DOI:
10.1039/C7RA01810J
(Paper)
RSC Adv., 2017,
7, 32989-33000
Aromatic poly(ether ester)s derived from a naturally occurring building block nipagin and linear aliphatic α,ω-diols†
Received
13th February 2017
, Accepted 23rd June 2017
First published on 29th June 2017
Abstract
Nipagin, the methyl ester of p-hydroxybenzoic acid, which is present naturally in campanulaceae and ericaceous plants, was used to prepare poly(ether ester)s. In this study, two nipagin-based aromatic dimethyl esters were synthesized via one-step simple nucleophilic substitutions with methyl chloroacetate and 1,4-dibromobutane, respectively. Subsequently, two series of poly(ether ester)s, PN1-ωs and PN2-ωs, were synthesized using linear aliphatic α,ω-diols (n = 2, 3, 4, 6, 10, 12) after optimizing the reaction conditions, like feed ratio, reaction catalyst, time and temperature. The synthesized poly(ether ester)s exhibit weight-average molecular weights (Mws) in the range of 9300–32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 g mol−1, together with dispersity (D) values between 1.5 and 1.8. 1H NMR, 13C NMR and FTIR spectroscopy were used to characterize their chemical structures. The thermal properties were studied by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). The results show that the nipagin-based poly(ether ester)s exhibit excellent thermal stability with the initial decomposition temperature above 350 °C and a two-step degradation mechanism. Glass transition (Tg), melting and crystallization temperatures (Tm, Tc), and the corresponding melting and crystallization enthalpies (ΔHm, ΔHc) are closely related to the lengths of the α,ω-diols. The Tg values show a decreasing trend with a gradual increase in α,ω-diol length in both PN1-ωs and PN2-ωs. The crystallization behaviors were also studied by isothermal crystallization and wide-angle X-ray diffraction (WAXD). The crystallizability proves to be enhanced with a gradual increase in α,ω-diol length in each series. The mechanical properties were studied using tensile assays and dynamic mechanical analysis (DMA), which show that the nipagin-based poly(ether ester)s feature excellent mechanical properties with elastic modulus and tensile strength values reaching 600–800 MPa and 10–35 MPa, respectively. Finally, the nipagin-based poly(ether ester)s feature excellent comprehensive performance and the renewable nipagin will broaden the thermoplastic spectrum and have practical applications in the field of polymeric materials.
100 g mol−1, together with dispersity (D) values between 1.5 and 1.8. 1H NMR, 13C NMR and FTIR spectroscopy were used to characterize their chemical structures. The thermal properties were studied by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). The results show that the nipagin-based poly(ether ester)s exhibit excellent thermal stability with the initial decomposition temperature above 350 °C and a two-step degradation mechanism. Glass transition (Tg), melting and crystallization temperatures (Tm, Tc), and the corresponding melting and crystallization enthalpies (ΔHm, ΔHc) are closely related to the lengths of the α,ω-diols. The Tg values show a decreasing trend with a gradual increase in α,ω-diol length in both PN1-ωs and PN2-ωs. The crystallization behaviors were also studied by isothermal crystallization and wide-angle X-ray diffraction (WAXD). The crystallizability proves to be enhanced with a gradual increase in α,ω-diol length in each series. The mechanical properties were studied using tensile assays and dynamic mechanical analysis (DMA), which show that the nipagin-based poly(ether ester)s feature excellent mechanical properties with elastic modulus and tensile strength values reaching 600–800 MPa and 10–35 MPa, respectively. Finally, the nipagin-based poly(ether ester)s feature excellent comprehensive performance and the renewable nipagin will broaden the thermoplastic spectrum and have practical applications in the field of polymeric materials.
Introduction
There are growing concerns regarding the use of petroleum-based resources due to their dwindling amounts and limited availability. Hence, there is a great demand for other replaceable resources.1,2 This has inspired scientists to find other feasible alternatives from naturally occurring sustainable biomass to replace the presently used petrochemicals. Researchers from both polymer and materials science fields have put in their best efforts into solving these problems in the past few decades.3,4 Amongst the most researched renewable resources, abundantly available carbohydrates and vegetable oils have attracted significant attention from both academia and industry for the production of valuable chemicals, energy fuels, and polymeric materials.5,6 However, there are still many shortcomings in the polymers synthesized directly from carbohydrates and vegetable oils, such as limited thermal stability due to the presence of various reactive functional groups along the molecular chains.7 The other drawback is the low reactivity of secondary hydroxyl groups in carbohydrate-derived monomers.8 Furthermore, the mechanical properties of the materials synthesized from these bio-based resources are far from satisfactory compared to petroleum-based materials like PET and PBT.9 All of these factors limit the use of carbohydrates and vegetable oils in practical applications. Hence, it is necessary to further explore renewable substitutes for fossil resources in order to produce structural materials with excellent thermal and mechanical properties and scale up the production to an industrial level eventually.10,11
4-Hydroxybenzoic acid, as a renewable feedstock exists naturally in campanulaceae and ericaceous plants.12 It is industrially produced by the carboxylation of potassium phenoxide using CO2 under pressure.13 The target compound can also be obtained by the isomerization of salicylic acid, but this process is expensive.14 Additionally, 4-aminophenol is initially diazotized and then subjected to Sandmeyer reaction to obtain the desired 4-hydroxybenzoic acid.15 4-Hydroxybenzoic acid is a widely used reactant in organic synthesis, especially for the synthesis of parabens including nipagin, ethylparaben, isopropylparaben to heptylparaben, etc.16 Almost all of these parabens can be used as additives in soy sauces, vinegar-cured food, as well as chilled beverages. They can also act as antiseptic and antifungal agents in cosmetics and pharmaceuticals.17,18 Due to the presence of a phenolic hydroxyl group, nipagin exhibits stronger antibacterial property that is more effective than benzoic acid and sorbic acid. The antibacterial action involves the destruction of cytomembranes of microorganisms, followed by the denaturation of intracellular proteins, and then results in the inhibition of activities of respiratory enzymes and enzymes involved in the electron-transport system in microbial cells.19 Apart from applications mentioned above, the bio-based parabens are also used for the synthesis of various multifunctional polymeric materials, such as epoxy resins and liquid crystal materials.20,21 It has been proved that bisphenol A features hormone-like properties and concerns have been raised regarding its potential health hazards to the infants when used in consumer products.22 As a consequence, bisphenol A (BPA)-containing nursing products have been banned in all countries since year 2011 and gradually replaced by bisphenol S (BPS) and bisphenol F (BPS)-based products.23 With the aim to replace bisphenol A-based epoxy resins with safer alternatives, Mülhaupt et al. studied bio-based epoxy resins derived from the multifunctional 4-hydroxybenzoates.24 These polymers with special properties, such as high Tg, stiffness and strength, together with excellent chemical and environmental stability, were synthesized to meet the requirements for application as structural adhesives. Parabens were also utilized as building blocks of liquid crystal materials. M. Blanca Ros et al. used 4-hydroxybenzoic acid for the synthesis of bent-shaped liquid crystal materials by copper-catalyzed azide–alkyne click reaction in order to achieve innovative and compact supramolecular organizations.25 Furthermore, Mülhaupt also reported the synthesis of liquid rubber and toughening agents in epoxy resin formulations originating from ethyl-4-hydroxybenzoate.26
Although the wide applications of parabens in the field of epoxy resins and liquid crystal materials have received extensive attention, poly(ether ester) materials from renewable parabens have been rarely reported. In the present study, two nipagin-based dimethyl esters N1 and N2 were synthesized via one-step simple nucleophilic substitutions with methyl chloroacetate and 1,4-dibromobutane, respectively. Subsequently, two series of aromatic poly(ether ester)s, PN1-ωs and PN2-ωs, were synthesized by polymerization of N1 and N2 with renewable linear aliphatic α,ω-diols (n = 2, 3, 4, 6, 10, 12). Herein, the 1,4-dibromobutane bridged N2 features a highly symmetrical structure, and consequently the thermal, crystalline and mechanical properties of PN2-ωs are significantly influenced by this structural characteristic. The dependence of properties of both PN1-ωs and PN2-ωs on the α,ω-diol length and presences of phenoxy-ether linkages was emphatically investigated. The main purpose of this study is to prepare renewable structural materials from naturally occurring resources in order to reduce the utilization of fossil resources in our effort.
Experimental section
Chemical reagents and materials
Nipagin (99%), methyl chloroacetate (98%), and the catalysts tetrabutyl titanate (TBT) (99%), titanium tetraisopropanolate (TIP) (99%), antimony acetate (99%) and zinc acetate (99%) were purchased from Sigma-Aldrich. 1,2-ethylene glycol (E, 99.5%), 1,3-propanediol (P, 99%), 1,4-butanediol (B, 99%), 1,6-hexanediol (H, 98%), 1,10-decanediol (D, 98%), and 1,12-dodecanediol (DD, 98%) were obtained from Shanghai Aladdin Chemical Reagent Co. 1,4-dibromobutane (98%) and other frequently used chemical reagents are of high purity grade and used as received without further purification. Silica-gel slices used for thin-layer chromatography (TLC) were purchased from Qingdao Haiyang Chemical Co. Ltd.
General instrumentation and methods
1H NMR and 13C NMR spectra were recorded in CDCl3 or CF3COOD solvents at 25 °C on a Bruker AVANCE III NMR spectrometer operating at 400 MHz and 100.6 MHz. Tetramethylsilane was used as the internal reference. Fourier transform infrared spectra (FTIR) were recorded using a Bio-Rad FTS6000 spectrophotometer at 25 °C. Polymer samples were prepared by grinding the polymers adequately with KBr powder, followed by compressing the mixture to form a pellet. Fourier transform high resolution mass spectra (FTMS) were recorded on a Varian 7.0T FTMS with positive electrospray ionization (+ESI). The molecular weights and dispersity (D) values of the obtained poly(ether ester)s were determined by size exclusion chromatography (SEC, Waters 2414 differential refraction detector) at 35 °C. CHCl3 and THF were used as the eluents at flow rates of 1.0 mL min−1. The average molecular weights were calibrated against monodisperse polystyrene (PS) standards. Thermogravimetric analysis (TGA) was carried out using a NETZSCH TG209 instrument. In a typical method, the polymer sample was heated from 25 to 800 °C under a nitrogen atmosphere at a rate of 10 °C min−1. The temperature at which 5% weight loss occurred, the temperature at which maximum rate of degradation occurred and residual weight (%) at 800 °C were recorded. Differential scanning calorimetry (DSC) analysis was carried out using a Mettler-Toledo DSC Q100 apparatus from TA instruments. DSC data were acquired from 5–10 mg samples at heating/cooling rates of 10 °C min−1 under a nitrogen flow rate of 20 mL min−1. Indium and zinc were used as the calibration standards for temperature and enthalpy. The glass transition temperatures were determined at a heating rate of 20 °C min−1 from rapidly melt-quenched polymer samples. The treatment of samples for isothermal crystallization was as the follows: the thermal history was erased by heating the sample 50 °C above its Tm and maintained at this temperature for 10 min, and then it was cooled at 50 °C min−1 to the specified crystallization temperature, where it was left to crystallize until saturation. Wide angle X-ray diffraction patterns were recorded on a D/max-2500 diffractometer using CuKα radiation with a wavelength of 0.1542 nm for powder samples obtained directly from synthesis. The tensile assays were carried out in triplicates on dog-bone-shaped specimens (12 × 2 × 0.5 mm3) at a stretching rate of 50 mm min−1 at 25 °C on a HZ1004A tensile testing machine. Before the assays, the samples of PN1-ωs and PN2-ωs were respectively heated to 40 °C and 85 °C for 30 min to eliminate the thermal history. Young's modulus, tensile strength, and elongation at break were obtained by averaging the data from the three parallel test specimens. Dynamic mechanical analysis (DMA) was carried out using a NETZSCH DMA242 analyzer in the controlled force-tension film mode with a preloaded force of 0.1 N, an amplitude of 10 mm, at a fixed frequency of 1 Hz in the −60 to 60 °C range and a heating rate of 3 °C min−1. The measurements were recorded in triplicates using rectangular specimens (length × width × thickness = 25 × 5 × 0.5 mm3). The films used for tensile assay and DMA were obtained from casting of chloroform solutions with concentrations of 0.1 g mL−1 for PN1-ωs and hot-press moulding for PN2-ωs.
Nipagin-based monomer synthesis
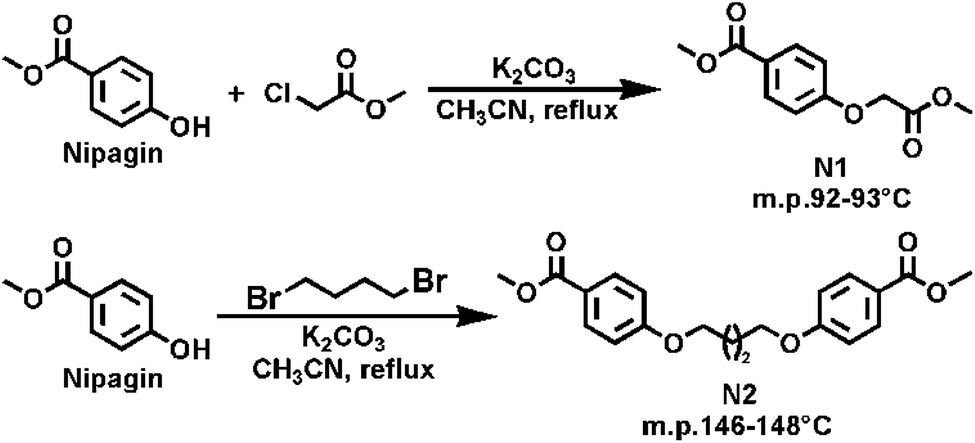
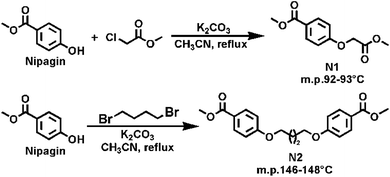
Synthesis of methyl 4-(2-methoxy-2-oxoethoxyl) benzoate (N1). Nipagin (6.14 g, 40 mmol), methyl chloroacetate (6.61 g, 60 mmol), anhydrous K2CO3 (8.34 g, 60 mmol), KI (0.33 g, 2 mmol), and 150 mL anhydrous CH3CN were simultaneously added into a 250 mL three-necked round bottom flask equipped with a magnetic stirrer (Scheme 1). Then the reaction mixture was refluxed at 82 °C for 8 hours under nitrogen atmosphere. The progress of the reaction was detected by TLC. After completion of the reaction, the suspension was cooled to room temperature and a white solid was filtered off. The filtrate was concentrated under vacuum to obtain a light yellow coloured solid. The above obtained filter cake was dissolved in distilled water and washed to remove the inorganic salts, and then the suspended solid was filtered off to obtain a light yellow solid. Finally, the two-step obtained light yellow solid were recrystallized twice from methanol to afford 8.51 g of N1 as a white crystalline solid in high-purity. 95% yield; mp. 92–93 °C. 1H NMR (CDCl3, 400 MHz): δ 8.04–7.97 (m, 2H, Ar-H), 6.95–6.89 (m, 2H, Ar-H), 4.69 (s, 2H, –O–CH2–CO–), 3.88 (s, 3H, ArCOO-CH3), 3.81 (s, 3H, –CH2COO–CH3) ppm; 13C NMR (CDCl3, 100.6 MHz): δ 168.64 (–OCH2–CO–), 166.39 (Ar-CO-) ppm; (Fig. S1 and S2†) HRMS (ESI) m/z calcd for C11H13O5 [M + H]+: 225.0763, found 225.0760, C11H12NaO5 [M + Na]+: 247.0582, found 247.0580.
 |
| | Scheme 1 Synthetic routes for the preparation of nipagin-based dimethyl esters N1 and N2. | |
Synthesis of 4,4′-[1,4-butanediyl-bisoxy]bis-1,1′-dimethyl benzoate (N2). Nipagin (6.45 g, 42 mmol), 1,4-dibromobutane (4.32 g, 20 mmol), anhydrous K2CO3 (5.85 g, 42 mmol), KI (0.16 g, 1 mmol), and 150 mL anhydrous CH3CN were added together in a 250 mL three-necked round bottom flask equipped with a magnetic stirrer (Scheme 1). Subsequently, the reaction mixture was refluxed at 82 °C for 24 hours under nitrogen atmosphere. The progress of the reaction was monitored by TLC. After completion of the reaction, the suspension was cooled to room temperature. A modified procedure was adopted for the work-up. First, the cooled suspension was immediately concentrated under vacuum to obtain a yellow solid. This solid was washed with distilled water to remove the inorganic salts. Then the insoluble solid was filtered off and extracted from chloroform repeatedly thrice. The combined chloroform extracts were collected and concentrated under vacuum to afford a white solid, which was subjected to recrystallization twice from methanol to obtain 6.59 g of N2 as a white solid with high-purity. 92% yield; mp. 146–148 °C. 1H NMR (CF3COOD, 400 MHz): δ 11.68 (solvent), 7.92–7.83 (m, 4H, Ar-H), 6.89–6.80 (m, 4H, Ar-H), 4.10–3.96 (m, 4H, ArO-CH2-), 3.88 (s, 6H, ArCO-CH3), 1.97–1.83 (m, 4H, ArOCH2-CH2-) ppm; 13C NMR (CF3COOD, 100.6 MHz): δ 170.35 (Ar-CO-), 163.26 (Ar-C), 160.96 (solvent), 160.53 (solvent), 160.10 (solvent), 159.67 (solvent), 131.71 (Ar-C), 121.00 (Ar-C), 118.57 (Ar-C), 115.75 (solvent), 114.11 (Ar-C), 112.93 (solvent), 110.11 (Ar-C), 67.74 (ArO-CH2-), 52.16 (ArCOO-CH3), 24.99 (ArOCH2-CH2-) ppm; (Fig. S3 and S4†) HRMS (ESI) m/z calcd for C20H23O6 [M + H]+: 359.1495, found 359.1492, C20H22NaO6 [M + Na]+: 381.1314, found 381.1311.
General procedure for the preparation of poly(ether ester)s
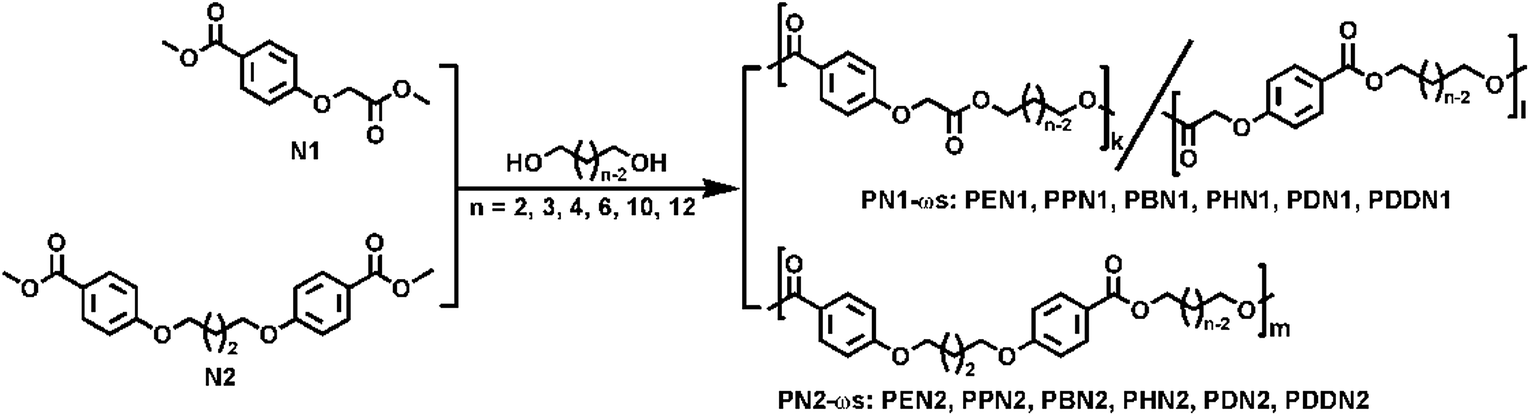
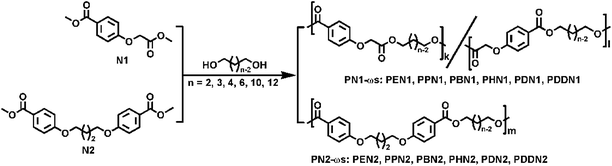
PN1-ωs and PN2-ωs were synthesized by reacting linear aliphatic α,ω-diols with N1 and N2, respectively. The polymerization reactions were carried out under the scale of grade g by a usual two-step melt polycondensation in a 50 mL Schleck round-bottomed flask equipped with a magnetic stirrer, a nitrogen inlet and a vacuum distillation outlet. The overhead stirring was replaced by the magnetic stirring, which has been verified to be easy-operation and ensure the ideal blend of monomers and oligomers and the final molecular weights.9 The detailed polymerization routes are shown in Scheme 2. For long-chain α,ω-diols (n = 6, 10, 12), a 1:1.05 molar ratio of dimethyl ester to α,ω-diol was sufficient to obtain the expected molecular weight and ensure hydroxyl termination. However, for short-chain α,ω-diols (n = 2, 3, 4), the polymerization process was a little problematic. Low boiling points of short-chain α,ω-diols make them very easy to volatilize and therefore an excess of α,ω-diol is required to compensate for the loss of short-chain α,ω-diols. Furthermore, the high crystallizability of N2 allowed the polycondensation stage just to a certain extent and hampered the progress of the reaction. Hence, the polymerization conditions for the poly(ether ester)s were individually optimized in order to obtain satisfactory molecular weights. The representative grade-screening of feed ratios of N1 to 1,2-ethylene glycol, catalysts, and reaction temperatures and the results of their effects on polymerization are all presented in Table 1. Based on these results, tetrabutyl titanate (TBT, 0.6% molar, relative to dimethyl ester) was selected as the optimal catalyst.
 |
| | Scheme 2 Synthetic routes for the preparation of PN1-ωs and PN2-ωs poly(ether ester)s. | |
Table 1 Screening of polymerization conditions of poly(ether ester)s based on PEN1 according to the entry number 1–12 successively
| Entry |
N1:ethylene glycol (mol:mol) |
Catalysta |
Transesterificationb (°C, h) |
Polycondensationb (°C, h) |
Mnc (g mol−1) |
Mwc (g mol−1) |
Dc |
Isolated yieldd |
| TBT = tetrabutyl titanate; TIP = titanium tetraisopropanolate; ATA = antimony acetate; ZAT = zinc acetate. Molar ratio of catalyst to N1 = 0.6%. Transesterification reactions carried out under low nitrogen flow; polycondensation reactions carried out under 0.03–0.06 mbar. SEC carried out in CHCl3 against PS standards. After purification by precipitating from methanol. |
| 1 |
1:2.0 |
TBT |
160, 2; 180, 2 |
200, 2; 220, 2 |
4600 |
5000 |
1.1 |
43% |
| 2 |
1:2.2 |
TBT |
160, 2; 180, 2 |
200, 2; 220, 2 |
4600 |
5100 |
1.1 |
64% |
| 3 |
1:2.5 |
TBT |
160, 2; 180, 2 |
200, 2; 220, 2 |
6500 |
8500 |
1.3 |
87% |
| 4 |
1:2.8 |
TBT |
160, 2; 180, 2 |
200, 2; 220, 2 |
5100 |
7100 |
1.4 |
77% |
| 5 |
1:3.0 |
TBT |
160, 2; 180, 2 |
200, 2; 220, 2 |
5200 |
6700 |
1.3 |
75% |
| 6 |
1:2.5 |
TIP |
160, 2; 180, 2 |
200, 2; 220, 2 |
4800 |
7200 |
1.5 |
78% |
| 7 |
1:2.5 |
ATA |
160, 2; 180, 2 |
200, 2; 220, 2 |
5900 |
8300 |
1.4 |
83% |
| 8 |
1:2.5 |
ZAT |
160, 2; 180, 2 |
200, 2; 220, 2 |
3400 |
5400 |
1.6 |
56% |
| 9 |
1:2.5 |
TBT |
160, 2; 180, 2; 200, 2 |
200, 2; 220, 2 |
5900 |
8900 |
1.5 |
75% |
| 10 |
1:2.5 |
TBT |
160, 2; 180, 2 |
200, 2; 220, 2; 240, 2 |
5800 |
9300 |
1.6 |
85% |
| 11 |
1:2.5 |
TBT |
160, 2; 180, 2 |
200, 2; 220, 2; 240, 3 |
5700 |
9100 |
1.6 |
86% |
| 12 |
1:2.5 |
TBT |
160, 2; 180, 2 |
200, 2; 220, 2; 240, 2; 250, 1 |
5900 |
8900 |
1.5 |
85% |
Before carrying out the transesterification reaction, the apparatus was purged with nitrogen for 10 minutes to ensure that no residual oxygen remained in the system, thus preventing oxidation during polymerization. Transesterification reactions were carried out at 160–200 °C for 4–6 hours under a low nitrogen flow. Polycondensation reactions were then carried out at 200–250 °C for 4–7 hours under a vacuum of 0.03–0.06 mbar. In case of each reaction, the reaction was continued till the viscosity of the mixture increased to such an extent that the stirrer was jammed, which indicated a good progress of the polymerization reaction. Subsequently, nitrogen was purged to prevent degradation of the product while the reaction mixture was cooled to room temperature. The crude product was dissolved in minimum amount of chloroform and precipitated in excess methanol in order to remove the oligomers and impurities. Finally, the polymers were obtained after filtration, followed by thorough washing with methanol, and dried in vacuo. The product coding is adopted the combination of acronyms of dimethyl ester monomer and α,ω-diol. For example, PEN1 denotes the poly(ether ester) that N1 was polymerized with 1,2-ethylene glycol, and by this analogue. The characterization data for PBN1 and PDN2 is presented along with the other obtained polymers, which have similar abbreviations and their corresponding spectra can be seen in Fig. S5–S12.†
PBN1. 1H NMR (CDCl3, 400 MHz): δ 8.17–7.82 (m, 2H, Ar-H), 7.10–6.78 (m, 2H, Ar-H), 4.70 (s, 2H, –O–CH2–CO–), 4.50–4.03 (m, 4H, –COO–CH2–), 2.10–1.63 (m, 4H,–OCH2–CH2–) ppm; 13C NMR (CDCl3, 100.6 MHz): δ 168.26 (–OCH2–CO–), 165.87 (Ar-CO-), 161.44 (Ar-C), 131.52 (Ar-C), 123.60 (Ar-C), 114.23 (Ar-C), 65.02 (–O–CH2–CO–), 64.90–64.01 (m, –COO–CH2–), 25.50–24.95 (m, –OCH2–CH2–) ppm; FTIR: 2957, 1757 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1706 (CO), 1605, 1510, 1275, 1173, 1078, 942, 850, 769, 696 cm−1.
O), 1706 (CO), 1605, 1510, 1275, 1173, 1078, 942, 850, 769, 696 cm−1.
PDN2. 1H NMR (CF3COOD, 400 MHz): δ 11.66 (solvent), 8.47–7.86 (m, 4H, Ar-H), 7.43–6.80 (m, 4H, Ar-H), 4.64–4.32 (m, 4H, –COO–CH2–), 4.42–4.12 (m, 4H, ArO-CH2-), 2.26–2.18 (m, 4H, ArOCH2-CH2-), 2.03–1.81 (m, 4H, –COOCH2–CH2–), 1.72–1.29 (m, 12H, –COO(CH2)2–(CH2)3–) ppm; 13C NMR (CF3COOD, 100.6 MHz): δ 170.37 (Ar-CO-), 163.20 (Ar-C), 161.90 (solvent), 161.47 (solvent), 161.03 (solvent), 160.60 (solvent), 131.83 (Ar-C), 121.74 (Ar-C), 118.58 (Ar-C), 115.76 (solvent), 114.41 (Ar-C), 112.94 (solvent), 110.13 (Ar-C), 68.10 (ArO-CH2-), 66.73 (–COO–CH2–), 28.95, 28.74, 27.96, 25.44, 25.04 (ArOCH2-CH2-) ppm; FTIR: 2927, 2855, 1706 (CO), 1604, 1510, 1251, 1170, 1105, 1009, 975, 849, 771, 696 cm−1.
Results and discussion
Synthesis and molecular characterization of the poly(ether ester)s
The melting points of N1 and N2 are 92–93 °C and 146–148 °C, respectively. Especially the Tm of N2 was very close to that of dimethyl terephthalate (DMT) (139–141 °C). DMT is a commonly-used synthon for the manufacturing of engineering plastics (i.e. PET, PBT). Hence, N1 and N2 possess inherent characteristics that make them well suitable as excellent renewable compensates for DMT during the production of thermoplastic polymeric materials. In this study, the polymerization reaction was carried out in the absence of solvents in order to simulate the conditions of the industrial production of polyesters as much as possible.
As mentioned above, the polymerization of short-chain α,ω-diols (n = 2, 3, 4) was not so smooth because of their low boiling points. More efforts will be required to optimize the polymerization conditions for these α,ω-diols. Keeping the reaction conditions (catalyst: TBT, transesterification at 160 °C for 2 h, 180 °C for 2 h; polycondensation at 200 °C for 2 h, 220 °C for 2 h) unchanged, the optimized feed ratios of dimethyl ester to diol were found to be 1:1.8 and 1:1.3 for PPN1 and PBN1, respectively. The corresponding feed ratios were also optimum for PPN2 and PBN2. However, regarding PEN1 the entire range of practical feed ratios from 1:2 to 1:3 failed to give a satisfactory molecular weight. Although the feed ratio of 1:2.5 was the best compared to others (entry 1–5, Table 1), the desired molecular weight could not be obtained. Consequently, the preferable 1:2.5 was chosen for further optimization of the polymerization conditions. It is well known that a catalyst plays an important role in chemical reactions. Therefore, the most suitable catalyst was needed to screen from the four different types of catalysts (entry 3 and 6–8, Table 1). Herein, ATA was found to have a catalytic activity comparable to TBT and superior to the other two catalysts (TIP and ZAT). Hence, the cheaper and environmentally friendly TBT was chosen as the optimum one. Subsequently, the temperatures and time periods for the transesterification and polycondensation steps were further optimized (entry 9–12, Table 1). The important inferences drawn from the optimization process are that increasing the temperature for polycondensation was favorable for the reaction. However, excessively high temperatures and long times led to the degradation and staining of products due to the oxidation of phenoxy-linkages and decomposition of TBT.9 The molecular weight of PEN1 could not be further improved in despite of our best. The low molecular weight was probably the cause of high stiffness of PEN1, leading to the jamming of magnetic stirrer in the later stages of polymerization. In case of PEN2, the results were more disappointing. Apart from high stiffness, the strong crystallizability also prevented the molecular weight from reaching a high value (Table 2). Soon after the beginning of polycondensation stage, a large amount of white solids appeared and prevent the proceeding of reaction. Furthermore, the strong crystallizability of PEN2 makes it insoluble in chloroform. Trifluoroacetic acid, hexafluoroisopropanol, or other excellent solvents for polyesters are probably able to dissolve PEN2 completely.27 Other polymerization methods (overheading stirring, elevating pressure during transesterification stage) have also been carried out in our group for increasing the molecular weight as much as possible.
Table 2 Feed ratio, molecular weight, dispersity and isolated yield of PN1-ωs and PN2-ωs
| Poly(ether ester) |
Diester:diol (mol:mol) |
Mn (g mol−1) |
Mw (g mol−1) |
D |
Isolated yieldd |
| SEC carried out in CHCl3 relative to PS calibrations. SEC carried out in THF relative to PS calibrations. Insoluble in CHCl3 and THF. After purification by precipitating from methanol. |
| PEN1a |
1:2.5 |
5800 |
9300 |
1.6 |
85% |
| PPN1b |
1:1.8 |
10300 |
17500 |
1.7 |
86% |
| PBN1b |
1:1.3 |
11300 |
20700 |
1.8 |
85% |
| PHN1b |
1:1.05 |
14900 |
26300 |
1.8 |
92% |
| PDN1b |
1:1.05 |
16900 |
30300 |
1.8 |
94% |
| PDDN1b |
1:1.05 |
16900 |
31600 |
1.8 |
95% |
| PEN2c |
1:2.5 |
— |
— |
— |
84% |
| PPN2a |
1:1.8 |
9600 |
17600 |
1.8 |
87% |
| PBN2a |
1:1.3 |
10500 |
18400 |
1.8 |
88% |
| PHN2a |
1:1.05 |
14200 |
24300 |
1.7 |
94% |
| PDN2a |
1:1.05 |
21700 |
32100 |
1.5 |
95% |
| PDD2a |
1:1.05 |
16300 |
30100 |
1.8 |
96% |
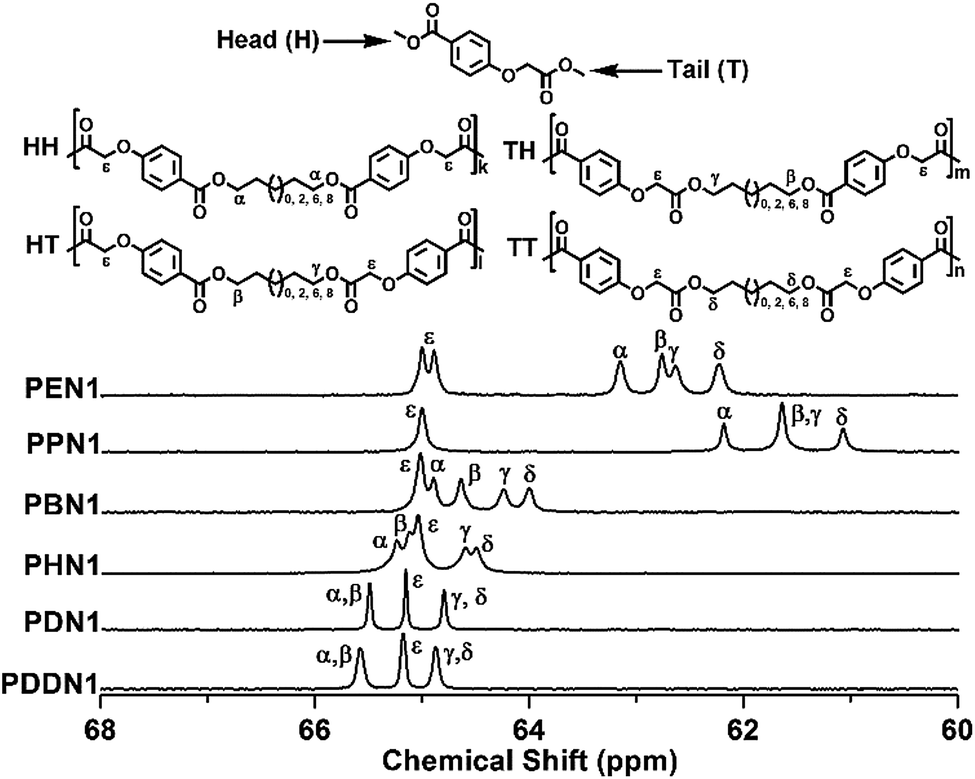
The chemical structures of the two series of poly(ether ester)s were determined by 1H NMR (Fig. S7 and S9†), 13C NMR (Fig. S8 and S10†), and FTIR spectroscopy (Fig. S11 and S12†). It should be noted that the signals attributed to the methylenes adjacent to the hydroxy-oxygens in the 1H NMR spectra of PN1-ωs, are split into a triplet for PEN1, PPN1, and PBN1, and gradually transform into two triplets for PHN1, PDN1, and PDDN1. This difference arises due to the inequivalent nature of the two types of carbonyls in N1 units and this inequivalence appears to become more obvious with the increase in α,ω-diol length. However, this nature was absent in case of PN2-ωs. The methylenes adjacent to the hydroxy-oxygens in the spectra of PN2-ωs, split into a single triplet in the entire series from PEN1 to PDDN1, since the two carbonyls in N2 were completely equivalent.
The inequivalent nature of the two carbonyls in PN1-ωs is evident not only in the 1H NMR, but also in the 13C NMR spectra. In this study, quantitative 13C NMR spectroscopy was used for the microstructure analysis of the poly(ether ester)s. As previously reported, apart from the substituted aromatic carbons, the methylene carbons adjacent to any asymmetrical factor also become sensitive to sequence distributions at the dyad level.28 The splitting patterns of the methylene carbons attached to the hydroxy-oxygens along the main-chains of PN1-ωs are illustrated in Fig. 1. Since the N1 unit lacks symmetry, the signals assigned to the methylene carbons connected to the hydroxy-oxygens, appear to split into a total of four peaks due to four types of dyads (HH, HT, TH, and TT) are possible. The four types of dyads generated due to the orientations of N1 were incorporated into the polymeric chains adopting an arbitrary manner. The chemical shifts of the methylene carbons show certain amount of shifting towards the down field region as a whole depending on the increase in the α,ω-diol length. These shifts are also a result of the changes in inductive and electronic effects as the variations of α,ω-diol lengths. As shown in Fig. 1, the peak ε corresponding to the methylene carbon attached to the phenolic oxygen is an exclusive one, which maintained its position at about 65.2 ppm due to the minor influence of inductive and electronic effects towards α,ω-diol length varying. The shorter the length of the α,ω-diol, the more obvious the inequivalence of two carbonyls in PN1-ωs. α versus β and γ versus δ inherently belong to the same type of carbons. However, when α,ω-diol lengths become shorter (n = 2, 3, 4), the difference between α and β carbons is clear and the corresponding signals are clearly resolved due to the uncoordinated inductive and electronic effects of different carbonyls. The same reasoning can be applied to γ and δ carbons as well. On the other hand, when the α,ω-diol length gradually increases, the inductive and electronic effects due to the different carbonyls decease progressively. As a consequence, the differences of both α versus β and γ versus δ gradually decrease with the same trend for both inductive and electronic effects. This results in the fusion of the four peaks into one peak as seen in PDN1 and PDDBN1. Such a situation does not apply for PN2-ωs since N2 contains two magnetically equivalent ester carbonyls as mentioned above (Fig. S13†). A large difference of chemical shift is also probably due to the varying degree of inductive and electronic effects based on the α,ω-diol length.
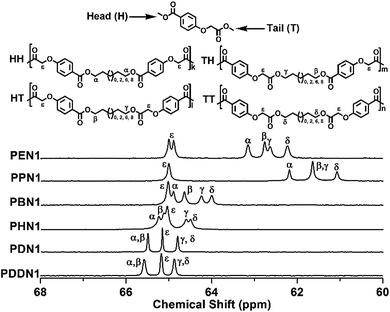 |
| | Fig. 1 The splitting situations of the methylenes adjacent to the hydroxy-oxygen in PN1-ωs with the indications of the dyads to which they are assigned. | |
Thermal properties
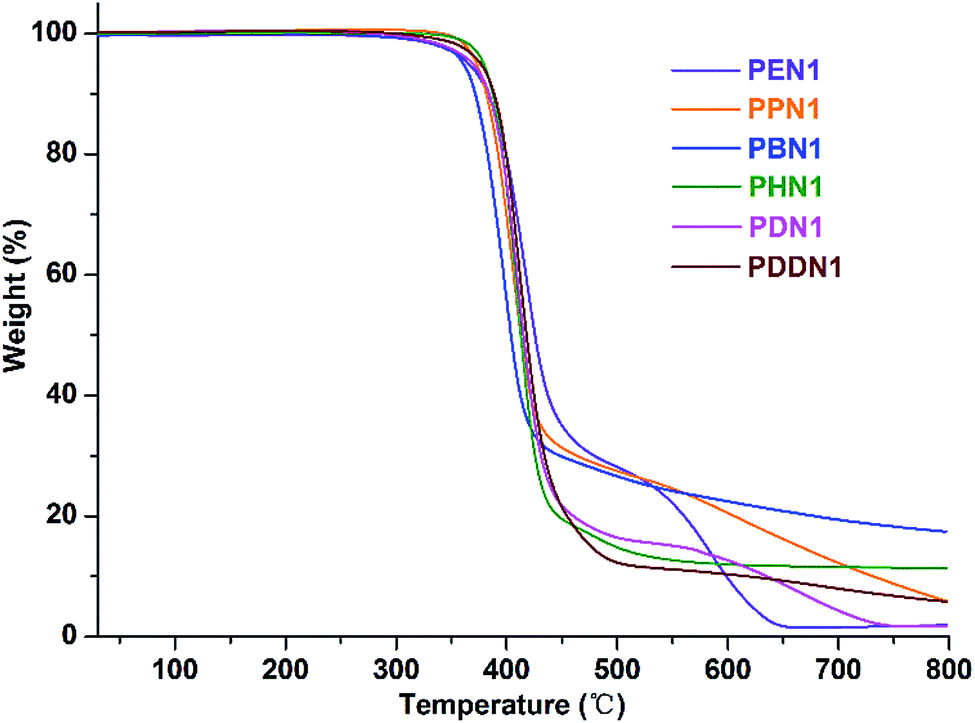
The thermal behaviours of the two series of nipagin-based poly(ether ester)s PN1-ωs and PN2-ωs were studied by TGA and DSC under inert atmosphere. The TGA and derivative curves are depicted in Fig. 2 and S14–S16,† and the thermal parameters are gathered in Table 3. The weight loss profiles revealed a thermal degradation mechanism involving one-main step or two steps occurring at 400–450 °C and 550–700 °C, respectively. Except for PBN1, all the other polymers in PN1-ωs and the whole series of PN2-ωs, exhibit a more or less two-step degradation pattern. The two-step degradation mechanism can be explained as follows: first, the pyrolysis of ester groups along the polyester main chains resulted in the release of molecular fragments to bring about the first-step of degradation. Straight after, more stable structures were formed with the continuous increase of temperature and thus caused the second degradation.29 Negligible residual weights at 800 °C are observed for PN1-ωs and PN2-ωs samples except for PBN1 and PHN1, which have final residues amounting to 11.34% and 17.60% of their initial weights, respectively.
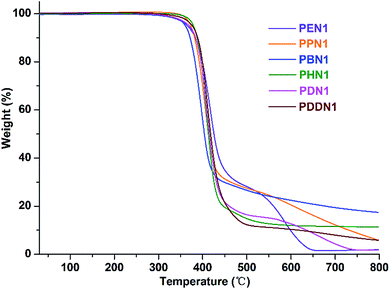 |
| | Fig. 2 TGA curves of PN1-ωs recorded from 25–800 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere. | |
Table 3 Thermal property parameters of PN1-ωs and PN2-ωs
| Poly(ether ester) |
TGA |
DSC |
| T5%a (°C) |
Tdb (°C) |
Wc (%) |
First coolinge |
Second heatinge |
| Tgd (°C) |
Tc (°C) |
ΔHc (J g−1) |
Tg (°C) |
Tc (°C) |
ΔHc (J g−1) |
Tm (°C) |
ΔHm (J g−1) |
| Temperature at which 5% weight loss was observed. Temperature for maximum degradation rate. Remaining weight at 800 °C. Glass transition temperatures taken as the inflection points of the heating DSC traces of melt-quenched samples recorded at 20 °C min−1. Glass transition, melting, and crystallization temperatures (Tg, Tm, Tc) and melting and crystallization enthalpies (ΔHm, ΔHc) calculated from the second heating DSC traces of precipitated samples at heating/cooling rates of 10 °C min−1. Mn and Mw of PDT are 15200 and 26800, respectively. |
| PEN1 |
365 |
417/586 |
2.0 |
54.8 |
— |
— |
55.7 |
— |
— |
— |
— |
| PPN1 |
372 |
406/611 |
5.9 |
39.4 |
— |
— |
40.0 |
— |
— |
— |
— |
| PBN1 |
361 |
398 |
17.6 |
28.5 |
— |
— |
29.3 |
— |
— |
— |
— |
| PHN1 |
379 |
409/480 |
11.3 |
12.6 |
— |
— |
13.1 |
— |
— |
— |
— |
| PDN1 |
367 |
409/658 |
1.6 |
−8.0 |
— |
— |
−8.3 |
6.2 |
20.4 |
65.1 |
41.7 |
| PDDN1 |
375 |
411/656 |
5.6 |
−12.9 |
35.8 |
63.9 |
— |
— |
— |
Tm1 = 67.5, Tm2 = 76.2 |
ΔHm1 = 16.5, ΔHm2 = 47.8 |
| PEN2 |
411 |
459/656 |
1.5 |
62.3 |
193.7 |
154.6 |
— |
209.4 |
10.5 |
Tm1 = 214.3, Tm2 = 221.9 |
ΔHm1 = 48.6, ΔHm2 = 96.5 |
| PPN2 |
386 |
418/646 |
4.9 |
45.9 |
169.8 |
|
— |
— |
— |
Tm1 = 178.6, Tm2 = 187.8 |
ΔHm1 = 36.2, ΔHm2 = 116.8 |
| PBN2 |
350 |
446/669 |
7.5 |
35.6 |
146.0 |
152.1 |
— |
— |
— |
Tm1 = 157.5, Tm2 = 165.8 |
ΔHm1 = 25.2, ΔHm2 = 127.4 |
| PHN2 |
395 |
432/614 |
2.1 |
32.2 |
131.0 |
163.8 |
— |
— |
— |
Tm1 = 152.4, Tm2 = 160.3 |
ΔHm1 = 16.5, ΔHm2 = 147.8 |
| PDN2 |
395 |
424/633 |
2.8 |
16.1 |
103.3 |
169.5 |
— |
— |
— |
Tm1 = 131.5, Tm2 = 136.8 |
ΔHm1 = 5.9, ΔHm2 = 164.2 |
| PDDN2 |
402 |
436/672 |
9.0 |
10.2 |
101.0 |
187.2 |
— |
— |
— |
131.7 |
187.8 |
| PDTf |
381 |
405 |
3.0 |
−0.2 |
|
|
— |
— |
— |
Tm1 = 120.0, Tm2 = 127.5 |
ΔHm1 = 8.9, ΔHm2 = 40.7 |
Furthermore, the negligible residual weights indicate that the lost components are probably due to the release of gases like CO and CO2, which were generated from the thermal pyrolysis and oxidation of poly(ether ester) main chains. The combined thermal data indicate that the poly(ether ester)s derived from the renewable nipagin feature a more than satisfactory resistance to heat with the onset decomposition temperature of about 350 °C. Although a direct correlation of decomposition temperature with the length of α,ω-diol is not evident in the two series, the thermal stability could also be comparatively evaluated from the ranges of the values observed from each set of the poly(ether ester)s. The resistance to heat is undoubtedly closely related to the molecular weight and the length of α,ω-diol in case of both PN1-ωs and PN2-ωs. Specifically, heat-resistivity increases with the molecular weight and decreases with the α,ω-diol length.30,31 The thermal stabilities of PN2-ωs are generally 15–30 °C higher than that of PN1-ωs when the molecular weights and α,ω-diol lengths are similar. It has been proved that the molecular structure is also crucial to thermal stability.7 As mentioned in the above section, N1 lacks structural symmetry, whereas N2 is highly symmetrical. Consequently, PN2-ωs feature superior thermal stability as compared to PN1-ωs. By comparing the data of PDN1 with the DMT-based polydecylene terephthalate (PDT),32 (the data of PDT here is original), it is noteworthy that the incorporation of phenoxy-linkages in N1 units has no significant influence on the thermal stability of PDN1 with an enhanced Td value and the T5% just decreasing 14 °C. Due to the high crystallizability, PDN2 features the superior thermal property than PDT. The valuable conclusion drawn from the TGA study is that the renewable nipagin-based poly(ether ester)s feature excellent thermal stabilities even if the insertion of phenoxy-linkages that make them have the potential characteristics for use as structural materials.
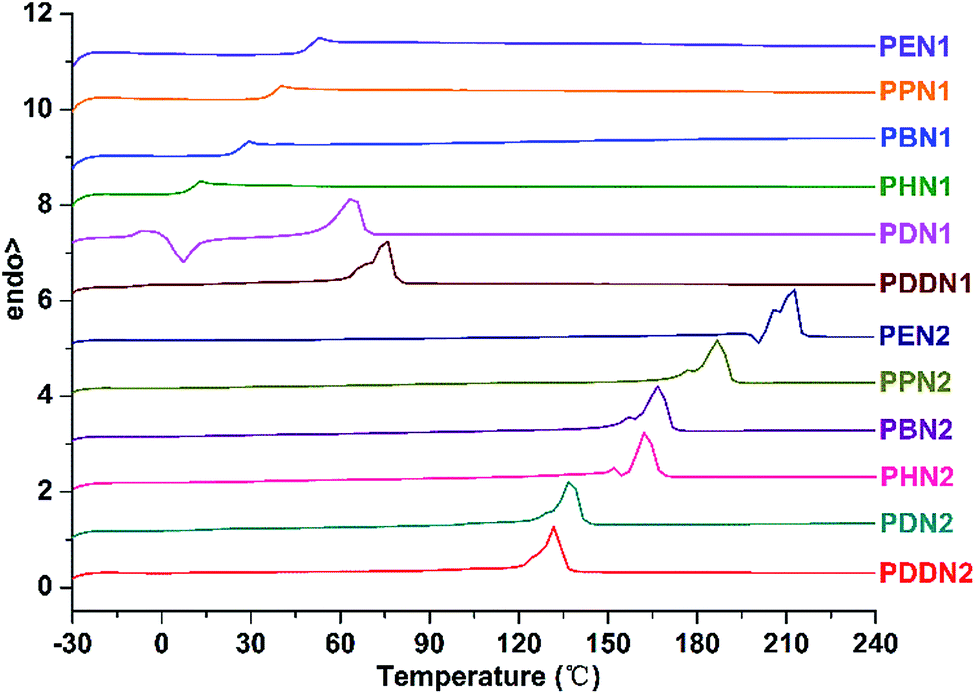
DSC analysis also demonstrates the differences in thermal properties between PN1-ωs and PN2-ωs. The DSC curves of the two series of poly(ether ester)s are depicted in Fig. 3, and the corresponding glass transition, melting and crystallization data are presented in Table 3. In the series of PN1-ωs, only when the α,ω-diols have lengths with n greater than or equal to 10, namely PDN1 and PDDN1, exhibit semi-crystalline properties. All the other poly(ether ester)s appear to be amorphous at the given heating/cooling rate of 10 °C min−1. Furthermore, the Tg values of PN1-ωs exhibit a tendency to decrease with the increase in α,ω-diol length. It can be explained on the basis that the density of rigid aromatic rings along the poly(ether ester) chains progressively decreases with the concomitant increase in α,ω-diol length.30,31 However, PDDN1 and the entire series of PN2-ωs present inconspicuous glass transition temperature on the second DSC heating run. Due to the high crystallizability, the glass transition temperatures of PN2-ωs will not be clearly emerged in the DSC curves under the condition of no melt-quench for the samples. However, obvious Tg can be observed after the samples were melt-quenched. The Tg values for PN2-ωs obtained from melt-quenched samples also exhibit a tendency to decrease with the increase in α,ω-diol length as observed for PN1-ωs (Fig. S17 and S18†).
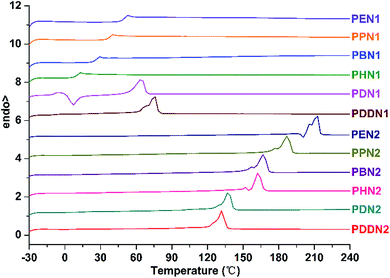 |
| | Fig. 3 Second heating DSC curves of PN1-ωs and PN2-ωs after precipitation from methanol carried out from −30 to 240 °C at a heating/cooling rate of 10 °C min−1. | |
It has been previously reported that the crystallizability of DMT-based aromatic polyesters (i.e. PET, PPT, PBT) is gradually enhanced with the increase in α,ω-diol length.33 The DSC analysis of PN1-ωs also arrives at this conclusion. PDN1 shows a cold crystallization peak and a following melting peak on the second heating run, whereas a single melting peak is observed in the case of PDDN1, indicating that PDN1 is more difficult to crystallize from the melt compared to PDDN1. The dependence of crystallizability on the α,ω-diol length is more obvious in PN2-ωs series. Firstly, no cold crystallization peak is observed from the second heating runs of PN2-ωs. Secondly, the two melting peaks fused into one gradually and the melting enthalpy increases gradually with the increase in α,ω-diol length. All these results demonstrate that the crystallizability is progressively enhanced with the increase in α,ω-diol length in each series and that the PN2-ωs have stronger crystallizability than PN1-ωs due to the more symmetric structural characteristic of N2 unit. However, the melting point of PN2-ωs decreases gradually with the increase in α,ω-diol length. This is largely attributed to a steady increase in the aliphatic methylene sections, which result in a decrease in the density of aromatic rings and thus caused a decrease in melting point in spite of the enhanced crystallizability due to the gradual increase in α,ω-diol length.
Powder X-ray diffraction analysis
The preliminary X-ray diffraction analysis of PN1-ωs and PN2-ωs is fully consistent with the results afforded by DSC analysis. The powder WAXD profiles recorded for PN1-ωs and PN2-ωs are depicted in Fig. S19 and S20† and the corresponding data are gathered in Tables S1 and S2.† The presence of sharp diffraction peaks reveals the semi-crystalline nature for all the poly(ether ester)s. However, the diffraction angles (2θ) and intensities of the peaks in each of the patterns have large discrepancies, which indicate that each of the poly(ether ester) species possesses a different crystalline structure.30,31 Regarding the intensities of corresponding peaks in PN1-ωs and PN2-ωs series, PN2-ωs show stronger crystallizability than PN1-ωs in accordance with DSC analysis. Nevertheless, certain differences still exist between them. For example, the second DSC traces of PN1-ωs (n = 2, 3, 4, 6) show independent Tg and no endothermic melting peaks, which prove that PEN1, PPN1, PBN1, and PHN1 could not crystallize from the melt during the first cooling run or the second heating run at the given heating/cooling rate of 10 °C min−1. However, the WAXS profiles of PN1-ωs (n = 2, 3, 4, 6) exhibit certain discrete scattering peaks, indicating that the samples coming directly from precipitation actually are able to crystallize and lower heating/cooling rates may facilitate the samples to crystallize from the melt or the glassy state. As explained for DSC analysis, the low crystallizability of PN1-ωs is attributed to the asymmetrical structural characteristic of N1. Furthermore, the WAXD profiles of PHN1 and PHN2 are not well resolved, demonstrating the length of 1,6-hexandiol is unfavourable for the crystal packaging. On the contrary, the diffraction patterns of PBN1 and PBN2 show well discrete diffraction peaks since 1,4-butanediol is favourable for crystal packaging. The more prominent scattering of PPN2 compared to PPN1 also prove the higher crystallizability of PN2-ωs compared to PN1-ωs, which complies with the results of DSC as well.
Isothermal crystallization
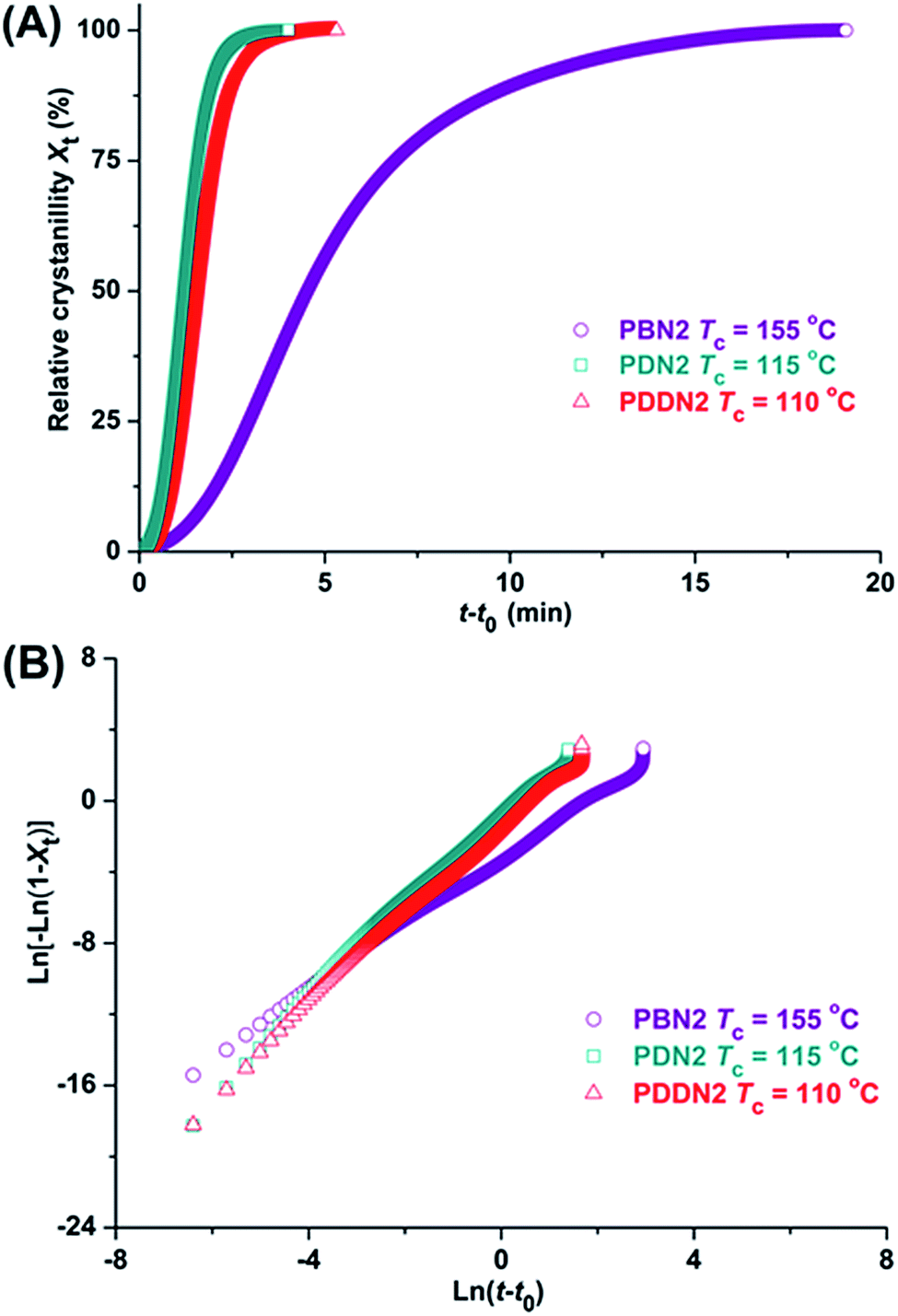
The first cooling DSC traces obtained from molten samples prove that PDDN1 and the whole series of PN2-ωs are able to crystallize from the melt. After crystallization at a constant cooling rate of 10 °C min−1, all the poly(ether ester)s recovered about 80–95% of their initial crystallinities and show the approximate melting temperatures compared to those displayed by their initial samples. Given the close relationship between crystallizability from the melt and material processing, as well as considering the nipagin-based poly(ether ester)s for quantitative evaluation of the influence of α,ω-diol length on crystallization, the isothermal crystallization was carried out at temperatures ranging from 55 to 200 °C. Unfortunately, isothermal crystallization could not be carried out at exactly the same crystallization temperature due to the large differences in crystallization rates of the polymers. Nevertheless, the selection of crystallization temperature should be as close as possible and subsequently valuable conclusions could be drawn. The evolution of the relative crystallinity, Xt versus crystallization time t, together with their respective Avrami ln[−ln(1 − Xt)] versus ln(t − t0) plots for illustration are depicted in Fig. 4 and S21.† Kinetic data, including the observed times of onset t0 and crystallization half-times t1/2, as well as the corresponding Avrami parameters afforded by this analysis, are presented in Table 4. Due to large differences in crystallization temperatures, it is difficult to draw a specific conclusion. Actually, the low crystallization temperature and melting enthalpy have already indicated the inferior crystallizability of PDDN1. Furthermore, as expected, PN2-ωs display stronger crystallizability than PN1-ωs and the crystallization rates exhibit an increasing tendency with the increase in α,ω-diol length, in spite of having different crystallization temperatures. Avrami exponents ns oscillate in the range of 1.67–2.63, which indicate a complex axialitic-spherulitic crystallization mechanism that may be adopted for the poly(ether ester) samples. The double-logarithmic plots indicate that primary and secondary crystallizations took place at the selected time intervals. The early stages of crystallization matched excellently with the Avrami model. However, the double-logarithmic plots gradually deviated from the Avrami equation, which was probably due to the secondary crystallization that caused by the collision of crystals.
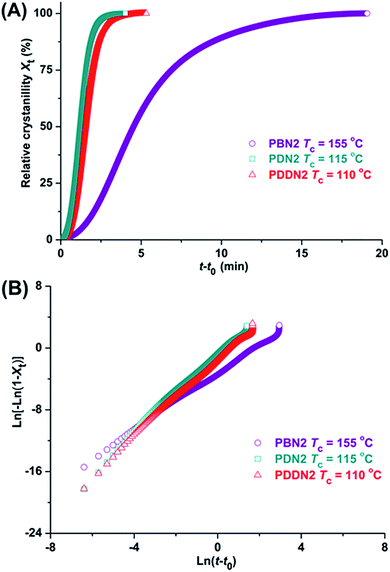 |
| | Fig. 4 Isothermal crystallization of PBN2, PDN2 and PDDN2 at the indicated temperatures. Relative crystallinity versus time plots (A), and ln[−ln(1 − Xt)] versus ln(t − t0) plots (B). | |
Table 4 Isothermal crystallization data of PDDN1, PEN2, PBN2, PHN2, PDN2, PDDN2
| Poly(ether ester) |
Tca (°C) |
ΔTa (°C) |
t0b (min) |
t1/2c (min) |
nd |
–lnke |
Tmf (°C) |
| Tc: isothermal crystallization temperature, ΔT = Tm − Tc. Onset crystallization time. Crystallization half-time. Avrami exponent. Kinetic rate constant k (min−1). Melting temperature after crystallization. |
| PDDN1 |
55 |
21.2 |
0.48 |
4.82 |
2.63 |
4.00 |
76.9 |
| PEN2 |
200 |
21.9 |
0.27 |
1.35 |
1.67 |
0.56 |
222.4 |
| PBN2 |
155 |
10.8 |
0.28 |
4.81 |
2.03 |
3.45 |
166.3 |
| PHN2 |
145 |
15.3 |
0.64 |
5.05 |
2.50 |
4.09 |
160.7 |
| PDN2 |
115 |
21.8 |
0.23 |
1.42 |
2.23 |
0.83 |
137.6 |
| PDDN2 |
110 |
21.7 |
0.25 |
1.77 |
2.56 |
1.54 |
132.5 |
Stress–strain behaviour
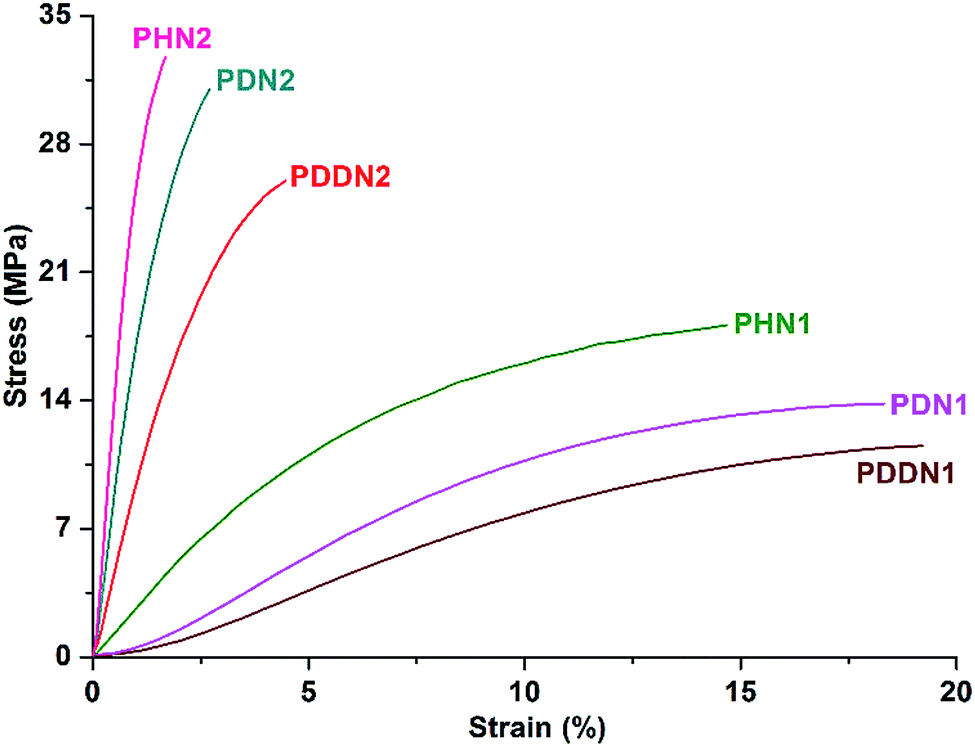
In order to investigate the mechanical properties of the nipagin-based poly(ether ester)s and also the dependence of these properties on α,ω-diol lengths, preliminary evaluation of the mechanical properties was performed by tensile assays and dynamic mechanical analysis. The films of PHN1, PDN1 and PDDN1 used for mechanical property analysis were cast from their respective chloroform solutions. Meanwhile, the films for PHN2, PDN2, and PDDN2 were made from the hot-press molding due to their highly fragile and crystallization. The stress–strain curves of the poly(ether ester) specimens measured are shown in Fig. 5 and the mechanical property parameters are presented in Table 5. PHN2, PDN2, and PDDN2 exhibit the typical characteristics of a brittle material, whereas PHN1, PDN1, and PDDN1 feature soft and weak characteristic properties originating from their inferior crystallizabilities due to the asymmetric nature of N1. Young's modulus and tensile strength show a nearly steady decreasing trend for both PN1-ωs and PN2-ωs with the increase in the α,ω-diol length in their respective series.30,31 However, the elongations at break show a reverse trend as that of Young's modulus and tensile strength. It is observed that the elongations at break increase gradually with the increases of α,ω-diol lengths. With the similar length of α,ω-diol, PN2-ωs feature higher elastic modulus and tensile strength compared to PN1-ωs. Meanwhile, the elongations at break are opposite to above. This is undoubtedly due to the excellent symmetry in the structure of N2 unit as compared to that of N1 unit, which results in PN2-ωs having higher crystallinity than PN1-ωs. From the data in Table 5 we can observe that the nipagin-based poly(ether ester)s especially for PN2-ωs have excellent mechanical properties with Young's modulus and tensile strength values ranging from 600 to 800 MPa and 10 to 35 MPa, respectively. Furthermore, the incorporation of phenoxy-linkages has significantly enhanced influence on the toughness of materials by comparing the data of PDN1 with the petroleum-based PDT,32 (the data of PDT here is original). Due to the high crystallizability, PDN2 exhibit the superior Young's modulus and tensile strength than PDT. The valuable conclusion drawn from tensile assays is that the nipagin-based materials have the excellent mechanical properties (i.e. Young's modulus, tensile strength) so that these polymers have the potential for use as structural materials.
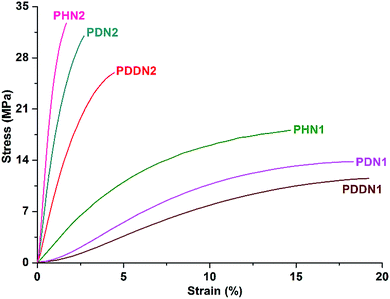 |
| | Fig. 5 Stress–strain curves for the nipagin-based poly(ether ester)s PN1-ωs and PN2-ωs. | |
Table 5 Mechanical and dynamic mechanical properties of the poly(ether ester)s
| Poly(ether ester) |
Mechanical properties |
Dynamic mechanical properties |
| Elastic modulus (MPa) |
Tensile strength (MPa) |
Elongation at break (%) |
Tg (°C) |
Storage modulus (MPa, 20 °C) |
Tanδmax |
| PHN1 |
696 ± 24 |
17.2 ± 1.5 |
14.5 ± 2.2 |
10.6 |
350 ± 25 |
0.15 |
| PHN2 |
785 ± 28 |
33.2 ± 2.5 |
1.8 ± 0.3 |
22.4 |
2450 ± 60 |
0.13 |
| PDN1 |
645 ± 25 |
12.3 ± 1.2 |
18.2 ± 2.5 |
−8.2 |
65 ± 8 |
0.14 |
| PDN2 |
746 ± 15 |
30.8 ± 2.1 |
3.1 ± 0.6 |
15.6 |
1050 ± 50 |
0.12 |
| PDDN1 |
623 ± 18 |
10.2 ± 0.9 |
19.8 ± 2.8 |
−10.8 |
52 ± 6 |
0.11 |
| PDDN2 |
725 ± 16 |
25.2 ± 1.8 |
4.2 ± 0.5 |
10.5 |
120 ± 25 |
0.09 |
| PDT |
488 ± 24 |
16.8 ± 2.8 |
5.0 ± 1 |
−0.2 |
56 ± 6 |
0.11 |
Dynamic mechanical analysis
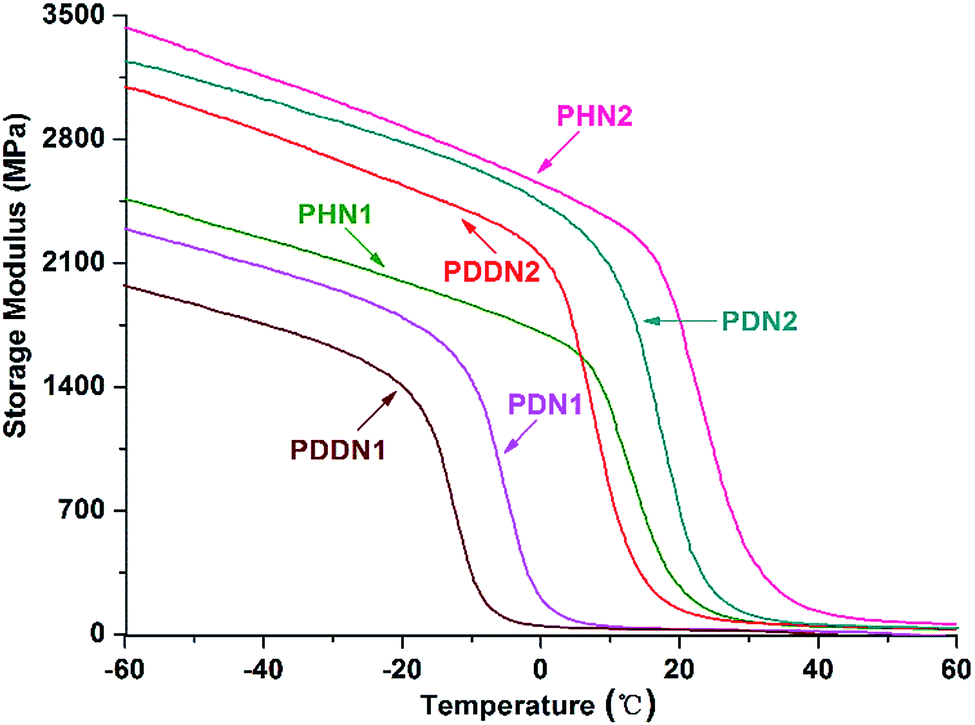
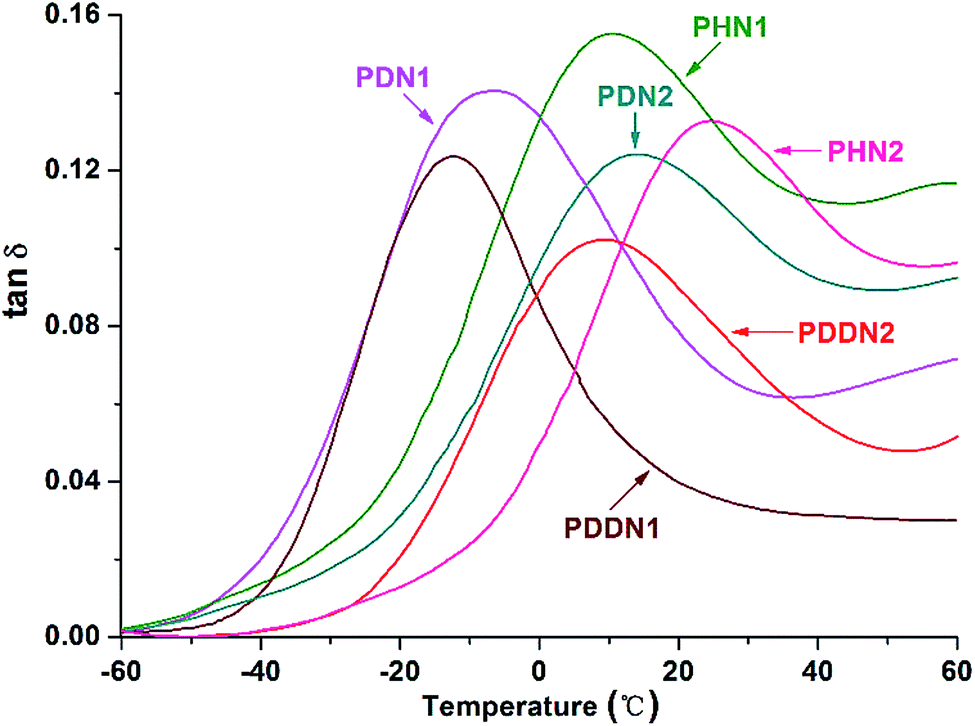
In order to evaluate the viscoelasticity of the nipagin-based poly(ether ester)s under the conditions of alternating stress or strain, dynamic mechanical analysis was carried out in the tensile mode using three parallel rectangular specimens. The dynamic storage tensile moduli of the poly(ether ester)s as a function of temperature plots are shown in Fig. 6. While comparing the storage tensile moduli at the initial (−60 °C) and the final (60 °C) temperatures, a decrease in the values is observed with the gradual increase in α,ω-diol length in each series. Additionally, the values for PN2-ωs are greater than that for PN1-ωs. This trend can be predicted as the storage modulus generally increases with the aromatic density and crystallinity.34 When the α,ω-diol lengths of PN1-ωs and PN2-ωs are similar and equal to or more than 6 (n = 6, 10, 12), PN2-ωs have the higher aromatic ring density and hence the higher storage modulus. Furthermore, as mentioned above, PN2-ωs are highly symmetrical and hence have high crystallinity. A drastic decrease of storage modulus during glass transition is observed and this indicates that the poly(ether ester)s are greatly influenced by the small portions of amorphous segments packed into the ordered crystalline phases during the course of glass transition and these packed segments could not maintain an adequate storage modulus. The viscoelasticity was also investigated by analyzing the tanδ value, which represents the ratio of loss modulus (E′′) to storage modulus (E′). It can be seen from Fig. 7 and Table 5 that the tanδmax values for PN2-ωs are lower than those for PN1-ωs as a whole, which indicate more energy is needed to deform the PN2-ωs specimens elastically recovered than that is dissipated on internal friction of molecular system. This result is also consistent with the DSC analysis. It should be noted that the tanδmax value for PHN2 is close to that of PDDN1 due to the similar crystallinity. The tanδmax values gradually move towards the direction of high temperatures with the decreases in α,ω-diol lengths in each series, suggesting a gradual increase in Tg values with the increase in rigid aromatic rings densities.
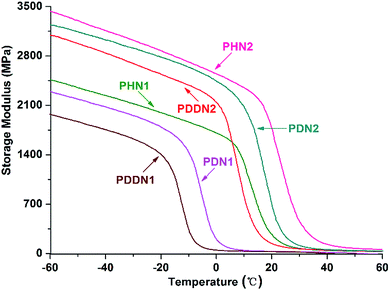 |
| | Fig. 6 Storage modulus as a function of temperature for PN1-ωs and PN2-ωs poly(ether ester)s. | |
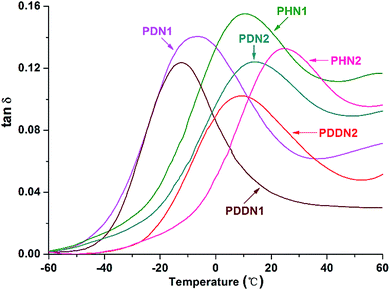 |
| | Fig. 7 Tanδ as a function of temperature for PN1-ωs and PN2-ωs poly(ether ester)s. | |
Conclusions
With an aim to find the novel functional structural materials, and also limit the utilization of fossil resources as much as possible, renewable nipagin-derived dimethyl esters N1 and N2 were prepared via one-step simple nucleophilic substitutions with methyl chloroacetate and 1,4-dibromobutane, respectively. Subsequently, two series of aromatic poly(ether ester)s were prepared from the nipagin-based N1, N2 and linear aliphatic α,ω-diols. The formed poly(ether ester)s exhibit a more than satisfactory resistance to heat with the onset decomposition temperature of about 350 °C. The incorporation of phenoxy-linkages in N1 units has no significant influence on the thermal stability of PDN1 with an enhanced Td value and the T5% just decreasing 14 °C compared to the DMT-based PDT. The Tg values of PN1-ωs and PN2-ωs exhibit a tendency to decrease with the gradual increase in α,ω-diol length in their respective series. The results from differential scanning calorimetry, wide angle X-ray diffraction and isothermal crystallization demonstrate that PN2-ωs have the higher crystallizabilities than PN1-ωs. Additionally, the crystallizability is enhanced with the gradual increase of α,ω-diol length in each series. The nipagin-based poly(ether ester)s have excellent mechanical properties with Young's modulus and tensile strength ranging from 600 to 800 MPa and 10 to 35 MPa, respectively. Furthermore, the incorporation of phenoxy-linkages has significantly enhanced influence on the toughness of materials by comparing the data of PDN1 with the DMT-based PDT. Due to the high crystallizability, PDN2 has the superior thermal stability and mechanical properties than PDT. In conclusion, the nipagin-based poly(ether ester)s have the potential characteristics that make them the excellent structural materials, such as plastic bottles and medical apparatus and instruments.
Acknowledgements
This work was funded by NSFC (51203079), the Natural Science Foundation of Tianjin (14JCYBJC18100), PCSIRT (IRT1257), and NFFTBS (J1103306).
References
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- G. Q. Chen and M. K. Patel, Chem. Rev., 2012, 112, 2082–2099 CrossRef CAS PubMed.
- U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schafer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 CrossRef CAS PubMed.
- C. Lavilla and S. Munoz-Guerra, Green Chem., 2013, 15, 144–151 RSC.
- J. Wu, P. Eduard, S. Thiyagarajan, L. Jasinska-Walc, A. Rozanski, C. F. Guerra, B. A. J. Noordover, J. van Haveren, D. S. van Es and C. E. Koning, Macromolecules, 2012, 45, 5069–5080 CrossRef CAS.
- F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J. P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 CrossRef CAS.
- K. L. Hu, D. P. Zhao, G. L. Wu and J. B. Ma, Polym. Chem., 2015, 6, 7138–7148 RSC.
- M. Colonna, C. Berti, M. Fiorini, E. Binassi, M. Mazzacurati, M. Vannini and S. Karanam, Green Chem., 2011, 13, 2543–2548 RSC.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G. J. M. Gruter, J. F. J. Coelhob and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- S. Tomizawa, J. A. Chuah, K. Matsumoto, Y. Doi and K. Numata, ACS Sustainable Chem. Eng., 2014, 2, 1106–1113 CrossRef CAS.
- T. Iijima and T. Yamaguchi, Tetrahedron Lett., 2007, 48, 5309–5311 CrossRef CAS.
- J. Hayashida and V. H. Rawal, Angew. Chem., Int. Ed., 2008, 47, 4373–4376 CrossRef CAS PubMed.
- W. B. Xu, Q. H. Xu and J. Z. Li, Org. Chem. Front., 2015, 2, 231–235 RSC.
- D. Bledzka, J. Gromadzinska and W. Wasowicz, Environ. Int., 2014, 67, 27–42 CrossRef CAS PubMed.
- Y. Guo and K. Kannan, Environ. Sci. Technol., 2013, 47, 14442–14449 CrossRef CAS PubMed.
- N. Cabaleiro, I. de la Calle, C. Bendicho and I. Lavilla, TrAC, Trends Anal. Chem., 2014, 57, 34–46 CrossRef CAS.
- R. J. Fussell, M. G. Lopez, D. N. Mortimer, S. Wright, M. Sehnalova, C. J. Sinclair, A. Fernandes and M. Sharman, J. Agric. Food Chem., 2014, 62, 3651–3659 CrossRef CAS PubMed.
- Y. Q. Tian, E. Akiyama and Y. Nagase, J. Mater. Chem., 2003, 13, 1253–1258 RSC.
- J. Y. Lee and J. S. Jang, Polymer, 2006, 47, 3036–3042 CrossRef CAS.
- L. Wang, Y. H. Wu, W. Zhang and K. Kannan, Environ. Sci. Technol., 2013, 47, 2069–2076 CrossRef CAS PubMed.
- A. Maiorana, S. Spinella and R. A. Gross, Biomacromolecules, 2015, 16, 1021–1031 CrossRef CAS PubMed.
- D. Fourcade, B. S. Ritter, P. Walter, R. Schonfeld and R. Mulhaupt, Green Chem., 2013, 15, 910–918 RSC.
- N. Gimeno, R. Martin-Rapun, S. Rodriguez-Conde, J. L. Serrano, C. L. Folcia, M. A. Pericas and M. B. Ros, J. Mater. Chem., 2012, 22, 16791–16800 RSC.
- J. Frohlich, H. Kautz, R. Thomann, H. Frey and R. Mulhaupt, Polymer, 2004, 45, 2155–2164 CrossRef.
- C. Lavilla, A. M. de Ilarduya, A. Alla, M. G. Garcia-Martin, J. A. Galbis and S. Munoz-Guerra, Macromolecules, 2012, 45, 8257–8266 CrossRef CAS.
- C. C. Pang, J. Zhang, Q. F. Zhang, G. L. Wu, Y. N. Wang and J. B. Ma, Polym. Chem., 2015, 6, 797–804 RSC.
- O. Persenaire, M. Alexandre, P. Degee and P. Dubois, Biomacromolecules, 2001, 2, 288–294 CrossRef CAS PubMed.
- J. E. Báez, P. Galindo-Iranzo and Á. Marcos-Fernández, International Journal of Polymer Analisys and Characterization, 2016, 21, 149155 Search PubMed.
- J. E. Báez, Á. Marcos-Fernández, A. Martínez-Richa and P. Galindo-Iranzo, Polym.-Plast. Technol. Eng., 2017, 56, 889898 CrossRef.
- K. L. Hu, D. P. Zhao, G. L. Wu and J. B. Ma, Polym. Chem., 2016, 7, 1096–1110 RSC.
- E. Gubbels, L. Jasinska-Walc, D. H. Merino, H. Goossens and C. Koning, Macromolecules, 2013, 46, 3975–3984 CrossRef CAS.
- Z. Zhang, Q. L. Wu, K. L. Song, S. X. Ren, T. Z. Lei and Q. G. Zhang, ACS Sustainable Chem. Eng., 2015, 3, 574–582 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and Tables S1 and S2. See DOI: 10.1039/c7ra01810j |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a and
Jianbiao Ma*b
*a and
Jianbiao Ma*b