DOI:
10.1039/C7RA01678F
(Paper)
RSC Adv., 2017,
7, 19904-19911
Graphene material preparation through thermal treatment of graphite oxide electrochemically synthesized in aqueous sulfuric acid
Received
9th February 2017
, Accepted 30th March 2017
First published on 4th April 2017
Abstract
The present work demonstrates a simple and low-cost method to produce bulk quantities of graphene material through the thermal treatment of graphite oxide (GO). GO of a high oxidation degree was synthesized by electrochemical overoxidation of natural graphite in 11 M H2SO4 using linear sweep voltammetry (LSV) technique. The thus synthesized GO was thermally exfoliated-reduced at 500 °C in air, giving the final product – thermally reduced graphite oxide (TRGO). It should be emphasized that the process of TRGO formation from electrochemically obtained GO is for the first time described in the present work. Due to shock treatment, the BET specific surface area of TRGO increased from 4 to 455 m2 g−1. Additionally, a decrease in the concentration of oxygen functionalities was also observed. Thermal stability of electrochemically synthesized GO was investigated by thermogravimetric analysis (TGA). In order to characterize the synthesized TRGO, investigations by X-ray diffraction (XRD), Raman spectroscopy, X-ray photoelectron spectroscopy (XPS) and scanning electron microscopy (SEM) were performed.
1. Introduction
Graphene materials belong to the wide family of graphene nanomaterials containing graphene layers, such as multilayered materials, materials synthesized from graphite or graphene oxide (GO) and chemically modified forms of graphene oxide and reduced graphene oxide (RGO).1 Due to their interesting electrical, thermal and mechanical properties, the interest in graphene materials appears to be enormous.2–5
A variety of methods have been employed to produce graphene materials, however exfoliation by mechanical (ball milling, micromechanical cleavage, sonication)5 as well as electrochemical treatment6–8 appear to be most often used. Graphene materials can also be synthesized by other methods, such as chemical or electrochemical reduction of graphene oxide,2,7 thermal exfoliation of graphite/graphene oxide,9–11 unzipping of carbon nanotubes,12 chemical vapour deposition,13 or plasma assisted method.14 Properties of graphene materials being strongly related to the method used as well as conditions applied, determine its practical application, i.e. energy storage devices (capacitors, batteries), catalysts support, sensors, composite materials and conductive inks.2,4,5,15,16
Taking into account the scale of the considered process of graphene formation, a special attention should be paid to the thermal exfoliation–reduction of GO. Owing to thus method large quantities of graphene material can be synthesized.11,17 Thermal treatment of GO reduces the concentration of oxygen functional groups, in parallel the volume of formed TRGO drastically increases comparing to the starting GO.9 GO being the precursor for thermal reduction process is commonly synthesized by chemical methods.9,10,17 However GO prepared by electrochemical overoxidation of graphite can be also used.11,18 GO can be thermally reduced by shock treatment at the temperatures higher than 1000 °C in air or argon atmosphere19–22 or by gradual heating under the inert gas flow (nitrogen or argon).9,23 To remove all oxygen functionalities from the graphitic structure, thermal treatment of GO should be carried out in the presence of hydrogen.17 On much less scale TRGO is produced by microwave assisted treatment of GO.24 The oxidation stage as well as the stage of graphene layer separation are influenced by the employed conditions (atmosphere, temperature, treatment time).14–22,24
In this work, we present a simple, low-cost and scalable method enabling production the large amounts of graphene material through the thermal exfoliation–reduction of GO. Contrary to the most of research works, where as a precursor the GO synthesized by chemical methods is used, in our work the GO precursor characterized by the high content of oxygen functionalities was synthesized by electrochemical overoxidation of graphite flakes in aqueous H2SO4 solution. Contrary to the chemical methods, GO gathered by electrochemical methods is not contaminated by oxidant and by-products of the conducted oxidation. According to our best knowledge, the formation of graphene material from GO previously prepared by deep anodic overoxidation of graphite in aqueous H2SO4 is for the first time described in our present work. Additionally the amount of used reagents is significantly reduced. Process of thermal exfoliation–reduction of GO was carried out at elevated temperature under presence of air. In order to acquire information on the chemical composition of the synthesized carbon materials as well as their physicochemical features X-ray diffraction (XRD), scanning electron spectroscopy (SEM), Raman spectroscopy, and X-ray photoelectron spectroscopy (XPS) were performed. The choice of exfoliation temperature was proceeded by the thermogravimetric analysis (TGA).
2. Material and methods
2.1. Materials
Natural graphite flakes (purity 99.5%, flake size 170–283 μm) were purchased from Graphit Kropfmühl, Germany. Platinum mesh (purity 99.9%) and platinum wire (purity 99.9%, 1 mm diameter) were purchased from Goodfellow, United Kingdom. Sulfuric acid (purity 95%) was purchased from Avantor, Poland.
2.2. Electrochemical oxidation of graphite
GO was synthesized by deep anodic oxidation of natural graphite flakes closed in platinum mesh. Natural graphite flakes played a role of the working electrode. Platinum wire was a counter electrode. Working electrode as well as counter electrode was immersed in 11 M H2SO4. Electrochemical synthesis were carried out in three-electrode system using linear sweep voltammetry method (LSV). As a reference electrode Hg/Hg2SO4/1 M H2SO4 was used. Measurements were performed in potential range from working electrode rest potential (ER) to 1.6 V with scan rate 0.01 mV s−1.
2.3. Thermal exfoliation–reduction of GO
GO was thermally reduced through the thermal treatment, accompanied by its exfoliation, giving final product – TRGO. Quartz crucible filled with GO was rapidly inserted into the muffle furnace adjusted to the 500 °C. The process of thermal exfoliation–reduction was carried out under air atmosphere for 4 min. The as prepared TRGO was cooled in air to ambient temperature.
2.4. Instrumentation
Process of graphite electrooxidation was carried out by linear sweep voltammetry using Autolab PGSTAT 302N potentiostat/galvanostat. Thermal treatment of GO was performed in muffle furnace Lentron. The porous structure of synthesized carbon materials was analyzed from nitrogen adsorption/desorption measurements using Autosorb iQ at 77 K. Specific surface area was calculated by Brunauer–Emmett–Teller method (BET). Thermal stability of GO was characterized by thermogravimetric analysis (TGA) using Setaram apparatus. During this measurements the sample was heated up to 800 °C with a heating rate equal to 5 °C min−1. Morphological characterization of GO and TRGO was performed by scanning electron microscopy (SEM) using S-3400N Hitachi microscope and high resolution transmission electron microscopy (HRTEM) (FEI S/TEM TITAN 80-300 microscope). X-ray diffraction (XRD) analysis was performed by PANalytical diffractometer using Cu Kα radiation (1.54 Å) with 2 theta scan range from 4° to 60° and step size of 0.04°. Raman spectroscopy measurements were recorded by inVia Renishaw micro-Raman system with an argon laser, emitting 514.5 nm wavelength. XPS spectra were recorded on VSW spectrometer equipped with hemispherical analyzer. Mg Kα X-ray radiation with 200 W energy was used as the excitation source.
3. Results and discussion
3.1. Material preparation
3.1.1. Synthesis of GO. Voltammetric curve illustrating the electrochemical oxidation of natural graphite to GO is shown in Fig. 1. The process of graphite electrochemical transformation into GO can be divided into two parts. Within the first one, graphite is anodically polarized giving the C+x lattice. At the potential of about 0.65 V, the H2SO4 molecules and HSO4− ions start to fill the interlayer spaces between the graphene layers. This process is called intercalation, whereas the product of this reaction is known as graphite intercalation compound (GIC).25 The process of H2SO4 intercalation into the graphite is finished at potential about 1.15 V yielding 2-stage graphite intercalation compound with sulfuric acid (H2SO4-GIC). It means that each neighboring layers of intercalate are isolated by two graphene layers.26 The second part of electrochemical oxidation in the range of the potentials 1.15 and 1.5 V is depicted as huge asymmetric peak with number of maxima. Within this stage of the conducted, the water molecules underwent co-intercalation into the H2SO4-GIC. The interactions of H2O molecules with graphite matrix and the oxygen functionalities formation on graphene surface result in form of final product – GO.27 Asymmetric shape of the above mentioned anodic peak as well as current oscillations indicate the multistep character of GIC transformation into the GO.18 The explanation of this phenomenon can be based on Beck et al. investigations concerning potential oscillations during galvanostatic oxidation of graphite.27 In this work, natural graphite flakes were pressed into thin sheet and then electrochemically oxidized by galvanostatic technique. Electrochemical process was carried out in aqueous solution of sulfuric acid (concentration varied from 9 to 18 M) by employing constant current densities ranged from 1 to 20 mA cm−2. In our case, the probable mechanism of oscillation reactions is as follow. When the critical potential is reached, the co-intercalation of water molecules followed by their transformation into vicinal OH groups takes place. This electrochemical behavior is demonstrated by the rise in recorded current. On further oxidation, the formation of carbonyl and/or epoxy groups from vicinal OH groups occurs. In consequence, the current in turn is falling down (see Fig. 1).
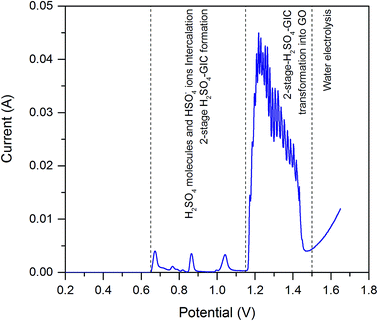 |
| | Fig. 1 Linear sweep voltammetry curve recorded for graphite in 11 M H2SO4. Potential range: ER → 1.6 V. Scan rate: 0.01 mV s−1. | |
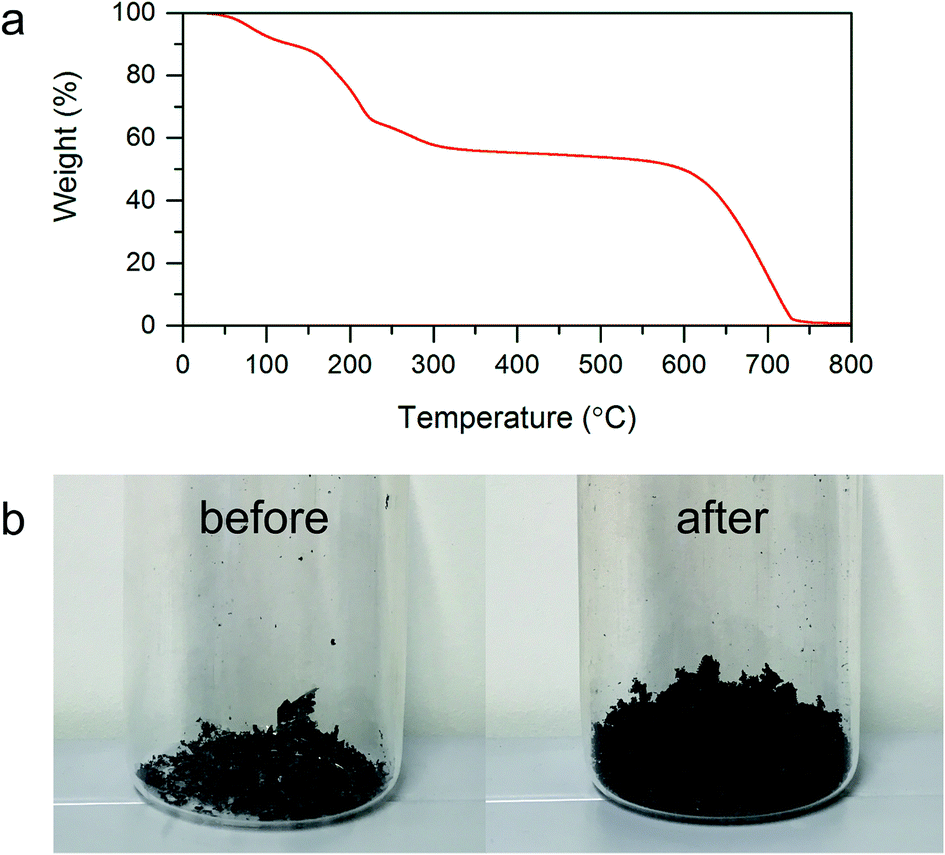
3.1.2. Thermal treatment of GO. Within the subsequent step of investigations of the as prepared GO underwent thermal treatment to obtain TRGO. In order to determine the appropriate temperature of GO reduction–exfoliation, the investigations of thermal stability of synthesized GO were performed using thermal gravimetric analysis (TGA). Thermogravimetric curve presented in Fig. 2a is characterized by two major weight drops. The first effect observed between the temperatures of 100 and 300 °C is associated with the thermal removal of oxygen functionalities chemically bonded with graphene layers.9,22,28 The second significant loss of sample weight begins at the temperature of around 550 °C. This effect corresponds to the GO thermal decomposition. In order to achieve the appropriate separation of graphene layers, the temperature of shock thermal GO exfoliation–reduction was equal to 500 °C. The selected temperature ensured an abrupt removal of oxygen functional groups with simultaneous GO exfoliation yielding graphene material as well as avoided its thermal degradation. The changes in sample volume resulted in shock thermal treatment of GO can be observed in Fig. 2b depicting digital photos of GO before and after the exfoliation–reduction process.
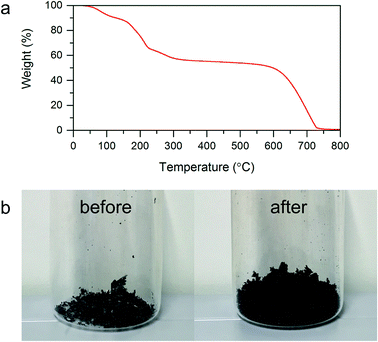 |
| | Fig. 2 (a) Thermogravimetric curve recorded for GO under an air atmosphere with heating range equal 5 °C min−1. (b) Digital photo GO before and after thermal exfoliation–reduction process. | |
3.2. Materials characterization
3.2.1. SEM and TEM observations. Morphology of the synthesized carbon material can be observed in Fig. 3. As seen from images in Fig. 3a and b, electrochemical intercalation of H2SO4 molecules and HSO4− ions followed by the co-intercalation of water into the GIC resulting its transformation into GO brought about significant deformation of graphitic structure. Process of deep anodic oxidation of graphite in aqueous H2SO4 causes delamination of carbon material. Due to oxidative treatment, graphene layers of GO become creased and wrinkled (see Fig. 3a). However, it should be noted that some residual layered structures can be also observed (see Fig. 3b). The influence of shock thermal treatment of GO on its morphology can be evaluated from the SEM images shown in Fig. 3c and d. Most of graphene layers pertaining to TRGO are damaged, twisted and crumbled. Such a behavior justify the assumption that during the shock thermal treatment of GO, its decomposition followed by the abrupt eruption of gaseous products (CO, CO2) occurs. In consequence, a significant disordering of graphitic structure accompanied by the partial separation of graphene layers are taking place.
 |
| | Fig. 3 SEM micrographs recorded for (a and b) GO and (c and d) TRGO under different magnification. | |
TEM images of GO and TRGO recorded under different magnifications support the above mentioned supposition (see Fig. 4). TEM micrograph of GO confirms its wrinkled structure (Fig. 4a) while HRTEM image discloses that layered structure partially preserved after the deep oxidation of graphite in sulfuric acid (Fig. 4b). It is worth to note that these observations are consistent with the results of XRD analysis. TEM image of TRGO reveals typical deformation of graphene sheets which become practically transparent for electron beam (Fig. 4c). Considerably larger area of a high transparency of graphene layers observed for the reduced graphite oxide (Fig. 4a) contrary to graphite oxide (Fig. 4c) indicates that thermal reduction leads to the partial delamination of graphene layers. HRTEM micrograph of synthesized TRGO (Fig. 4d) confirms the presence of few-layer graphene within its structure.
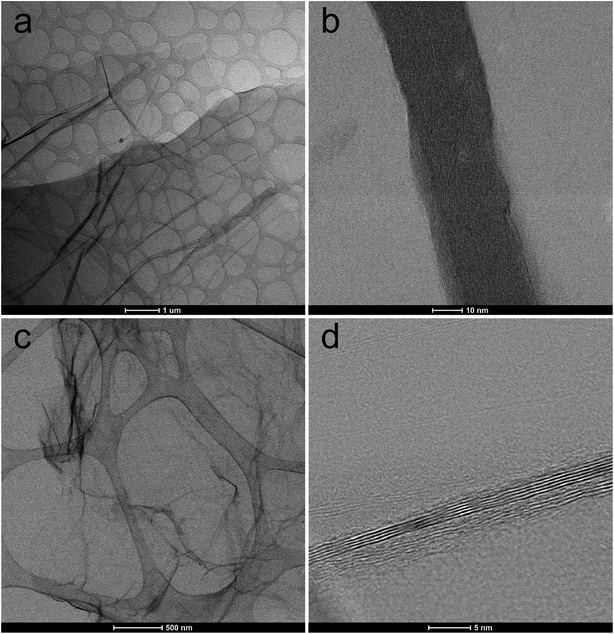 |
| | Fig. 4 TEM micrographs recorded for (a and b) GO and (c and d) TRGO under different magnification. | |
Besides the above mentioned results, the increased concentration of edges as well as surface defects are also noted. These effects are partially confirmed by the results of specific surface area measurements.
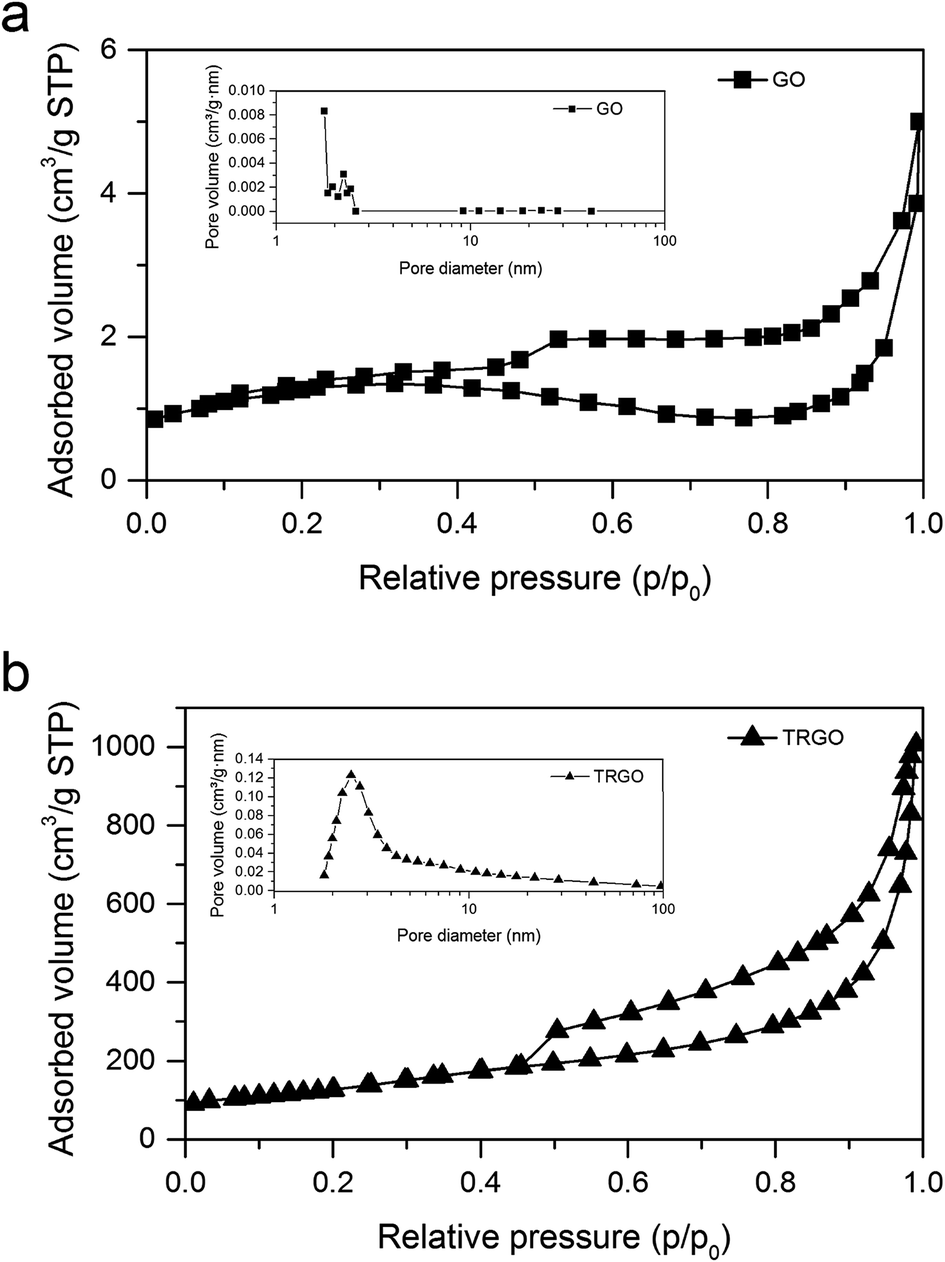
3.2.2. BET specific surface area measurements. Nitrogen adsorption–desorption isotherms and pore size distribution of GO an TRGO are displayed in Fig. 5. The additional information associated with the development of specific surface area and pore volumes of synthesized graphitic materials are shown in Table 1. Taking into consideration that BET surface area of GO synthesized by Hummers method varies from 20 to 50 m2 g−1,4,29 it can be concluded that GO prepared through the electrochemical method is characterized by the significantly less developed specific surface area (4 m2 g−1). This discrepancy can be explained by the fact that an increase in BET surface area is related with the worsening of graphene layer ordering. Hence, it can be stated that graphitic structure of GO synthesized by electrochemical method is preserved in higher degree as compared to GO prepared by Hummers method. Due to thermal treatment of GO, BET surface area increased essentially from 4 for GO to 455 m2 g−1 for TRGO. Total pore volume (VT) increases drastically after thermal reduction–exfoliation process from 0.0056 to 1.57 cm3 g−1 for GO and TRGO, respectively. A small adsorption uptake of nitrogen at relative pressure lower than 0.4 indicates a mesoporous character of TRGO.30 The mesopores diameter of reduced graphene material calculated by BJH method varies from 2 to 3.5 nm with average size of about 2.5 nm. These results justify the statement that the pore structure of TRGO from electrochemically obtained GO is very closed to that noted for the TRGO synthetized on the basis of Hummers method. From the literature it is known that depending on exfoliation conditions such as temperature, atmosphere, period time, the specific surface area of TRGO ranges from 300 to 800 m2 g−1 while average pore size from 2 to 20 nm.4,22,29,31–33
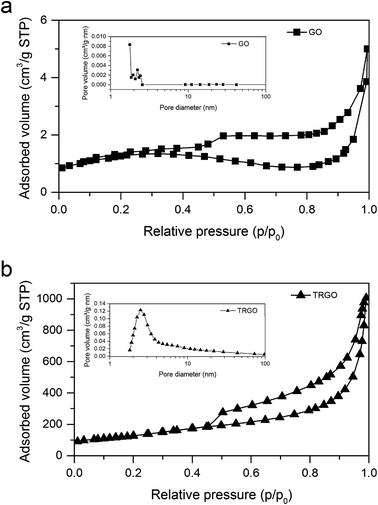 |
| | Fig. 5 Nitrogen adsorption–desorption isotherms at 77 K and BJH adsorption pore size distribution for (a) GO and (b) TRGO. | |
Table 1 Pore structure characteristics for GO and TRGOa
| Sample |
S BET (m2 g−1) |
V T (cm3 g−1) |
V micro (cm3 g−1) |
V meso (cm3 g−1) |
| S BET – specific surface area; VT – total pore volume; Vmicro – micropore volume; Vmeso – mesopore volume. |
| GO |
4 |
0.0056 |
0.00046 |
0.00096 |
| TRGO |
455 |
1.57 |
0.0028 |
0.21 |
3.2.3. XRD analysis. To prove the changes in GO structure due to its thermal treatment yielding TRGO, the XRD measurements were performed. The results of the XRD analysis of graphite, GO and TRGO are depicted in Fig. 6. Diffraction pattern of graphite (see Fig. 6a) reveals two characteristic peaks. The first one with maximum 26.6° 2 theta corresponds to the d spacing 3.35 Å and the latter one with maximum 54.8° 2 theta is associated with d spacing 1.68 Å.34 XRD pattern recorded for GO (Fig. 6b) demonstrates changes in graphite structure affected by the deep anodic oxidation of graphite in 11 M H2SO4. Formation of oxygen functionalities on the surface of graphene layers increased d spacing from 3.35 to 8.05 Å. Such a behavior clearly reveals the GO formation. A relatively weak peaks with maxima 22.1 and 26.6° 2 theta can be likely ascribed to the disordered character of graphitic structure caused by oxidation process as well as the presence of small parts of unoxidized graphite.35,36 On the other hand, shock thermal treatment of GO brought about decomposition of oxygen functional groups followed by the rapid removal of generated gaseous products. The above mentioned results justify the statement that during the process of TRGO formation, the exfoliation with simultaneous reduction of GO occur. A broad diffraction peak with maximum 18.9° 2 theta corresponding to 3.78 Å d spacing can be observed for TRGO pattern (see Fig. 6c). In can be concluded that synthesized TRGO consists of non-crystalline graphene stacks with random interlayer spaces.9,22,36
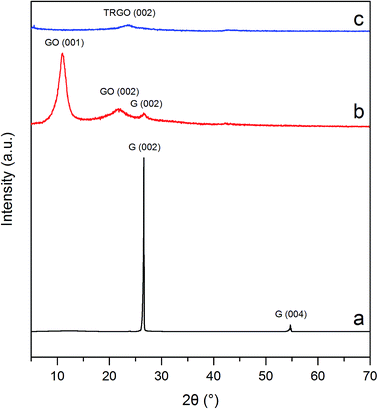 |
| | Fig. 6 XRD patterns of (a) graphite, (b) electrochemically synthesized GO and (c) TRGO. | |
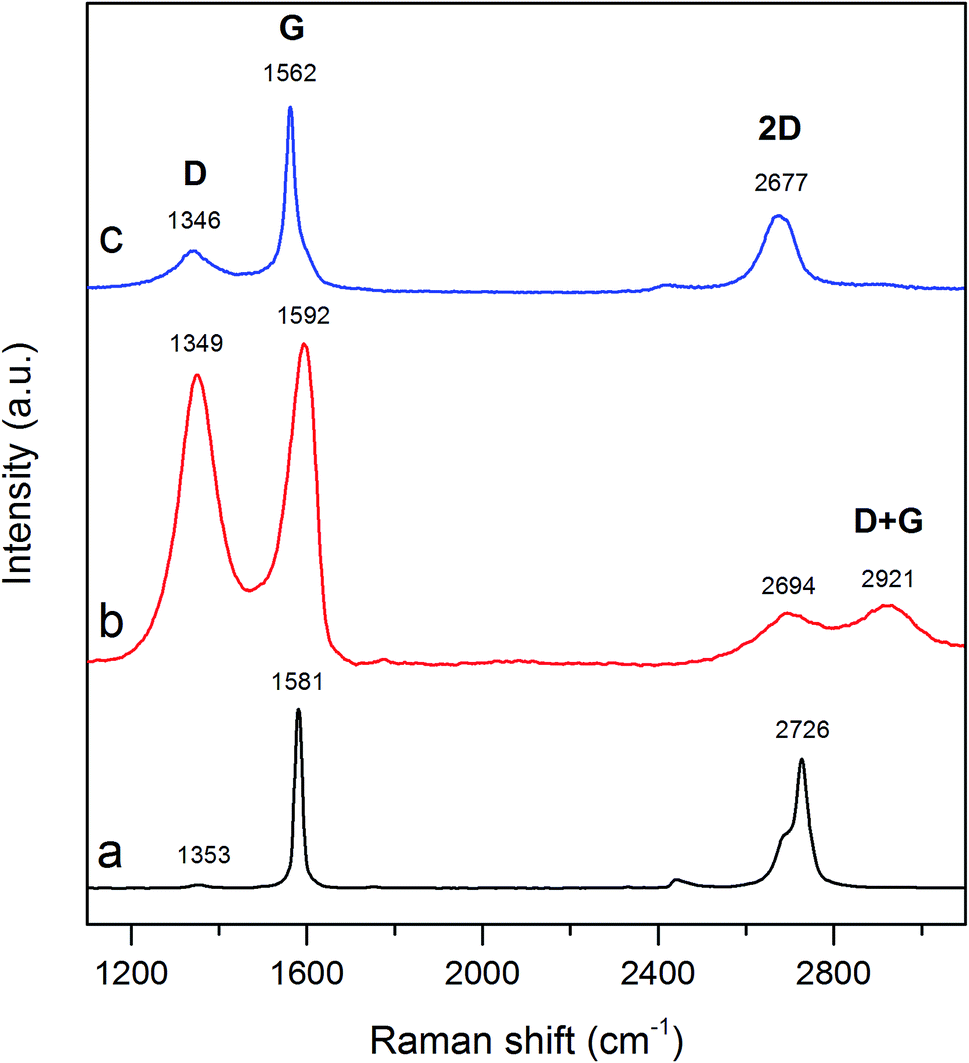
3.2.4. Raman spectroscopy analysis. To acquire information on the degree of structure disordering, the synthesized carbon material was investigated by Raman spectroscopy method. Raman spectra of graphite, GO and TRGO are shown in Fig. 7. As can be seen on spectrum of graphite (Fig. 7a), there are three characteristic bands, positioned at 1353, 1581 and 2726 cm−1. The first one (D band) is related with the structural imperfections i.e. defects, edges as well as oxygen functionalities bonded with carbon surface.37,38 The second one (G band) is attributed to the first-order scattering of the E2g mode.39,40 The last band being associated with second-order of D band (2D band) is splitted into two bands. Profile of 2D band undoubtedly pertains to the bulk graphite.39,41 A low value of ID/IG ratio (ID – intensity of D band, IG – intensity of G band) for graphite (0.02) confirms the appearance of well-defined graphitic structure. Raman spectrum gathered for GO (Fig. 7b) consists of four characteristic bands with maxima at 1349, 1592, 2694 and 2921 cm−1. Electrochemical oxidation of graphite increased the concentration of oxygen functional groups on the graphene surface thus imposing the conversion of sp2 C–C bonds into sp3 C–C bonds. In consequence, ID/IG ratio enhanced significantly to 0.90. The transformation of graphite into GO accompanied by the deformation of graphitic structure made the 2D band wider and weaker.42 However, it should be noted that D + G band being typical for GO structure is also noted (Fig. 7b).43,44 Raman spectrum of electrochemically synthesized GO is very closed to that recorded for GO synthesized by Hummers method.37,42–44 After heat treatment of GO (Fig. 7c), G band shifts to 1562 cm−1 and becomes slightly sharper compared to that recorded for GO (Fig. 7b). On the other hand, the intensity of D band of TRGO is low and its maximum is shifted to 1346 cm−1. The ID/IG ratio in turn decreases reaching 0.22. It stems from the thermal reduction of GO during which the sp2-hybridized C–C bonds are recovered.45,46 The increment in intensity of 2D band accompanied by the shift of its location (2677 cm−1) suggest the presence of single-layer graphene within synthesized material.42,47 On the other hand, broad shape of 2D band (Fig. 7c) implies the coexistence of single-layer graphene and multi-layer graphene in synthesized TRGO.
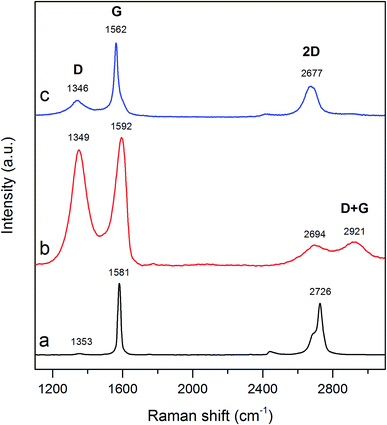 |
| | Fig. 7 Raman spectra recorded for (a) graphite, (b) GO and (c) TRGO. | |
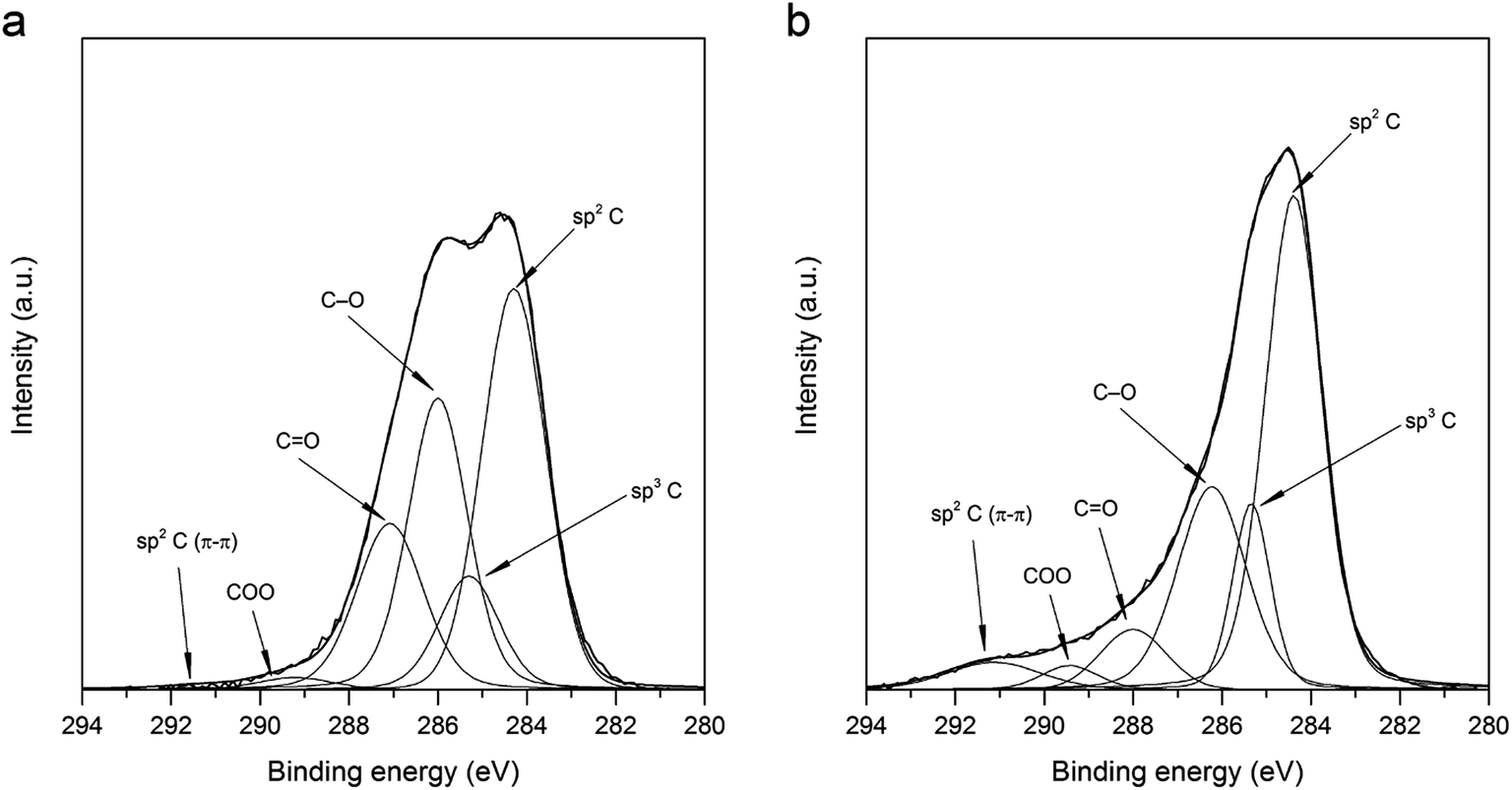
3.2.5. XPS measurements. Influence of shock thermal treatment on chemical composition of GO surface was investigated by XPS analysis. Fig. 8 exhibits deconvoluted XPS spectra of C1s region recorded for GO (Fig. 8a) and TRGO (Fig. 8b). The C/O atomic ratio for GO synthesized by electrochemical treatment of graphite in 11 M H2SO4 is equal to 3.3. It indicates that oxidation degree is slightly lower as compared to that calculated for GO synthesized by Hummers method (C/O equal to around 2).9,43,45,47 Due to thermal treatment, the total oxygen content decreases significantly from 23.1 for GO to 9.58% for TRGO (Table 2). The release of oxygen functionalities during thermal treatment of GO is accompanied by the considerable deformation of its structure. The scale of structure modification justifies the statement that during the regarded treatment process of exfoliation occurs. Thermal reduction of GO causes the decomposition of carbonyl and hydroxyl/epoxy groups, illustrated by the decrease in intensity of peaks at 287.6 and 286.1 eV for TRGO (Fig. 8b) as compared to GO (Fig. 8a).48–51 However, a small increase in concentration of carboxyl groups is noted. It can be assumed that the presence of air during the thermal treatment of GO led to the formation of some new oxygen groups (carboxyl) or transformation of non-carboxyl functionalities into the carboxyl groups. It should be emphasized that the regarded treatment increases contribution of sp2 C noticeably from 39.73 for GO (Fig. 8a) to 49.42% for TRGO (Fig. 8b). This effect is agreed with the results of Raman spectroscopy investigations, revealing that thermal reduction of GO leads to the recovering of sp2-hybridized carbon structures.
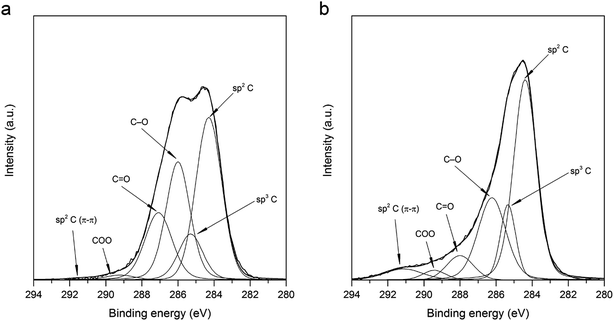 |
| | Fig. 8 Deconvoluted C1s XPS spectra recorded for (a) GO (a) and (b) TRGO. | |
Table 2 The results of C1s XPS spectra for GO and TRGO
| |
Atomic% |
| GO |
TRGO |
| C |
76.90 |
90.42 |
| O |
23.10 |
9.58 |
| Surface groups |
Peak (eV) |
Content (%) |
Peak (eV) |
Content (%) |
| sp2 C (π–π) |
291.4 |
0.22 |
291.1 |
4.57 |
| COO |
289.2 |
1.52 |
289.4 |
2.41 |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
287.1 |
18.47 |
288.0 |
6.54 |
| C–O |
286.0 |
28.73 |
286.2 |
24.63 |
| sp3 C |
285.3 |
11.33 |
285.3 |
12.43 |
| sp2 C |
284.3 |
39.73 |
284.4 |
49.42 |
4. Conclusions
Our work describes simple and low-cost method to produce a bulk quantities of graphene material. The investigated process comprises of electrochemical oxidation of graphite in 11 M H2SO4 yielded GO and subsequent its thermal treatment giving final product thermal reduced graphite oxide (TRGO). Electrochemically synthesized GO is characterized by the lower oxidation degree as compared to the GO synthesized by Hummers method. Shock thermal treatment of GO caused significant reduction of oxygen functionalities with parallel delamination of graphene layers. Additionally, the structural properties of synthesized graphene material has been significantly modified compared to host material. TRGO is characterized by the relatively high specific surface area, increased amount of mesopores and enhanced concentration of edges as well as surface defects. The above mentioned features are very close to these noted for TRGO prepared from GO synthesized by Hummers method. It indicates that GO synthesized by anodic oxidation in 11 M H2SO4 can be used to produce TRGO instead of GO synthesized by Hummers method.
Acknowledgements
This work was financially supported by the National Science Centre of Poland (3/31/PNCN/0371).
References
- A. Bianco, H. M. Chen, T. Enoki, Y. Gogotsi and R. H. Hurt, Carbon, 2013, 63, 1 CrossRef.
- Y. Yang, C. Han, B. Jiang, J. Iocozzia, C. He, D. Shi, T. Jiang and Z. Lin, Mater. Sci. Eng., R, 2016, 102, 1 CrossRef.
- H. Hu, Z. Zhao, Q. Zhou, Y. Gogotsi and J. Qiu, Carbon, 2012, 50, 3267 CrossRef.
- Y. M. Shulga, S. A. Baskakov, E. I. Knerelman, G. I. Davidova, E. R. Badamshina, N. Y. Shulga, E. A. Skryleva, A. L. Agapov, D. N. Voylov, A. P. Sokolov and V. M. Martynenko, RSC Adv., 2014, 4, 587 RSC.
- M. Yi and Z. Shen, J. Mater. Chem. A, 2015, 3, 11700 Search PubMed.
- P. Poizot, B. Humbert, C. P. Ewels, J. Y. Mevellec, N. Stephant and J. Simonet, Carbon, 2016, 107, 823 CrossRef.
- S. Y. Toh, K. S. Loh, S. K. Kamarudin and W. R. W. Daud, Chem. Eng. J., 2014, 251, 422 CrossRef.
- S. Shahrokhian, R. Mohammadi and M. K. Amini, Electrochim. Acta, 2016, 206, 317 CrossRef.
- C. Botas, P. Álvarez, C. Blanco, R. Santamaría, M. Granda, M. D. Gutiérrez, F. Rodríguez-Reinoso and R. Menéndez, Carbon, 2013, 52, 476 CrossRef.
- X. Chen, D. Meng, B. Wang, B. W. Li, W. Li, C. W. Bielawski and R. S. Ruoff, Carbon, 2016, 101, 71 CrossRef.
- B. Gurzęda, P. Florczak, M. Wiesner, M. Kempiński, S. Jurga and P. Krawczyk, RSC Adv., 2016, 6, 63058 RSC.
- S. Salimian and M. E. A. Araghi, Carbon, 2016, 107, 754 CrossRef.
- S. D. Costa, J. Ek Weis, O. Frank, M. Fridrichová and M. Kalbac, RSC Adv., 2016, 76, 72859 RSC.
- S. W. Lee, C. Mattevi, M. Chhowalla and R. M. Sankaran, J. Phys. Chem. Lett., 2012, 3, 772 CrossRef PubMed.
- H. Gao and H. Duan, Biosens. Bioelectron., 2015, 65, 404 CrossRef PubMed.
- E. B. Secor, B. Y. Ahn, T. Z. Gao, J. A. Lewis and M. C. Hersam, Adv. Mater., 2016, 27, 6683 CrossRef PubMed.
- D. Zhan, Z. Ni, W. Chen, L. Sun, Z. Luo, L. Lai, T. Yu, A. T. S. Wee and Z. Shen, Carbon, 2011, 49, 1362 CrossRef.
- B. Gurzęda, P. Florczak, M. Kempiński, B. Peplińska, P. Krawczyk and S. Jurga, Carbon, 2016, 100, 540 CrossRef.
- M. M. Storm, M. Overgaard, R. Younesi, N. E. A. Reeler, T. Vosch, U. G. Nielsen, K. Edström and P. Norby, Carbon, 2016, 85, 233 CrossRef.
- S. R. C. Vivekchand, C. S. Rout, K. S. Subrahmanyam, A. Govindarajm and C. N. R. Rao, J. Chem. Sci., 2008, 120, 9 CrossRef.
- O. Akhavan, Carbon, 2010, 48, 509 CrossRef.
- M. J. McAllister, J. L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud’homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396 CrossRef.
- C. Punckt, M. A. Pope, Y. M. Liu and I. A. Aksay, J. Electrochem. Soc., 2016, 163, 491 CrossRef.
- Q. Y. Yan, Q. Liu and J. Wang, Ceram. Int., 2016, 42, 3007 CrossRef.
- J. M. Skowroński, J. Appl. Electrochem., 1994, 24, 245 CrossRef.
- D. D. L. Chung, J. Electron. Mater., 1978, 7, 189 CrossRef.
- F. Beck, J. Jiang and H. Krohn, J. Electroanal. Chem., 1995, 389, 161 CrossRef.
- T. Kuila, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Nanoscale, 2013, 5, 52 RSC.
- C. M. Chen, Q. Zhang, M. G. Yang, C. H. Huang, Y. G. Yang and M. Z. Wang, Carbon, 2012, 50, 3572 CrossRef.
- Y. Gao, H. Huang, W. Tang, X. Liu, X. Yang and J. Zhang, Microporous Mesoporous Mater., 2015, 217, 210 CrossRef.
- H. Xian, T. Peng, H. Sun and J. Wang, Nano-Micro Lett., 2015, 7, 17 CrossRef.
- Q. Du, M. Zheng, L. Zhang, Y. Wang, J. Chen, L. Xue, W. Dai, G. Ji and J. Cao, Electrochim. Acta, 2010, 55, 3897 CrossRef.
- P. Lian, X. Zhu, S. Liang, Z. Li, W. Yang and H. Wang, Electrochim. Acta, 2010, 55, 3909 CrossRef.
- Z. Q. Li, C. J. Lu, Z. P. Xia, Y. Zhou and Z. Luo, Carbon, 2007, 45, 1686 CrossRef.
- T. N. Blanton and D. Majumdar, Powder Diffr., 2013, 28, 68 CrossRef.
- S. Gadipelli and Z. X. Guo, Prog. Mater. Sci., 2015, 69, 1 CrossRef.
- A. Kaniyoor, T. T. Baby and S. Ramaprabhu, J. Mater. Chem., 2010, 20, 8467 RSC.
- D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. A. Field, C. A. Ventrice Jr and R. S. Ruoff, Carbon, 2009, 47, 145 CrossRef.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51 CrossRef.
- M. Ramm, M. Ata, T. Gross and W. Unger, Appl. Phys. A: Mater. Sci. Process., 2000, 70, 387 CrossRef.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47 CrossRef.
- K. Krishnamoorthy, M. Veerapandian, K. Yun and S. J. Kim, Carbon, 2013, 53, 38 CrossRef.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Proud’homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36 CrossRef PubMed.
- A. M. Dimiev and J. M. Tour, ACS Nano, 2014, 8, 3060 CrossRef PubMed.
- Z. González, C. Botas, P. Álvarez, S. Roldán, S. Blanco, R. Santamaría, M. Granda and R. Menéndez, Carbon, 2012, 50, 828 CrossRef.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef.
- C. Liu, G. Hu and H. Gao, J. Supercrit. Fluids, 2012, 63, 99 CrossRef.
- T. D. Dao and H. M. Jeong, Mater. Res. Bull., 2015, 70, 651 CrossRef.
- Z. J. Fan, W. Kai, J. Yan, T. Wei, L. J. Zhi, J. Feng, Y. Ren, L. P. Song and F. Wei, ACS Nano, 2011, 5, 191 CrossRef PubMed.
- E. Paparazzo, Carbon, 2013, 63, 578 CrossRef.
- Y. Matsumoto, H. Tateishi, M. Koinuma, Y. Kamei, C. Ogata, K. Gezuhara, K. Hatakeyama, S. Hayami, T. Taniguchi and A. Funatsu, J. Electroanal. Chem., 2013, 704, 233 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a