
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous fabrication of multi-stimuli responsive graphene oxide composite hydrogel fibres by microfluidics†
Li Penga,
Yan Liu ab,
Jinghua Gonga,
Kaihuan Zhang*b and
Jinghong Ma*a
ab,
Jinghua Gonga,
Kaihuan Zhang*b and
Jinghong Ma*a
aState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Material Science and Engineering, Donghua University, C472, 201620, Shanghai, China. E-mail: mjh68@dhu.edu.cn
bMaterials Science and Technology of Polymers, MESA+ Institute of Nanotechnology, University of Twente, P. O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: k.zhang-1@utwente.nl
First published on 31st March 2017
Abstract
Microfluidics appeared in the 1990s as a promising technology and has received considerable attention in developing stimuli-responsive hydrogel fibres in microscale for tissue engineering and actuation devices. In this work, thermo- and electro-responsive graphene oxide/poly(N-isopropylacrylamide)/sodium alginate (GO/PNIPAM/SA) hydrogel fibres were prepared via microfluidics and off-chip free radical polymerization. The composite hydrogel fibres were characterised using FTIR, SEM, and DSC. The thermo-triggered volume-phase transition and electrically triggered bending behaviours were also investigated. The results show that the hydrogel fibres have porous internal structures and the pore size becomes smaller with the increase of GO content due to the hydrogen bonding between the amide groups of PNIPAM chains and oxygen-containing groups on the GO nanosheets. Besides this, the incorporation of increased GO content enlarges the swelling ratio of the hydrogel fibre. The hydrogel fibres also exhibit bending behaviour under the non-contact direct current electric field.
1 Introduction
An electroactive polymer artificial muscle, with the property of deformation in response to an electrical stimulus, can directly convert electrical energy into mechanical energy.1 Different kinds of artificial muscle materials based on dielectric elastomers, ionic polymer–metal composites, conductive polymers, carbon nanotubes, and polymer gels have been extensively studied in recent years. Polymer gels have attracted considerable attention because of their significant deformation under low driving voltage (deformation includes swelling, shrinking, and bending).2 For example, poly(2-acrylamido-2-methylpropane sulfonic acid) (PAMPS) hydrogel moves forward with a worm-like motion in water by periodically bending and stretching under an electric field.3 Poly(vinyl alcohol)–poly(sodium acrylate) (PVA–PAA) composite gel was designed to construct a robot hand with four gel fingers capable of grasping a fragile egg, an artificial fish with a gel tail waving in response to electrical stimuli in electrolyte solutions, and even a microrobot hand with two gel fingers working in the air.4,5 The migration or diffusion of ions leads to the slow response rate.6,7 Therefore, the application of polymer gels in artificial muscle materials is limited because of the relatively slow response rate and poor mechanical properties.The response rate of gels is proportional to the linear dimension.8 Gel fibres owning one-dimensional geometry can significantly improve the response rate. Moreover, the long, thin gel fibre is more flexible to be assembled into an artificial muscle system. The traditional hydrogel fibres are fabricated by cutting hydrogel film, wet spinning,9 or electrospinning.10 However, there still are some defects such as the poor mechanical property and the inability to control the shape of fibres. Recently, microfluidic spinning is emerging as a promising method to generate hydrogel fibre at the microscale.11 In microfluidic channel, the continuous fluid of a fibre precursor solution is capsulated by a second solution and then produce solid fibre after either a physical or chemical crosslinking or solidification.12 Microfluidic-spun fibers have been produced with a variety of shapes, e.g., cylinder,13 ribbon,14 hollow tube,15,16 and anisotropic structures17,18 by using different materials involving alginate,19,20 PLGA,21 and chitosan.22 The diameter can be easily adjusted, depending on flow rates, viscosities of the fluids, and microfluidic dimensions. Furthermore, regenerative biomaterials such as protein, drugs, and cells can be loaded into the fibres with bioactivity. Therefore, the simple and cost-effective fibre fabrication technology has been widely used in tissue engineering. However, the responsive hydrogel fibre as artificial muscle materials is rarely reported.
The hydrogel can also be doped with conducting fillers such as graphite, graphene, and carbon nanotubes to improve the response rate under electric field. Poly(acrylamide-co-acrylic acid)/graphite hydrogel with the highest weight percentage of graphite showed the highest bending deformation within the given time interval.23 Na-MWCNT/PVA composite hydrogel strip bent faster than the corresponding Na-PAA/PVA hydrogel strip.24 Graphene oxide (GO), the precursor of graphene, is considered as an ideal material for enhancing the electrical and mechanical properties of composite hydrogel because of its good dispersion in water. Graphene oxide/poly(acrylamide) (GO/PAM) and graphene oxide/polyacrylamide-co-poly(acrylic acid) (GO/PAM-co-PAA) composite hydrogels were also investigated and illustrated the incorporation of GO can significantly increase the compressive strength of the hydrogel and the bending angle under an electric field.25 GO/PAA hydrogel exhibited a much higher electroresponsive rate compared with the PAA hydrogel, especially under stronger electric fields.26
In this paper, graphene oxide composite hydrogel fibres based on crosslinked poly(N-isopropylacrylamide) (PNIPAM) and sodium alginate (SA) were prepared by a microfluidic spinning process, and GO was utilised as a filler to improve the response rate under an electric field. The structure, morphology, swelling property and electric field response of the hydrogel fibre were investigated.
2 Experimental
2.1 Materials
N-Isopropylacrylamide (NIPAM, TCI Co., Ltd.) was purified by recrystallization from toluene/cyclohexane (6/4, v/v) to remove inhibitor before use. Potassium persulfate (KPS, Shanghai Chemical Co., Ltd.) was purified by recrystallization in deionized water. Graphene oxide (GO) was purchased from Nanjing XFNANO Materials Tech Co., Ltd. Sodium alginate (SA) and N,N,N′,N′-tetramethylethylenediamine (TEMED) were obtained from Aladdin Reagent Co., Ltd. N,N′-Methylene-bis-acrylamide (BIS) and benzoin dimethyl ether (BDK) were purchased from Sigma-Aldrich. Calcium chloride (CaCl2) was purchased from Shanghai Chemical Co., Ltd. Poly(dimethylsiloxane) (PDMS) prepolymer and curing agent were obtained from Dow Corning.2.2 Microfluidic device
The coaxial laminar flow microfluidic device was designed and assembled as reported by Beebe et al.13 A glass capillary (inner diameter 0.6 mm, outer diameter 0.9 mm) was fixed in the middle of a plastic Petri dish and used to mold the main flow channel. A mixture of PDMS prepolymer and curing agent was prepared in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w) ratio and poured in the Petri dish, and then cured for 2 h at 80 °C. After removing the Petri dish and extracting the capillary, the cured PDMS substrate with a centre channel was cut into a cuboid.
:1 (w/w) ratio and poured in the Petri dish, and then cured for 2 h at 80 °C. After removing the Petri dish and extracting the capillary, the cured PDMS substrate with a centre channel was cut into a cuboid.
Another glass capillary for core fluid was inserted from the left side of the PDMS substrate into the centre channel and a rectangular capillary (square inner diameter 1.0 mm, square outer diameter 1.4 mm) was inserted from the right side as an outlet (Fig. 1a). A hole for the sheath flow is cored out using a 10 gauge needle.
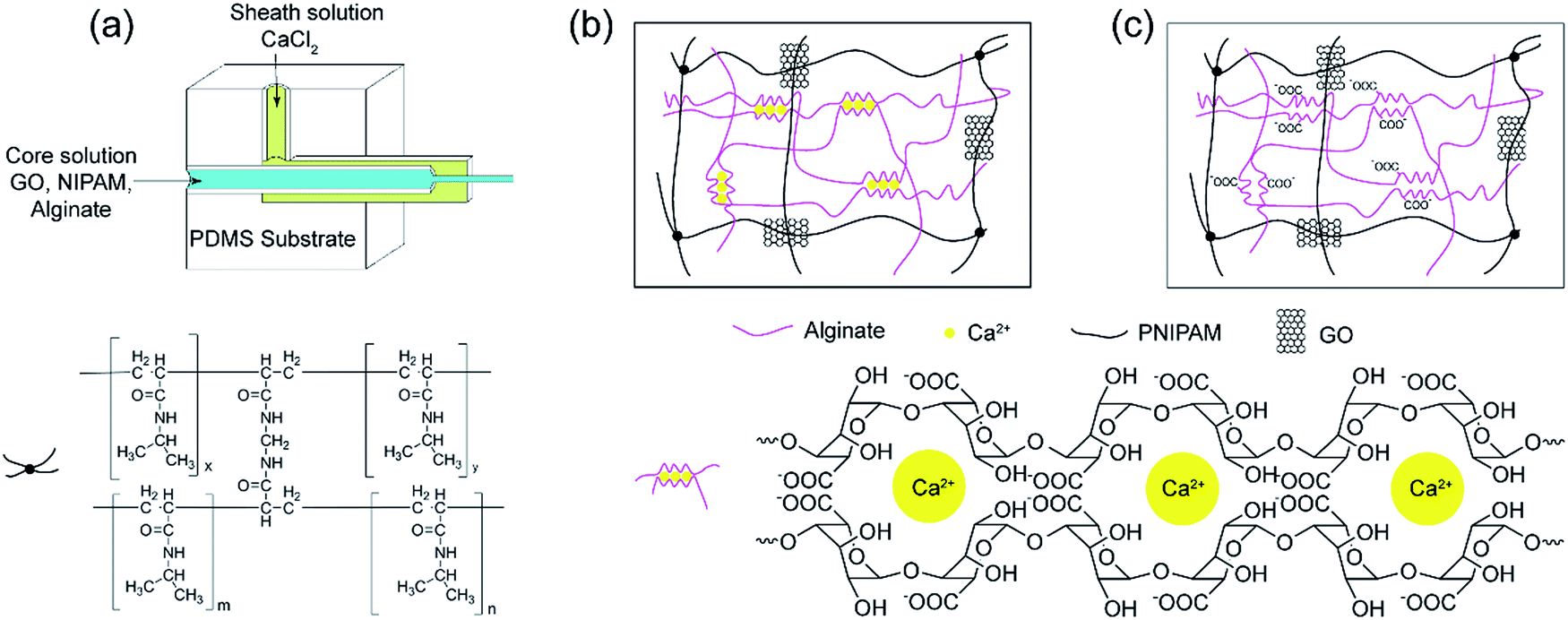 | ||
| Fig. 1 The preparation process of GNA hydrogel fibre. (a) Schematic of the coaxial microfluidic device. (b) GNAC hydrogel fibre composed of crosslinked PNIPAM and Ca-alginate by interpenetrating polymer network (IPN). (c) GNA hydrogel composed of crosslinked PNIPAM and linear alginate by semi-IPN. | ||
2.3 Preparation of GNA hydrogel fibre
Different mass of GO powder was ultrasonicated in deionized water (9.5 mL), and the pH value was adjusted to 7.0 using NaOH. Then 1.5 g NIPAM, 0.03 g BIS and 0.1 g SA were added and dissolved under stirring in an ice-water bath. Finally, initiator KPS (0.01 g in 0.5 mL deionized water) and catalyst TEMED (10 μL) were added. The solution for core fluid and 200 mM CaCl2 solution for sheath fluid were introduced into microfluidic device by syringe pumps, respectively. By using this microfluidic spinning process, the as-spun fibre was formed in the outlet pipe and collected on a rotating aluminium cylinder. Then the as-spun fibre was placed in soybean oil containing photoinitiator BDK (1%, w/v) and irradiated under UV light for 30 min, subsequently polymerised at 20 °C for 20 h to prepare GO/PNIPAM/calcium alginate hydrogel fibre (GnNAC hydrogel fibre). Finally, the hydrogel fibre was decalcified by the saturated Na2SO4 solution to obtain GO/PNIPAM/sodium alginate hydrogel fibre (GnNA hydrogel fibre). The number n represents the concentration of GO contents (mg mL−1). The swollen hydrogel fibres were quickly frozen in liquid nitrogen and then freeze-dried for 12 h. The morphology of the hydrogel fibre was observed with scanning electron microscope (SEM, SU8010, Hitachi). The hydrogel fibres were also analysed by FTIR (Nicolet 6700, Thermo Fisher, KBr) and EDS (Inca X-Max, Oxford Instruments).2.4 Mechanical property
Tensile stress measurements were performed on GNAC hydrogel fibres (diameter, 600 μm) using an Instron 5900 testing machine at room temperature. The distance between the two clamps is 20 mm and the crosshead speed is 20 mm min−1.2.5 Equilibrium swelling ratio
The GNA hydrogel fibres (length: 3 m, diameter: 600 μm) were immersed into deionized water to approach the equilibrium state at 20 °C. After wiping away the excess water on the surface with filter paper, the mass of the swollen hydrogel fibre was measured at a various temperature or time intervals. The swelling ratio (SR) of the hydrogel fibre was calculated as follows:| SR = (Ws − Wd)/Wd |
2.6 DSC analysis
The volume-phase transition temperature (VPTT) of the GNA hydrogel fibre was analysed using a differential scanning calorimeter (DSC1, Mettler Toledo) from 20 °C to 45 °C at a constant heating rate of 1 °C min−1 under a nitrogen atmosphere with a flow rate of 50 mL min−1.2.7 Electro-responsive behaviour
The GNA hydrogel fibre with diameter 600 μm was immersed in a 0.1 M Na2SO4 aqueous solution to reach swelling equilibrium and cut into 1 cm in length. The same solution was poured into a plastic container equipped with two parallel graphite electrodes (60 mm apart). The hydrogel fibre was placed in the centre of the electric filed (Fig. 2). A 20 V direct current (dc) voltage was applied between the electrodes. The bending deformation of the hydrogel fibre was recorded by the camera.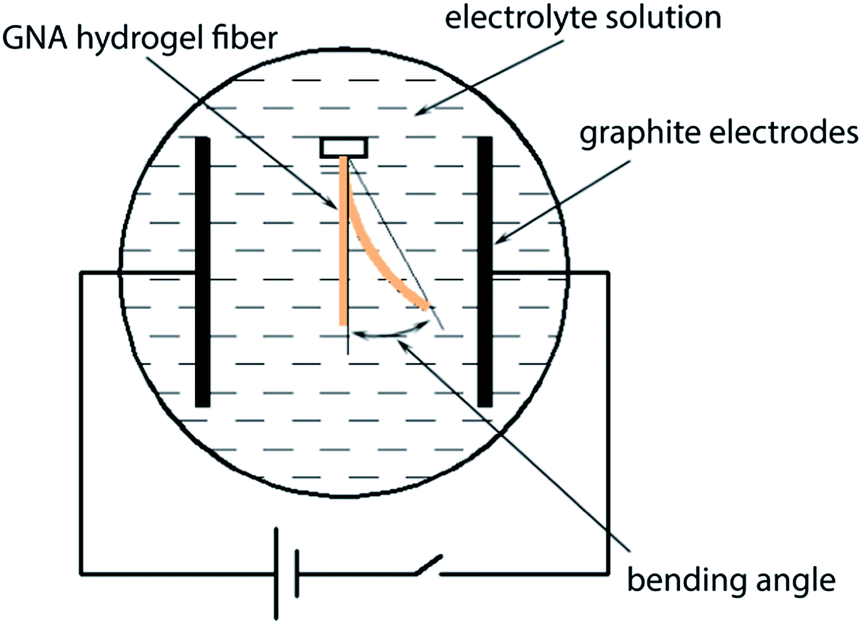 | ||
| Fig. 2 The experimental diagram for testing the bending behaviour of GNA hydrogel fibre. | ||
3 Results and discussion
3.1 Preparation and characterization of GNA hydrogel fibre
The preparation process of the as-spun fibre is shown in Fig. S1.† The incorporation of alginate in core solution increases the viscosity, and ensures a stable laminar flow.27 Under a confined microfluidic geometry and interfacial crosslinking, a stable coaxial flow was formed within the rectangular capillary where the core and sheath fluid merged. At the interface Ca2+ ions diffused into the core fluid quickly and the free alginate chains solidified by an ionic crosslinking. During the time that the as-spun fibre travelled inside of the outlet pipe, it formed spiral curls and caused clogging of the outlet pipe. It has been previously reported the device was put vertically in microfluidic spinning.19 However, in our work, a winding device was employed to solve this problem and the microfluidic device remained horizontal all the time. Meanwhile, it is more convenient to collect the as-spun fibre in a continuous way.The as-spun fibre consisted of Ca-alginate, GO, NIPAM and the initiator and crosslinker. Then NIPAM monomer was subsequently polymerised and crosslinked in the presence of a pre-formed network comprising ionic crosslinked alginate. The reaction took more than 20 h at 20 °C. Therefore, the as-spun fibre was collected and placed in soybean oil to prevent the evaporation of water. Besides, to avoid NIPAM diffusing outward from the as-spun fibre and influencing subsequent polymerization reaction, photo-initiator BDK was dissolved in soybean oil. Under UV light, the NIPAM monomer on the fibre surface was immobilised quickly through a photo-polymerization, thus inhibiting the subsequent diffusion of NIPAM. The prepared GNAC hydrogel fibres can exist stably in acid or alkali solution, implying the successful formation of an interpenetrating polymer network (IPN) composed of crosslinked PNIPAM and Ca-alginate. Finally, GNA semi-IPN hydrogel fibre was obtained by the dissociation of Ca2+ form the Ca-alginate network in saturated Na2SO4 solution.
The hydrogel fibre was characterised by FTIR. As shown in Fig. 3, characteristic absorption bands of PNIPAM at 1641 and 1536 cm−1 for amide I (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and amide II (N–H) can be observed in FTIR spectra of the hydrogel fibres, which provides the evidence of the formation of PNIPAM in free radical polymerization reaction.27,28 In the FTIR spectrum of G0NAC hydrogel fibre, the characteristic peak at 3434 cm−1 for O–H stretching vibration becomes broader, which is caused by the oxygen atoms from the guluronate chains of alginate involved in the coordination of Ca2+ ions.29 Thus, G0NAC hydrogel fibre is composed of both PNIPAM and Ca-alginate by IPN. After the dissociation of Ca2+ from Ca-alginate network, a semi-IPN composed of PNIPAM and linear SA remained in the hydrogel fibre. In the FTIR spectra of GnNA (n = 2, 4, 6) hydrogel fibres, most of the bands belonging to GO become weak or even disappear. However, the band for N–H stretching vibration shifts to a higher wavenumber compared with that of G0NA hydrogel fibre, which is attributed to the formation of hydrogen bonding between the N–H bond of PNIPAM and the O–H bond of GO.30 The hydrogen bonding and embedded GO nanosheet can significantly enhance the mechanical properties of the hydrogel.27,31,32
O) and amide II (N–H) can be observed in FTIR spectra of the hydrogel fibres, which provides the evidence of the formation of PNIPAM in free radical polymerization reaction.27,28 In the FTIR spectrum of G0NAC hydrogel fibre, the characteristic peak at 3434 cm−1 for O–H stretching vibration becomes broader, which is caused by the oxygen atoms from the guluronate chains of alginate involved in the coordination of Ca2+ ions.29 Thus, G0NAC hydrogel fibre is composed of both PNIPAM and Ca-alginate by IPN. After the dissociation of Ca2+ from Ca-alginate network, a semi-IPN composed of PNIPAM and linear SA remained in the hydrogel fibre. In the FTIR spectra of GnNA (n = 2, 4, 6) hydrogel fibres, most of the bands belonging to GO become weak or even disappear. However, the band for N–H stretching vibration shifts to a higher wavenumber compared with that of G0NA hydrogel fibre, which is attributed to the formation of hydrogen bonding between the N–H bond of PNIPAM and the O–H bond of GO.30 The hydrogen bonding and embedded GO nanosheet can significantly enhance the mechanical properties of the hydrogel.27,31,32
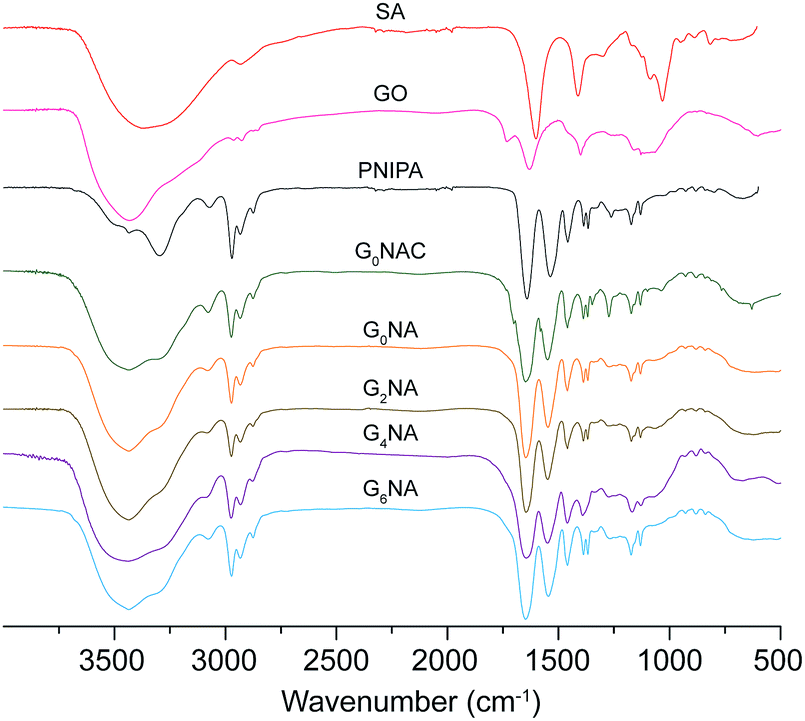 | ||
| Fig. 3 FTIR spectra for SA, GO, PNIPAM, G0NAC and GNA hydrogel fibres. | ||
The morphologies of the freeze-dried hydrogel fibres were observed by SEM. As shown in Fig. 4a–d, GNAC hydrogel fibres show porous structure. The pore sizes are heterogeneous, ranging from hundreds of nanometers to several micrometres, which is induced by crosslinking guluronate units in SA with the inward diffusion of Ca2+ to form a buckled structure, that so-called “egg-box” junction.33 Besides, the nanometre-sized pores increase in density with increasing the GO content, which is due to the coordination of Ca2+ with oxygen-containing groups of GO.34 However, the bindings would influence the electro-response behaviour of the hydrogel fibre. Therefore, GNA hydrogel fibre was finally obtained by the removal of the Ca2+ ions. As reported by Gombotz, Ca2+ crosslinked alginate gel can be degraded by a high concentration of ions such as Na+, Mg2+.35 So saturated Na2SO4 was used to dissociate Ca-alginate network, thereby releasing Ca2+ and meanwhile forming calcium sulphate precipitate. Moreover, the stacked alginate chains were released again, resulting in the uniform network structure (Fig. 4e–h). The GNA hydrogel fibre is composed of crosslinked PNIPAM and linear SA by semi-IPN. Besides, the pore size becomes smaller with the increase of GO content, implying a denser network structure of the hydrogel fibre. The dense network can be attributed to the hydrogen bonding interaction between the N–H bond of PNIPAM and the O–H bond of GO nanosheet. As a result, the intercalated GO increases the mechanical property by acting as a cross-linker.36
 | ||
| Fig. 4 SEM images of (a) G0NAC, (b) G2NAC, (c) G4NAC, (d) G6NAC, (e) G0NA, (f) G2NA, (g) G4NA and (h) G6NA hydrogel fibres. Scale bar, 10 μm. | ||
The decalcification of the hydrogel fibre was characterised by EDS analysis (Fig. S2†). EDS spectra of G0NA hydrogel fibre show the disappearance of high-intensity calcium peak compared with that of G0NAC hydrogel fibre, indicating the successful removal of Ca2+ from GNAC hydrogel fibre.
3.2 Influence of flow rate on diameter of the hydrogel fibre
The diameter of the G0NAC hydrogel fibre fabricated under various flow conditions was measured by optical microscope. As shown in Fig. 5, under a fixed sheath flow rate, the diameter of the hydrogel fibre increases almost linearly with increasing the core flow rate. Similarly, the decrease in sheath flow rate can also increase the diameter of the hydrogel fibre. But the core flow rate plays a dominant role here. The coaxial microfluidic device is capable of creating a continuous hydrogel fibre with lengths up to several meters. The illustration in Fig. 5 shows a hydrogel fibre with a length more than 3 m collected on an aluminium cylinder.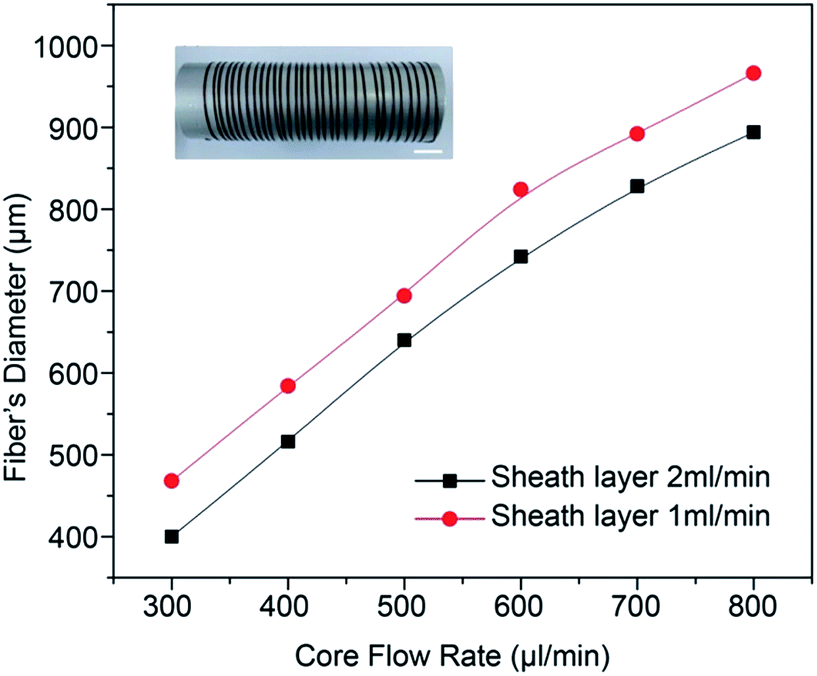 | ||
| Fig. 5 The GNAC hydrogel fibre diameter as a function of the core and sheath flow rate. Scale bar, 1 cm. | ||
3.3 Mechanical property
The mechanical property is important for the application of the hydrogel fibre in artificial muscle materials. Fig. 6 shows the typical strain–stress curves of GNAC hydrogel fibres. The strength and the modulus of the hydrogel fibre increase by embedding GO into the hydrogel matrix.37 The incorporation of GO improves the mechanical property of the hydrogel fibre by introducing hydrogen bonding between the N–H bond of PNIPAM and the O–H bond of GO.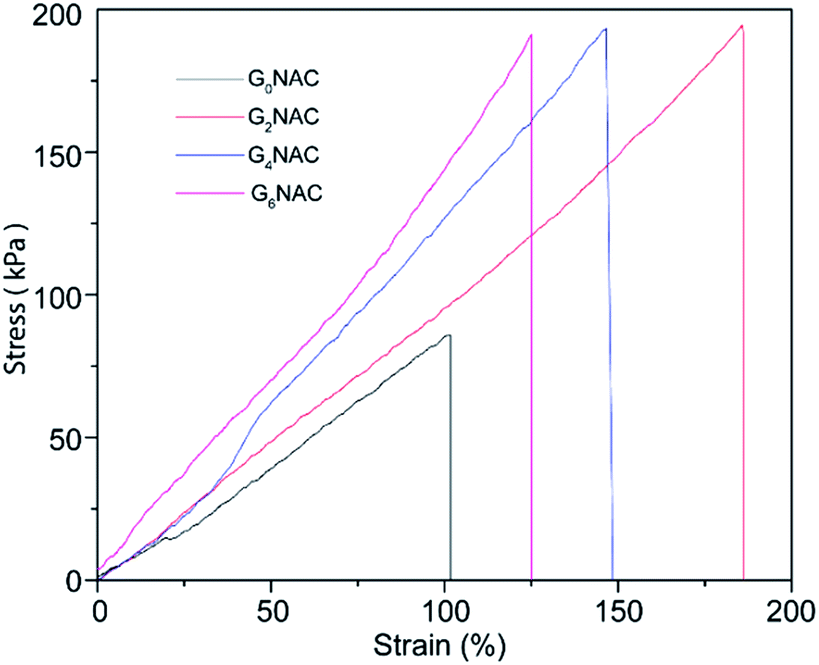 | ||
| Fig. 6 Tensile strain–stress curves for GNAC hydrogel fibres. | ||
3.4 Swelling behaviour
The swelling behaviour was investigated to further reveal the structure of GNA hydrogel fibre. As shown in Fig. 7, the swelling ratios of GNA hydrogel fibres increase dramatically at the early stage, and then reach an equilibrium. Compared with the G0NA hydrogel, the incorporation of GO obviously shortens the time needed for equilibrium and improves the equilibrium swelling ratio of the hydrogel fibre. The reason is that the incorporation of GO increases the overall hydrophilic groups in the hydrogel matrix that more water content can be held in the polymer network, resulting in the increased equilibrium swelling ratio.34,38 However, the equilibrium swelling ratio decreases with further increase of GO content, which could be attributed to the relatively compact polymer network structure.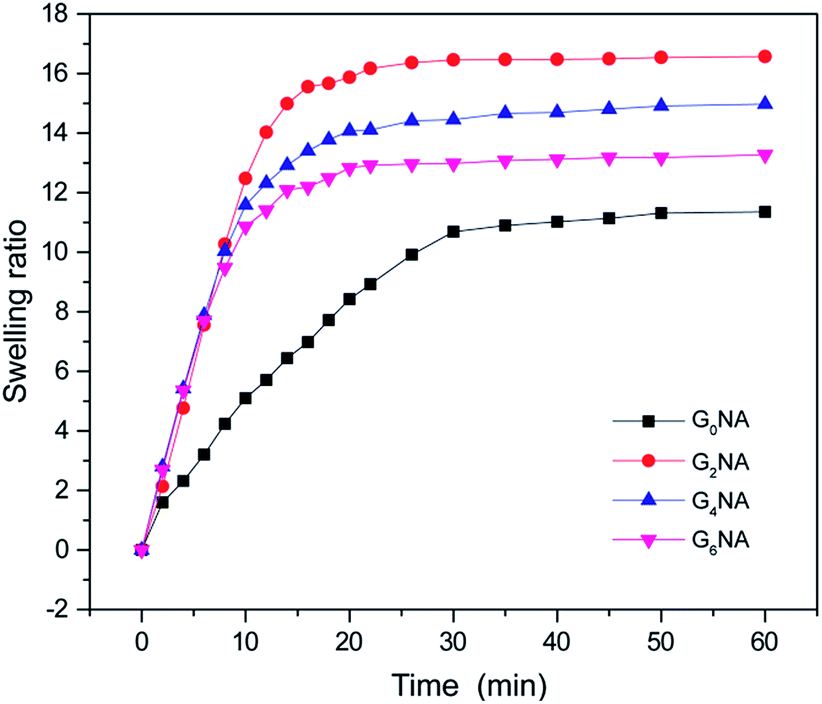 | ||
| Fig. 7 Swelling ratio of GNA hydrogel fibres in deionized water at 20 °C. | ||
3.5 Thermo-responsive behaviour
The swelling ratios of GNA hydrogel fibres were also investigated as a function of temperature in deionized water. As shown in Fig. 8a, a sharp decrease of the swelling ratio can be observed around 34 °C, resulting from the coil-to-globule transition of PNIPAM. This indicates GNA hydrogel fibre still retains the temperature response of PNIPAM network.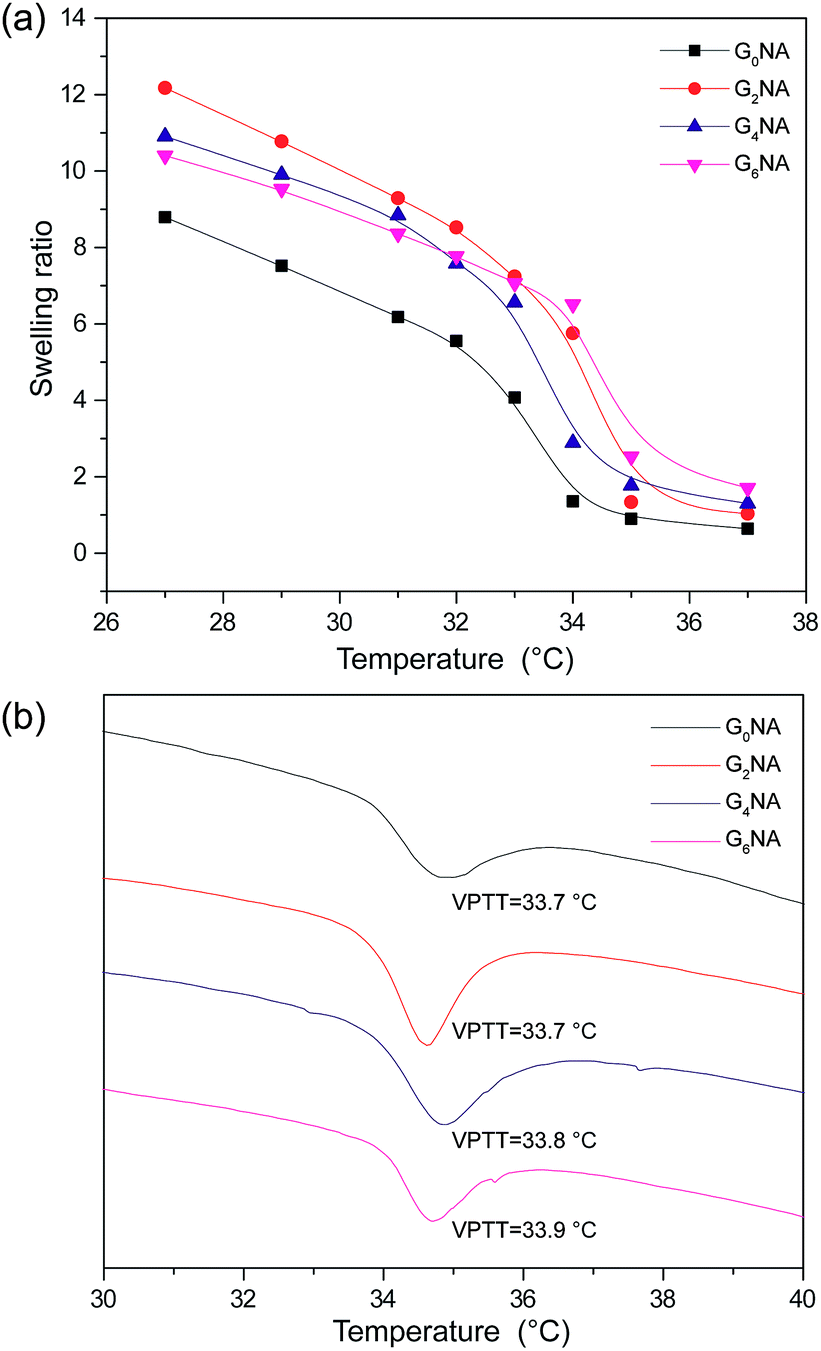 | ||
| Fig. 8 (a) Swelling ratio of GNA hydrogel fibres as a function of temperature in deionized water. (b) DSC thermogram of GNA hydrogel fibres. | ||
The VPTT of GNA hydrogel fibres was also measured by DSC. The result shows the incorporation of GO has limited influence on the VPTT of the hydrogel fibre because of no chemical bond formed between PNIPAM network and GO nanosheet (Fig. 8b).
3.6 Electro-responsive behaviour
Polyelectrolyte hydrogels exhibit bending behaviour under a non-contact direct current electric field, and the bending mechanism can usually be explained by the osmotic pressure.6 When an electric field is applied between the electrodes, the polyion, an ionic group in the polymer network, remains immobile, while the counterion of the polyion moves toward the counter electrode. Also, the free ions in the surrounding solution move toward their counter electrode and diffuse into the hydrogel. Consequently, an ionic concentration gradient occurs along the direction of the electric field, which results in the difference in osmotic pressure within the hydrogel, and it is the driving force of bending. For the polyanionic hydrogel, the osmotic pressure near the anode side increases with time and becomes larger than that of the cathode side. Thus, the hydrogel swells on the anode side and shrinks on the cathode side, resulting in bending toward the cathode.GNA hydrogel fibres are polyanionic hydrogel because SA is an anionic polyelectrolyte. It is interesting that GNA hydrogel fibres first bend towards the cathode, and then bend back to the centre along with the shrinkage in 0.1 M Na2SO4 solution under a dc voltage of 20 V. The reason is that GNA hydrogel fibres are different from the typical polyanionic hydrogels. They have not only anionic polyelectrolyte but also have temperature-responsive polymer. Thus, the elevated temperature of electrolyte solution during experiments should also be considered. The temperature of the electrolyte solution was measured at different times after applying a dc voltage of 20 V. As shown in Fig. S3,† the temperature of the electrolyte solution is maintained below 34 °C in 120 s, corresponding to the VPTT of hydrogel fibres. Before 120 s, the bending deformation towards the cathode is mainly caused by an osmotic pressure difference, resulting in the anisotropic structure that the swelling ratio on the anode side is higher than that on the cathode side. After that the hydrogel fibre on the anode side shrinks faster than that on the cathode side along with the increase in temperature of the electrolyte solution, resulting in the reverse bending of the hydrogel fibre and also shrinkage in length.
When investigating the influence of GO content on the bending behaviours of the hydrogel fibres, the initial temperature of the electrolyte solution and environmental temperature, the volume of the electrolyte solution and the electrolytic cell have been fixed at same parameters. Fig. 9a and b shows the bending angle of GNA hydrogel fibres within 120 s in 0.1 M Na2SO4 solution under a dc voltage of 20 V. It is clearly observed that the bending angle and response rate increase with increasing the GO content. GO, and SA both contain carboxylate groups. The hydrogel fibre with higher GO content shows the larger bending angle within the given time interval. Besides, the GO content in the composite hydrogel is also regarded as the electrostatic double layer that the electron would move and get polarised, which generates a secondary electric field that combines with and strengths the first field.26 Therefore, the incorporation of GO can significantly improve the electro-response of the hydrogel fibre in an dc electric field.
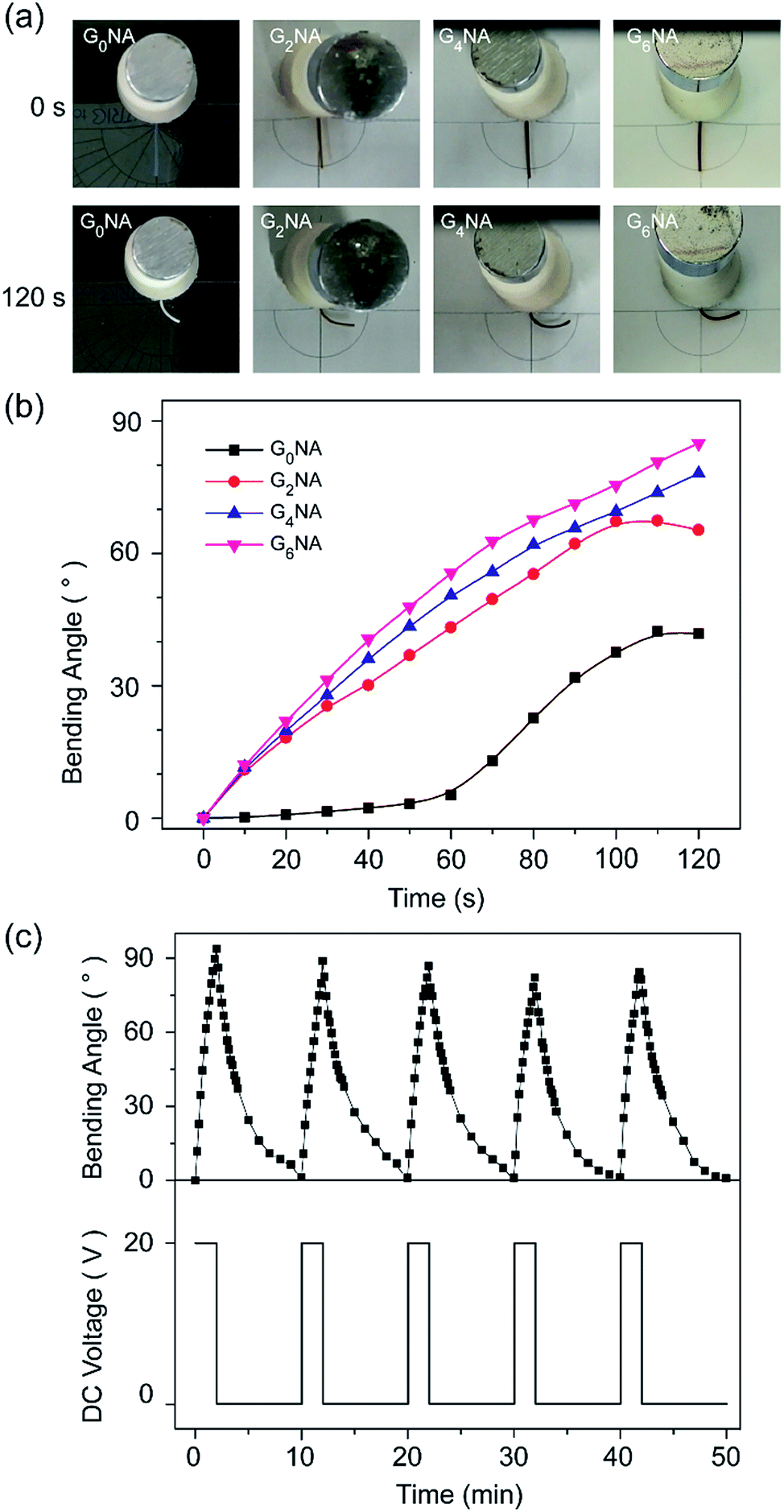 | ||
| Fig. 9 (a) Photographs of GNA hydrogel fibres at 0 and 120 s (the right side is cathode), (b) the bending behaviour of GNA hydrogel fibres with 600 μm diameter within 120 s, and (c) reversible bending behaviour of the G6NA hydrogel fibre in 0.1 M Na2SO4 solution under a dc voltage of 20 V. | ||
The reversible bending behaviour of the G6NA hydrogel fibre was investigated by alternately applying an electric voltage of 20 V for 120 s and then removing the voltage. Fig. 9c shows the hydrogel fibre bends and returns to the initial position as the electric field is turned on and off in several cycles. The deformation behaviour of each cycle is similar, which indicates an excellent reversibility.39
4 Conclusions
GNA hydrogel fibre was successfully prepared from a microfluidic spinning process and off-chip free radical polymerization. The GO/PNIPAM/SA composite hydrogel fibres exhibit thermo-response. Meanwhile, the incorporation of GO improves the swelling property, mechanical property and electro-response property of the hydrogel fibre. Besides, the hydrogel fibres also exhibit good reversible bending behaviour. This suggests that the hydrogel fibre has potential in the application of artificial muscles. Due to the potential photothermal energy transformation of GO nanosheet, GNA hydrogel fibre can also respond to near-infrared light irradiation. Thus, we expect the multi-stimuli responsive graphene oxide composite hydrogel fibres can be further exploited in the fabrication of artificial muscle with a photothermal response.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 11179027). YL thanks the Chinese Scholarship Council for the scholarship support. KZ have been supported by the Netherlands Organization for Scientific Research (NWO).References
- P. Brochu and Q. Pei, Macromol. Rapid Commun., 2010, 31, 10–36 CrossRef CAS PubMed.
- D. Kaneko, J. P. Gong and Y. Osada, J. Mater. Chem., 2002, 12, 2169–2177 RSC.
- Y. Osada, H. Okuzaki and H. Hori, Nature, 1992, 355, 242–244 CrossRef CAS.
- T. Shiga, Y. Hirose, A. Okada and T. Kurauchi, J. Mater. Sci., 1994, 29, 5715–5718 CrossRef CAS.
- T. Shiga, Y. Hirose, A. Okada and T. Kurauchi, J. Intell. Mater. Syst. Struct., 1993, 4, 553–557 CrossRef.
- T. Shiga and T. Kurauchi, J. Appl. Polym. Sci., 1990, 39, 2305–2320 CrossRef CAS.
- M. Doi, M. Matsumoto and Y. Hirose, Macromolecules, 1992, 25, 5504–5511 CrossRef CAS.
- T. Tanaka and D. J. Fillmore, J. Chem. Phys., 1979, 70, 1214–1218 CrossRef CAS.
- J. Fei, Z. Zhang and L. Gu, Polym. Int., 2002, 51, 502–509 CrossRef CAS.
- H. Chen and Y. L. Hsieh, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 6331–6339 CrossRef CAS.
- B. G. Chung, K.-H. Lee, A. Khademhosseini and S.-H. Lee, Lab Chip, 2012, 12, 45–59 RSC.
- M. A. Daniele, D. A. Boyd, A. A. Adams and F. S. Ligler, Adv. Healthcare Mater., 2015, 4, 11–28 CrossRef CAS PubMed.
- W. Jeong, J. Kim, S. Kim, S. Lee, G. Mensing and D. J. Beebe, Lab Chip, 2004, 4, 576–580 RSC.
- Z. Bai, J. M. M. Reyes, R. Montazami and N. Hashemi, J. Mater. Chem. A, 2014, 2, 4878–4884 CAS.
- K. H. Lee, S. J. Shin, Y. Park and S. H. Lee, Small, 2009, 5, 1264–1268 CrossRef CAS PubMed.
- Z.-J. Meng, W. Wang, R. Xie, X.-J. Ju, Z. Liu and L.-Y. Chu, Lab Chip, 2016, 16, 2673–2681 RSC.
- M. Yamada, R. Utoh, K. Ohashi, K. Tatsumi, M. Yamato, T. Okano and M. Seki, Biomaterials, 2012, 33, 8304–8315 CrossRef CAS PubMed.
- M. Yamada, S. Sugaya, Y. Naganuma and M. Seki, Soft Matter, 2012, 8, 3122–3130 RSC.
- S.-J. Shin, J.-Y. Park, J.-Y. Lee, H. Park, Y.-D. Park, K.-B. Lee, C.-M. Whang and S.-H. Lee, Langmuir, 2007, 23, 9104–9108 CrossRef CAS PubMed.
- E. Kang, S.-J. Shin, K. H. Lee and S.-H. Lee, Lab Chip, 2010, 10, 1856–1861 RSC.
- C. M. Hwang, A. Khademhosseini, Y. Park, K. Sun and S.-H. Lee, Langmuir, 2008, 24, 6845–6851 CrossRef CAS PubMed.
- K. H. Lee, S. J. Shin, C.-B. Kim, J. K. Kim, Y. W. Cho, B. G. Chung and S.-H. Lee, Lab Chip, 2010, 10, 1328–1334 RSC.
- M. Boruah, P. Phukon and S. Dolui, Polym. Compos., 2014, 35, 27–36 CrossRef CAS.
- J. Shi, Z.-X. Guo, B. Zhan, H. Luo, Y. Li and D. Zhu, J. Phys. Chem. B, 2005, 109, 14789–14791 CrossRef CAS PubMed.
- N. Zhang, R. Li, L. Zhang, H. Chen, W. Wang, Y. Liu, T. Wu, X. Wang, W. Wang and Y. Li, Soft Matter, 2011, 7, 7231–7239 RSC.
- Z. Tai, J. Yang, Y. Qi, X. Yan and Q. Xue, RSC Adv., 2013, 3, 12751–12757 RSC.
- Y. Liu, K. Zhang, J. Ma and G. J. Vancso, ACS Appl. Mater. Interfaces, 2017, 9, 901–908 CAS.
- J. Ma, Y. Xu, B. Fan and B. Liang, Eur. Polym. J., 2007, 43, 2221–2228 CrossRef CAS.
- D. A. Rees and E. J. Welsh, Angew. Chem., Int. Ed., 1977, 16, 214–224 CrossRef.
- J. Fan, Z. Shi, M. Lian, H. Li and J. Yin, J. Mater. Chem. A, 2013, 1, 7433–7443 CAS.
- J. T. Zhang, R. Bhat and K. D. Jandt, Acta Biomater., 2009, 5, 488–497 CrossRef CAS PubMed.
- L. W. Xia, R. Xie, X. J. Ju, W. Wang, Q. Chen and L. Y. Chu, Nat. Commun., 2013, 4, 2226–2236 Search PubMed.
- I. Braccini and S. Pérez, Biomacromolecules, 2001, 2, 1089–1096 CrossRef CAS PubMed.
- H. P. Cong, P. Wang and S. H. Yu, Small, 2014, 10, 448–453 CrossRef CAS PubMed.
- W. R. Gombotz and S. F. Wee, Adv. Drug Delivery Rev., 2012, 64, 194–205 CrossRef.
- X. Ma, Y. Li, W. Wang, Q. Ji and Y. Xia, Eur. Polym. J., 2013, 49, 389–396 CrossRef CAS.
- J. Shen, B. Yan, T. Li, Y. Long, N. Li and M. Ye, Composites, Part A, 2012, 43, 1476–1481 CrossRef CAS.
- H. Zhang, D. Zhai and Y. He, RSC Adv., 2014, 4, 44600–44609 RSC.
- J. Shang, Z. Shao and X. Chen, Biomacromolecules, 2008, 9, 1208–1213 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01750b |
| This journal is © The Royal Society of Chemistry 2017 |