
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA DFT study on the reaction mechanism between tetrachloro-o-benzoquinone and H2O2 and an alternative reaction approach to produce the hydroxyl radical†
Ping Li *a,
Chao Guoa,
Wenling Fenga,
Qiao Sunb and
Weihua Wang*a
*a,
Chao Guoa,
Wenling Fenga,
Qiao Sunb and
Weihua Wang*a
aKey Laboratory of Life-Organic Analysis, School of Chemistry and Chemical Engineering, Qufu Normal University, Qufu, 273165, P. R. China. E-mail: lignip@163.com; wwh78@163.com
bCollaborative Innovation Center of Radiation Medicine of Jiangsu Higher Education Institutions, School for Radiological and Interdisciplinary Sciences, Soochow University, Suzhou, 215123, P. R. China
First published on 26th April 2017
Abstract
The formation of hydroxyl and alkoxyl radicals in the reaction of halogenated quinones and organic hydroperoxides can be used to elucidate the potential carcinogenicity of polyhalogenated aromatic environmental pollutants. To further enrich the understanding of the reactivity of the halogenated quinones with organic hydroperoxides, in this study, the reaction mechanism of tetrachloro-o-benzoquinone (o-TCBQ) with H2O2 has been systematically investigated at the B3LYP/6-311++G** level. It was found that a molecular complex was formed as the first step of the title reaction. After that, the nucleophilic attack of H2O2 on o-TCBQ occurs to produce an unstable intermediate containing an O–O bond. Subsequently, the unstable intermediate decomposes homolytically via the cleavage of the O–O bond, resulting in the formation of the OH radical. Note that explicit water molecules play an important positive role in the nucleophilic attack process. The nucleophilic attack process is the rate-determining step in the whole reaction. Moreover, selected substitution effects on the title reaction have also been studied. In addition, as an alternative reaction approach, it was found that the formed unstable intermediate containing an O–O bond mentioned above can be produced directly from the nucleophilic attack of the anionic form of H2O2 on o-TCBQ in the absence of explicit water molecules.
1. Introduction
As a class of toxic intermediates, halogenated quinones can initiate many harmful effects in vivo, such as acute hepatotoxicity, nephrotoxicity, and carcinogenesis.1,2 Some of them, such as tetrachloro-p-benzoquinone (p-TCBQ), tetrachloro-o-benzoquinone (o-TCBQ), and 2,5-dichloro-1,4-benzoquinone (DCBQ), are the major genotoxic and carcinogenic quinoid metabolites of widely used chlorinated phenol pesticides, like pentachlorophenol (PCP), where PCP has been classified as a group 2B environmental carcinogen by the International Agency for Research on Cancer (IARC) and a group B2 carcinogen by the Environmental Protection Agency (EPA). In addition, halogenated quinones can also be produced during the oxidation or destruction of halophenols and other polyhalogenated persistent organic pollutants (POPs).3–5 Moreover, some halobenzoquinones have been identified as an emerging class of disinfection byproducts in drinking water.6–8Recently, more and more studies have shown that the chlorinated benzoquinones, such as p-TCBQ and DCBQ, can react with the organic hydroperoxides to produce the hydroxyl radical, organic alkoxyl radicals, and quinone ketoxy radicals under mild conditions experimentally.9–13 These produced active radicals can be used to partially elucidate the potential carcinogenicity of polyhalogenated aromatic environmental pollutants since they can cause the oxidative damage to the DNA, protein, and lipids.14
As for p-TCBQ, its interaction and reaction with organic hydroperoxides have been systematically investigated theoretically.15,16 It was found that explicit water molecules can interact with p-TCBQ through weak intermolecular H-bonds and play an important role in the formation of those radicals. As the isomer of p-TCBQ and another quinoid metabolites of PCP, however, o-TCBQ has been paid less attentions although o-TCBQ has been involved in the toxicity of the bleached kraft chlorination effluent many years ago.17–22 Do o-TCBQ and its derivatives have similar chemical reactivity to p-TCBQ to produce the hydroxyl and alkoxyl radicals? Can the anion of H2O2 react with the o-TCBQ since the acid–base dissociation equilibrium of H2O2 exits and the anion behaves as a good nucleophile. Unfortunately, these questions remain unclear to the best of our knowledge. A theoretical investigation on the title reaction appears to be highly desirable in the lack of the relevant experimental studies.
Therefore, in this study, the reaction mechanism of o-TCBQ with H2O2 has been systematically investigated employing the density functional theory (DFT) to address those above questions. Moreover, the substitution effects on the title reaction have also been explored. Expectedly, the present results not only can provide helpful information to better understand the potential carcinogenicity of polyhalogenated aromatic environmental pollutants, but also can enrich the knowledge of the reactivity of halogenated quinones with the organic hydroperoxides.
2. Computational methods
All the species in the whole reactions have been fully optimized using the density functional theory at the B3LYP/6-311++G** level, where the reliability and efficiency of the method in predicting the geometries and properties has been verified by a number of systems including the halogenated quinones and the homolytical cleavage of the O–O bond.23–39 Subsequently, vibrational frequency analysis has been performed at the same level to identify the nature of the optimized structures. In addition, intrinsic reaction coordinate (IRC)40,41 calculations were performed to further verify that the calculated transition states indeed connected the reactants and products.To investigate the bulk solvent effects on the nucleophilic attack of H2O2 to o-TCBQ, the polarizable continuum model (PCM)42,43 was employed in aqueous solution within the framework of self-consistent reaction filed (SCRF) theory. To explore the positive role of water molecule in the reaction, different numbers of explicit waters ranging from one to three have been introduced to assist the proton transfer process.
To clarify the formation and the nature of the intermolecular H-bonds formed in the reaction process, atoms in molecules (AIM) theory was employed on the basis of the optimized structures. In the AIM analyses,44 the intermolecular interaction between atoms was indicated by the presence of the bond critical point (BCP). From the magnitude of the electron density (ρbcp) at the BCP, one can estimate the strength of the interactions. Similarly, the ring structures were characterized by the location of a ring critical point (RCP). Moreover, the nature of the H-bond interaction can be predicted from the topological parameters at the BCP. For example, the positive values of the Laplacian of electron density (∇2ρbcp) and energy density (Hbcp) suggest that the interaction should be predominated by the electrostatic interaction. However, the positive ∇2ρbcp and negative Hbcp suggest that the interaction is partly covalent in nature.45–47
All the calculations have been carried out using Gaussian 09 program.48
3. Results and discussion
3.1 The reaction of o-TCBQ with H2O2
Promoted by the reaction mechanism of p-TCBQ with H2O2,9,15 the proposed reaction mechanism for the title reaction has been given in Scheme 1. Firstly, the direct nucleophilic attack process has been investigated. As a result, eight reaction modes, named as modes 1/1′, 2/2′, 3/3′, and 4/4′, have been constructed considerations of the symmetry of o-TCBQ and the different orientations of the hydrogen atoms of H2O2. Correspondingly, the respective transition state has been obtained as shown in Fig. 1, where the relevant molecular complexes (MC), transition states (TS), and intermediates (IM) have been given in Fig. S1 of the ESI† for reference. As given in Table 1, the energy barriers for the available attack modes range from 44.30 to 49.27 kcal mol−1 relative to the separated reactants. Similar to p-TCBQ,15 these high energy barriers suggest that the direct nucleophilic processes are difficult to proceed under mild conditions.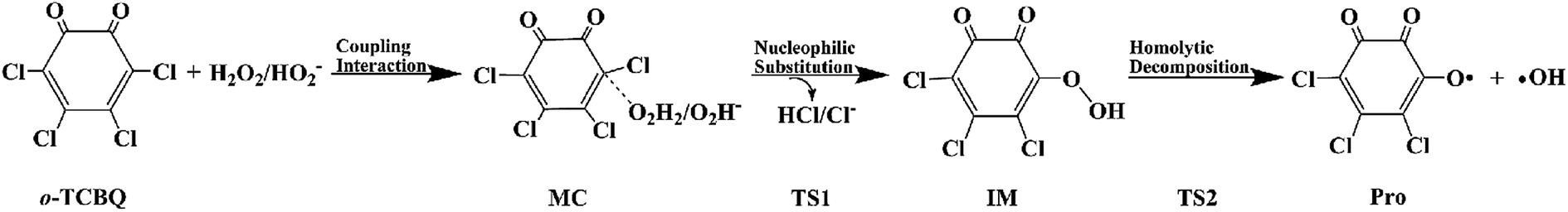 | ||
| Scheme 1 The proposed reaction mechanism for the reaction of o-TCBQ with H2O2/HO2−. | ||
 | ||
| Fig. 1 Calculated transition states in the direct nucleophilic attack process of H2O2 to o-TCBQ. The selected interatomic distances are given in Å. | ||
| Attack modes | ΔE1 | ΔE2 |
|---|---|---|
| a All the units are in kcal mol−1. ΔE1 and ΔE2 refer to the results in the gas phase and in aqueous solution, respectively. | ||
| Mode 1/1′ | 45.62/45.58 | 45.60/41.84 |
| Mode 2/2′ | 45.66/44.30 | 48.19/43.62 |
| Mode 3/3′ | 49.27/47.56 | 49.04/44.73 |
| Mode 4/4′ | 48.13/44.80 | 47.14/41.55 |
Subsequently, the bulk solvent effects on the above nucleophilic attack processes have been considered in aqueous solution employing the PCM model since the reaction takes place in solution generally. As a result, it was found that all the energy barriers have been decreased by about 0.03–3.74 kcal mol−1 except for the attack mode 2, where there is an increment of 2.53 kcal mol−1 for the attack mode 2. However, as given in Table 1, the energy barriers are still too high to overcome for the reaction overall. Motivated by the reaction of p-TCBQ with the assistance of the explicit water molecules,15 the different numbers of water molecules ranging from one to three have been introduced to promote the reaction in the following discussions.
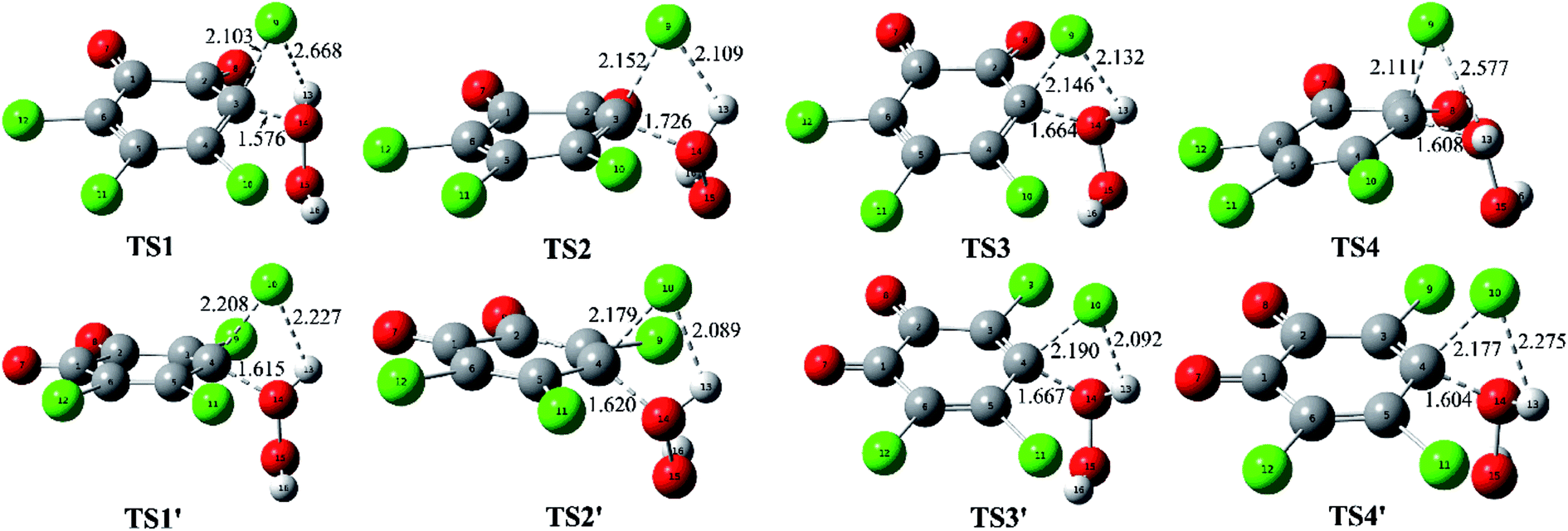
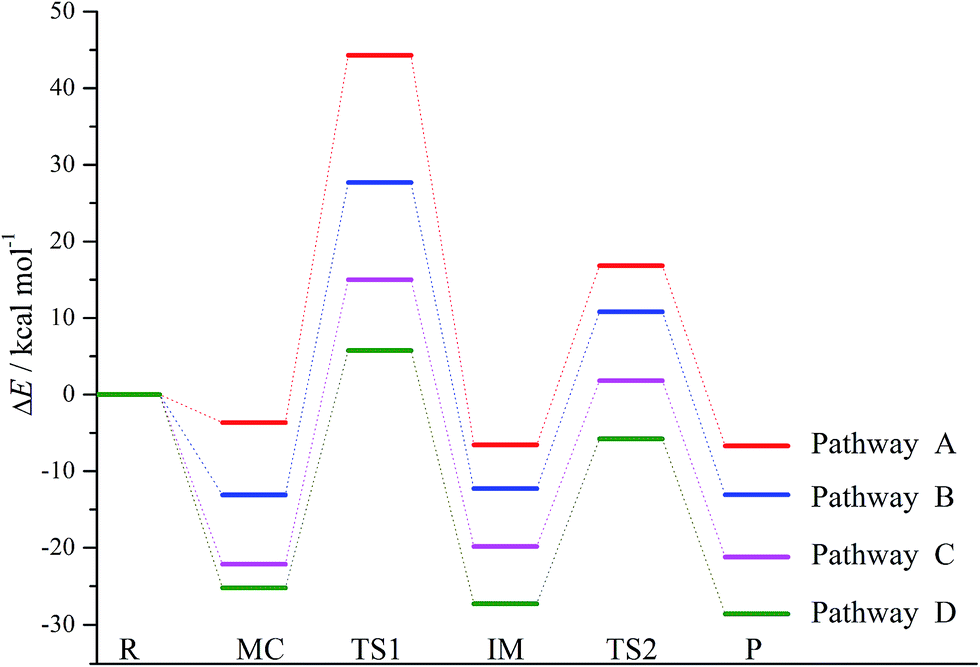
For simplicity, the symbols MC(nw), IM(nw), TSx(nw), and Pro(nw) have been employed to stand for the optimized molecular complexes, intermediates, transition states, and products, where x and nw refer to the sequence of the formed species and the numbers of the water molecules involved, respectively. For example, TS1(1w)/TS2(2w) refer to the first/second transition states in the reaction pathways involving one/two water molecules. In view of the fact that attack mode 2′ has the lowest energy barrier, so the following nucleophilic attack modes in the presence of explicit water molecules are constructed on the basis of the mode 2′, where four reaction pathways involving zero, one, two, and three water molecules have been named as pathways A, B, C, and D, respectively.
 | ||
| Fig. 2 Optimized molecular complexes (MC), transition states (TS), intermediates (IM), and products (Pro) for the available reaction pathways. The selected interatomic distances are given in Å. | ||
For the formed molecular complex MC(0w) in the absence of water molecules, the both O atoms of H2O2 molecule interact with the two C atoms of o-TCBQ, which can be confirmed by the presence of the BCP of the C⋯O bond as shown in Fig. S2 of the ESI.† From the large contact distances between H2O2 and o-TCBQ, one can say that the interaction between two species should be weak. Actually, as given in Table 2, MC(0w) has been stabilized by about 3.65 kcal mol−1 relative to that of the reactants.
| Pathways | Parameters | MC(nw) | TS1(nw) | IM(nw) | TS2(nw) | Pro + ˙OH |
|---|---|---|---|---|---|---|
| a All the units are in kcal mol−1. The data in parentheses refer to the results relative to the IM(nw). | ||||||
| A | ΔE | −3.65 | 44.30 | −6.56 | 16.82(23.38) | −6.70 |
| ΔH | −3.21 | 43.46 | −6.14 | 17.18 | −5.42 | |
| B | ΔE | −13.10 | 27.69 | −12.27 | 10.81(23.08) | −13.09 |
| ΔH | −13.42 | 26.11 | −12.40 | 10.67 | −12.39 | |
| C | ΔE | −22.14 | 15.01 | −19.83 | 1.81(21.64) | −21.21 |
| ΔH | −23.16 | 12.92 | −20.61 | 0.92 | −21.16 | |
| D | ΔE | −25.24 | 5.76 | −27.29 | −5.79(21.50) | −28.62 |
| ΔH | −26.81 | 2.63 | −29.41 | −8.20 | −30.16 | |
As for the other MCs in the presence of water molecules, as displayed in Fig. S2 of the ESI,† all of them are characterized by the intermolecular H-bonds from the locations of the BCPs between the selected O and H atoms. As a general rule, the introduced water molecules interact with the H2O2 and o-TCBQ via the intermolecular H-bonds. Moreover, H2O2 acts as a proton donor in the formed hydrated complexes except for in MC(3w). Here, H2O2 acts as proton donor and acceptor simultaneously in MC(3w). Moreover, the strengths of the H-bonds involving H2O2 as the proton donor are stronger than those of the H-bonds involving H2O2 as the proton acceptor, suggesting that H2O2 is a good proton donor. This point can be reflected from the shorter H-bond distances and larger electron density at the BCPs of the corresponding H-bonds involving H2O2 as the proton donor. Actually, this phenomenon has also been observed in the interaction of H2O2 with p-TCBQ.16
Compared with the intermolecular H-bonds formed between H2O and o-TCBQ, the shorter H-bond distances between H2O and H2O2 suggest that the adjacent water molecules tend to interact with H2O2 rather than o-TCBQ in realistic system, which is more obvious in MC(3w). Actually, as given in Table S1 of the ESI,† topological analyses suggest that the intermolecular H-bonds between H2O and H2O2 as well as the H-bonds between water molecules possess partly covalent character. As for the intermolecular H-bonds between H2O and o-TCBQ, they should be predominated by the electrostatic interactions.
As presented in Table 2, the molecular complexes involving water molecules have been stabilized by about 13.10, 22.14, and 25.24 kcal mol−1 in MC(1w), MC(2w), and MC(3w), which are much larger than that of MC(0w). At the same time, all the formation processes of these MCs are exothermic from the negative enthalpy changes ranging from −3.21 to −26.81 kcal mol−1. Moreover, the released heat increases with the increasing of the numbers of the water molecules, which is necessary for the following nucleophilic attack process probably.
As for the nucleophilic attack processes in the presence of water molecules, the water molecules directly or indirectly participate in the proton transfer process. For example, the water molecule accepts the proton of H2O2 and gives its own proton to the Cl atom of o-TCBQ simultaneously in the assistance of one water molecule. Similarly, the same is also true if a second water molecule is introduced, where the second water molecule accepts the proton of the first water molecule and gives its proton to the Cl atom of o-TCBQ. Therefore, the introduced water molecules above play the bridge role in the proton transfer process. However, if a third water molecule is introduced, the proton transfer occurs from H2O2 to its nearest water molecule in TS1(3w), resulting in the formation of the hydronium ion. Further IRC calculations suggest that the other two water molecules do not directly participate in the proton transfer process although they play an important role in decreasing the energy barrier as mentioned below.
As mentioned above, the Cl atom will be dissociated from the C atom of o-TCBQ in the nucleophilic attack process. So, the degree of the dissociation of Cl atom in the transition state is an important index to influence the magnitude of the energy barrier, where the degree of the dissociation of Cl atom can be reflected from the distance of the dissociated Cl atom and its linked C atom (RC⋯Cl). Namely, the larger RC⋯Cl implies the larger energy barrier required to overcome. Analyses of the different transition states suggest that the RC⋯Cl decreases upon introduction of water molecules and decreases with the increasing of the numbers of water molecules. For example, the RC⋯Cl is 2.179, 2.056, 1.882, and 1.845 Å in TS1(0w), TS1(1w), TS1(2w), and TS1(3w), respectively. Thus, one can predict that the corresponding energy barrier should decrease with the increasing of the numbers of water molecules, which is consistent with the calculated energy barrier below.
As shown in Table 2, the original energy barrier in the nucleophilic attack processes has been decreased significantly with the assistance of water molecules, where all the energy barriers are relative to the separated reactants. For example, the energy barrier has been decreased by 16.61 to 27.69 kcal mol−1 upon introduction of one water molecule. Moreover, the energy barrier has been further reduced to 15.01 and 5.76 kcal mol−1 in the presence of two and three water molecules, respectively. Note that these energy barriers can be comparable to those of the reaction of p-TCBQ with H2O2 in the assistance of water molecules.15 Therefore, similar to p-TCBQ, the nucleophilic attack of H2O2 to o-TCBQ can take place in the assistance of explicit water molecules, exhibiting the important positive role of water molecules.
To further evaluate the strength of the O–O bond in different IM(nw) species, its vertical and adiabatic bond dissociation enthalpies (BDEs) have been calculated as well as those of the H2O2 for comparison. Here, the adiabatic BDE is calculated as the enthalpy difference between the optimized two free radicals dissociated and the IM(nw) species. Similarly, the vertical BDE is calculated as the energy difference between two free radicals without considering structural relaxation and the IM(nw) species. As presented in Table 3, it was found that the vertical BDE of O–O bond in IM(nw) species has been decreased significantly by about 30 kcal mol−1 relative to that of H2O2. Further consideration of the structural relaxation, the homolytical decomposition of O–O bond is a thermal neutral process, which can be confirmed by the calculated small adiabatic BDEs.
| Species | H2O2 | IM(0w) | IM(1w) | IM(2w) | IM(3w) |
|---|---|---|---|---|---|
| a All the units are in kcal mol−1. The data behind the slash refer to adiabatic BDEs. | |||||
| BDE | 48.62/44.08 | 19.59/0.72 | 18.80/0.01 | 18.33/−0.63 | 18.27/−0.75 |
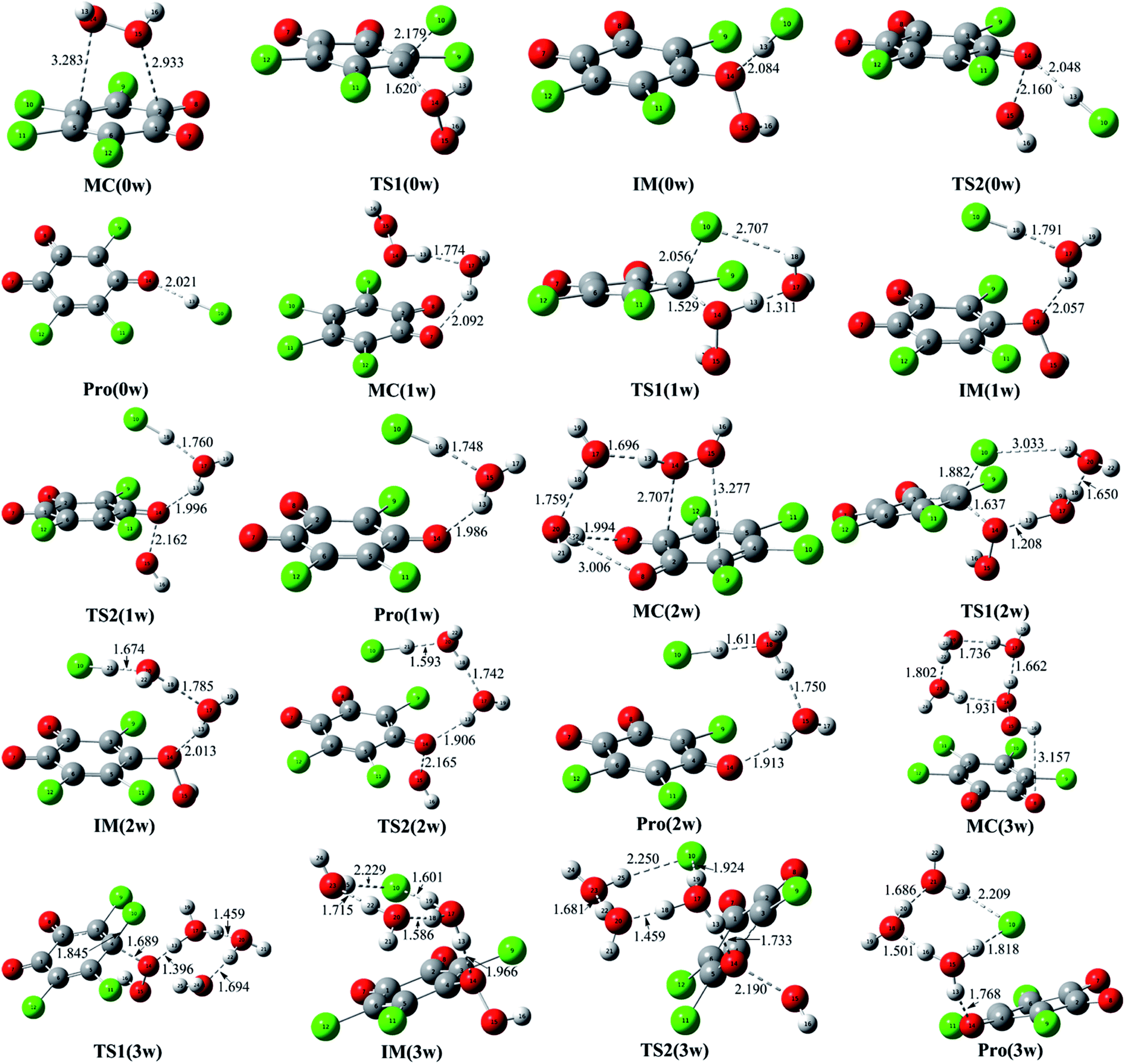
Moreover, all the transition states associated with the dissociation of the O–O bond have been shown in Fig. 2. As presented in Table 2, the calculated energy barrier is 23.38 kcal mol−1 in the absence of the water molecules relative to IM(0w). Moreover, the energy barrier has been further reduced to 23.08, 21.64, and 21.50 kcal mol−1 in the presences of one, two, and three water molecules, respectively. Actually, these energy barriers become small if the heat of reaction released from the formation of the MC is considered. For example, the energy barrier is 1.81 kcal mol−1 relative to the separated reactants in the presence of two water molecules. Therefore, it is possible for these IM(nw) species to decompose homolytically to produce the hydroxyl radical.
As displayed in Fig. 3, the whole reaction profiles for the formation of the hydroxyl radical have been summarized. Obviously, the nucleophilic attack process should be the rate-determining step due to its high energy barrier relative to that of the formation process of the hydroxyl radical. Overall, the whole reaction processes to produce the free radicals are exothermic from the negative values of the enthalpy changes as presented in Table 2.
 | ||
| Fig. 3 The reaction profiles for the reaction between o-TCBQ and H2O2. The symbols R and P refer to the separated reactants and products including Pro and hydroxyl radical, respectively. | ||
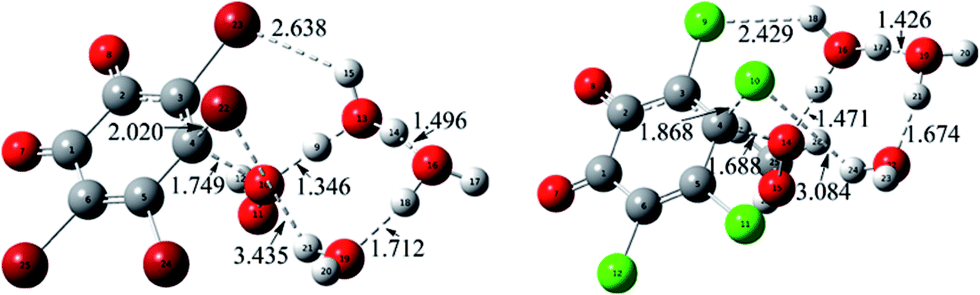 | ||
| Fig. 4 Calculated transition states in the nucleophilic attack processes for the reactions of Br- and CH3-substituted cases. The selected distances are given in Å. | ||
3.2 The reaction of o-TCBQ with HO2− anion
Considering the existence of the HO2− anion in system due to the acid–base dissociation equilibrium of H2O2 and the nature of the HO2− anion as a good nucleophile, one can say that the direct reaction between o-TCBQ and HO2− is also possible. To testify this point, the reaction of o-TCBQ with HO2− has been investigated. As shown in Fig. 5, similar to the reaction of o-TCBQ with H2O2 mentioned above, a molecular complex has been located firstly, which is lower in energy by 53.51 and 21.76 kcal mol−1 in the gas phase and in aqueous solution relative to the reactants. Subsequently, nucleophilic attack of HO2− to o-TCBQ occurs, leading to the formation of the unstable intermediate containing O–O bond. Expectedly, the following process is similar to that of the reaction of neutral H2O2 with o-TCBQ. However, unlike the reaction involving neutral H2O2, the corresponding transition state TS′ in the nucleophilic attack process is lower in energy by about 50.25 and 14.72 kcal mol−1 relative to the reactants in the gas phase and in aqueous solution, respectively. Correspondingly, the energy barrier is 3.26 and 7.04 kcal mol−1 relative to the corresponding MC in the gas phase and in aqueous solution, suggesting that the reaction involving HO2− is easy to occur. Therefore, compared with the above reaction mechanism involving neutral H2O2, the explicit water molecules are not necessary for the formation of the unstable intermediate containing O–O bond here. Certainly, other more sophisticated theoretical calculations are required to further confirm this alternative reaction approach since it is difficult to detect those intermediates experimentally due to their short half-lives and low concentrations.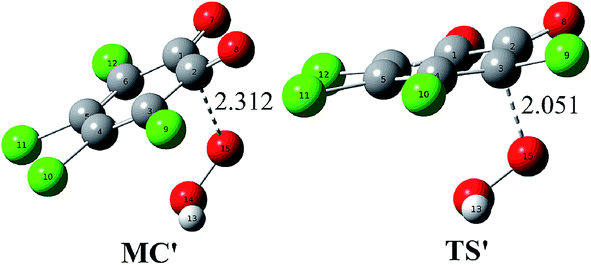 | ||
| Fig. 5 Optimized molecular complex (MC′) and transition state (TS′) in the reaction of o-TCBQ with HO2−. The selected interatomic distances are given in Å. | ||
4. Conclusions
In this study, the reaction mechanism between o-TCBQ and H2O2 has been systematically investigated at the B3LYP/6-311++G** level. It was found that o-TCBQ can react with H2O2 and its alkyl derivatives through two reaction approaches. One is the reaction of o-TCBQ with neutral H2O2 with the assistance of the explicit water molecules. The other one is the reaction of o-TCBQ with the anionic form of H2O2. In both reaction approaches, a molecular complex has been formed firstly followed by the nucleophilic attack process, resulting in the formation of an unstable intermediate containing O–O bond. After that, the homolytical decomposition of the O–O bond can lead to the formation of the hydroxyl or alkoxyl radicals. Moreover, the nucleophilic attack process is the rate-determining step in the whole reaction. Probably, those formed active radicals can be used to partially elucidate the potential carcinogenicity of polyhalogenated aromatic environmental pollutants. Hopefully, the present results can further enrich the knowledge of the reactivity of the halogenated quinones in the presence of organic hydroperoxides.Acknowledgements
This work is supported by NSFC (21577076, 21303093, and 21003082) and the NSF of Shandong Province (ZR2014BM020).References
- J. L. Bolton, M. A. Trush, T. M. Penning, G. Dryhurst and T. J. Monks, Chem. Res. Toxicol., 2000, 13, 135–160 CrossRef CAS PubMed.
- Y. Song, B. A. Wagner, J. R. Witmer, H. J. Lehmler and G. R. Buettner, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9725–9730 CrossRef CAS PubMed.
- B. Meunier, Science, 2002, 296, 270–271 CrossRef CAS PubMed.
- S. S. Gupta, M. Stadler, C. A. Noser, A. Ghosh, B. Steinhoff, D. Lenoir, C. P. Horwitz, K. Schramm and T. J. Collins, Science, 2002, 296, 326–328 CrossRef PubMed.
- A. Sorokin, J. L. Seris and B. Meunier, Science, 1995, 268, 1163–1166 CAS.
- Y. L. Zhao, F. Qin, J. M. Boyd, J. Anichina and X. F. Li, Anal. Chem., 2010, 82, 4599–4605 CrossRef CAS PubMed.
- F. Qin, Y. Y. Zhao, Y. L. Zhao, J. M. Boyd, W. J. Zhou and X. F. Li, Angew. Chem., Int. Ed., 2010, 49, 790–792 CrossRef CAS PubMed.
- J. H. Li, W. Wang, B. Moe, H. L. Wang and X. F. Li, Chem. Res. Toxicol., 2015, 28, 306–318 CrossRef CAS PubMed.
- B. Z. Zhu, B. Kalyanaraman and G. B. Jiang, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17575–17578 CrossRef CAS PubMed.
- B. Z. Zhu, L. Mao, C. H. Huang, H. Qin, R. M. Fan, B. Kalyanaraman and J. G. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16046–16051 CrossRef CAS PubMed.
- B. Z. Zhu, H. T. Zhao, B. Kalyanaraman, J. Liu, G. Q. Shan, Y. G. Du and B. Frei, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3698–3702 CrossRef CAS PubMed.
- B. Z. Zhu, G. Q. Shan, C. H. Huang, B. Kalyanaraman, L. Mao and Y. G. Du, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11466–11471 CrossRef CAS PubMed.
- C. H. Huang, F. R. Ren, G. Q. Shan, H. Qin, L. Mao and B. Z. Zhu, Chem. Res. Toxicol., 2015, 28, 831–837 CrossRef CAS PubMed.
- R. Yin, D. Zhang, Y. Song, B. Z. Zhu and H. Wang, Sci. Rep., 2013, 3, 1269 CrossRef PubMed.
- P. Li, W. H. Wang, Q. Sun, Z. Li, A. J. Du, S. W. Bi and Y. Zhao, ChemPhysChem, 2013, 14, 2737–2743 CrossRef CAS PubMed.
- P. Li, W. H. Wang, S. W. Bi and H. T. Sun, Struct. Chem., 2013, 24, 1253–1264 CrossRef CAS.
- B. S. Das, S. G. Reid, J. L. Betts and K. Patrick, J. Fish. Res. Board Can., 1969, 26, 3055–3067 CrossRef CAS.
- X. W. Guo and H. Mayr, J. Am. Chem. Soc., 2014, 136, 11499–11512 CrossRef CAS PubMed.
- S. Maddila, V. D. B. C. Dasireddy and S. B. Jonnalagadda, Appl. Catal., B, 2013, 138–139, 149–160 CrossRef CAS.
- M. Freytag, P. G. Jones, R. Schmutzler and M. Yoshifuji, Heteroat. Chem., 2001, 12, 300–308 CrossRef CAS.
- M. Kot and W. Zaborska, J. Enzyme Inhib. Med. Chem., 2006, 21, 537–542 CrossRef CAS PubMed.
- H. R. Zare, M. Eslami, M. Namazian and M. L. Coote, J. Phys. Chem. B, 2009, 113, 8080–8085 CrossRef CAS PubMed.
- Y. Y. Zhao, F. M. Tao and E. Y. Zeng, J. Phys. Chem. A, 2007, 111, 11638–11644 CrossRef CAS PubMed.
- M. Melicherčík, L. F. Pašteka, P. Neogrády and M. Urban, J. Phys. Chem. A, 2012, 116, 2343–2351 CrossRef PubMed.
- A. Gupta, H. M. Jaeger, K. R. Compaan and H. F. Schaefer III, J. Phys. Chem. B, 2012, 116, 5579–5587 CrossRef CAS PubMed.
- L. M. Wang and A. L. Tang, Chemosphere, 2011, 82, 782–785 CrossRef CAS PubMed.
- X. B. Wang, Q. Fu and J. L. Yang, J. Phys. Chem. A, 2010, 114, 9083–9089 CrossRef CAS PubMed.
- P. Li, Z. T. Shen, W. H. Wang, Z. Y. Ma, S. W. Bi, H. T. Sun and Y. X. Bu, Phys. Chem. Chem. Phys., 2010, 12, 5256–5267 RSC.
- C. Guo, W. H. Wang, W. L. Feng and P. Li, RSC Adv., 2017, 7, 12775–12782 RSC.
- W. L. Feng, C. Ren, W. H. Wang, C. Guo, Q. Sun and P. Li, Theor. Chem. Acc., 2016, 135, 190 CrossRef.
- W. L. Feng, C. Ren, W. H. Wang, C. Guo, Q. Sun and P. Li, RSC Adv., 2016, 6, 48099–48108 RSC.
- W. H. Wang, X. X. Zhang, P. Li, Q. Sun, Z. Li, C. Ren and C. Guo, J. Phys. Chem. A, 2015, 119, 796–805 CrossRef CAS PubMed.
- S. L. Zhuang, H. F. Wang, K. K. Ding, J. Y. Wang, L. M. Pan, Y. L. Lu, Q. J. Liu and C. L. Zhang, Chemosphere, 2016, 144, 1050–1059 CrossRef CAS PubMed.
- S. L. Zhuang, X. Lv, L. M. Pan, L. P. Lu, Z. W. Ge, J. Y. Wang, J. P. Wang, J. S. Liu, W. P. Liu and C. L. Zhang, Environ. Pollut., 2017, 220, 616–624 CrossRef CAS PubMed.
- P. Li, Z. Y. Ma, W. H. Wang, Y. Z. Zhai, H. T. Sun, S. W. Bi and Y. X. Bu, Phys. Chem. Chem. Phys., 2011, 13, 941–953 RSC.
- P. Li, Z. Y. Ma, W. H. Wang, R. Song, Y. Z. Zhai, S. W. Bi, H. T. Sun and Y. X. Bu, Phys. Chem. Chem. Phys., 2011, 13, 5931–5939 RSC.
- L. V. Liu, S. Hong, J. Cho, W. Nam and E. I. Solomon, J. Am. Chem. Soc., 2013, 135, 3286–3299 CrossRef CAS PubMed.
- A. Bassan, M. R. A. Blomberg, P. E. M. Siegbahn and L. Que Jr, J. Am. Chem. Soc., 2002, 124, 11056–11063 CrossRef CAS PubMed.
- M. L. Kuznetsov, Y. N. Kozlov, D. Mandelli, A. J. L. Pombeiro and G. B. Shul'pin, Inorg. Chem., 2011, 50, 3996–4005 CrossRef CAS PubMed.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- R. F. W. Bader, Atoms in molecules: a quantum theory, Oxford University Press, Oxford, UK, 1990 Search PubMed.
- L. F. Pacios, J. Phys. Chem. A, 2004, 108, 1177–1188 CrossRef CAS.
- I. Rozas, I. Alkorta and J. Elguero, J. Am. Chem. Soc., 2000, 122, 11154–11161 CrossRef CAS.
- W. D. Arnold and E. Oldfield, J. Am. Chem. Soc., 2000, 122, 12835–12841 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2013.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01878a |
| This journal is © The Royal Society of Chemistry 2017 |