg of MatB protein, and 10 mg of NbMaT1 protein in 30 mL of potassium phosphate reaction buffer (100 mM, pH 7.5). After incubation at 30 °C for 12 hours, the reaction was terminated by adding 60 mL of methanol. The obtained supernatant was filtered and evaporated to dryness under reduced pressure. The resulting residue was re-dissolved in 5 mL of methanol before being separated by reverse phase semi-preparative HPLC, which was performed using a YMC-Pack ODS-A HPLC column (10 mm I.D. × 250 mm, 5 μm) at the flow rate of 3 mL min−1. The mobile phase was water (A) and acetonitrile (B). The gradient elution A:B (v/v) was as follows: 0 min (85:15), ∼5 min (60:40), ∼15 min (20:80), and ∼20 min (0:100). The obtained products were confirmed by 1H and 13C nuclear magnetic resonance spectroscopy, HSQC (heteronuclear single-quantum correlation) and HMBC (heteronuclear multiple bond correlation) analyses.
2.8. 1H and 13C NMR data for the new malonylated product of icariin (9)
The NMR spectra were obtained using a Varian INOVA-500 spectrometer (Varian Corporation, Palo Alto, CA, USA) operating at 500 MHz for 1H and 125 MHz for 13C.![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3), 25.88 (C-14), 22.73 (C-11), 18.32 (C-15), 17.68 (C-6′′).
H3), 25.88 (C-14), 22.73 (C-11), 18.32 (C-15), 17.68 (C-6′′).3. Results and discussion
3.1. Gene cloning, heterogeneous expression, and bioinformatic analyses of NbMaT1
The coding region of NbMaT1 (GenBank accession KY563646) was amplified by RT-PCR from the cDNA of N. benthamiana. For gene expression, the 1362 bp coding sequence of NbMaT1 was cloned into the expression vector pET-28a. Soluble proteins were obtained from the transformants of E. coli cells after overnight induction with 0.5 mm IPTG at 23 °C and were subjected to purification with Ni-NTA agarose. His6–NbMaT1 was purified to apparent homogeneity, as judged by SDS-PAGE. The observed molecular mass reasonably corresponded to the calculated mass of 50.91 kDa for His6–NbMaT1 (Fig. S23†). A protein yield of 3.02 mg L−1 of the pure recombinant NbMaT1 was obtained.Bioinformatic analyses are reported to be useful tools to predict the possible substrates for enzymes that have not been biochemically characterized.31,32 The deduced amino acid sequence of NbMaT1 was subsequently subjected to the multiple alignment analyses with other reported BAHD acyltransferases. As shown in Fig. S1,† in addition to the conserved motif HXXXD and DFGWG, which were common to the BAHD acyltransferase family, NbMaT1 also contained the YFGNC(A) sequence, which was a specific conserved motif of the subfamily termed as anthocyanin malonyltransferase.22–24,33–35 Moreover, sequence alignment showed the highest homology (86% identity) of NbMaT1 with NtMaT1, which was identified as the malonyltransferase of flavonoid and naphthol glucosides from N. tabacum. Further, phylogenetic tree analysis also indicated that NbMaT1 may be an orthologous of NtMaT1 (Fig. 1). Thus, all the bioinformatic results hinted that NbMaT1 might be type of malonyltransferase.
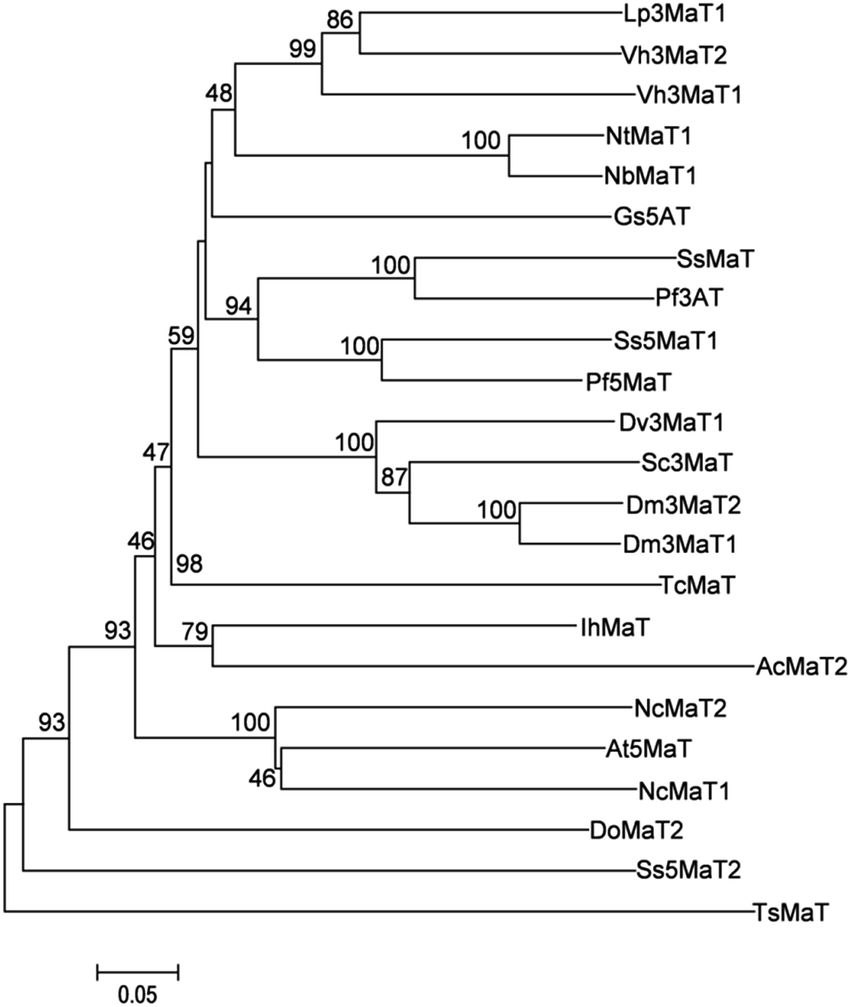 | ||
| Fig. 1 The phylogenetic relationships between NbMaT1 and the related acyl transferases from plants. The results were calculated using MEGA version 6 software. Method: neighbor-joining, bootstrap: 1000. The branch lengths represent the relative genetic distances. The protein sequences and corresponding accession numbers that were used for this comparison are as follows: Lp3MaT1 (AAS77404); Vh3MaT2 (AAS77403); Vh3MaT1 (AAS77402); NtMaT1 (BAD93691); NbMaT1 (KY563646); Gs5AT (BAD44688); SsMaT (AAR28757); Pf3AT (BAA93475); Ss5MaT1 (AAL50566); Pf5MaT (AAL50565); Dv3MaT1 (AAO12206); Sc3MaT (AAO38058); Dm3MaT2 (AAQ63616); Dm3MaT1 (AAQ63615); TcMaT (EOX96706); IhMaT (AIA22175); AcMaT2 (OAY66923); NcMaT2 (JAU99726); At5MaT (NP_189600); NcMaT1 (JAU75409); DoMaT2 (OEL31624); Ss5MaT2 (AAR26385); TsMaT (GAT33610). | ||
3.2. Enzymatic assays of NbMaT1
An acceptor library with structurally diverse drug-like glycoside scaffolds was established to investigate the substrate selectivity of NbMaT1. In total, 16 different members (their structures are shown in Fig. 2), including five flavonol glycosides (quercetin 3-O-β-D-glucoside-7-O-α-L-rhamnoside 1; isoquercitrin 2; quercetin 3-O-β-D-xylopyranoside 3; hyperoside 4; and icariin 9), two flavone glycosides (swertisin 5 and vitexin 6), two dihydroflavone glycosides (naringin 7 and liquiritin 8), five isoflavone glycosides (sophoricoside 10; glycitin 11; tectoridin 12; ononin 13; and calycosin 7-O-β-D-glucopyranoside 14), one phenylethyl chromone glycoside 15, and one coumarin glycoside 16 were used for the enzymatic assays with standard malonyl-CoA as the malonyl donor.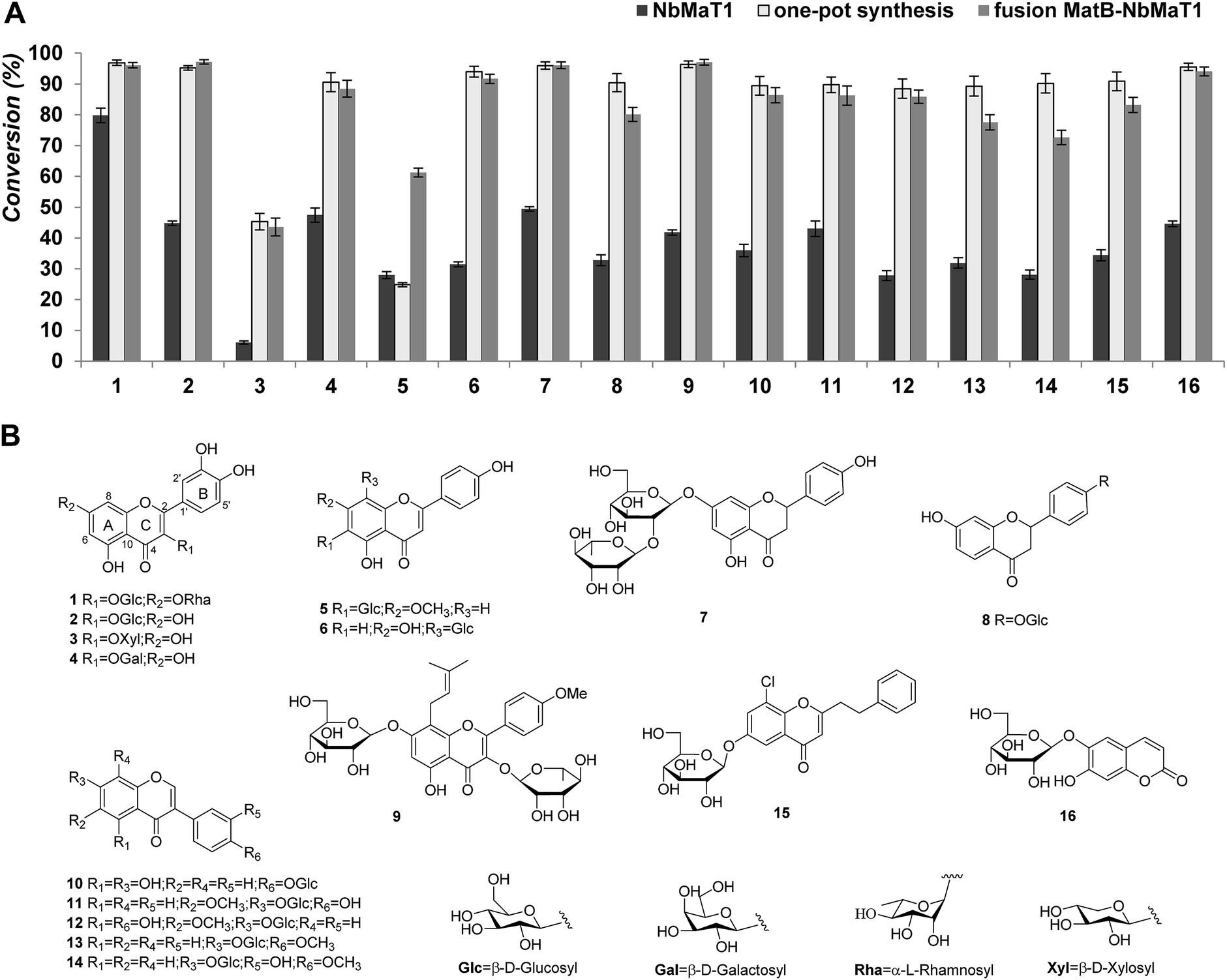 | ||
| Fig. 2 Exploring the substrate tolerance of NbMaT1 using malonyl-CoA as an acyl donor. (A) The conversion of the malonylated products catalyzed by NbMaT1. The members are listed with the number that corresponds to the structures listed in part (B). The columns in dark color represent the conversion percentage catalysed by NbMaT1; the lighter color represents the conversion percentage catalysed by the fusion protein MatB–NbMaT1, whereas the lightest color represents the conversion percentage catalysed by the one-pot synthesis system. (B) The structures of all the tested acyl acceptors. | ||
After 12 hours of incubation, all the reaction mixtures were processed further to remove the residual proteins and inorganic salts before being submitted to HPLC analysis. HPLC-UV analysis revealed that NbMaT1 could catalyse the malonylation of all the 16 tested glycosides belonging to the 4 structural types. Taking substrate 1 as an example, the enzymatic assay of 1 showed a product peak (1a) with relatively lower polarity on HPLC (Fig. S3†). Through high resolution quadruple time-of-flight electrospray ionization mass spectrometry (HR-QTOF-ESI-MS) analyses, it was observed that peak 1a exhibited an [M − H]− at m/z 695.1426 with the predicted formula of C30H32O19, which is higher than that of substrate 1 by m/z 86 and consistent with the theoretical formula of the malonylated product at m/z 695.1465 [M − H]−, suggesting the occurrence of the malonylation reaction. All the other predicted malonylated products were also further identified by HR-QTOF-ESI-MS. The molecular mass found for the novel peaks exactly matched with their calculated mass (Table 1).
| Substrate (no.) | Substrate RT (min) | Product RT (min) | Product yielda (%) | Product yieldb (%) | Product yieldc (%) | Measured m/z | Predicted formula | Theoretical m/z | Error (ppm) |
|---|---|---|---|---|---|---|---|---|---|
| a The product yield catalyzed by NbMaT1.b The product yield catalyzed by the one-pot synthesis.c The product yield catalyzed by the fusion protein MatB–NbMaT1.d The amount of the product was calculated from the product peak integral, assuming that the extinction coefficient of the product (malonyl glycoside) was the same as that of the substrate (glycoside), by HPLC. | |||||||||
| 1 | 21.5 | 22.0 | 79.75 | 96.89 | 96.06 | 695.1426 [M − H]− | C30H32O19 | 695.1465 | −5.61 |
| 2 | 23.7 | 24.4 | 44.79 | 95.22 | 97.14 | 551.1027 [M + H]+ | C24H22O15 | 551.1031 | −0.73 |
| 3 | 24.5 | 26.3 | 6.07 | 45.35 | 43.58 | 521.0897 [M + H]+ | C23H20O14 | 521.0926 | −5.57 |
| 4 | 23.9 | 24.8 | 47.45 | 90.60 | 88.47 | 549.0853 [M − H]− | C24H22O15 | 549.0886 | −6.01 |
| 5 | 23.7 | 25.1 | 27.95 | 24.83 | 61.24 | 533.1278 [M + H]+ | C25H24O13 | 533.1290 | −2.25 |
| 6 | 23.3 | 24.5 | 31.46 | 93.96 | 91.69 | 519.1127 [M + H]+ | C24H22O13 | 519.1133 | −1.16 |
| 7 | 26.4 | 25.5 | 49.40 | 95.98 | 96.07 | 665.1774 [M − H]− | C30H34O17 | 665.1723 | 7.67 |
| 8 | 23.8 | 28.0 | 32.79 | 90.41 | 80.10 | 505.1295 [M + H]+ | C24H24O12 | 505.1341 | −9.11 |
| 9 | 27.3 | 28.2 | 41.79 | 96.39 | 97.06 | 761.2299 [M − H]− | C36H42O18 | 761.2298 | 0.13 |
| 10 | 25.2 | 26.8 | 35.94 | 89.40 | 86.35 | 517.0968 [M − H]− | C24H22O13 | 517.0988 | −3.87 |
| 11 | 22.5 | 24.3 | 43.04 | 89.73 | 86.25 | 533.1286 [M + H]+ | C25H24O13 | 533.1290 | −0.75 |
| 12 | 24.3 | 26.0 | 27.82 | 88.46 | 85.85 | 549.1232 [M + H]+ | C25H24O14 | 549.1239 | −1.27 |
| 13 | 26.3 | 28.0 | 31.88 | 89.26 | 77.54 | 517.1341 [M + H]+ | C25H24O12 | 517.1341 | 0.00 |
| 14 | 23.1 | 25.0 | 28.08 | 90.22 | 72.64 | 533.1296 [M + H]+ | C25H24O13 | 533.1290 | 1.13 |
| 15 | 28.9 | 29.4 | 34.39 | 90.87 | 83.18 | 549.1126 [M + H]+ | C26H25O11Cl | 549.1158 | −5.83 |
| 16 | 17.0 | 21.8 | 44.61 | 95.54 | 94.06 | 427.0872 [M + H]+ | C18H18O12 | 427.0871 | 0.23 |
Although NbMaT1 shares a close phylogenetic relationship with NtMaT1, their catalytic behaviors are different and NbMaT1 exhibited more significant substrates promiscuities than NtMaT1 by contrast. First, NbMaT1 can catalyse the malonylation of flavonoid 7-O-glucosides and flavonoid 3-O-glucosides, which were the substrates of NtMaT1. However, in addition to these compounds, dihydroflavone glycosides and isoflavone glycosides were also accepted by NbMaT1 as positive substrates. Second, the sugar moieties malonylated by NbMaT1 were substituted at different positions in the flavonoid skeleton such as the C-6 (substrate 5), C-7 (substrate 7, 9, 11–14), C-8 (substrate 6) sites on ring A, and the C-4′ (substrate 8, 10) site on ring B as well as the C-3 (substrate 1–4) site on ring C. Third, in addition to the most commonly reported glucosyl moiety, flavonoid glycosides substituted with xylosyl (substrate 3) and galactosyl group (substrate 4) were also accepted by NbMaT1, which have rarely been reported earlier. Even for the glucosyl moiety, both O-glucosides (the most common glucosylation type) and C-glucoside (substrate 5 and 6) can be malonylated by NbMaT1. Moreover, the malonylation of NbMaT1 was not influenced by the number of substituted sugar moieties because malonylation of both mono- and di-glycosides (substrates 1, 7, and 9) was successfully carried out. Finally, our enzymatic assays also revealed that NbMaT1 can catalyse the malonylation of phenylethyl chromone glycosides (substrate 15); the enzymatic malonylation of these types of glycosides is novel.
Furthermore, note that many of the tested substrates were leading compounds with various bioactivities in drug research. For instance, isoquercitrin (2) has significant antihypertensive effects; swertisin (5) exhibits obvious memory repairment activity; and vitexin (6) and icariin (9) have been recognized to exhibit a wide range of pharmacological effects including but not limited to anti-oxidant, anti-cancer, anti-inflammatory, antihyperalgesic, neuroprotective effects, and so on. Malonylation of these compounds usually results in the formation of diverse acylated glycosides, which usually leads to improved lipid solubility, stability, and enhanced bioavailability. Thus, NbMaT1 may be accepted as a promising enzyme for the creation of structurally diverse malonylated glycosides for the discovery of bioactive leading compounds.
3.3. The enzymatic synthesis of the malonyl-CoA by malonyl-CoA synthetase MatB
Malonyl-CoA is an active acyl donor of malonyltransferase, which is commercially available but is expensive and chemically unstable. In vivo, malonyl-CoA is biosynthesized by malonyl-CoA synthetase. To obtain sufficient malonyl-CoA for the subsequent acyl enzymatic reactions as well as the biosynthesis pathway construction, we amplified the coding region of a known malonyl-CoA synthetase MatB (At3g16170, GenBank accession AY250842) based on the reported sequence using the cDNA of A. thaliana as the template by RT-PCR. The obtained 1635 bp open reading frame was then inserted into the pET28a vector before being expressed in an E. coli strain. Recombinant soluble proteins were obtained after overnight induction with 0.5 mM IPTG at 23 °C and further purified by Ni-NTA-agarose affinity chromatography to apparent homogeneity by SDS-PAGE. The observed molecular mass of MatB was 60 kDa, which was in good agreement with the calculated mass of 60.07 kDa (Fig. S23†).A chemoenzymatic synthesis of malonyl-CoA was performed by MatB using malonic acid and CoA as the substrates and ATP as the co-factor. The overall reaction comprised two sequential steps. First, malonyl-AMP formation: malonic acid + ATP → malonyl-AMP + PPi (adenylation step). Second, CoA transfer to form malonyl-CoA: malonyl-AMP + CoA → malonyl-CoA + AMP (thioesterification step). The obtained fractions containing the malonyl-CoA esters were assayed by HPLC-UV and compared with the authentic standards. As shown in Fig. S2,† the reaction product (peak a) exhibited a UV absorption spectrum and retention time identical to those of the standard malonyl-CoA under the same analysis conditions. These characteristics confirmed the production of malonyl-CoA in our in vitro chemoenzymatic synthesis system.
3.4. Enzymatic assays by one-pot reaction system and the fusion protein MatB–NbMaT1
In consideration of the desired malonylation promiscuities of NbMaT1, it was expected that this enzyme possesses potential applications for drug preparation. However, acyltransferases are not very convenient for industrial application, even for a large-scale synthesis in the laboratory because they strictly require the corresponding acyl-coenzyme A as the acyl donor, which must be used in a stoichiometric amount. In reference to NbMaT1, malonyl-CoA is its active acyl donor whose high price and limited source restricts the further application of NbMaT1 to a great extent. From many successive reports, it was observed that one-pot synthesis is an alternative strategy to improve the efficiency of reactions, whereby all the reactants are subjected with the catalyst(s) in just one reactor. Since malonyl-CoA can be biosynthesized using the obtained malonyl-CoA synthetase MatB, the one-pot biosynthesis system using malonic acid, CoA, ATP, and the malonyl acceptor was conducted with both MatB and NbMaT1. In this system, malonic acid can be activated to form malonyl-CoA thioester through the ATP-dependent MatB and directly submitted to the malonyl acceptor by NbMaT1, which results in the reaction equilibrium being efficiently shifted towards the formation of the end-products. The abovementioned substrates were subjected to the one-pot enzymatic synthesis, and the HPLC-UV-HR-MS analyses showed that all the 16 tested compounds could also be catalysed to obtain the same malonylated product obtained using NbMaT1 and standard malonyl-CoA alone. Moreover, it was observed that most of the tested substrates exhibited a dramatically increased conversion rate when compared with those of the single enzymatic reaction. For example, the product yield of substrate 13 and 15 were increased to more than three times that of the original (from 27.82% to 88.46% and 28.08% to 90.22%, respectively). In addition, 14 of the substrates (with the exception of 3 and 5) gave a high yield of more than 85% (Table 1).In addition, on the basis of one-pot synthesis investigation, to further simplify the protein purification procedure, a fusion protein was constructed by removing the stop codon of MatB and the insertion of a three amino acid linker (Gly-Ser-Gly) between the open reading frames of the two genes to obtain a polypeptide MatB–NbMaT1 with functional properties derived from each of the original proteins. The enzymatic assays revealed that the fusion MatB–NbMaT1 exhibited comparable malonylation activity in contrast with the one-pot synthesis system, which was much higher than that observed using NbMaT1 alone (Fig. 2). Since in both cases, the enzymes behave as a complex where not only the active sites of the biocatalysts are very close but also the product of one enzyme is the substrate of the next. As a result of this, the reactions using the fusion protein or one-pot system can possibly avoid secondary reactions while effectively increasing the global rate of the process to achieve an increased yield of the malonylated products.
To sum up, the one-pot synthesis system and the fusion protein MatB–NbMaT1 significantly shortened the number of steps required overall, saving time and resources, avoiding a lengthy separation process and purification of the intermediate compounds while remarkably improving the malonylated product yield with notable importance in both the foundation investigations and industrial applications.
3.5. Preparation of the enzymatic product of icariin by NbMaT1
Our extensive enzymatic reactions revealed the wide substrate selectivity of NbMaT1 as a malonyltransferase. To further reveal the catalytic characteristic of NbMaT1 and determine the substitution site of the malonyl group, a large scale preparation of the enzymatic product was performed using the one-pot reaction system. For comprehensive consideration of structure and bioactivity, a broad and significant pharmacological effective substrate icariin (9) with both rhamnopyranosyl group and glucosyl group substituted at different positions in the flavonol scaffold was chosen as an malonyl acceptor. One malonylated product (9a) was obtained and structurally analyzed by HR-MS, 1H, 13C-NMR and 2D-NMR spectroscopy (HMBC and HSQC).In the negative HR-QTOF-ESI-MS, 9a exhibited an [M − H]− at m/z 761.2299, which is higher than that of 9 by m/z 86. Three singlets at δ 162.05, 161.03, and 64.42 in the 13C NMR spectrum of 9a as well as two doublets at δ 3.59 and 3.53 in its 1H NMR spectrum indicate that this metabolite contains a malonyl group. The carbon and hydrogen signals were accurately assigned by comprehensive analysis of the 2D-NMR spectra. The chemical shift of C-6′′′ (δ 65.05) is lower than that of 9 (δ 60.71) by 4.3 ppm, indicating that the malonyl group is attached at C-6 of glucose through an ester bond. A characteristic downfield shift of H-6′′′a (δ 4.49) and H-6′′′b (δ 4.33) in 9a when compared with the corresponding signals in 9 (at δ 3.71 and 3.56, respectively) was also observed. Therefore, compound 9a was characterized as 8-prenyl-kaempferol-4′-methylether-3-O-rhamnoside-7-O-(6-O-malonyl)-β-D-glucopyranoside (Fig. 3). The malonyl group was specifically transferred onto the 6-OH group in the glucose moiety of 9 by NbMaT1.
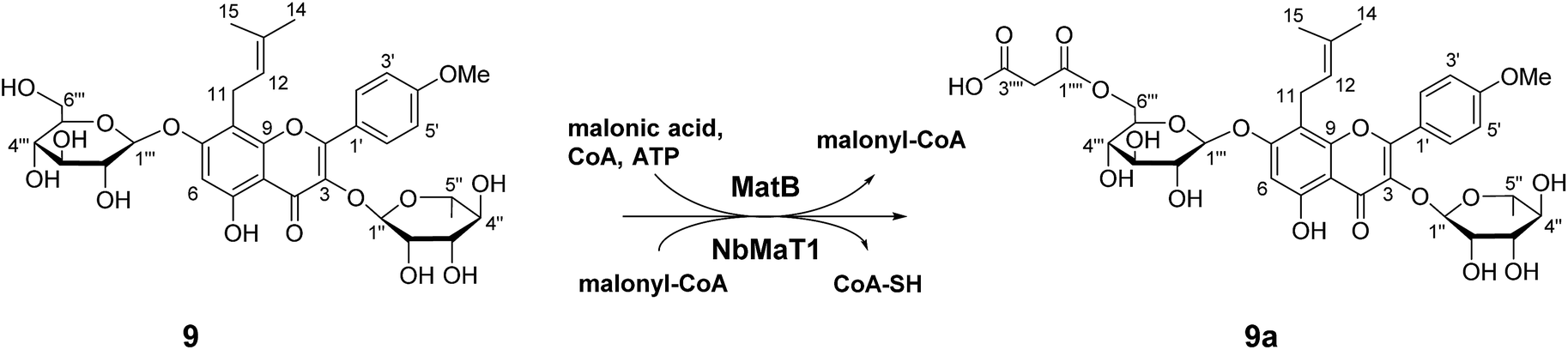 | ||
| Fig. 3 The one-pot enzymatic malonylation of icariin (9) by NbMaT1 and MatB. | ||
4. Conclusions
In summary, as a newly identified malonyltransferase of the plant BAHD family, NbMaT1, exhibited remarkable substrate tolerance towards diverse natural products with different saccharide groups substituted at various positions of the skeleton of flavonoids, coumarins, and phenylethyl chromones. Coupled with malonyl-CoA synthetase MatB, the one-pot synthesis and the fusion protein MatB–NbMaT1 were constructed to afford an effective malonylation system for further application. NbMaT1 is expected to be an effective tool and potentially general functional part for the chemoenzymatic synthesis pathway construction of diverse malonylated glycosides derivatives for leading compound discovery in drug research.Acknowledgements
We are thankful to the National Natural Science Foundation of China (Grant No. 81402809) for the financial support.References
- O. D. Cunningham and R. Edwards, Phytochemistry, 2008, 69, 2016 CrossRef CAS PubMed.
- K. Vladimir and M. Ludmila, Curr. Med. Chem., 2001, 8, 1303 CrossRef.
- S. Riva, J. Mol. Catal. B: Enzym., 2002, 19–20, 43 CrossRef CAS.
- F. Mellou, D. Lazari, H. Skaltsa, A. D. Tselepis, F. N. Kolisis and H. Stamatis, J. Biotechnol., 2005, 116, 295 CrossRef CAS PubMed.
- J. H. Salem, I. Chevalot, C. Harscoat-Schiavo, C. Paris, M. Fick and C. Humeau, Food Chem., 2011, 124, 486 CrossRef.
- M. P. Salas, G. Céliz, H. Geronazzo, M. Daz and S. L. Resnik, Food Chem., 2011, 124, 1411 CrossRef CAS.
- S. Tommasini, D. Raneri, R. Ficarra, M. L. Calabrò, R. Stancanelli and P. Ficarra, J. Pharm. Biomed. Anal., 2004, 35, 379 CrossRef CAS PubMed.
- J. H. Salem, C. Humeau, I. Chevalot, C. Harscoat-Schiavo, R. Vanderesse, F. Blanchard and M. Fick, Process Biochem., 2010, 45, 382 CrossRef CAS.
- F. Mellou, H. Loutrari, H. Stamatis, C. Roussos and F. N. Kolisis, Process Biochem., 2006, 41, 2029 CrossRef CAS.
- M. Ardhaoui, A. Falcimaigne, S. Ognier, J. M. Engasser, P. Moussou, G. Pauly and M. Ghoul, J. Biotechnol., 2004, 110, 265 CrossRef CAS PubMed.
- G. Taguchi, T. Ubukata, H. Nozue, Y. Kobayashi, M. Takahi, H. Yamamoto and N. Hayashida, Plant J., 2010, 63, 1031 CrossRef CAS PubMed.
- U. Matern, C. Reichenbach and W. Heller, Planta, 1986, 167, 183 CrossRef CAS PubMed.
- C. Gao, P. Mayon, D. A. MacManus and E. N. Vulfson, Biotechnol. Bioeng., 2000, 71, 235 CrossRef CAS PubMed.
- H. P. V. Rupasinghe, Tetrahedron Lett., 2013, 54, 1933 CrossRef.
- C. L. Zhao, Y. Q. Yu, Z. J. Chen, G. S. Wen, F. G. Wei, Q. Zheng, C. D. Wang and X. L. Xiao, Food Chem., 2017, 214, 119 CrossRef CAS PubMed.
- M. M. Giusti and R. E. Wrolstad, Biochem. Eng. J., 2003, 14, 217 CrossRef CAS.
- D. E. Stevenson, R. Wibisono, D. J. Jensen, R. A. Stanley and J. M. Cooney, Enzyme Microb. Technol., 2006, 39, 1236 CrossRef CAS.
- J. C. D'Auria, Curr. Opin. Plant Biol., 2006, 9, 331 CrossRef PubMed.
- B. St-Pierre and V. De Luca, Recent Adv. Phytochem., 2000, 34, 285 CAS.
- B. St-Pierre, P. Laflamme, A. M. Alarco and V. De Luca, Plant J., 1998, 14, 703 CrossRef CAS PubMed.
- X. Ma, J. Koepke, S. Panjikar, G. Fritzsch and J. Stöckigt, J. Biol. Chem., 2005, 280, 13576 CrossRef CAS PubMed.
- B. A. Manjasetty, X. H. Yu, S. Panjikar, G. Taguchi, M. R. Chance and C. J. Liu, Planta, 2012, 236, 781 CrossRef CAS PubMed.
- X. H. Yu, J. Y. Gou and C. J. Liu, Plant Mol. Biol., 2009, 70, 421 CrossRef CAS PubMed.
- H. Suzuki, S. Sawada, K. Watanabe, S. Nagae, M. A. Yamaguchi, T. Nakayama and T. Nishino, Plant J., 2004, 38, 994 CrossRef CAS PubMed.
- H. Chen, H. U. Kim, H. Weng and J. Browse, Plant Cell, 2011, 23, 2247 CrossRef CAS PubMed.
- K. Ziegler, K. Braun, A. Böckler and G. Fuchs, Arch. Microbiol., 1987, 149, 62 CrossRef.
- J. H. An and Y. S. Kim, Eur. J. Biochem., 1998, 257, 395 CAS.
- X. Guan and B. J. Nikolau, Plant J., 2016, 85, 581 CrossRef CAS PubMed.
- H. M. Koo and Y. S. Kim, Arch. Biochem. Biophys., 2000, 378, 167 CrossRef CAS PubMed.
- N. L. Pohl, M. Hans, H. Y. Lee, Y. S. Kim, D. E. Cane and C. Khosla, J. Am. Chem. Soc., 2001, 123, 5822 CrossRef CAS PubMed.
- C. Stewart, B. C. Kang, K. Liu, M. Mazourek, S. L. Moore, E. Y. Yoo, B. D. Kim, L. Paran and M. M. Jahn, Plant J., 2005, 42, 675 CrossRef CAS PubMed.
- J. Beekwilder, M. Alvarez-Huerta, E. Neef, F. W. Verstappen, H. J. Bouwmeester and A. Aharoni, Plant Physiol., 2004, 135, 1865 CrossRef CAS PubMed.
- X. H. Yu, M. H. Chen and C. J. Liu, Plant J., 2008, 55, 382 CrossRef CAS PubMed.
- J. C. D'Auria, M. Reichelt, K. Luck, A. Svatoš and J. Gershenzon, FEBS Lett., 2007, 581, 872 CrossRef PubMed.
- H. Unno, F. Ichimaida, H. Suzuki, S. Takahashi, Y. Tanaka, A. Saito, T. Nishino, M. Kusunoki and T. Nakayama, J. Biol. Chem., 2007, 282, 15812 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01940h |
| This journal is © The Royal Society of Chemistry 2017 |