
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA halotolerant aldose reductase from Debaryomyces nepalensis: gene isolation, overexpression and biochemical characterization†
Bhaskar Paidimuddala,
Gopala Krishna Aradhyam and
Sathyanarayana N. Gummadi *
*
Applied and Industrial Microbiology Laboratory, Department of Biotechnology, Bhupat and Jyoti Mehta School of Biosciences, Indian Institute of Technology Madras, Chennai 600 036, India. E-mail: gummadi@iitm.ac.in; Fax: +91-44-2257-4102; Tel: +91-44-2257-4114
First published on 7th April 2017
Abstract
Aldose reductase (AR) catalyzes the conversion of aldoses to their corresponding polyols in yeasts and filamentous fungi. ARs have the potential to be exploited for the enzymatic production of xylitol, thus the identification and characterization of ARs from novel strains have gained interest. In this study, we chose the novel yeast Debaryomyces nepalensis as a source for an AR gene. For the first time, here we isolated the AR gene from D. nepalensis (DnAR) that encodes a protein of 320 amino acids with a predicted molecular weight of 36.7 kDa using the RACE technique. It was heterologously expressed in Escherichia coli as a His-tagged fusion protein and purified. The enzyme showed strict NADPH dependence and broad substrate specificity with high catalytic efficiency for arabinose, xylose and 3-nitro benzaldehyde. Remarkably, it was active and stable in the presence of high concentrations of salts (KCl/NaCl), thus exhibiting halotolerance. It showed 75% and 45% activity at 0.5 and 1 M concentration of salts respectively. Enzyme half-lifetime at 1 M KCl and 1 M NaCl was found to be 30 h and 16.5 h respectively. Furthermore, to explore the structural basis of its halotolerance, we built a homology model of DnAR. Surprisingly, we found that the existence of a uniform negative electrostatic potential over the protein surface, which is one of the known mechanisms governing protein halotolerance. Therefore, DnAR could be exploited as a biocatalyst to develop an enzyme based bioprocess for xylitol production from lignocelluloses. Moreover, this is the first report providing the genetic sequence and biochemical characteristics of a halotolerant aldose reductase.
1. Introduction
Aldose reductase (AR) (EC 1.1.1.21) is a member of the aldo-keto reductase (AKR) super family of enzymes, which catalyses the reduction of aldehydes to their corresponding alcohols.1 ARs are highly conserved and found in animals, plants and microbes.2 Though broad substrate specificity is the signature property of ARs, the exact biological function differs among the organisms.2,3 In animals and plants, AR preferably catalyses the conversion of glucose to sorbitol with strict dependence on NADPH.3,4 AR in humans has been implicated in the onset of diabetic complications by forming excessive polyols in the hyperglycemic state in tissues.4 Due to this reason, numerous reports on the development of therapeutic AR inhibitors have been published.5,6 In contrast to human AR, the yeast AR is involved in the reduction of aldopentoses such as xylose, arabinose and erythrose with concomitant oxidation of either NADPH or NADH.7 Therefore, yeast ARs have the potential for the exploitation of lignocellulosic xylose to produce xylitol, a pentose alcohol.8,9Xylitol is a multi-beneficial natural sugar substitute with potential applications in sugar-free foods and drinks, oral hygiene, pharma and cosmetic products.10 Currently, xylitol production at industrial scale involves chemical hydrogenation of xylose extracted from lignocellulose under extreme conditions of pressure and temperature with toxic catalysts.11 Although microbial production of xylitol is advantageous as compared to chemical processes in terms of cost and environmental impact, catabolite repression and downstream processing from by-products (glycerol, arabitol and ethanol) makes it expensive.12 Since only one enzyme AR, requires for the production of xylitol, the development of enzymatic bioprocess could surpass the limitations of current processes.8,9 Therefore, ARs have gained interest, leading to the characterization of putative ARs and identification of new ARs having remarkable properties such as halotolerance and thermotolerance which help sustain industrial conditions as well as salts contaminated lignocellulose hydrolysates.
For this study, we chose Debaryomyces nepalensis NCYC 3413, a non-pathogenic saccharomycetes yeast which was previously isolated from rotten apple.13 It has been shown to survive in a medium containing pectin as the sole source of carbon and can overcome osmotic stress by producing polyols.14 D. nepalensis could utilize both hexoses and pentoses, and xylitol was found to be a major product when grown in the medium containing xylose, suggesting the presence of aldose metabolizing enzymes.15 Nevertheless, earlier we purified a protein with xylose reductase activity from D. nepalensis, hence it has been proven as a good candidate strain to take for AR gene fishing.16 However, lack of information on its genome sequence was eliminating the possibilities of specific genes isolation and their cloning.
In the present study, we attempted AR gene isolation from D. nepalensis by designing the primers based on genome sequence of its close relatives such as D. hansenii and Candida species. Later, the obtained partial AR gene sequence was taken to design the primers for the full-length gene isolation by rapid amplification of cDNA ends (RACE), so that we successfully isolated novel AR gene from D. nepalensis (DnAR). Next, it was overexpressed as a His-tagged fusion protein in E. coli Rosetta, purified and its biochemical properties were determined. The enzyme showed strict dependence on cosubstrate NADPH, broad substrate specificity and halotolerance. In addition, we developed a homology model for DnAR in order to understand the molecular basis of its halotolerance and found that its protein surface contains uniform negative electrostatic potential, a signature property of halotolerant proteins.
2. Results
2.1. Molecular cloning of D. nepalensis AR gene
Using the primers F1 and R1, an amplicon of 0.85 kb size was obtained from D. nepalensis gDNA (Fig. 1A). Upon sequence alignment, it showed high homology with other aldose (xylose) reductase genes from various yeasts, which confirmed the successful isolation of partial DnAR gene. The incomplete ORF of DnAR (61–918 bp region of full-length gene) was used for designing gene-specific primers to amplify the missing terminals by RACE. The complete ORF of DnAR gene (963 bp) was determined by overlapping of the resultant sequences of PCR products from 5′ RACE (0.63 kb) and 3′ RACE (0.84 kb) (Fig. 1B). Using the primers F2 and R2 designed based on the RACE results, the full-length DnAR gene was amplified from cDNA as well as from gDNA (Fig. 1C).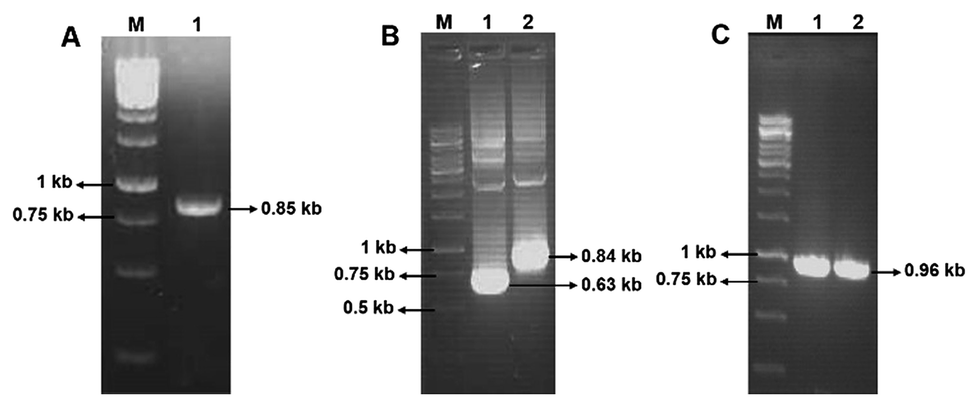 | ||
| Fig. 1 Isolation of D. nepalensis AR gene. In all three, lane 1 indicates 1 kb DNA ladder. (A) Lane 2: an amplicon of the partial DnAR gene from the genomic DNA; (B) lane 2: PCR amplification of 5′ RACE product and lane 3: 3′ RACE product within a TA cloning vector; (C) lane 2: full-length gene isolated from genomic DNA using the primers designed from RACE products and lane 3: PCR amplified full length gene within an expression vector pET28a-DnAR. The exact size of the PCR products indicated was determined by sequencing. | ||
The DnAR gene amplified from both gDNA and cDNA was sequenced and its deduced amino acid sequence is shown in Fig. 2. The sequence results revealed that the gene was uninterrupted by introns. DnAR showed 70% sequence similarity with aldose (xylose) reductase from C. tenuis (CtXR) hence the coordinates of CtXR crystal structure (PDB ID: 1K8C) was used as template to create the homology model of DnAR using SWISS MODEL automated mode. It was found that the TIM barrel [(β/α)8 barrel] fold, residues involving catalysis substrate binding and important residues mediating cosubstrate specificity are conserved in the model (Fig. 3).
 | ||
| Fig. 2 Nucleotide and deduced amino acid sequences of the D. nepalensis AR gene. The deduced amino acid sequence is given below the nucleotide sequence. An asterisk indicates the termination codon. The primers used for partial gene isolation (F1, R1), 5′ RACE (SP1-3), 3′ RACE (SP4) and full length gene isolation (F2, R2) are highlighted with arrows which showing annealing position and direction of extension. JustBio hosted tool Translator was used for representation. | ||
 | ||
| Fig. 3 Homology model of DnAR built with SWISS MODEL. The DnAR model (cyan) and the aldose (xylose) reductase crystal structure of C. tenuis (PDB ID: 1K8C) (gray) upon superimposition showed 70% structural similarity (RMSD = ∼2.5 A°). The bound cofactor NADPH (green) and catalytic triad (red) and important residues conferring cofactor specificity (magenta) in the model are visualized using PyMOL. | ||
2.2. Heterologous expression and purification
The plasmid pET28a-DnAR encoding N-terminal His6-tagged DnAR was expressed in E. coli Rosetta under IPTG induction. The overexpressed protein constitutes 60% of the total cellular soluble protein. The cell-free lysate soluble fraction showed AR activity when xylose and NADPH were used as substrate and cosubstrate respectively, confirming the successful heterologous expression of DnAR. Next, the resultant recombinant protein was purified in a single step with an immobilized metal ion affinity chromatography (IMAC) purification using Ni2+ chelating column. The molecular weight of purified DnAR (with tag) was ∼38.7 kDa, which is consistent with its predicted molecular weight (Fig. 4).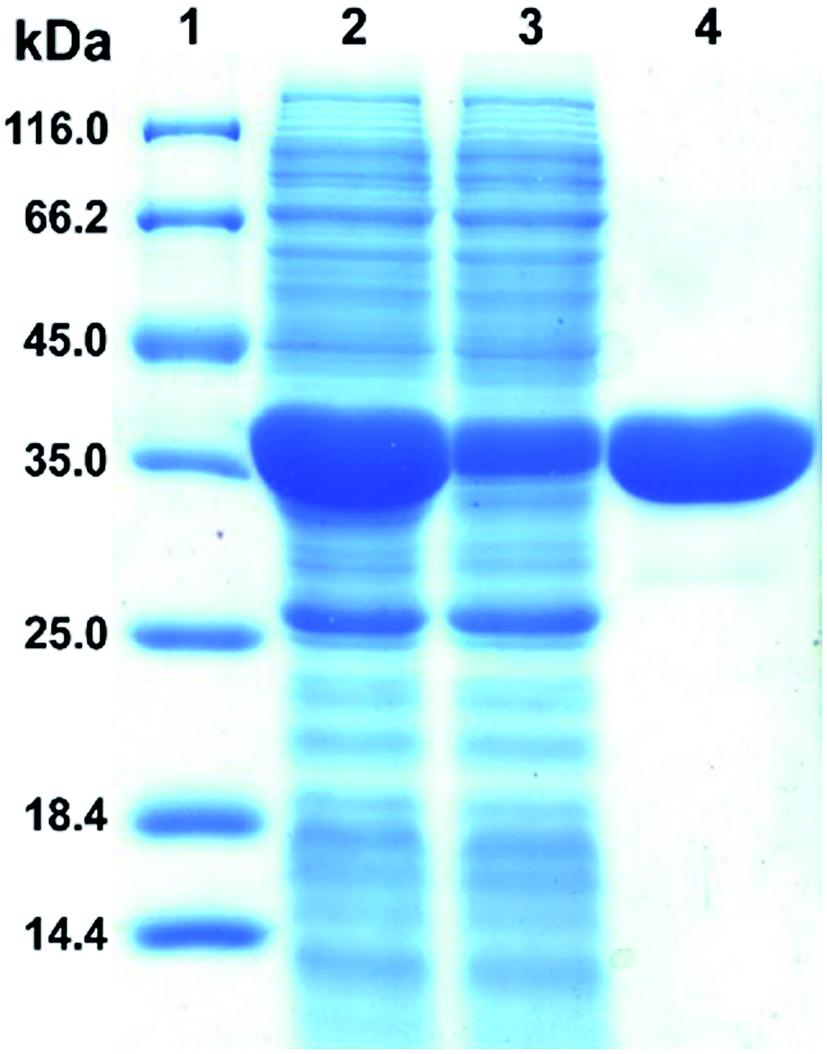 | ||
| Fig. 4 Purification of heterologously expressed DnAR-His6 tagged fusion protein. Lane 1: molecular weight (in kDa) marker, lane 2: soluble fraction of the induced cells, lane 3: flow-through during the purification and lane 4: final purified AR. The samples run on a 12% acrylamide gel stained with coomassie brilliant blue R-250. It was performed in three biological replicates. | ||
The activity yield obtained by this purification strategy was found to be 51% (3.3 U mg−1) (Table 1). To confirm if the overexpressed protein is AR or not, the purified protein was trypsin digested and subjected to LC-MS/MS analysis. The digested peptide fragments of DnAR matched with parts of protein sequences of ARs from different organisms (Fig. S1†). Moreover, the differences in residues between the previously characterized native xylose reductase and present recombinant DnAR confirmed that this DnAR is one of the isomeric forms of AR from D. nepalensis.
| Step | Total protein (mg) | Total activity (U) | Specific activity (U mg−1) | Purification fold | Yield (%) |
|---|---|---|---|---|---|
| Crude | 51.3 | 26 | 0.5 | 1 | 100 |
| Purified DnAR | 4.0 | 13.3 | 3.3 | 6.6 | 51.2 |
2.3. Effect of pH, temperature and metal ions on DnAR activity
The effect of pH on DnAR activity was studied by measuring the activity at various pHs ranging between 4.0 and 10.0 by keeping temperature as constant. Maximum specific activity of 80 ± 2 U mg−1 was obtained at pH 7.0 and the enzyme demonstrated activity in a broad pH range by retaining 60% activity at pH 6.0 and 55% activity at 8.0. At pH 5.0 and 9.0 only 30% of the maximum activity was observed (Fig. 5A). The enzyme activity increased from 10 to 45 °C; maximum activity was shown at 45 °C and beyond 50 °C a drastic drop in enzyme activity was observed (Fig. 5B). | ||
| Fig. 5 Effect of pH (A), temperature (B) and divalent metal ions on DnAR activity (C). AR assay was performed in the reaction mixture containing 50 mM sodium phosphate buffer (pH 7.0), 300 mM xylose, 0.3 mM NADPH and 0.3 μg protein at optimal conditions. Results are representative of three analytical replicates and the error bars indicate ± SD. | ||
The effect of metal ions on DnAR activity was studied at three different concentrations (0.5, 1.0 and 2.0 mM) of various metal ions. Ca2+ and Mn2+ up to 2 mM concentration did not significantly affect the activity (75% of residual activity), whereas 0.5 mM Cu2+ completely inhibited the enzyme activity (Fig. 5C). Fe2+ and Ni2+ showed 50% reduction in activity at 2 mM whereas 80% reduction in activity was observed when 2 mM Mg2+ was used in the reaction mixture. In the presence of Zn2+, DnAR activity was reduced to 60% of its original activity at 0.5 mM and did not show further reduction at higher concentrations of Zn2+ (Fig. 5C).
2.4. Cosubstrate and substrate specificity
Cosubstrate specificity was tested by measuring the enzymatic activity in the presence of different concentrations ranging from 0 to 1 mM NADH and NADPH, and the results clearly showed that DnAR is specific only for NADPH. Further, the kinetic properties of the enzyme for cosubstrate were determined by performing kinetics as described in materials and methods. KM for NADPH was found to be 0.13 ± 0.01 mM and kcat/KM was 728 ± 12 s−1 mM−1. The DnAR showed broad substrate specificity, which is a key property of the enzymes belonging to the AKR family (Table 2). Xylose, arabinose, ribose, galactose, glucose, fructose and mannose were tested as substrates for DnAR with NADPH as a cofactor. Among the tested, xylose (KM = 106 ± 12 mM) and arabinose (KM = 110 ± 3 mM) showed higher affinity to the enzyme. However, DnAR showed higher kcat for arabinose (163 ± 1 s−1) when compared to xylose (108 ± 3 s−1). It was also found that the catalytic efficiency (kcat/KM) for arabinose is 46% higher than that for xylose. Moreover, DnAR was also reduced another pentose ribose and hexoses such as glucose, galactose and fructose (Table 2). But DnAR did not show any activity towards mannose (data not shown). Among the non-sugar carbonyl substrates tested for activity, the enzyme showed higher catalytic efficiency with 3-nitro benzaldehyde and a lower with glyoxal (Table 2).| Substrate | KM (mM) | kcat (s−1) | kcat/KM (mM−1 s−1) |
|---|---|---|---|
| Xylose | 106 ± 12 | 108 ± 4 | 1.0 ± 0.3 |
| Arabinose | 110 ± 3.0 | 163 ± 1 | 1.5 ± 0.3 |
| Ribose | 377 ± 21 | 92 ± 3 | 0.24 ± 0.1 |
| Galactose | 572 ± 82 | 106 ± 9 | 0.2 ± 0.1 |
| Glucose | 889 ± 75 | 61 ± 2 | 0.07 ± 0.0 |
| Fructose | 1269 ± 231 | 11 ± 1 | 0.01 ± 0.0 |
| Glyoxal | 70 ± 2.0 | 41 ± 1 | 0.58 ± 0.5 |
| Isophthalaldehyde | 2.0 ± 0.2 | 28 ± 0.5 | 12.0 ± 2.5 |
| Benzaldehyde | 3.3 ± 0.4 | 11 ± 0.5 | 3.3 ± 1.2 |
| Pyridine 3-carboxyaldehyde | 4.5 ± 0.5 | 7 ± 0.5 | 1.6 ± 1.0 |
| 3-Nitro benzaldehyde | 1.7 ± 0.4 | 40 ± 0.5 | 24 ± 1.2 |
2.5. Halotolerance of DnAR
The recombinant DnAR activity was determined in the presence of various concentrations of NaCl and KCl ranging between 0 and 2 M at optimal pH 7.0 and optimal temperature 45 °C (Fig. 6A). The enzyme exhibited specific activity 110 ± 3 U mg−1 (75% of original activity) and 65 ± 2 U mg−1 (45% of original activity) at 0.5 and 1 M KCl respectively. However, in the presence of 0.5 M and 1 M NaCl, the enzyme showed specific activity 95 ± 4 U mg−1 (65% of original activity) and 45 ± 2 U mg−1 (30% of original activity) respectively. | ||
| Fig. 6 Halotolerance of recombinant DnAR. (A) The enzyme activity in the presence of salts at 45 °C determined at standard assay conditions; (B) stability of the enzyme in the presence of the salts up to 2 M at 30 °C. Results are representative of three analytical replicates and the error bars indicate ± SD. | ||
Even at 2 M, the KCl enzyme showed specific activity 20 ± 1 U mg−1 (Fig. 6A). Moreover, DnAR kinetics using xylose as substrate were also performed in the presence of salts and it was observed that kcat/KM was nearly 0.75 ± 0.3 mM−1 s−1 (75%) and 0.33 ± 0.2 mM−1 s−1 (33%) in the presence of 0.5 M and 1 M concentration of both salts respectively (Table 3). The stability of the enzyme was studied in the presence of 1 M and 2 M NaCl and KCl at 30 °C. The residual activity at various time points was measured and the data were fitted to the first-order deactivation kinetics and the half-life of enzyme was determined. DnAR exhibited half-life time of 30 ± 2 h in the absence of salts (control). In the presence of 1 M KCl, no change in the half-life time was observed when compared to the control, whereas the half-life was reduced to 16.5 ± 3 h in the presence of 1 M NaCl (Fig. 6B). It was found that NaCl affected both the activity and the stability of the enzyme more than KCl at all concentrations. Moreover, the surface electrostatic properties of DnAR when compared to non-halotolerant aldose (xylose) reductase from C. tenuis revealed the presence of more negative surface electrostatic potential over the surface of DnAR than the CtXR (Fig. 7).
| Salt | KM (mM) | kcat (s−1) | kcat/KM (mM−1 s−1) | % catalytic efficiency |
|---|---|---|---|---|
| Control | 106 ± 12 | 108 ± 4 | 1.03 ± 0.3 | 100 |
| 0.5 M KCl | 171 ± 10 | 133 ± 3 | 0.78 ± 0.3 | 76 |
| 0.5 M NaCl | 149 ± 16 | 112 ± 5 | 0.75 ± 0.3 | 73 |
| 1.0 M KCl | 227 ± 23 | 86 ± 5 | 0.38 ± 0.2 | 37 |
| 1.0 M NaCl | 200 ± 30 | 60 ± 3 | 0.30 ± 0.1 | 29 |
 | ||
| Fig. 7 Surface electrostatic potentials of halotolerant DnAR and aldose (xylose) reductase from non-halotolerant C. tenuis (PDB ID: 1K8C). The first, second, third and fourth row were seen from the right, left, front and back of the structures, respectively. The red surface was corresponded to negatively charged residues and the blue surface corresponded to positively charged residues. The potentials were contoured to −1.0 kT per electron (red) and +1.0 kT per electron (blue). | ||
2.6. Circular dichroism studies
The secondary structural elements of DnAR were analyzed in the presence of 0–2 M concentrations of salts (NaCl/KCl) by far-UV CD spectroscopy. At low wavelengths (<205) high salt concentration in the samples obstructed the CD signals.17 However, the larger ellipticity of the two negative peaks was observed at 208 nm and 222 nm, which corresponded to α-helix. In the presence of 2 M NaCl and KCl, no major changes in the secondary structure of the protein were observed with respect to control (Fig. 8A and B). The effect of salts on the thermal unfolding of DnAR was investigated in the presence of salts up to 2 M. As shown in Fig. 9, the thermal melting curves in the presence of salts showed a decrease in Tm of protein by 8 °C and 10 °C in the presence of KCl and NaCl respectively as compared to the control (Fig. 9A–C). In spite of that, we observed 3 °C difference in Tm between 1 M and 2 M of KCl (Fig. 9B) whereas no significant changes in Tm were observed between 1 M and 2 M NaCl (Fig. 9C).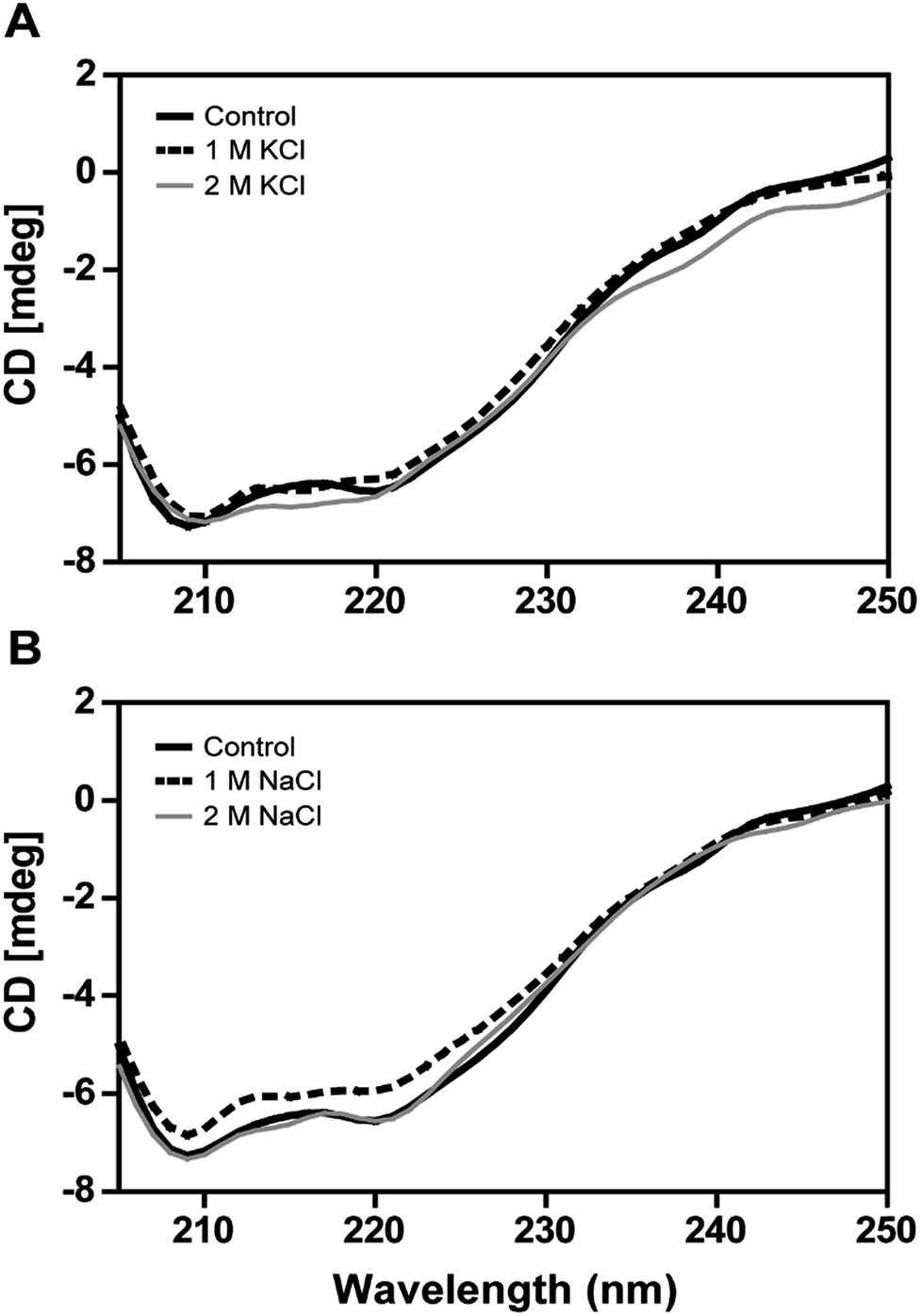 | ||
| Fig. 8 Far-UV CD spectra of DnAR in the presence of salts. (A) CD spectra of DnAR in the presence of 0–2 M KCl; (B) CD spectra of DnAR in the presence of 0–2 M NaCl. The CD spectra were measured using a protein concentration of 3 μM in 10 mM sodium phosphate buffer (pH 7.0) at ambient temperature. The CD signals at low wavelengths (<205 nm) were obstructed by high concentration of salt (not shown in the figure). The experiment was performed in three analytical replicates and mean values were taken for representation. | ||
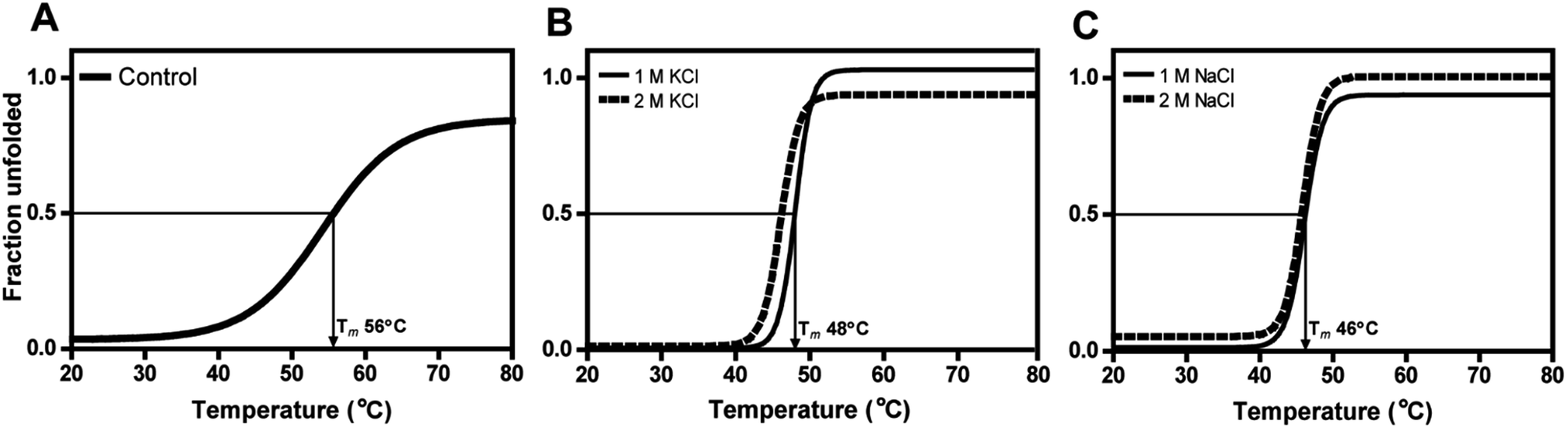 | ||
| Fig. 9 Thermal unfolding of DnAR in the presence of high concentration of salts. (A) Control; (B) effect of KCl; (C) effect of NaCl on the thermal unfolding of the enzyme. Fraction unfolded was calculated using Gibbs–Helmholtz equation upon fitting the CD data. Boltzmann sigmoidal curve fitting function of Origin Lab software (v8.0773) was used for data representation. | ||
3. Discussion
Microbial or enzymatic conversion of cellulosic and lignocellulosic biomass to ethanol, xylitol and other industrially important products is prominent in the research field of bioenergy and biorefinery. One of the major problems in this field is the utilization of both hexoses and pentoses, and various efforts have been made to engineer the existing strains18,19 and in screening novel organisms capable of utilizing both hexoses and pentoses.20,21 We previously isolated halotolerant yeast D. nepalensis from rotten apple, which is capable of utilizing both hexoses and pentoses and tolerate both NaCl and KCl at 2 M.14,15 The strain was capable of converting xylose to xylitol with a yield of 0.6 g g−1,15 which intrigued to isolate the enzyme responsible for xylitol production from this strain.The aldose reductase gene isolated from D. nepalensis NCYC 3413 has a length of 963 bp and encodes a protein containing 320 amino acids. DnAR exists as a dimer with each monomeric subunit of 36.7 kDa. The size of the monomeric subunit of DnAR is matching with ARs from other organisms where it ranges from 35 to 40 kDa.2,4,7 The DnAR has a conserved TIM barrel fold, a structural property of the enzymes belonging to the AKR super family (Fig. 3). It shares other common features of this family of enzymes such as dependence on nicotinamide cofactors, hydrophobic substrate binding pocket and broad substrate specificity.1–3 The multiple sequence alignment of DnAR protein sequence with reported aldose (xylose) reductases showed conservation of residues those involving in catalysis, substrate and cosubstrate binding (Fig. S1†).
The DnAR gene was overexpressed in E. coli Rosetta and final yield of the purified DnAR obtained by the single-step affinity chromatography was 48 mg g−1 dry weight of E. coli (Table 1). DnAR showed strict NADPH specificity as a signature property of ARs which distinct it from typical xylose reductases which are specific to either NADPH or NADH.2,4,22 DnAR exhibited broad substrate specificity like other AKR proteins.2,4,7 Similar to AR from C. boidinii22 the catalytic efficiency of DnAR with arabinose was higher than xylose. It is another feature that separate it from typical xylose reductases (usually XRs are highly specific to xylose).22–24 Contrastingly, AR from C. tenuis showed maximum catalytic efficiency with D-erythrose.7 DnAR was also reduced hexoses namely glucose and fructose to a certain extent (Table 2). Nevertheless, toxic non-sugar substrates were also reduced to a significant extent by the enzyme similar to other ARs (Table 2).
The activity of enzymes can be influenced by the presence of metal ions, so in this study we tested several divalent metal ions for the activity of DnAR. We found that the DnAR exhibited moderate tolerance towards most of the metal ions (Fig. 5C). Nevertheless, the observed tolerance was higher than the C. tropicalis aldose (xylose) reductase which was known to be a highly active enzyme (kcat/KM = 7.62 s−1 mM−1).23 Conversely, DnAR was susceptible to Cu2+ toxicity as like enzymes from C. tropicalis and C. parapsilosis.23,32 Cu2+ is known to exert its effect by induction of site specific oxidation in human aldose reductase25 and probably the same mechanism might be applicable for DnAR.
The halophilic enzymes require salt to show optimal activity whereas halotolerant enzymes do not require salts for optimal activity.26 Similar to other halotolerant enzymes, DnAR showed optimal activity in the absence of salt and its activity decreased gradually upon increasing salt concentrations up to 2 M. In the presence of 0.5 M KCl and 0.5 M NaCl, DnAR exhibited retention of 76% and 73% of catalytic efficiency respectively at 45 °C. However, it showed only 37% and 29% retention of catalytic efficiency in the presence of 1 M KCl and NaCl respectively (Table 3). The reduction in activity can be probably attributed to the prevention of coenzyme release from the cofactor binding pocket of the enzyme in the presence of salts as indicated by a decrease in kcat/KM values for xylose (Table 3). Nevertheless, DnAR exhibited remarkable salt stability as shown in Fig. 6. The half-life of the enzyme in 1 M KCl was found to be 30 ± 2 h (same as control) and 16 ± 3 h in the presence of 1 M NaCl at 30 °C (Fig. 6B). The effect of NaCl was larger than that with KCl, which is similar to the other halotolerant proteins.27,28 C. tropicalis xylose reductase (having high catalytic efficiency) was highly unstable in the presence of salts. Even at low salt concentrations (0.1 mM NaCl), the activity of C. tropicalis enzyme was reduced to 25% upon incubation for 1 h.29 The effect of NaCl on the activity and stability of DnAR was greater than with KCl at all the tested concentrations. The CD spectra in the presence of salts suggested the existence of regular secondary structure of DnAR even at 2 M (Fig. 8). However, in the presence of 2 M salt, Tm of DnAR was reduced to 8–10 °C, which can be attributed to the mild enhancement in thermal unfolding at high salt concentrations (Fig. 9). The salt stability of DnAR was high when compared to the halotolerant enzyme Hsp90 from Dunaliella salina, which has been shown to exhibit conformational stability up to 1 M NaCl.30 The nitrate reductase (a member of the DMSO reductase family) from the halotolerant cyanobacterium Aphanothece halophytica has been shown to completely lose activity at ≥ 0.3 M NaCl31 whereas DnAR lost activity at very high concentrations of NaCl (>2 M) (Fig. 6A). Nevertheless, the negative surface electrostatic potential (red color) of DnAR was found to be higher than the non-halotolerant CtXR (Fig. 7). Similarly, the halotolerant esterase from a marine bacterium Pelagibacterium halotolerans has been shown to have more negative surface electrostatic potential than the non-halotolerant esterase from Alicyclobacillus acidocaldarius.32 A halotolerant carbonic anhydrase has been shown to have uniform negative surface electrostatic potential as seen in DnAR.41 The presence of high number of acidic amino acids on the surface of the protein imparts negative surface electrostatic potential, thereby confers halotolerane to the protein.26,32,33
4. Conclusions
We isolated novel aldose reductase gene from the halotolerant yeast D. nepalensis, cloned, overexpressed and its biochemical properties were determined. To the best of our knowledge, DnAR showed peculiar property of halotolerance, high catalytic efficiency and broad substrate specificity. Based on our results, we suggest that this enzyme can be used as a potential biocatalyst for upcoming in vitro xylitol production from lignocellulose hydrolysates. Further mutational and crystallographic studies will give more insights into the structure and function relationship of DnAR.5. Materials and methods
5.1. Materials
The strain Debaryomyces nepalensis NCYC 3413 was used as a source of AR gene. E. coli DH5α and E. coli Rosetta (both Novagen, USA) were used as cloning and host strains, respectively. The plasmid pGEM-T easy vector (Promega, Germany) was used as a cloning/sequencing vector. The vector pET28a(+) (Novagen, USA) carrying a kanamycin resistance marker and a cleavable His-tag was used for protein expression. Phusion DNA polymerase, dNTPs, various restriction enzymes and T4 DNA ligase (New England Biolabs, UK) were used for cloning experiments. TRIzol reagent (Life technologies US) was used for RNA isolation. 5′/3′ RACE second-generation kit and expand high fidelity PCR system (both Roche Diagnostics GmbH, Manheim, Germany) were used for isolation of full-length DnAR gene. NADH and NADPH (both ≥97% pure) (Sigma, USA) were used for enzyme activity measurements. Ni-NTA Agarose (Qiagen, Germany) was used as affinity matrix. Thrombin (Sigma, USA) was used for cleaving His-tag from DnAR-His6 fusion protein.5.2. Microorganisms and growth conditions
D. nepalensis NCYC 3413, previously isolated in our lab, was maintained on solid YPP (yeast extract 10 g l−1, peptone 20 g l−1, pectin 5 g l−1 and agar 20 g l−1) plates at 30 °C.15 The liquid media YPX (yeast extract 10 g l−1, peptone 20 g l−1 and xylose 20 g l−1) with a loopful of inoculum was incubated at 30 °C, 180 rpm for 12 h, then harvested and gDNA and total RNA were isolated from the harvested cells. E. coli DH5α was used as the plasmid host. E. coli Rosetta was used for a heterologous expression of DnAR. E. coli strains were grown in Luria–Bertani medium under antibiotic control as per supplier's instructions.5.3. Molecular cloning of D. nepalensis AR
D. nepalensis gDNA was isolated by phenol chloroform method.34 The partial DnAR gene was amplified by PCR using Taq DNA polymerase and two primers, namely, F1 (5′-GTGGACAATGCCACTTGTGC-3′) and R1 (5′-GACCAAGACTTGAG ATTCAACAAT-3′), designed based on highly conserved regions of amino acid sequence found upon alignment of aldose (xylose) reductases from D. hansenii, C. tenuis, C. tropicalis, S. passalidarum, M. guilliermondii and C. parapsilosis. The DnAR complete open reading frame (ORF) was determined by 5′ and 3′ RACE using 5′/3′ RACE second-generation kit (Roche Diagnostics GmbH, Manheim, Germany). RNA was isolated by TRIzol method and was used as a template. The obtained partial DnAR gene from gDNA was used for designing primers for RACE. For 5′ RACE, the designed gene-specific primers were SP1 (5′-GAGATGGGCTTCTCAAAGAGATATTGCTATCATTCC-3′), SP2 (1st nested) (5′-TTGGAATTGAAGCATTCTAAGGCATTGGATACTCC-3′) and SP3 (2nd nested) (5′-GAGTATGTTCAATCCCAAGGCATTGC TATTACCGC-3′). For 3′ RACE, SP4 (5′-CGATGCCGCAATGGACT ACGGTAACTG-3′) was used as the gene-specific primer. The obtained 5′ and 3′ RACE PCR products were cloned into pGEM-T easy vector. The full-length DnAR gene was then amplified using Taq DNA polymerase by overlap PCR with 5′ and 3′ RACE PCR products or directly from gDNA using two primers, namely, F2 (5′-GGAATTCCATATGATGTCAATCAAATTAAACTCTGG-3′) with NdeI site (underlined) and R2 (5′-GAATAGAATCCCTATCTTTGC CTAAGAATTCCGG-3′) with EcoRI site (underlined). After that, the complete DnAR ORF was cloned into the expression vector pET28a(+), designated as pET28a-DnAR and sequenced. Standard protocols for nucleic acid manipulation and molecular cloning were used.34 DNA sequencing was performed at Eurofins Analytical Services India Pvt Ltd using Sanger sequencing method. Three clones were sequenced using both the vector-specific primers (T7 FP/RP) to ensure the correctness of AR sequence.5.4. Heterologous expression of DnAR in E. coli Rosetta
Plasmid pET28a-DnAR was transformed into E. coli Rosetta (expression host) competent cells by heat shock. One of the colonies was inoculated into LB medium for preparing glycerol stocks and pre culture. Two percent of inoculum from pre culture was transferred to the main culture and incubated till OD600 reached 0.7–0.8. After that, IPTG was added to a final concentration of 0.3 mM for induction. LB medium containing 34 μg ml−1 chloramphenicol and 50 μg ml−1 kanamycin was used for pre and main cultures. The pre culture was incubated at 37 °C, 180 rpm for 12 h. The main culture was incubated at 37 °C, 180 rpm until induction, after the induction temperature was reduced to 30 °C and kept for 8 h. The cells were harvested by centrifugation at 8000×g for 5 min at 4 °C, resuspended in the lysis buffer [20 mM Tris–HCl (pH 7.5), 200 mM NaCl, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM dithiothreitol (DTT)] and were then lysed by sonication (Sonic Vibra-cell (USA), 1 cm diameter probe, 37% vibration amplitude, pulse 2 s on/5 s off for 5 min, 20 ml cell suspension at 4 °C). The supernatant and pellet fractions were separated by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g for 20 min at 4 °C. Supernatant from sonication was directly taken whereas pellet was resuspended in 4% SDS (same volume as supernatant), heated at 95 °C for 5 min and then taken for checking expression by SDS-PAGE.35
000×g for 20 min at 4 °C. Supernatant from sonication was directly taken whereas pellet was resuspended in 4% SDS (same volume as supernatant), heated at 95 °C for 5 min and then taken for checking expression by SDS-PAGE.35
5.5. Purification of recombinant DnAR
The soluble fraction (10 ml) was loaded onto a Ni2+ affinity column (2 ml Ni-NTA agarose), previously equilibrated with Tris-buffer saline (TBS) (20 mM Tris–HCl (pH 7.5), 200 mM NaCl) containing 10 mM imidazole and allowed to pass through at a flow rate of ∼0.25 ml min−1. The column was washed with 50 bed volumes of TBS containing 20 mM imidazole, at a flow rate of ∼1 ml min−1 and bound protein was then eluted by a gradient of 100, 200 and 300 mM imidazole in TBS buffer (each 5 ml) at a flow rate of ∼0.25 ml min−1. All of the fractions (flow through, 20, 100, 200, 300 mM imidazole elutions) flowing through the column were analyzed by SDS-PAGE. The fractions containing pure DnAR (100, 200 mM imidazole elutions) were collected and subjected to dialysis against 20 mM Tris–HCl buffer (pH 7.5) and stored in −80 °C until use. Protein concentrations were estimated by bicinchoninic acid (BCA) assay with bovine serum albumin as a standard.365.6. Aldose reductase assay and kinetic studies
Unless stated otherwise, the recombinant DnAR activity was determined spectrophotometrically (Perkin Elmer Lamda 25 UV/VIS spectrophotometer, USA) by monitoring the change in A340 in 500 μl reaction mixture containing 50 mM sodium phosphate buffer (pH 7.0), 0.3 mM NADPH as a cofactor and 300 mM xylose (a predominant pentose in lignocellulose) as a substrate at 45 °C. The change in absorbance at 340 nm was measured continuously at least for 1 min at every 1 s time point. It was ensured by the addition of a specific amount of enzyme that gives a linear curve. NADPH molar extinction coefficient 6.22 mM−1 cm−1 was used for calculation of enzyme activity. One enzyme unit (U) was defined as the amount of enzyme that caused the oxidation of 1 μmol of NADPH per minute. Heat inactivated enzyme and 50 mM sodium phosphate buffer (pH 7.0) were taken for measuring auto hydrolysis of NADPH in the standard reaction mixture and considered as a reference. The kinetic parameters of cosubstrate NADPH were determined by measuring the initial velocities at constant xylose (160 mM) and different NADPH concentrations (0.01–0.6 mM). The substrate kinetic parameters were determined at constant NADPH (0.3 mM determined from previous experiments) and different substrate concentrations (5–800 mM). Sugar assayed were D-xylose, L-arabinose, D-ribose, D-galactose, D-glucose, D-fructose and D-mannose, whereas non-sugar carbonyl compounds assayed were gloxal, isophathaldehyde, benzaldehyde, pyridine 3-carboxyaldehyde and 3-nitro benzaldehyde. The obtained data were used to calculate the kinetic constants by fitting for the Michaelis–Menten equation using GraphPad Prism 5.5.7. Effect of pH, temperature and metal ions
The effect of pH on enzyme activity was determined at 30 °C by assaying at a pH range from 4 to 10 using standard buffers namely, 50 mM sodium acetate (pH 4.0–5.5); 50 mM potassium phosphate (pH 6.0–8.0); 50 mM Tris–HCl (pH 8.5); and 50 mM glycine–NaOH (pH 9.0–10). To study the effect of temperature, the experiment was performed at optimal pH (determined from pH effect studies) and the enzyme assays were performed, at various temperatures ranging between 10 and 60 °C. The effect of divalent metal ions (Ca2+, Cu2+, Fe2+, Mg2+, Mn2+, Ni2+ and Zn2+) was studied at different concentrations of metal ions (0.5, 1 and 2 mM) at optimal conditions and residual activities of recombinant DnAR were determined.5.8. Salt tolerance and stability
The effect of salts on DnAR activity was determined by measuring the enzyme activity in the presence of different concentrations of NaCl and KCl up to 2 M at standard assay conditions. The enzyme kinetics using D-xylose as substrate was also performed in the presence of 0.5 M and 1.0 M NaCl/KCl respectively. The obtained data were used to calculate the kinetic constants by fitting for the Michaelis–Menten equation using GraphPad Prism 5 as described above. Salt stability of DnAR was assessed at 30 °C by incubating enzyme in the presence of 1 and 2 M of NaCl/KCl. Aliquots of enzyme at different time points were taken and specific activities were measured under standard assay conditions. The half-life of the enzyme was calculated by considering first order deactivation kinetics as described previously.37 To avoid the cumulative effect of temperature and salts, the salt stability studies were performed at 30 °C which was close to room temperature.5.9. Far-UV CD spectra measurement in the presence of salts
The CD spectra were recorded using JASCO J-810 spectropolarimeter (Easton, MD) at 25 °C with a thermostat cell holder connected to a Peltier temperature controller. The instrument was calibrated with 10-camphor sulphonic acid before scanning the samples. A cuvette of 1 mm path length was used for scanning samples at a wavelength range of 250 to 200 nm to obtain CD spectra. The scanning was done at the scan speed of 10 nm min−1 with a bandwidth of 2 nm. Average of three scans was taken for analysis. CD spectra were recorded using 3 μM protein in 10 mM sodium phosphate buffer (pH 7.0) at 0–2 M salt (NaCl/KCl), whereas the thermal unfolding of DnAR was analyzed from 20 to 80 °C using 6 μM protein in 10 mM sodium phosphate buffer (pH 7.0) at 0–2 M salt (NaCl/KCl). For determination of the thermal unfolding, the change in molar ellipticity at 222 nm was recorded continuously while the temperature was increased at the rate of 1.0 °C min−1. Protein fraction unfolded was calculated using Gibbs–Helmholtz equation [ΔG = ΔH (1 − T/Tm) − ΔCP((Tm − T) + Tln(T/Tm)) where G is the Gibbs free energy, H the enthalpy, T the absolute temperature, Tm the T at which fraction folded is 0.5 and Cp the heat capacity] upon fitting the change of CD at a single wavelength as a function of temperature.38 The obtained data were analyzed using Boltzmann curve fitting function of Origin Lab software (v8.0773).
5.10. Homology modelling
A homology model of DnAR was built with SWISS-MODEL (v8.05) automated mode39 using the crystal structure of C. tenuis aldose (xylose) reductase (CtXR) bound to NADPH (PDB ID: 1K8C) as a template.40 The surface electrostatic potentials of DnAR model and CtXR crystal structure were calculated using Adaptive Poisson-Boltzmann Solver (APBS) tool41 in Chimera molecular modeling system with the PQR file generated using PDB2PQR online server.425.11. Nucleotide sequence accession number
The nucleotide sequence of AR gene from D. nepalensis has been submitted to GenBank under the accession number KT239024.Acknowledgements
This work is supported by the research grant from Department of Biotechnology, Govt. of India (BT/PR6157/PID/6/701/2012). BP acknowledges Indian Council of Medical Research for his fellowship. Authors acknowledge DST-FIST facility at IIT Madras for CD spectroscopy.References
- H. Andleeb, Y. Tehseen, S. J. Ali Shah, I. Khan, J. Iqbal and S. Hameed, RSC Adv., 2016, 6, 77688–77700 RSC.
- R. D. Mindnich and T. M. Penning, Hum. Genomics, 2009, 3, 362–370 CAS.
- P. F. Kador, M. Wyman and P. J. Oates, Prog. Retinal Eye Res., 2016, 54, 1–29 CrossRef CAS PubMed.
- S. G. Mundree, A. Whittaker, J. A. Thomson and J. M. Farrant, Planta, 2000, 211, 693–700 CrossRef CAS PubMed.
- Z. Han, X. Hao, Z. Gao, B. Ma and C. Zhu, RSC Adv., 2016, 6, 12761–12769 RSC.
- S. Parpart, A. Petrosyan, S. J. Ali Shah, R. A. Adewale, P. Ehlers, T. Grigoryan, A. F. Mkrtchyan, Z. Z. Mardiyan, A. J. Karapetyan, A. H. Tsaturyan, A. S. Saghyan, J. Iqbal and P. Langer, RSC Adv., 2015, 5, 107400–107412 RSC.
- W. Neuhauser, D. Haltrich, K. D. Kulbe and B. Nidetzky, Biochem. J., 1997, 15, 683–692 CrossRef.
- S. S. da Silva and A. K. Chandal, D-Xylitol, Spriger-Verlag, Berlin Heidelburg, 1st edn, 2012 Search PubMed.
- I. S. M. Rafiqul, A. M. M. Sakinah and A. W. Zularisam, Appl. Biochem. Biotechnol., 2015, 176, 1071–1083 CrossRef CAS PubMed.
- S. Ur-Rehman, Z. Mushtaq, T. Zahoor, A. Jamil and M. A. Murtaza, Crit. Rev. Food Sci. Nutr., 2015, 55, 1514–1528 CrossRef CAS PubMed.
- W. Dieters, Xylitol production from D-xylose, Swiss Patent, 560, 175, 1975.
- H. Jain and S. Mulay, Int. J. Food Sci. Nutr., 2014, 65, 135–143 CrossRef CAS PubMed.
- S. N. Gummadi and D. S. Kumar, Res. J. Microbiol., 2006, 1, 152–159 CrossRef CAS.
- S. Kumar and S. N. Gummadi, Extremophiles, 2009, 13, 793–805 CrossRef CAS PubMed.
- S. Kumar and S. N. Gummadi, Appl. Microbiol. Biotechnol., 2011, 89, 1405–1415 CrossRef CAS PubMed.
- S. Kumar and S. N. Gummadi, Bioresour. Technol., 2011, 102, 9710–9717 CrossRef CAS PubMed.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, 2005, 1751, 119–139 CrossRef CAS PubMed.
- S. Krahulec, B. Petschacher, M. Wallner, K. Longus, M. Klimacek and B. Nidetzky, Microb. Cell Fact., 2010, 9, 16 CrossRef PubMed.
- G. Guirimand, K. Sasaki, K. Inokuma, T. Bamba, T. Hasunuma and A. Kondo, Appl. Microbiol. Biotechnol., 2016, 100, 3477–3487 CrossRef CAS PubMed.
- M. G. Wiebe, Y. Nygård, M. Oja, M. Andberg, L. Ruohonen, A. Koivula, M. Penttilä and M. Toivari, Appl. Microbiol. Biotechnol., 2015, 99, 9439–9447 CrossRef CAS PubMed.
- L. Venkateswar Rao, J. K. Goli, J. Gentela and S. Koti, Bioresour. Technol., 2016, 213, 299–310 CrossRef CAS PubMed.
- M. H. Kang, H. Ni and T. W. Jeffries, Appl. Biochem. Biotechnol., 2003, 3, 105–108 Search PubMed.
- F. Zhang, D. Qiao, H. Xu, C. Liao, S. Li and Y. Cao, J. Microbiol., 2009, 47, 351–357 CrossRef CAS PubMed.
- J.-K. Lee, B.-S. Koo and S.-Y. Kim, Appl. Environ. Microbiol., 2003, 69, 6179–6188 CrossRef CAS PubMed.
- I. Cecconi, M. Moroni, P. G. Vilardo, M. Dal Monte, P. Borella, G. Rastelli, L. Costantino, D. Garland, D. Carper, J. M. Petrash, A. Del Corso and U. Mura, Biochemistry, 1998, 37, 14167–14174 CrossRef CAS PubMed.
- G. Graziano and A. Merlino, Biochim. Biophys. Acta, 2014, 1844, 850–858 CrossRef CAS PubMed.
- P. L. Wejse, K. Ingvorsen and K. K. Mortensen, Extremophiles, 2003, 7, 423–431 CrossRef CAS PubMed.
- C. Liang, Y. Xue, M. Fioroni, F. Rodríguez-Ropero, C. Zhou, U. Schwaneberg and Y. Ma, Appl. Microbiol. Biotechnol., 2011, 89, 315–326 CrossRef CAS PubMed.
- Y. Su, W. Li, W. Zhu, R. Yu, B. Fei, T. Wen, Y. Cao and D. Qiao, Afr. J. Biotechnol., 2010, 9, 4954–4965 CAS.
- X.-J. Chen, M.-J. Wu, Y. Jiang, Y. Yang and Y.-B. Yan, Int. J. Biol. Macromol., 2015, 75, 418–425 CrossRef CAS PubMed.
- S. Thaivanich and A. Incharoensakdi, World J. Microbiol. Biotechnol., 2007, 23, 85–92 CrossRef CAS.
- X. Jiang, Y. Huo, H. Cheng, X. Zhang, X. Zhu and M. Wu, Extremophiles, 2012, 16, 427–435 CrossRef CAS PubMed.
- L. Premkumar, H. M. Greenblatt, U. K. Bageshwar, T. Savchenko, I. Gokhman, J. L. Sussman and A. Zamir, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7493–7498 CrossRef CAS PubMed.
- J. Sambrook and D. W. Russell, Molecular cloning, Cold Spring Harbor, N.Y, 3rd edn, 2001 Search PubMed.
- D. W. Cleveland, S. G. Fischer, M. W. Kfracfmer and U. K. Laemmli, J. Biol. Chem., 1977, 252, 1102–1106 CAS.
- P. K. Smith, R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, B. J. Olson and D. C. Klenk, Anal. Biochem., 1985, 150, 76–85 CrossRef CAS PubMed.
- G. S. N. Naidu and T. Panda, Biochem. Eng. J., 2003, 16, 57–67 CrossRef CAS.
- N. J. Greenfield, Nat. Protoc., 2007, 1, 2527–2535 CrossRef PubMed.
- K. Arnold, L. Bordoli, J. Kopp and T. Schwede, Bioinformatics, 2006, 22, 195–201 CrossRef CAS PubMed.
- K. L. Kavanagh, M. Klimacek, B. Nidetzky and D. K. Wilson, Biochemistry, 2002, 41, 8785–8795 CrossRef CAS PubMed.
- N. A. Baker, D. Sept, S. Joseph, M. J. Holst and J. A. McCammon, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10037–10041 CrossRef CAS PubMed.
- T. J. Dolinsky, P. Czodrowski, H. Li, J. E. Nielsen, J. H. Jensen, G. Klebe and N. A. Baker, Nucleic Acids Res., 2007, 35, W522–W525 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01697b |
| This journal is © The Royal Society of Chemistry 2017 |