DOI:
10.1039/C7RA01727H
(Paper)
RSC Adv., 2017,
7, 20947-20959
Novel binder-free electrode materials for supercapacitors utilizing high surface area carbon nanofibers derived from immiscible polymer blends of PBI/6FDA-DAM:DABA†
Received
10th February 2017
, Accepted 2nd April 2017
First published on 12th April 2017
Abstract
Carbon nanofibers with high surface area have become promising electrode materials for supercapacitors because of their importance in increasing energy density. In this study, a high free volume polymer, 6FDA-DAM:DABA (6FDD) was blended with polybenzimidazole (PBI) in different ratios to obtain different compositions of PBI/6FDD immiscible polymer blends. Freestanding nanofiber mats were obtained via electrospinning using blend precursors dissolved in N,N-dimethylacetamide (DMAc). Subsequently, carbonization, followed by CO2 activation at 1000 °C was applied to convert the fiber mats into porous carbon nanofibers (CNFs). The addition of 6FDD shows significant effects on the microstructure and enhancement of the surface area of the CNFs. The obtained CNFs show specific surface area as high as 3010 m2 g−1 with pore sizes comparable to those of the electrolyte ions (PYR14TFSI). This provides good electrolyte accessibility to the pore of the carbon materials resulting in enhanced energy density compared to the CNFs obtained from pure PBI. Electrodes derived from PBI:6FDD (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30) exhibited outstanding supercapacitor performance in coin cells with a specific capacitance of 142 F g−1 at the scan rate of 10 mV s−1 and energy density of 67.5 W h kg−1 at 1 A g−1 (58 W h kg−1 at 10 A g−1) thus demonstrating promising electrochemical performance for high performance energy storage system.
:30) exhibited outstanding supercapacitor performance in coin cells with a specific capacitance of 142 F g−1 at the scan rate of 10 mV s−1 and energy density of 67.5 W h kg−1 at 1 A g−1 (58 W h kg−1 at 10 A g−1) thus demonstrating promising electrochemical performance for high performance energy storage system.
1. Introduction
Supercapacitors are energy storage devices that possess high power density and good cycle stability with long lifetimes.1,2 Currently, supercapacitors are being used in transportation, consumer electronics and industrial applications because of these excellent characteristics.3 Electrochemical double-layer capacitors (EDLCs) are one type of supercapacitor, and are most often comprised of two porous carbon electrodes.4 Carbon precursor materials possessing very high surface areas are highly attractive materials to use as electrodes for EDLCs because the amount of energy that is stored and released via electrostatic adsorption and desorption of electrolyte ions on electrode surface greatly depends on the electrode's surface area.5,6
Carbon nanofibers (CNFs) possess high surface areas and are an interesting alternative to the other forms of carbonaceous materials.7 Carbon nanofibers can be produced by electrospinning methods from a variety of polymers8 such as polyacrylonitrile (PAN),9,10 cellulose,11 phenol resin,12 pitch13 and different types of polyimides.14 Polybenzimidazole (PBI) is a promising CNF precursor polymer due to its outstanding thermal stability, high carbon yield and spinnability. Moreover, PBI is commercially available. Despite these potential advantages only a limited number of investigations using CNFs derived from PBI for electrode materials in EDLCs have been reported. Kim et al. prepared supercapacitor electrodes using PBI based carbon nanofibers activated by steam which exhibited surface areas of 500–1220 m2 g−1.15 Nanofibers activated at 800 °C showed a capacitance of 202 F g−1 in 1 M H2SO4 aqueous electrolyte solution. Due to high thermal stability, PBI electrospun fibers do not require the thermal stabilization in air before carbonization that is common for PAN-based materials, thus saving time and energy. Aromatic polyimides (PI) also have potential as carbon nanofiber precursor polymers for electrode materials because of their high thermal and mechanical stabilities.16 Li et al. reported the fabrication of CNF using a heterocyclic polyimide containing benzopyrrole and benzimidazole rings.17 A volumetric capacitance of 159.3 F cm−3 at 1 A g−1 in 1 M NaOH was reported. Much research has been carried out on producing different forms of carbon materials, including CNFs from PIs, but very few studies report the use of PI based CNFs as supercapacitor electrodes.16
Polymer blending is a very important technique for producing new carbon materials with useful properties beyond the range that can be obtained from a single polymer.18 The domain size distribution, morphology and miscibility are influenced by the composition of polymer blends. There are several methods to make CNFs from polymer blends.19 For example two immiscible polymers having different thermal stability, where one acts as a carbonizing polymer and the second acts as a sacrificial polymer which decomposes upon carbonization, can form pores in the fiber to increase surface area.20–22 In another case, blends have been prepared utilizing two polymers that both act as carbonizing polymers. This can increase specific properties of CNFs such as conductivity and porosity to tailor the pore size distribution of the resultant carbon fiber matrix. PBI/PI blends are examples where the specific PI component can be tuned to the application.23,24 Both miscible and immiscible PBI/PI blends, as well as carbonized blends of PBI/PI have been reported for gas separations.25–27 Jung et al. reported fabrication of porous CNFs from miscible PBI/Matrimid blends as electrode materials for supercapacitors. Steam activated CNFs (ACNFs) from PBI/Matrimid (75:25) showed aspecific capacitance of 126 F g−1, and energy and power densities of 49 W h kg−1 and 7 kW kg−1 in an ionic liquid electrolyte, EMITFSI.28 Recently, porous carbon fibers have been prepared from polybenzimidazole/poly-L-lactic acid (PBI/PLLA) blends. PLLA acts as a sacrificial polymer in the blends and lends increased porosity upon decomposition. Specific capacitance of 111.9 F g−1 and highest energy density and power density of 36.9 W h kg−1 and 7.0 kW kg−1, respectively, were reported for activated PBI/PLLA (8:2) in EMITFSI electrolyte.29 We are unaware of any reports describing the use of carbonized immiscible blends of PBI/PI for use as electrode materials for supercapacitors.
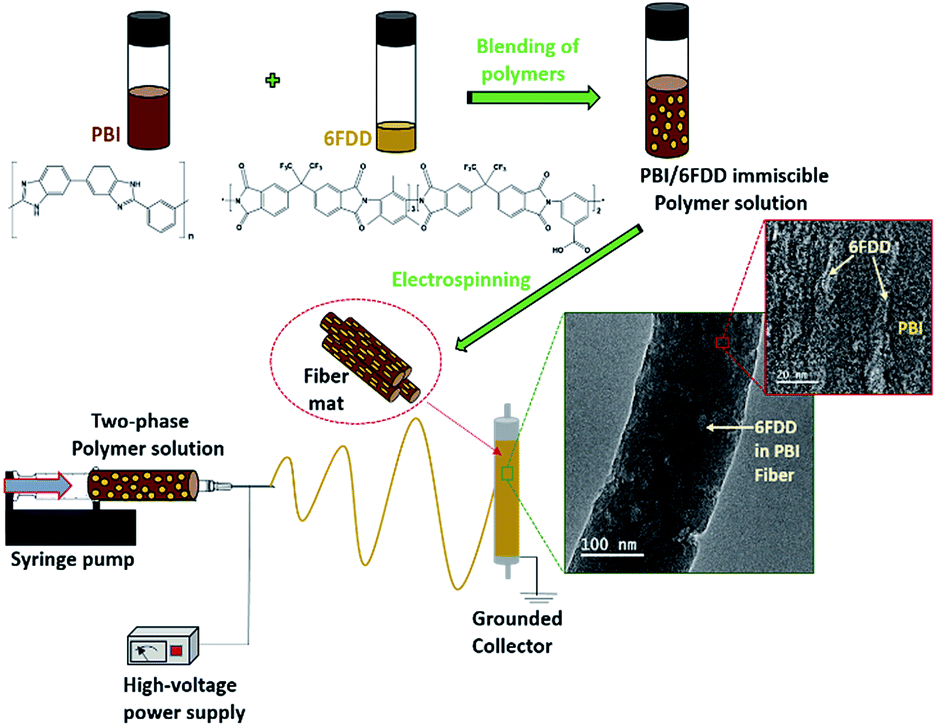
In this study, immiscible blends of PBI and 6FDA-DAM:DABA (3:2) with different ratios were used as carbon nanofiber precursors as shown in Fig. 1. PBI was selected as one carbonizing precursor polymer because of above mentioned reasons. The aromatic polyimide, 6FDA-DAM:DABA (3:2), composed of 4,4′-hexafluoroisopropylidene diphthalic anhydride (6FDA), 2,4,6-trimethyl-1,3-phenylenediamine (DAM) and 3,5-diaminobenzoic acid (DABA) [denoted as 6FDD] was selected as the second carbonizing polymer for blend preparation based on several considerations. One of the important properties of the 6FDD polymer is its high free volume (>0.18) due to the bulky hexafluoropropyl bridging moiety on its backbone which restricts chain mobility and interrupts chain packing.30,31 The use of this high free volume polymer leads to an increase in the surface area of the resulting fiber. The ability to crosslink due to decomposition of the pendant carboxylic acid on DABA moieties of the 6FDD polymer also facilitates the carbonization processes. During carbonization, crosslinking can lock the free volume of the precursor into the carbon fiber matrix.32 After mixing of two polymers, the blend solution was electrospun to obtain phase separated nanofiber as shown in the Fig. 1.
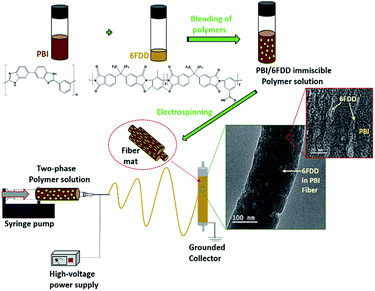 |
| | Fig. 1 Schematic illustration of the fabrication of the phase separated nanofibers using PBI/6FDD polymer blends and structures of PBI and 6FDD. | |
Here, we explore the possibility of using immiscible blends with two carbonizing polymers to achieve improved performance by controlling the microstructure. A uniform microstructure of 6FDD in PBI is important to tailor the micro/meso pore structure. 6FDD is more selective than PBI to CO2 activation because of its faster etching rate (vide infra). The surface area and electrochemical performance of the resulting CNFs were investigated as a function of 6FDD/PBI ratio.
2. Experimental
2.1 Materials
Polybenzimidazole (PBI) was purchased from PBI Performance Inc. (26 wt% in DMAc, 1.5% (w/w) LiCl, Mw 30000) and was used as received. 4,4-Hexafluoroisopropylidene diphthalic anhydride (6FDA, >99% purity) was purchased from Akron Polymer Systems Inc. and dried at 150 °C under vacuum prior to use. 2,4,6-Trimethyl-1,3-phenylenediamine (DAM, >97% purity) was purchased from TCI America. DAM was purified by vacuum sublimation. 3,5-Diaminobenzoic acid (DABA, 98% purity) was purchased from Sigma Aldrich and was purified by recrystallization from water. Anhydrous 1-methyl-2-pyrrolidone (NMP, 99.8% purity) was purchased from Sigma Aldrich and was dried over activated 4A molecular sieves purchased from Sigma Aldrich. Anhydrous N,N-dimethylacetamide (DMAc, 99.8% purity) was purchased from EMD chemicals and used for the electrospinning solutions. Electrochemical grade 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide (PYR14TFSI, 99.5% purity) was purchased from IoLiTec, Inc. and ethylene carbonate (EC), (99.5% purity) and propylene carbonate PC, (99.5% purity) were purchased from Sigma Aldrich, and were used without further purification.
2.2 Synthesis of 6FDA-DAM:DABA (6FDD)
The 6FDA-DAM:DABA copolyimide was synthesized using monomers 6FDA, DAM and DABA with 3:2 DAM:DABA ratio by following a reported procedure.33 Synthesis of 6FDA-DAM:DABA (3:2) via the poly(amic acid) is shown in Fig. S1.† First, a solution of 6FDA (2.000 g) in NMP (8.5 mL) was added to a solution of DABA (0.274 g) in NMP (2.5 mL) in a three-neck flask and stirred for 1 h at room temperature. A solution of DAM (0.406 g) in NMP (2.0 mL) was then added to the reaction flask and stirred at room temperature for 26 h. Next, the temperature was raised to 190 °C for 26 h and a Dean-Stark trap filled with 12 mL of o-dichlorobenzene was attached. After imidization, the polymer solution was precipitated in methanol and washed with methanol several times. The polymer was dried in a vacuum oven at 120 °C. Brown coloured polymer (91% yield) was obtained with Mw of 119 kDa and PDI of 2.5.
2.3 Preparation of CNF and ACNFs
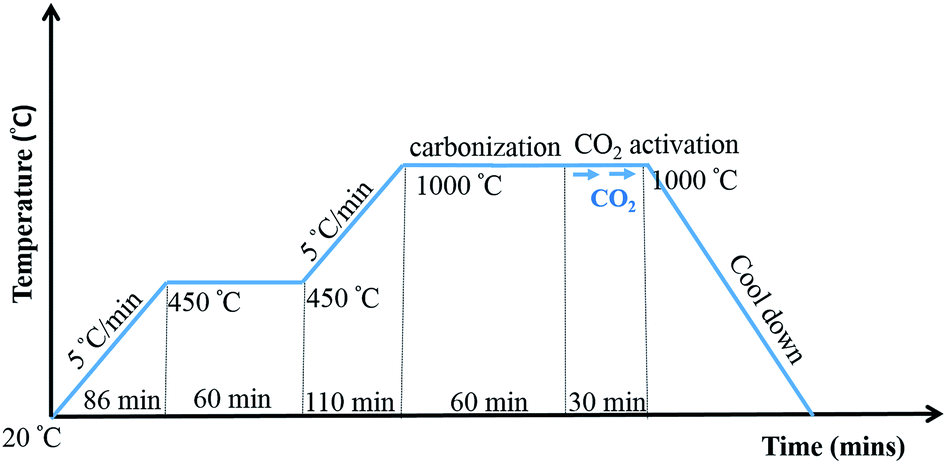
20 wt% of PBI/6FDA-DAM:DABA polymer blends in DMAc were used for electrospinning, First PBI and 6FDA-DAM:DABA were dissolved in DMAc separately by stirring at 80 °C for 4 h. Then 6FDD solutions were added drop wise to PBI solutions with stirring. The blend solutions were further stirred at 80 °C for 12 h before electrospinning. The polymer ratio of PBI/6FDD was selected as 90/10, 70/30 and 50/50 by weight and denoted as PBI:6FDD (90:10), PBI:6FDD (70:30) and PBI:6FDD (50:50), respectively. Also, 20 wt% of pure PBI solution was prepared according to the same procedure as a control to compare with the blend solutions. Then electrospinning was applied to obtain fibers. A 12 mL syringe was filled with 10 mL of the blend solution. An applied voltage of 18–20 kV (Gamma High Voltage Research, Inc., power supply) was used and solutions were fed through a 20G needle at a constant rate using a syringe pump. Parameters for the electrospinning were set as follows: flow rate of 0.3 mL h−1, tip-to-collector distance of 15 cm and rotating speed of grounded drum collector at 300 rpm. As-spun nanofibers were carbonized under helium atmosphere. First, the electrospun fiber samples were heated for 1 h at 450 °C and then the temperature was increased up to 1000 °C at a heating rate of 5 °C min−1. The carbonization process was performed at 1000 °C for 1 h and then CO2 was introduced during 30 min at this temperature to activate the samples and finally cooled to room temperature. The detailed carbonization and activation protocol is illustrated in Fig. 2.
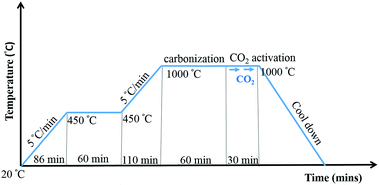 |
| | Fig. 2 Carbonization and CO2 activation protocol. | |
2.4 Characterization
The structure of synthesized 6FDA-DAM-DABA (3:2) was confirmed by proton NMR (1H NMR) and attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy. NMR spectra were recorded on a Bruker AVANCE III™ (500 MHz) in (CD3)2SO with TMS (1H) as internal standard. FTIR spectra of the synthesized polymer and as-spun samples were acquired using a Nicolet 360 FTIR spectrophotometer. The molecular weight of as-synthesized 6FDD was determined by gel permeation chromatography (Viscotek GPCmax, VE2001) using two ViscoGEL columns in series (I-MBHMW 3078, Viscotek) and a Triple Array Detector (Module TDA 320, Viscotek). THF was used as the mobile phase at a flow rate of 1 mL min−1 and polystyrene standards (Polymer Laboratories) were used for calibration. Scanning electron microscopy (SEM) was performed to characterize morphologies of samples using a Zeiss-LEO (Model 1530) Scanning electron microscope equipped with a field emission gun operating at 10 keV acceleration voltage. Gold coated samples were used for SEM imaging of the electrospun mats of the PBI and the blends whereas images of the carbonized samples were obtained without gold coating. The average fiber diameter of the electrospun and carbonized fiber are reported based on measurements of 100 fibers and diameter distributions of the samples were obtained using the publically available ImageJ software. Transmission electron microscope (TEM) images were obtained using a JEOL JEM-2100 TEM at 200 kV (JEOL Co Ltd). Samples were prepared by dispersing electrospun fibers in methanol and the fishing technique was used to obtain fibers on to the 300 mesh Cu lacey carbon grid. The microstructures of the polymer blends were observed by using the thin film membranes prepared by solvent-casting the same solutions used for electrospinning onto glass substrates, employing a Sheen (1133N) automatic casting table equipped with a doctor blade. SEMs of the cross sections were taken after selectively extracting 6FDD from these prepared membranes using THF as a solvent in a Soxhlet extractor. To determine the thermal stability and the carbon yield in all blend compositions, thermogravimetric analysis (TGA) was carried out with a TA Instruments SDT Q600 Analyzer by heating from room temperature to 1000 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere flow rate of 50 mL min−1. Raman spectra of the carbonized materials were recorded on a Nicolet Almega XR Raman spectrometer equipped with a 780 nm laser. Surface areas and pore properties of the carbon fibers were characterized using N2 adsorption/desorption isotherms measured at 77 K using a Quantachrome Autosorb-1 analyzer. Samples were degassed at 120 °C before being analyzed. The Brunauer–Emmett–Teller (BET) method was used to calculate surface area and Density Functional Theory (DFT) was used to obtain the pore size distribution. X-ray diffraction (XRD) patterns were collected by using a RigakuUltima IV X-ray diffractometer using Cu Kα radiation. X-Ray Photoelectron Spectroscopy (XPS) were recorded with a Physical Electronics Quantum 2000 scanning ESCA microprobe spectrometer equipped with a monochromatic Al Kα source and a concentric hemispherical electron energy analyzer under ultrahigh vacuum (10 −9 mbar). The samples were located at 45° with respect to the analyzer. Before the recordings of the XPS spectra, the samples were cleaned by an argon sputtering performed at 1 kV on a 1 mm2 surface area during 5 min. A pass energy of 23.50 eV and a resolution of 0.2 eV step over 12–20 eV regions were used during the analyses of the followings elements C 1s (280–295 eV), N 1s (394–406 eV), O 1s (526–539 eV), F 1s (679–699 eV), Si 2p (97–107 eV) and Ar 2p (235–255 eV).
2.5 Electrochemical tests
Supercapacitor test cells were assembled in a CR2032 coin cell configuration using two electrodes punched out from the carbonized mat. The coin cell assembly parts were obtained from Shenzhen Yongxingye Precision Machinery Mould Co. Ltd., China. Carbon coated aluminum was used as the current collectors on each side of electrodes that were separated by a Gore™ PTFE separator. The electrode performance was measured in an ionic liquid electrolyte mixture of 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide (PYR14TFSI), propylene carbonate (PC) and ethylene carbonate (EC), (PYR14TFSI:PC:EC) (3:3:2). All the coin cells were assembled inside a nitrogen filled glovebox (Vacuum Atmospheres Company). Coin cells were tested using an Arbin Supercapacitor Testing Station (SCTS). Cyclic voltammetry (CV) and constant current charge/discharge tests (CDC) were performed to calculate specific capacitance and energy density. CV was performed from −2 V to 2 V by varying the scan rate from 10 to 100 mV s−1. CDC was obtained between 3.5 V and 0 V. Electrochemical impedance spectroscopy was done at 0 V DC bias over the frequency range of 100 kHz to 10 mHz using a PARSTAT 2273 Galvanostat (Princeton Applied Research) operated with PowerSuite software.
3. Results and discussion
3.1 Characterization of the polymer
Characterization of the 6FDD polymer is included in ESI.† The molecular weight of the polymer (Mw) was 119 kDa with PDI of 2.5. The structure of the 6FDD was confirmed by 1H-NMR spectroscopy (Fig. S2(a)†), thermogravimetric analysis (TGA) (Fig. S2(b)†) and FT-IR spectroscopy (Fig. S2(c)†). The two absorption peaks appearing at 1780 cm−1 and 1720 cm−1 in the FT-IR spectrum of 6FDD (Fig. S2(c)†), ascribed to asymmetric and symmetric stretching vibrations of the imide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively, indicate complete conversion of the poly(amic acid) to the polyimide.34
O, respectively, indicate complete conversion of the poly(amic acid) to the polyimide.34
3.2 Characterization of the material
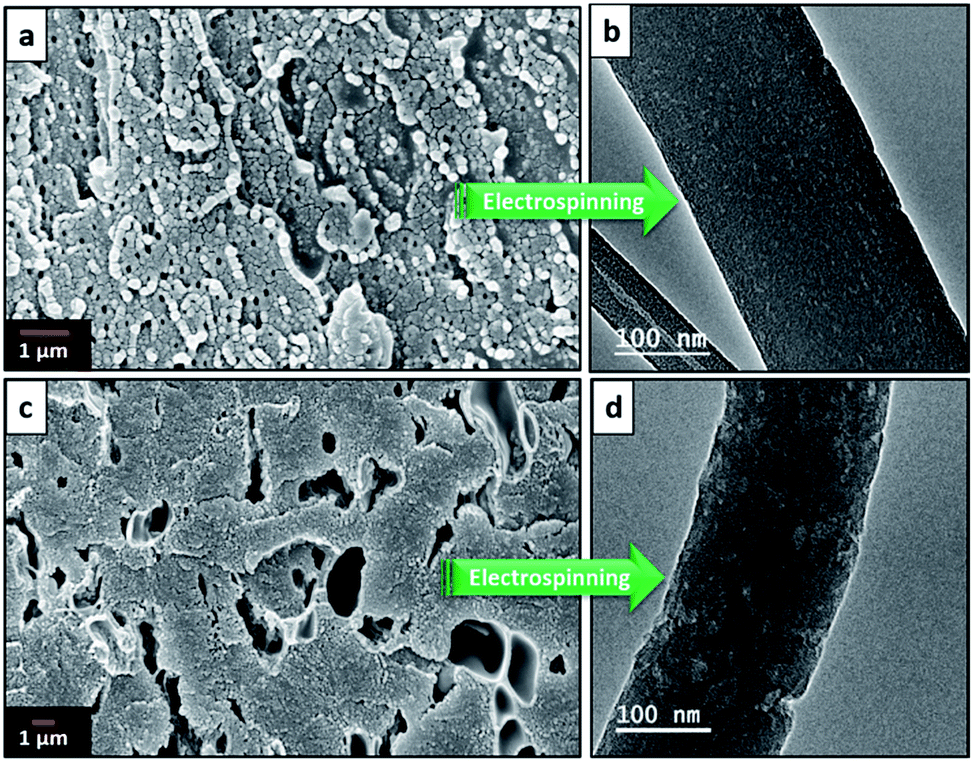
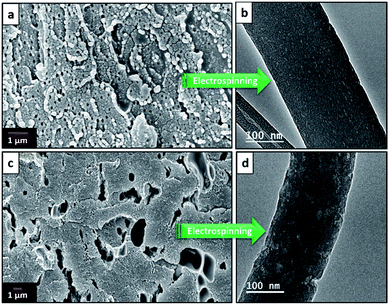
Electrospinning yielded free standing and flexible fiber mats of the precursor polymer blends. SEM images of as-spun fibers obtained for the different blend compositions are presented in Fig. 3. All of the as-spun mats exhibited long, continuous nanofibers. Table 1 shows average fiber diameters of as-spun samples and carbonized samples, respectively. The diameter of the fibers decreases with increasing 6FDD fraction. SEM of as spun PBI shows fibers consisting of both large and small diameters with a large standard deviation of the fiber diameter compared to the blends. After blending with 6FDD, fiber diameters become more uniform for all the compositions. Fig. 4 shows the SEM images of nanofibers carbonized and activated at 1000 °C. The fiber diameters became smaller after carbonization and the shapes of the CNFs are well maintained and retain the fibrous morphology. Diameters of the fibers decreases in the order PBI > PBI:6FDD (90:10) > PBI:6FDD (70:30) > PBI:6FDD (50:50) as shown in Table 1. Smaller diameter fibers are more advantageous to obtain good performance because of their higher surface area.
 |
| | Fig. 3 SEM images of electrospun (a) PBI, (b) PBI:6FDD (90:10), (c) PBI:6FDD (70:30) and (d) PBI:6FDD (50:50) nanofiber mats. | |
Table 1 Average fiber diameter of PBI and the PBI/6FDD blends
| Precursor |
As spun fiber diameter (nm) |
Carbonized fiber diameter (nm) |
| PBI |
385 ± 163 |
180 ± 73 |
| PBI:6FDD (90:10) |
306 ± 14 |
174 ± 26 |
| PBI:6FDD (70:30) |
288 ± 14 |
157 ± 10 |
| PBI:6FDD (50:50) |
263 ± 27 |
137 ± 11 |
 |
| | Fig. 4 SEM images of carbonized and activated (a) PBI, (b) PBI:6FDD (90:10), (c) PBI:6FDD (70:30) and (d) PBI:6FDD (50:50) nanofiber mats. | |
Fig. 5(a) and (c) show SEMs of the cross sections of PBI/6FDD of 70/30 and 50/50 blend films, respectively, after THF-extraction of 6FDD. PBI was the continuous phase and 6FDD was the dispersed phase. The size and shape of the dispersed minor phase in the immiscible blends of PBI/6FDD changed with the ratio of the two polymers with larger domain sizes appearing at the higher amounts of 6FDD. This trend can be explained by effect of enhanced coalescence of the 6FDD domains occurring during the phase separation process. The probability of collision between droplets would increase by increasing the composition of the dispersed phase.35 Because of that, at lower concentrations of 6FDD (30%), blending of two polymers resulted to a fine-size morphology with smaller domains compared to the 50% concentration of 6FDD. It is also possible that the higher amount of 6FDD leads to aggregation of 6FDD before it disperses within the PBI matrix during polymer mixing.
 |
| | Fig. 5 SEM images of the cross-section of the 6FDD extracted (a) PBI:6FDD (70:30) and (c) PBI:6FDD (50:50) films that were cast directly from electrospinning solutions and TEM images of electrospun fiber of (b) PBI:6FDD (70:30) and (d) PBI:6FDD (50:50). | |
This behaviour accounts for the different morphologies of the fibers after electrospinning of these blends as shown in TEM images of Fig. 5(b) and (d). The same trend of domain sizes was observed in the fiber as for the cast membranes but with somewhat more elongated domains after electrospinning. Fig. 6 shows the TEM images of the electrospun and carbonized fibers with different compositions of PBI/6FDD. In the TEM images, all the fibers from the blend solutions exhibited nanophase-separated domains, with isolated spheres of 6FDD dispersed in the continuous phase of PBI. Also, the 70/30 blend was found to have very uniform and smaller domains (area of 6FDD domain, 45 ± 15 nm2) in contrast to the 90/10 and 50/50 immiscible blends (area of 6FDD domain, 179 ± 149 nm2). The uniformity shown by 70/30 blend was important to tailor pore size distribution because the CO2 etch rate of the carbonized 6FDD at 1000 °C is approximately 20% faster than that of the carbonized PBI. The etch rates of the individual carbonized polymers of PBI and 6FDD were measured experimentally by separately activating thin film membranes of PBI and 6FDD for 30 min with CO2 following the same thermal treatment procedure as shown in Fig. 2 and then measuring mass before and after CO2 activation. Thus, the fine morphology of 70/30 blends ultimately leads to a balance of micro and meso pores on the activated carbon fiber. These different morphologies created by immiscible blends with various compositions greatly affect the resulting surface characteristics of fibers as confirmed by surface area analysis, and ultimately the electrochemical performance of electrodes made from these carbonized fibers.
 |
| | Fig. 6 TEM images of electrospun (a) PBI, (b) PBI:6FDD (90:10), (c) PBI:6FDD (70:30), (d) PBI:6FDD (50:50) nanofibers and carbonized and activated (e) PBI, (f) PBI:6FDD (90:10), (g) PBI:6FDD (70:30), (h) PBI:6FDD (50:50) nanofibers. | |
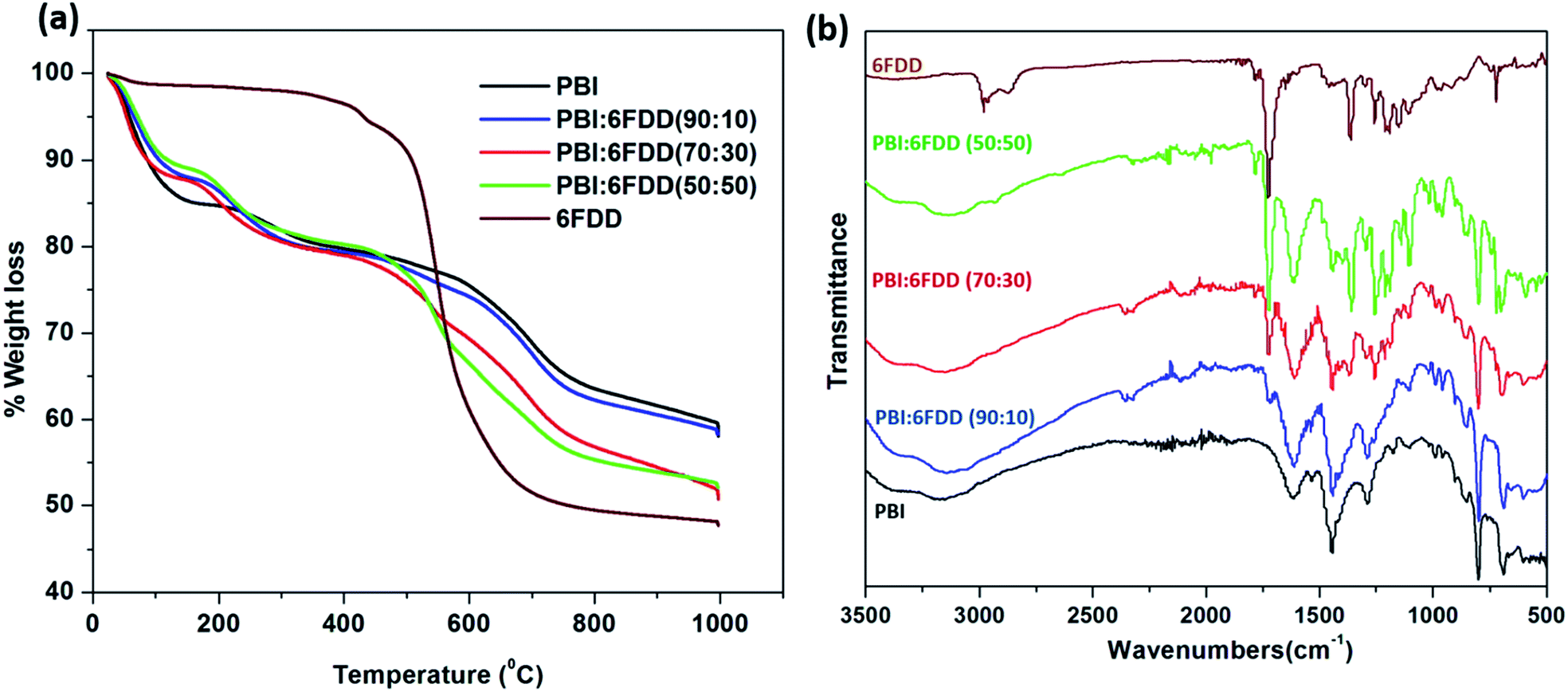
Fig. 7(a) shows weight-loss curves obtained from TGA in a N2 atmosphere for electrospun mats of pure PBI and PBI/6FDD blends. Pure 6FDD has a decomposition temperature around 490 °C. Three temperature regions at which mass losses occur can be clearly distinguished from the TGA profiles. These results are consistent with the previously reported thermal studies of polyimides by Kratochvil et al.32 The first weight loss up to 100 °C is associated with the evaporation of water and the second mass loss up to 250 °C is attributed to the evolution of trace amounts of residual solvent used for electrospinning. The significant amount of mass loss associated with main backbone decomposition of the polymer initiates at about 470 °C. The TGA profile of PBI:6FDD (50:50) blend, which has the highest percentage of 6FDD clearly shows a mass loss in this temperature region. Decarboxylation of DABA occurs over the temperature range of 350–390 °C but doesn't contribute to significant weight loss (normally around 3%). The carbon yields for PBI, PBI:6FDD (90:10), PBI:6FDD (70:30) and PBI:6FDD (50:50) at 1000 °C were 58.2%, 58.1%, 50.7% and 52.1%, respectively.
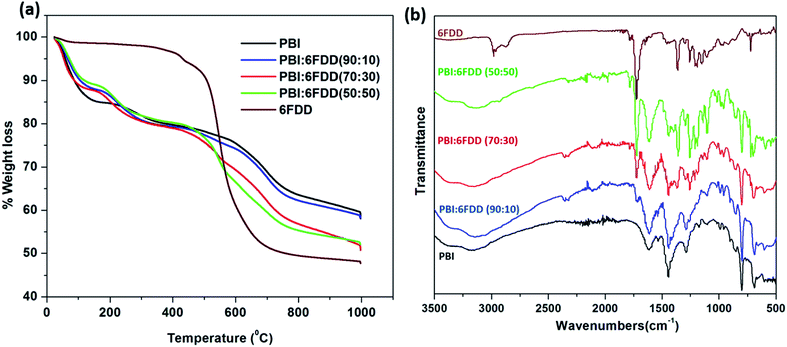 |
| | Fig. 7 TGA analysis (a) of as-spun mat of PBI and blends (b) and FTIR analysis. | |
FT-IR analysis shown in Fig. 7(b) confirms the blending of PBI and 6FDD. PBI and 6FDD show characteristic peaks at 3500–2500 cm−1 for the N–H bond and at 1630 cm−1 for the CN bond, respectively.36 The band appearing at 1460 cm−1 is assigned to the in-plane deformation peak of the imidazole ring. Pure 6FDD shows peaks at 1780 cm−1 and 1720 cm−1 that are assigned to the symmetric and asymmetric stretching vibrations of the imide CO, respectively. The FTIR spectra of the three blends show a combination of the peaks of the individual components in the blends. The intensities of peaks at 1780 and 1720 cm−1 increase with increasing composition of 6FDD in the blend from 10% to 50% and become dominant in the 50:50 blend.
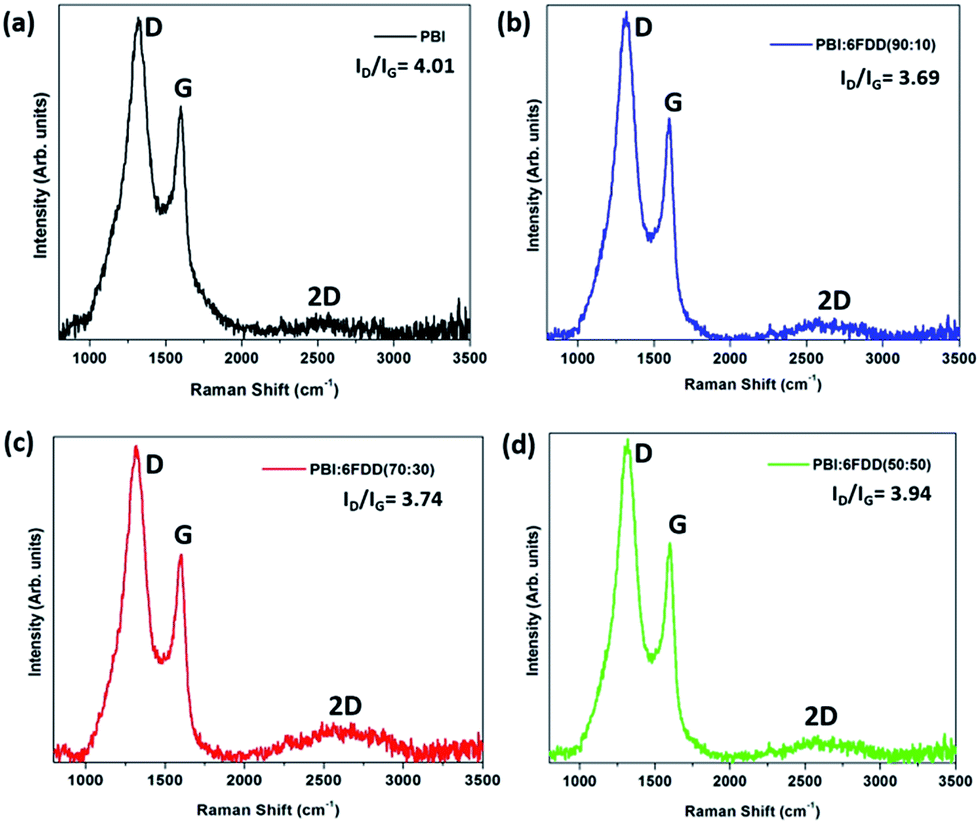
Fig. 8 shows the Raman spectra of carbonized samples. The first band around 1330 cm−1 (D-band) is due to presence of disordered carbon and the peak at 1590 cm−1 (G-band) is due to presence of highly oriented graphitic carbon.37 These two bands appear in all the spectra and confirms the formation of ordered graphitic structures. The relative intensity ratio of the D-band to the G-band, represented as ID/IG depends on the degree of graphitization and a lower ID/IG ratio indicates existence of the higher amount of ordered graphitic carbon in the sample. Lorentzian equation was used for peak fitting in order to calculate the ratios of the integrated area intensity of D band to G band (ID/IG) of all the spectra. OriginPro 8.0 software was used to deconvolute spectra into Lorentzian peaks and peak area obtained for D and G bands after fitting were used to calculate the ID/IG ratio.
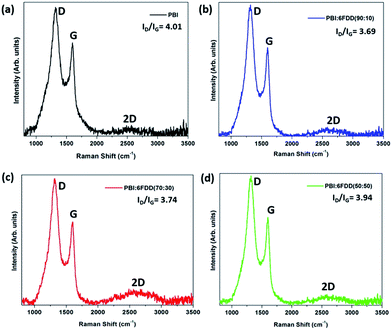 |
| | Fig. 8 Raman spectra of CNFs derived from (a) PBI, (b) PBI:6FDD (90:10), (c) PBI:6FDD (70:30) and (d) PBI:6FDD (50:50). | |
All blend samples showed ID/IG ratios in between 3.9–3.7, which are lower than pure PBI (4.01) suggesting that crosslinking of 6FDD facilitated the formation of more ordered graphitic structure during carbonization.38–40 Intense 2D bands observed around 2660 cm−1 for blends, and especially for 70/30 and 50/50 blends compared to the PBI, confirms the structural development of carbon materials after blending with 6FDD. The graphitic crystallite size (La) can be determine using Tuinstra–Koenig equation, La (nm) = 4.4/(ID/IG). The graphitic crystallite sizes (La) obtained for blends were ∼1.1 nm.41
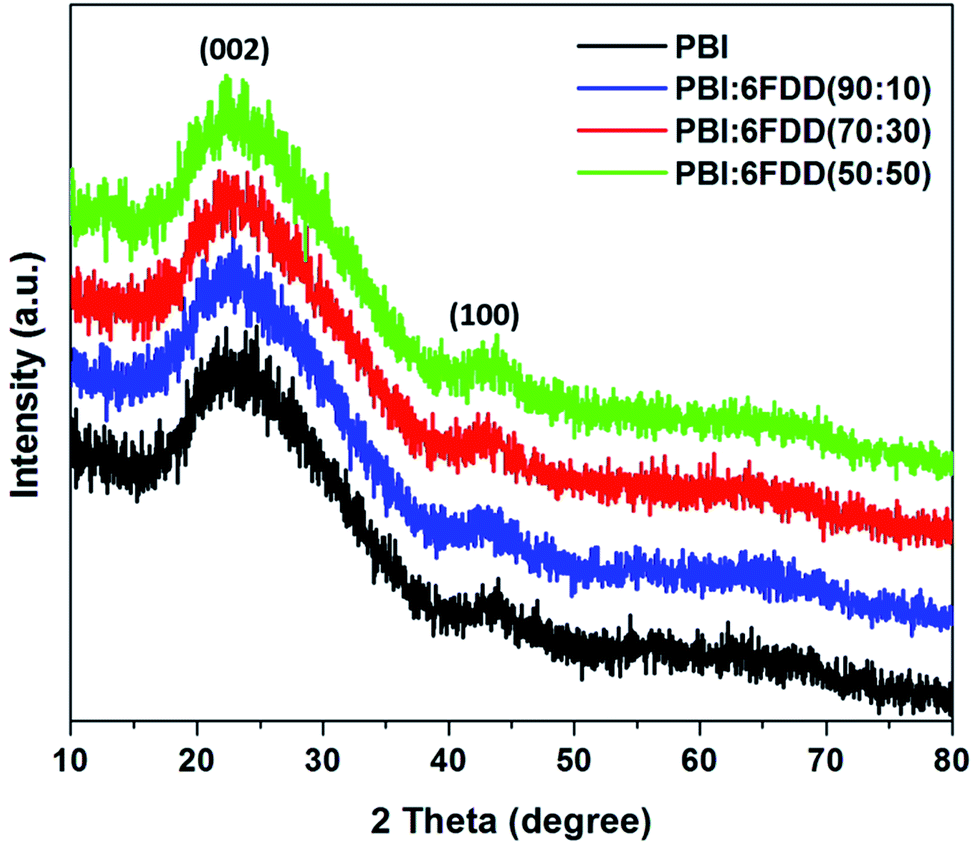
X-ray diffraction (XRD) patterns of all the carbonized samples were recorded as shown in Fig. 9 to further analyze structure and degree of graphitization of all samples. The XRD pattern of all the carbonized samples consists of a broad diffraction peak centered at around 24° (2θ) and a small peak around 44° (2θ), which correspond to (002) and (100) graphite planes of carbon, respectively and which confirms that all the samples have the graphitic structure.42,43
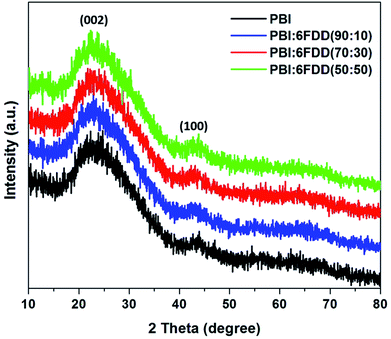 |
| | Fig. 9 XRD patterns of CNFs derived from PBI and PBI:6FDD blends. | |
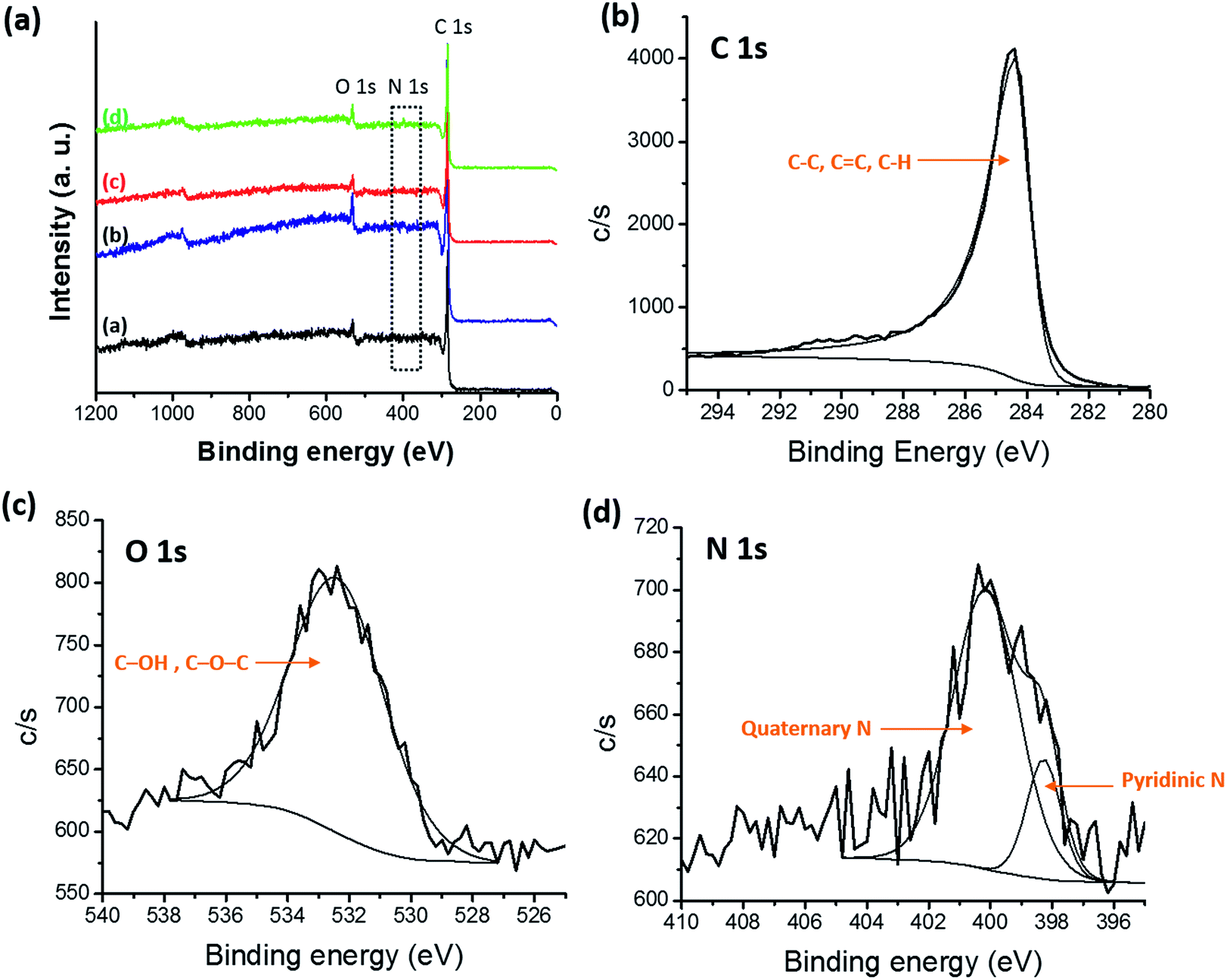
X-ray photoelectron spectroscopy (XPS) was performed to obtain surface elemental composition of carbonized samples and survey spectra were shown in Fig. 10(a). Three distinguishing peaks for C, N and O were observed in the survey spectra of all the samples and atomic percentages of C, O and N for all the carbonized samples were shown in Table 2. Trace amounts of fluorine (F) were detected in blends which originated from the CF3 moiety in 6FDA. Atomic percentage of F 1s was 0.09%, 0.05% and 0.31% for PBI:6FDD (90:10), PBI:6FDD (70:30) and PBI:6FDD (50:50), respectively. Also a trace amount of Si was detected which attributed to the quartz tube used for carbonization. The deconvoluted XPS spectra of C 1s, N 1s and O 1s for PBI:6FDD (70:30) are shown in the Fig. 10(b)–(d). Fig. S3–S5† show deconvoluted XPS spectra of C 1s, N 1s and O 1s for PBI, PBI:6FDD (90:10) and PBI:6FDD (50:50). High resolution XPS spectrum of C 1s for all the samples shows mainly a large peak at a binding energy of 284.3 eV for C–C peak for graphite (sp2 bonded carbon).44 The deconvoluted high resolution O 1s spectrum also mainly consist of one peak at the binding energy of 532.3 eV assigned to the C–OH or C–O–C.45 The deconvoluted N 1s spectrum has peaks at 398.2 eV and 401.1 eV, are attributed to pyridinic nitrogen and quaternary nitrogen atoms.43
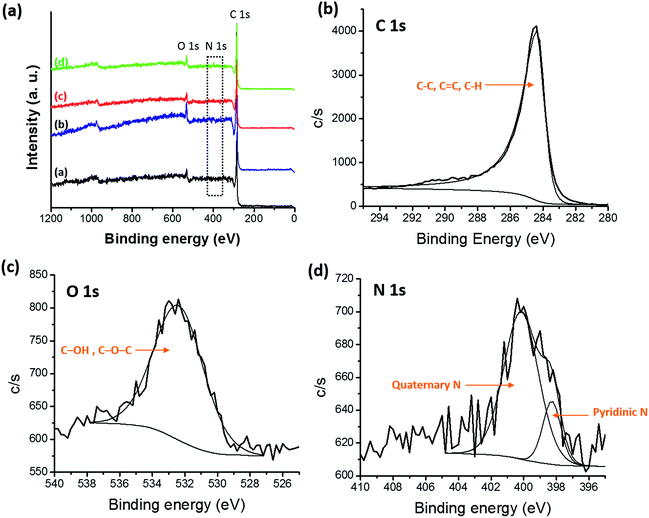 |
| | Fig. 10 (a) XPS survey spectra of all CNFs samples (inset, (a) PBI, (b) PBI:6FDD (90:10), PBI:6FDD (70:30) and (d) PBI:6FDD (50:50)), high-resolution deconvoluted (b) C 1s, (c) O 1s, and (d) N 1s, XPS spectra of CNFs derived from PBI:6FDD (70:30). | |
Table 2 Chemical composition of C 1s, N 1s and O 1s obtained from the XPS spectra
| Precursor |
C 1s (at%) |
O 1s (at%) |
N 1s (at%) |
| PBI |
94.69 |
3.09 |
1.25 |
| PBI:6FDD (90:10) |
95.15 |
3.04 |
1.60 |
| PBI:6FDD (70:30) |
95.55 |
2.96 |
1.41 |
| PBI:6FDD (50:50) |
93.76 |
3.58 |
1.79 |
3.3 Nitrogen adsorption and pore size analysis
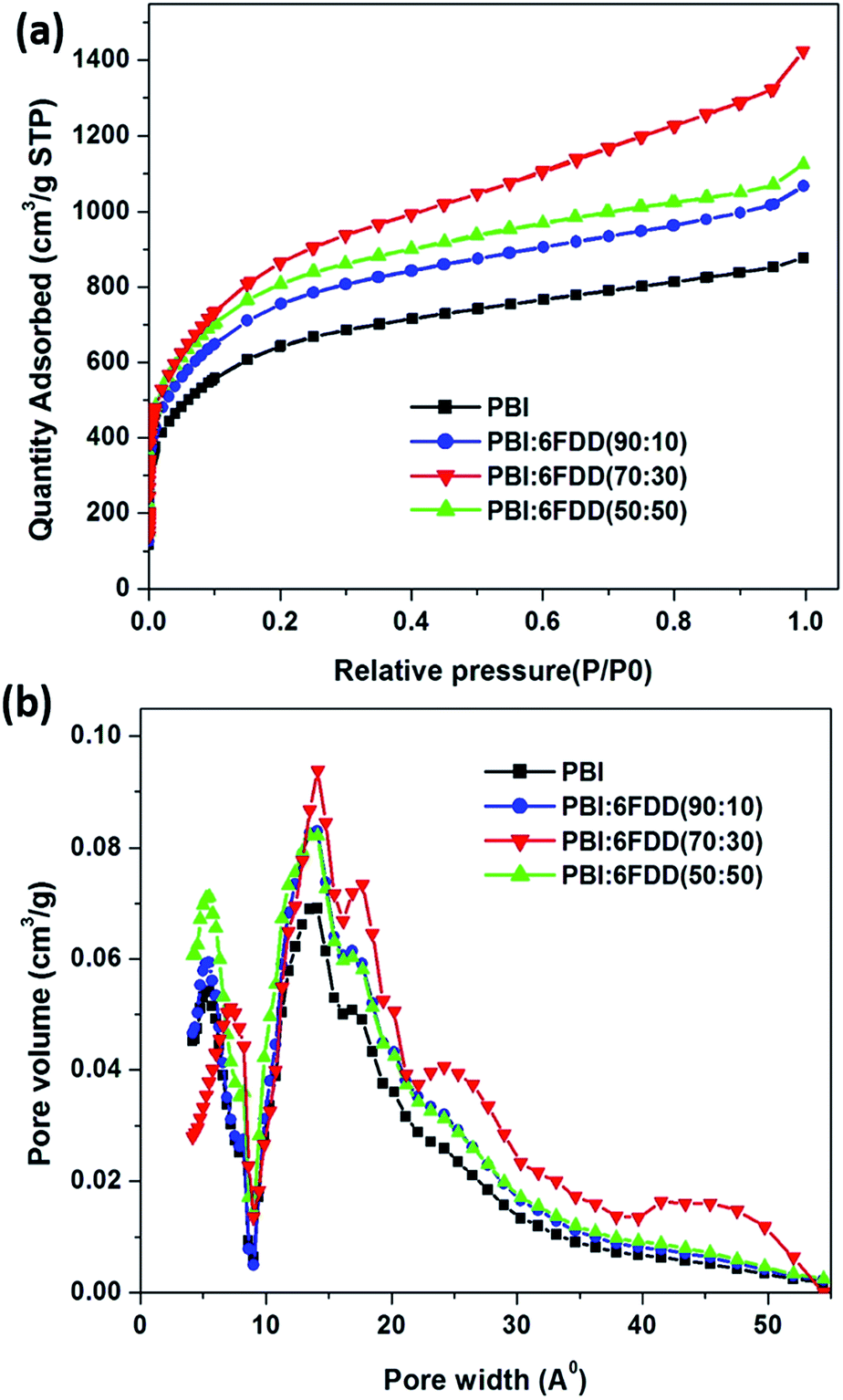
Nitrogen adsorption and pore size analysis were used to investigate the effect of blend compositions on the surface area properties and pore size distributions of the prepared CNFs. Isotherms for the carbon derived from the various precursors are shown in Fig. 11(a). The adsorption isotherms showed typical type I behavior representing the microporous adsorption with the absence of hysteresis loop. Fig. 11(b) shows the pore size distribution of activated CNFs. In order to derive maximum capacitance of EDLC, the pore sizes of electrode materials should be similar to the sizes of the electrolyte ions.46–48 Capacitance decreases when the pore size is too small to allow ions to enter the pores. The average pore sizes of the carbonized PBI/6FDD blends (1.3 nm) were close to the cation and anion size of the PYR14TFSI electrolyte which are reported as 1.1 nm and 0.7 nm, respectively.49 The matching of the pores of electrodes prepared from PBI/6FDD blends with ion size of electrolyte resulted in the significant increase in the capacitance obtained from the devices. Furthermore, there was a notable difference between the pore size distribution among the samples, with the 70:30 blend exhibiting a higher percentage of mesopores (42.8%) leading to a micro to mesopore ratio around 1.34. Micropores help to adsorb ions in to the pores to form the double layer whereas mesopores facilitate the ion diffusion to these micropores by providing low resistive pathways for electrolyte ions to pass through the porous carbon structure. Table 3 summarizes BET surface areas of CNF of pure PBI and blends with different compositions. The high free volume polymer, 6FDD, clearly contributed to increase in the surface area because all the CNFs prepared from PBI:6FDD blends exhibit higher surface areas (over 2795 m2 g−1) compared to the PBI CNFs. High free volume in the precursor matrix may contribute high porosity of the carbonized matrix. The highest surface area was obtained for PBI:6FDD (70:30) at 3010 m2 g −1 which is a 31% increase compared to that of pure PBI (2297 m2 g−1).
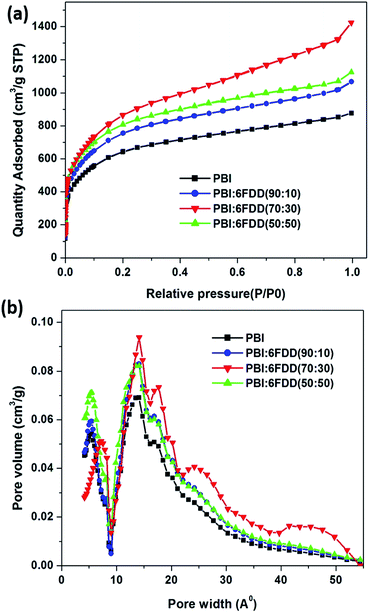 |
| | Fig. 11 (a) Nitrogen adsorption/desorption isotherms and (b) pore size distribution for CNFs derived from PBI and PBI:6FDD blends. | |
Table 3 Surface area and pore volume of PBI and PBI/6FDD based carbon nanofibers
| Precursor |
surface area (m2 g−1) |
Total pore volume (cm3 g−1) |
Vmicro (cm3 g−1) |
Vmeso (cm3 g−1) |
% micropores |
% mesopores |
Micropore/mesopore ratio |
| PBI |
2297 |
1.1009 |
0.7345 |
0.3664 |
66.7 |
33.3 |
2.01 |
| PBI:6FDD (90:10) |
2795 |
1.2998 |
0.8495 |
0.4503 |
65.3 |
34.7 |
1.89 |
| PBI:6FDD (70:30) |
3010 |
1.6077 |
0.9205 |
0.6872 |
57.2 |
42.8 |
1.34 |
| PBI:6FDD (50:50) |
2879 |
1.3913 |
0.9279 |
0.4634 |
66.7 |
33.3 |
2.00 |
3.4 Electrochemical properties of CNFs
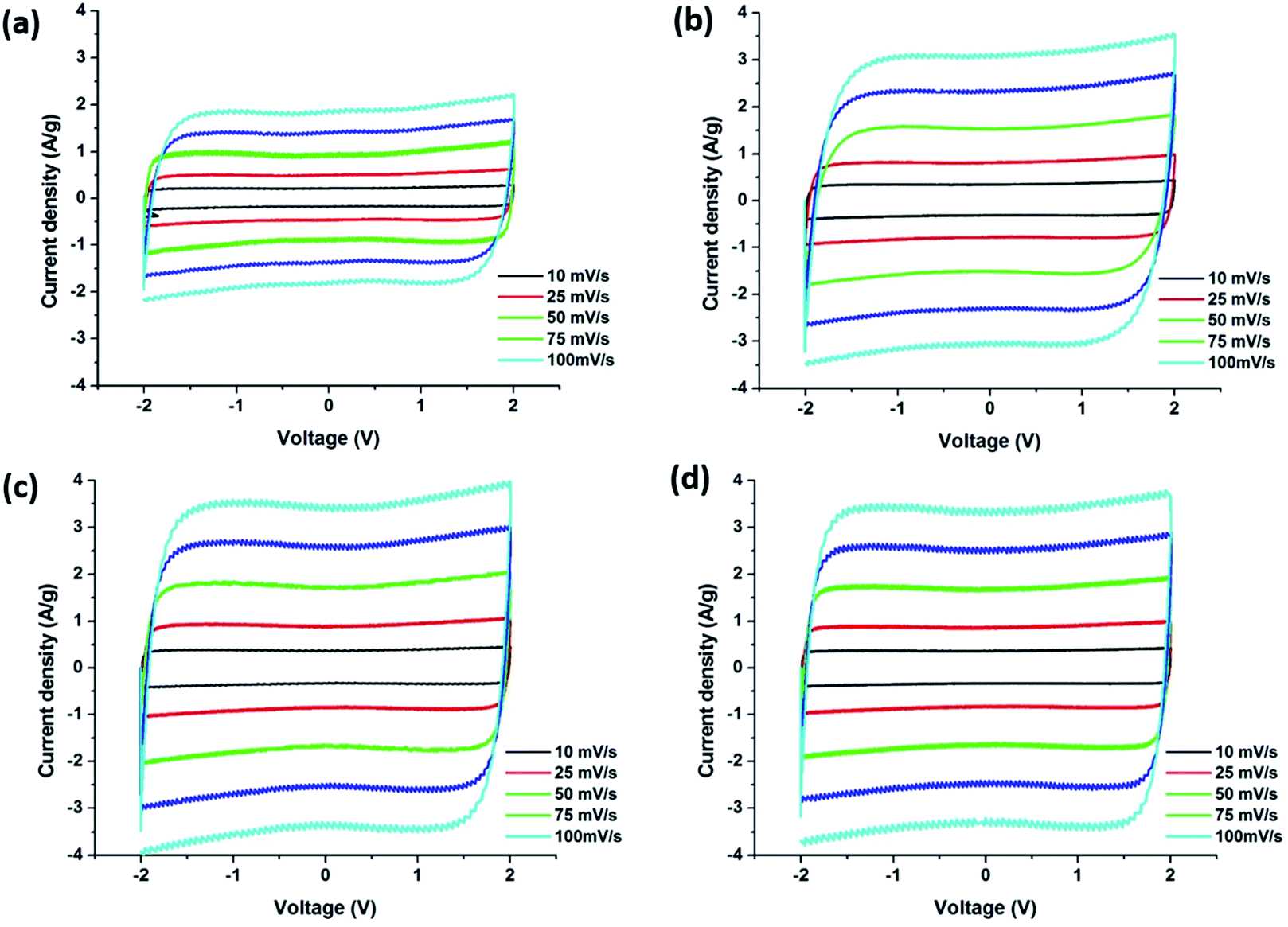
Fig. 12 displays the cyclic voltammetry (CV) curves obtained from the supercapacitors assembled with carbon precursor polymer blends that show rectangular shapes which indicates the ideal capacitive character of the EDLC. PBI:6FDD (70:30) and PBI:6FDD (50:50) also showed very box like CVs even at higher current densities when compared with pure PBI indicating fast charge transfer at the electrode/electrolyte interfaces. Rapid ion transport is facilitated by the significant amount of mesopores in the carbon materials especially in PBI:6FDD (70:30). Since the energy density of supercapacitors varies quadratically with voltage, we used the ionic liquid electrolyte, 1-butyl-1 methylpyrrolidiniumbis (trifluoromethylsulfonyl)imide (PYR14TFSI) that can operate at high voltage (∼4 V). The electrolyte used here is a mixture of (PYR14TFSI), propylene carbonate (PC) and ethylene carbonate (EC) in the volume ratio of 3:3:2 (PYR14TFSI:PC:EC). By mixing with the optimum ratio of PC and EC, the conductivity of PYR14TFSI (1.85 mS cm−1) was increased to 12.5 mS cm−1. This increased conductivity in electrolyte ions helped to decrease the charge transfer resistance between electrode and electrolyte and made the CV plot box-like even in the higher scan rates. The specific capacitance values obtained for the PBI:6FDD blends studied were higher than the pure PBI at all scan rates because of the high surface area of CNF derived from PBI:6FDD blends. The highest specific capacitance of PBI:6FDD (70:30) at 10 mV s−1 was calculated to be 142.8 F g−1 compared to 75.9 F g−1 for the pure PBI, as shown in Table 4. The capacitance increases linearly with the surface area. High performance was observed for PBI:6FDD (70:30) is due to high specific surface area and well balanced micro/mesoporosity of this precursor.
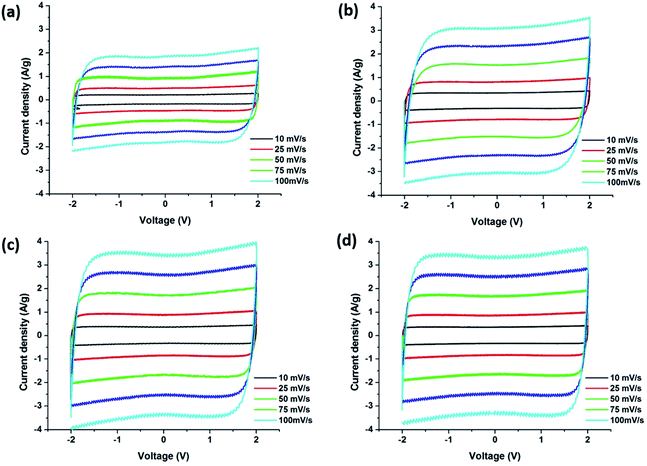 |
| | Fig. 12 Cyclic voltammograms of carbonized nanofibers derived (a) PBI, (b) PBI:6FDD (90:10), (c) PBI:6FDD (70:30) and (d) PBI:6FDD (50:50). | |
Table 4 Specific capacitance of the CNFs derived from the PBI and the PBI/6FDD blends at five different scan rates
| Scan rate (mV s−1) |
Specific capacitance (F g−1) |
| PBI |
PBI:6FDD (90:10) |
PBI:6FDD (70:30) |
PBI:6FDD (50:50) |
| 10 |
75.9 |
129.8 |
142.8 |
138.3 |
| 25 |
74.6 |
129.4 |
141.4 |
137.5 |
| 50 |
74.5 |
125.5 |
140.6 |
136.9 |
| 75 |
73.7 |
124.1 |
140.5 |
136.7 |
| 100 |
73.3 |
123.4 |
138.7 |
136.2 |
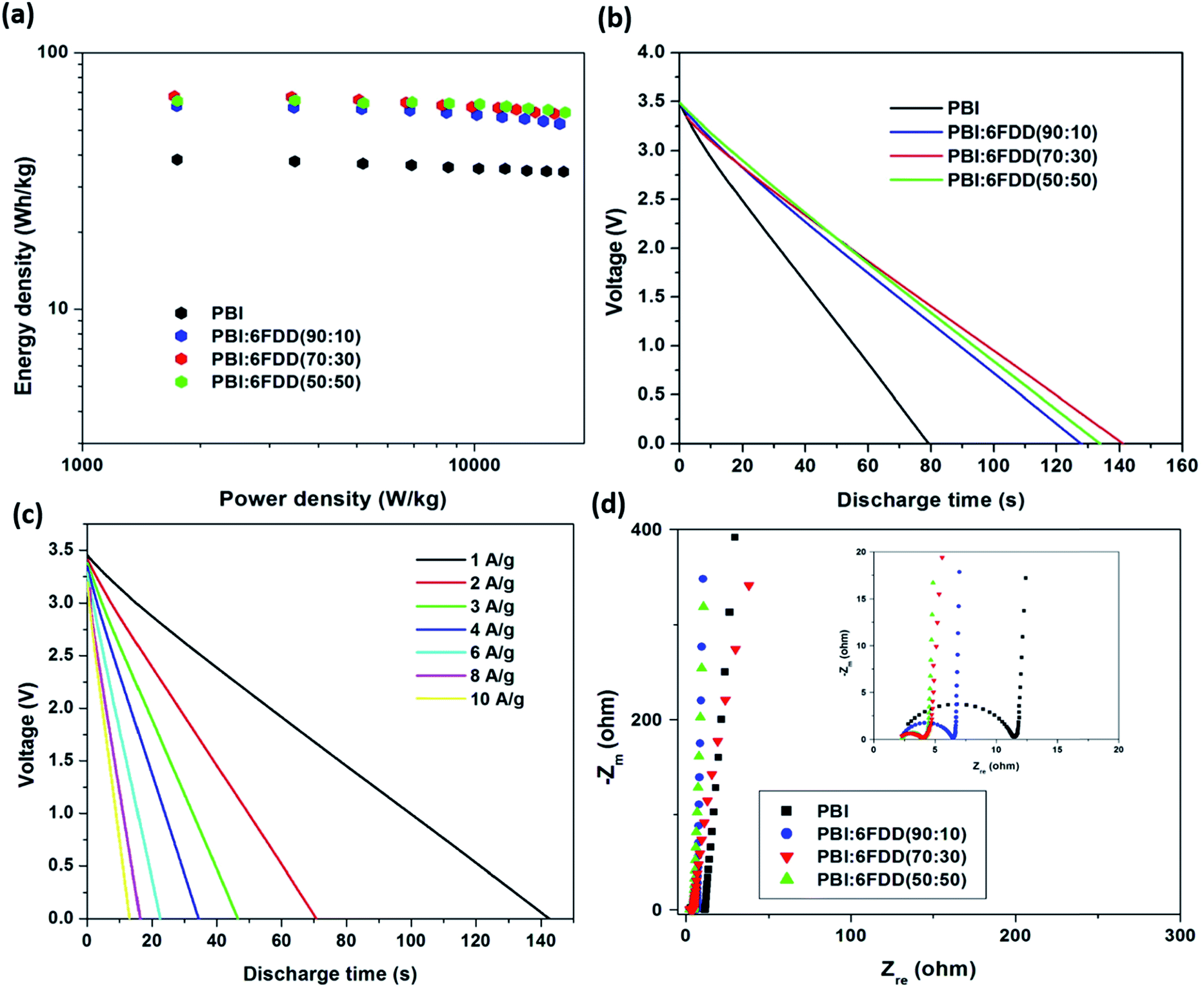
As shown in the Ragone plot (Fig. 13(a)) 85–90% of the energy density of the electrodes was retained when the power density increased 10-fold. The energy and power densities of the supercapacitors that were assembled with carbonized PBI/6FDD nanofibers at a current discharge density of 1 A g−1 and 10 A g−1 are listed in the Table 5. The energy density calculated for at 1 A g−1 was 67.5 W h kg−1 for PBI:6FDD (70:30) compared to that of PBI (38.4 W h kg−1) at similar power densities. Also the energy densities and power densities obtained for all the PBI/6FDD blends are much higher than the those reported for PBI blends with other polymers.28,29 Fig. 13(b) shows the galvanostatic discharging curves for carbonized blends and PBI at 1 A g−1. All the blend-derived fibers exhibited almost linear curves and there was significant difference between the discharge time between PBI and the PBI/6FDD blends, where the blends showed the longer discharge times. PBI:6FDD (70:30) shows the longest discharge time among the blends studied. Discharge curves of the PBI:6FDD (70:30) at different current densities are plotted in Fig. 13(c). Discharge curves of the PBI:6FDD (70:30) exhibited almost linear curves with no significant IR drop, which indicates a device with the good performance having high electrochemical stability and reversibility.
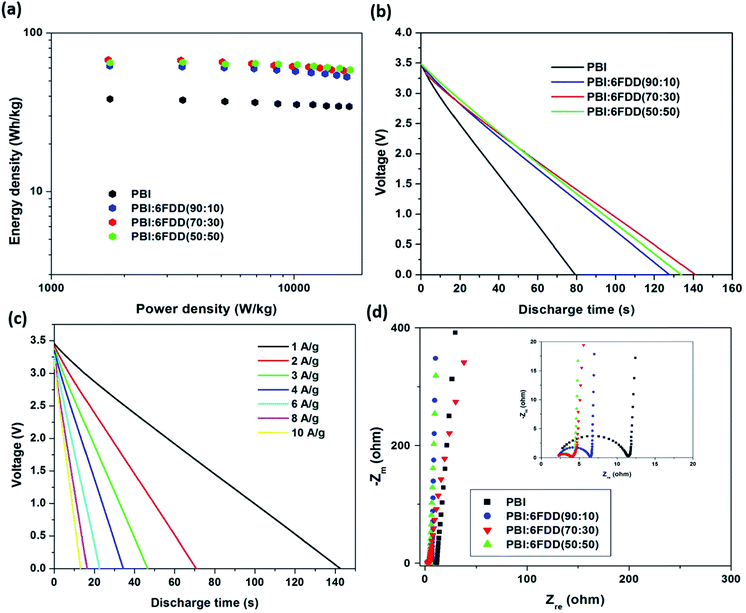 |
| | Fig. 13 (a) Ragone plots (b) galvanostatic discharging curves at a constant current density of 1 A g−1 (c) galvanostatic discharging curves for PBI:6FDD (70:30) at different current densities from 1 to 10 A g−1 (d) Nyquist plot of carbonized nanofibers derived from PBI and PBI:6FDD blends with different compositions. | |
Table 5 Energy densities and the power densities of the CNFs derived from PBI and the PBI/6FDD blends at current densities of 1 A g−1 and 10 A g−1
| Current density (A g−1) |
Parameters |
PBI |
PBI:6FDD (90:10) |
PBI:6FDD (70:30) |
PBI:6FDD (50:50) |
| 1 |
Energy density (W h kg−1) |
38.4 |
61.8 |
67.5 |
64.7 |
| Power density (W kg−1) |
1742.5 |
1738.3 |
1713.8 |
1743.5 |
| 10 |
Energy density (W h kg−1) |
34.4 |
52.8 |
57.8 |
58.3 |
| Power density (W kg−1) |
16830.7 |
16451.2 |
15930.6 |
16974.4 |
Fig. 13(d) displays the Nyquist plots of carbonized materials obtained from PBI and PBI/6FDD blends with different compositions. The impedance spectra for all the electrode materials displayed ideal capacitive behavior with a semicircle in the high frequency region and a straight line in the low-frequency region. The semicircle corresponds to the charge-transfer process while the linear region related to the diffusion of ions into the bulk of the electrode.50 The semicircle in the high-frequency region, is the sum of the resistance of the porous electrode and the interfacial contact resistance between the porous electrode and the current collector while the intercept on the real axis in the high-frequency region provides the resistance of the electrolyte.51 The diameter of the semicircles obtained for electrodes followed the order of PBI > PBI:6FDD (90:10) > PBI:6FDD (50:50) > PBI:6FDD (70:30) suggesting low charge-transfer resistance of those electrodes was obtained after blending with 6FDD. These enhancements could be due to the shorter ion diffusion routes formed by the mesopores in the pore structure of PBI:6FDD (70:30), which reduces the resistivity. Furthermore, electrochemical impedance spectroscopy analysis revealed that PBI/6FDD blend has a much smaller charge transfer resistance than PBI.
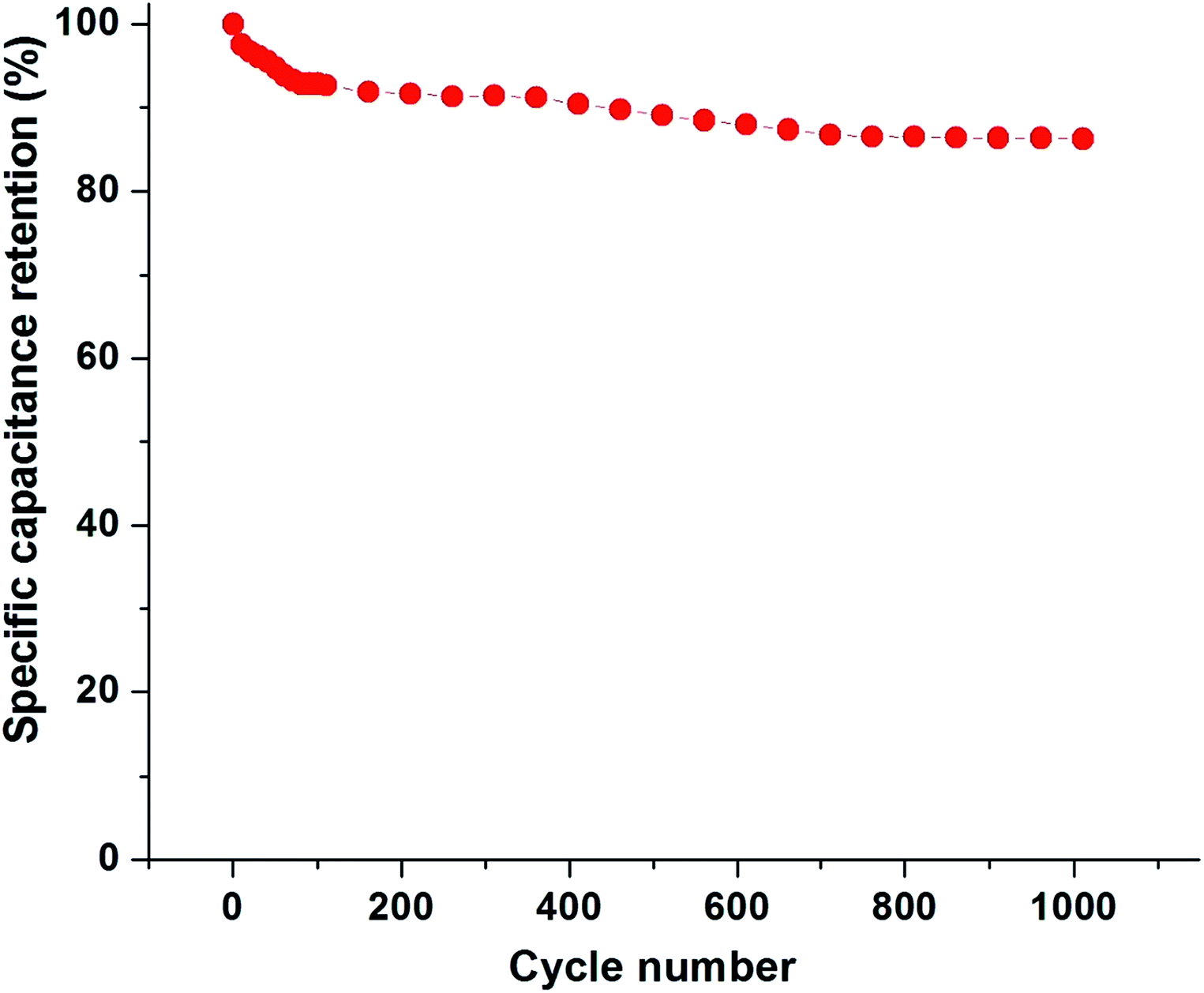
Fig. 14 shows the cycling stability of PBI:6FDD (70:30) at a current density of 1 Ag −1 in PYR14TFSI:PC:EC electrolyte. After the 100th cycle 92.8% of the specific capacitance was retained, which only dropped by 6.5% over the next 900 cycles. These results demonstrate that the activated carbon nanofibers derived from PBI:6FDD (70:30) have high electrochemical stability and reversibility during repetitive charge/discharge cycles in ionic liquid electrolyte system.
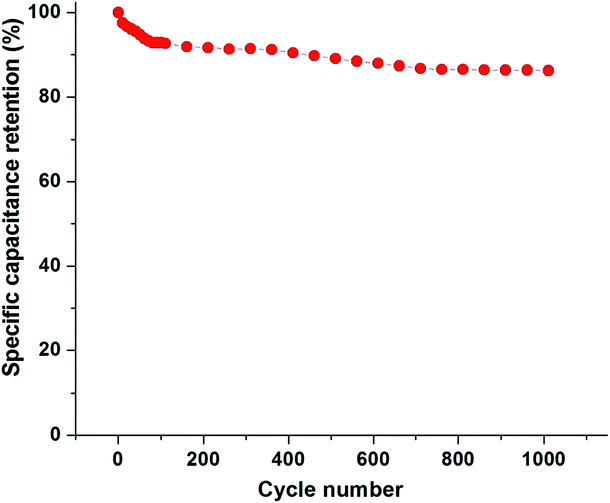 |
| | Fig. 14 Specific capacitance retention of PBI:6FDD (70:30) as a function of cycle number. | |
4. Conclusion
In summary, flexible and freestanding carbon nanofiber mats fabricated by electrospinning immiscible PBI/6FDD polymer blends, followed by subsequent thermal treatments, could be used as electrode materials for supercapacitors in the first time. The addition of 6FDD shows significant effects on the microstructure and surface areas of the CNFs. Binder-free electrodes were assembled into coin cells and tested. The highest capacitance obtained for the PBI:6FDD (70:30) was 142 F g−1 at 10 mV s−1 which is an 88% increase when compared with the electrode prepared from PBI-based CNF under the same conditions. Supercapacitors based on PBI:6FDD (70:30) exhibit a high energy density of 67.5 W h kg−1 at 1 A g−1. Improvements of the electrochemical performance are attributed to the following factors: more graphitic carbon produced by facilitated carbonization process due to crosslinking of 6FDD of PBI/6FDD blends; the large surface area CNFs derived from PBI/6FDD blends due to addition of high free volume polymer 6FDD; well developed and suitable pore structure of the carbon nanofibers derived from PBI/6FDD blends with the formation of well-balanced micro/mesoporosity with pore sizes very close to those of the electrolyte ions to create more access sites for charge transfer, especially for the PBI:6FDD (70:30) blend. PBI/6FDD blends can be directly carbonized without pre-stabilization, which adds to their attractiveness. This work highlights the potential applicability of immiscible polymer blends of PBI/6FDD to produce electrode materials for high performance energy storage systems.
Acknowledgements
The authors acknowledge the National Science Foundation (Grant no. IIP-1127564) for financial support. We also acknowledge the NSF (CHE-1126177) grant for providing the Bruker AVANCE III™ 500 NMR facility. We especially thank Dr Sumudu N. Wijenayake for providing the initial 6FDD sample. We also acknowledge to Dr Jean Francois Veyan (Department of Materials Science and Engineering, University of Texas at Dallas) for helping with the XPS analysis.
References
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- B. E. Conway, V. Birss and J. Wojtowicz, J. Power Sources, 1997, 66, 1–14 CrossRef CAS.
- Y. Z. Xie, Y. Liu, Y. D. Zhao, Y. H. Tsang, S. P. Lau, H. T. Huang and Y. Chai, J. Mater. Chem. A, 2014, 2, 9142 CAS.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS PubMed.
- K. Wang, M. Xu, X. Wang, Z. Gu, Q. H. Fan, W. Gibbons and J. Croat, RSC Adv., 2017, 7, 8236–8240 RSC.
- A. Zhang, A. Li, Y. Wang, M. Liu, H. Ma, Z. Song and J. Liu, RSC Adv., 2016, 6, 103843–103850 RSC.
- Q. Wang, J. Yan and Z. Fan, Energy Environ. Sci., 2016, 9, 729–762 CAS.
- M. Inagaki, Y. Yang and F. Kang, Adv. Mater., 2012, 24, 2547–2566 CrossRef CAS PubMed.
- L. Zhang, L. Han, S. Liu, C. Zhang and S. Liu, RSC Adv., 2015, 5, 107313–107317 RSC.
- S. Perananthan, J. S. Bonso and J. P. Ferraris, Carbon, 2016, 106, 20–27 CrossRef CAS.
- J. Cai, H. Niu, Z. Li, Y. Du, P. Cizek, Z. Xie, H. Xiong and T. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 14946–14953 CAS.
- C. Ma, Y. Li, J. Shi, Y. Song and L. Liu, Chem. Eng. J., 2014, 249, 216–225 CrossRef CAS.
- G. Gong and J. Wu, Chemistry to Applications, 2012, ch. 7, pp. 127–144 Search PubMed.
- C. Kim, Y. Choi, W. Lee and K. Yang, Electrochim. Acta, 2004, 50, 883–887 CrossRef CAS.
- C. Kim, J. Power Sources, 2005, 142, 382–388 CrossRef CAS.
- M. Inagaki, N. Ohta and Y. Hishiyama, Carbon, 2013, 61, 1–21 CrossRef CAS.
- Y. Li, J. Dong, J. Zhang, X. Zhao, P. Yu, L. Jin and Q. Zhang, Small, 2015, 11, 3476–3784 CrossRef CAS PubMed.
- D. Hulicova and A. Oya, Carbon, 2003, 41, 1443–1450 CrossRef CAS.
- J. Ozaki, N. Endo, W. Ohizumi, K. Igarashi, M. Nakahara and A. Oya, Carbon, 1997, 35, 1031–1033 CrossRef CAS.
- N. C. Abeykoon, J. S. Bonso and J. P. Ferraris, RSC Adv., 2015, 5, 19865–19873 RSC.
- E. Jo, J.-G. Yeo, D. K. Kim, J. S. Oh and C. K. Hong, Polym. Int., 2014, 63, 1471–1477 CrossRef CAS.
- X. Tian, N. Zhao, K. Wang, D. Xu, Y. Song, Q. Guoa and L. Liu, RSC Adv., 2015, 5, 40884–40891 RSC.
- S. S. Hosseini and T. S. Chung, J. Membr. Sci., 2009, 328, 174–185 CrossRef CAS.
- S. S. Hosseini, M. M. Teoh and T. S. Chung, Polymer, 2008, 49, 1594–1603 CrossRef CAS.
- N. P. Panapitiya, S. N. Wijenayake, Y. Huang, D. Bushdiecker, D. Nguyen, C. Ratanawanate, G. J. Kalaw, C. J. Gilpin, I. H. Musselman, K. J. Balkus and J. P. Ferraris, Polymer, 2014, 55, 2028–2034 CrossRef CAS.
- V. Pirouzfar, A. Z. Moghaddam, M. R. Omidkhah and S. S. Hosseini, J. Ind. Eng. Chem., 2014, 20, 1061–1070 CrossRef CAS.
- S. S. Hosseini, N. Penga and T. S. Chung, J. Membr. Sci., 2010, 349, 156–166 CrossRef CAS.
- K. Jung and J. P. Ferraris, Carbon, 2012, 50, 5309–5315 CrossRef CAS.
- K. Jung and J. P. Ferraris, Nanotechnology, 2016, 27(42), 425708 CrossRef PubMed.
- J. H. Kim, W. J. Koros and D. R. Paul, Polymer, 2006, 47, 3094–3103 CrossRef CAS.
- W. Qiu, L. Xu, C. Chen, D. R. Paul and W. J. Koros, Polymer, 2013, 54, 6226–6235 CrossRef CAS.
- A. M. Kratochvil and W. J. Koros, Macromolecules, 2008, 41, 7920–7927 CrossRef CAS.
- I. C. Omole, S. J. Miller and W. J. Koros, Macromolecules, 2008, 41, 6367–6375 CrossRef CAS.
- T. H. Le, Y. Yang, Z. Huang and F. Kang, J. Power Sources, 2015, 278, 683–692 CrossRef CAS.
- N. Grizzuti and O. Bifulco, Rheol. Acta, 1997, 36, 406–415 CrossRef CAS.
- P. A. Christensen and S. W. M. Jones, Polym. Degrad. Stab., 2014, 105, 211–217 CrossRef CAS.
- Y. Zhang, X. Liu, S. Wang, S. Dou and L. Li, J. Mater. Chem. A, 2016, 4, 10869–10877 CAS.
- W. Zhao, Y. Lu, J. Wang, Q. Chen, L. Zhou, J. Jiang and L. Chen, Polym. Degrad. Stab., 2016, 133, 16–26 CrossRef CAS.
- W. Liu, M. Wang, Z. Xing, Y. Qi and G. Wu, Radiat. Phys. Chem., 2012, 81(6), 622–627 CrossRef CAS.
- K. Kim, H. Lee, J. An, B. Kim, B. Min, S. Kang, K. An and B. Kim, Carbon letters, 2015, 16(3), 147–170 CrossRef.
- M. Hoefer and P. R. Bandaru, Appl. Phys. Lett., 2009, 95(18), 183108 CrossRef.
- X. Yang, J. Yu, W. Zhang and G. Zhang, RSC Adv., 2017, 7(25), 15096–15101 RSC.
- A. Choudhury, J. Kim, S. S. Mahapatra, K. S. Yang and D. J. Yang, ACS Sustainable Chem. Eng., 2017, 5(3), 2109–2118 CrossRef CAS.
- X. Li, Y. Zhao, Y. Bai, X. Zhao, R. Wang, Y. Huang, Q. Liang and Z. Huang, Electrochim. Acta, 2017, 230, 445–453 CrossRef CAS.
- S. Biniak, G. Szymanski, J. Siedlewski and A. Swiatkowski, Carbon, 1997, 35, 1799–1810 CrossRef CAS.
- M. Lazzari, M. Mastragostino, A. G. Pandolfo, V. Ruiz and F. Soavi, J. Electrochem.
Soc., 2011, 158, 22–25 CrossRef.
- C. H. Kim, J. Wee, Y. A. Kim, K. S. Yang and C. Yang, J. Mater. Chem. A, 2016, 4, 4763–4770 CAS.
- E. Raymundo-Pinero, K. Kierzek, J. Machnikowski and F. Beguin, Carbon, 2006, 44, 2498–2507 CrossRef CAS.
- S. Pohlmann, B. Lobato, T. A. Centeno and A. Balducci, Phys. Chem. Chem. Phys., 2013, 15, 17287–17294 RSC.
- K. Subramani, N. Sudhan, R. Divyaa and M. Sathish, RSC Adv., 2017, 7, 6648–6659 RSC.
- C. Lei, F. Markoulidis, Z. Ashitaka and C. Lekakou, Electrochim. Acta, 2013, 92, 183–187 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: (1) Synthesis of 6FDA-DAM:DABA, (2) characterization of 6FDA-DAM:DABA (a) NMR, (b) TGA and (c) FT-IR. (3), (4) and (5) XPS analysis of precursor. See DOI: 10.1039/c7ra01727h |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab