
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkaline-promoted Ni based ordered mesoporous catalysts with enhanced low-temperature catalytic activity toward CO2 methanation†
Leilei Xu *a,
Fagen Wangb,
Mindong Chen*a,
Haoming Yanga,
Dongyang Niea,
Lu Qia and
Xinbo Liana
*a,
Fagen Wangb,
Mindong Chen*a,
Haoming Yanga,
Dongyang Niea,
Lu Qia and
Xinbo Liana
aCollaborative Innovation Center of the Atmospheric Environment and Equipment Technology, School of Environmental Science and Engineering, Nanjing University of Information Science & Technology, Jiangsu Key Laboratory of Atmospheric Environment Monitoring and Pollution Control, 210044, Nanjing, China. E-mail: leileixu88@gmail.com; chenmdnuist@163.com; Tel: +86-25-58731089
bSchool of Chemistry and Chemical Engineering, Jiangsu University, 301 Xuefu Road, Zhenjiang 212013, P. R. China
First published on 27th March 2017
Abstract
For CO2 methanation reaction, a Mg species is often utilized as the alkaline promotor for Ni based catalysts to enhance the low-temperature catalytic activity. Herein, based on a pioneer ordered mesoporous NiO–Al2O3 catalyst, a Mg alkaline promotor had been incorporated into the ordered mesoporous framework via a one-pot evaporation induced self-assembly (EISA) strategy. As a result, the ordered mesoporous NiO–MgO–Al2O3 composite oxides with Mg/Al molar ratios in a wide range (0–10%) were successfully fabricated and directly utilized as the catalysts for CO2 methanation reaction. These mesoporous catalysts were carefully characterized by X-ray diffraction, N2 adsorption–desorption, transmission electron microscopy, selected area electron diffraction, energy dispersive spectrometer, X-ray photoelectron spectroscopy, H2 temperature-programmed reduction, and CO2 temperature-programmed desorption measurements. It was found that the ordered mesoporous materials with large specific surface areas (180.8–232.8 m2 g−1), big pore volumes (0.37–0.43 cm3 g−1), and narrow pore size distributions (around 9.5 nm) could be successfully retained after the calcination at 700 °C. The highly dispersed Ni species were strongly interacted with the mesoporous framework in the form of NiAl2O4 spinel. The incorporation of the Mg progressively increased the surface basicity of these catalysts, which could intensify the chemisorption and activation of CO2 during the CO2 methanation reaction. Therefore, the low-temperature catalytic activity was significantly enhanced. The “volcano-shape curve” relationship between the Mg/Al molar ratio and catalytic activity had been interestingly observed, suggesting only appropriate surface basicity could obtain the optimum catalytic activity. Besides, there was no evident deactivation over these mesoporous catalysts after 50 h long-term stability tests due to the confinement effect of the mesoporous framework. Therefore, the present ordered mesoporous NiO–MgO–Al2O3 materials could be considered as a series of potential catalyst candidates for CO2 methanation.
1. Introduction
In recent years, the increasing amount of CO2 emission by the combustion of fossil fuels contributes to around 82% of the greenhouse gases.1,2 As a result, the global warming problem gradually becomes a threat to the global climate, which can cause the rise of sea levels, melting in glaciers, and other environmental problems.3–5 Meanwhile, CO2 is also considered as a raw material for many chemical processes due to its available, renewable, and nontoxic features.6–8 The conversion of CO2 into useful low-carbon fuels (such as methanol, dimethyl ether, methane, etc.) via hydrogenation processes are the main approaches for its large-scale resource utilization.2 Although methanol and dimethyl ether are promising fuels, the production of them from CO2 hydrogenation often demands high reaction pressure around 5 MPa.9 Besides, the CO2 conversions of these processes are also relatively low (<20%) and it is still difficult to realize the complete conversion of CO2.10 Compared with these processes, the CO2 hydrogenation to methane (also called Sabatier reaction) possesses following advantages. The synthetic natural gas (SNG, CH4) can be directly injected and transported by the existing pipeline system.11–13 Furthermore, this reaction can be carried out under atmospheric pressure and the production of CH4 is favorable at low temperature according to its thermodynamic feature.2,7,14 Furthermore, it also has potential industrial application prospect once the hydrogen can be derived from the sustainable sources, such as biomass, photocatalytic water splitting, etc.2 The NASA is also greatly interested with this reaction in view of the future manned space colonization on Mars.7,15 The CO2 (95%) atmosphere of Mars together with the hydrogen from the earth can be converted into methane and water via this process, providing the fuel and life-support system for the astronauts.2,7 Therefore, CO2 methanation has recently been received more and more attention.The metals from group VIII, such as Ru, Rh, Pd, Ni, and Co, are effective catalysts for CO2 methanation based on previous literatures.16–23 Among them, the Ni based catalysts are of great interest and have been extensively investigated due to their low cost and facile availability.24–27 However, compared with noble metal catalysts, the Ni based catalysts display poorer catalytic activity, especially at low reaction temperature. Although the low temperature is beneficial to the achievement of high CO2 conversion and CH4 selectivity according to thermodynamic calculation, the full reduction of CO2 into CH4 is still difficult at low temperature over Ni based catalysts. Because this reaction is an eight-electron process with significant kinetic barrier.7,28 In order to obtain acceptable CO2 conversion rate, the catalyst is usually required to decrease the activation energy. Therefore, the development of Ni based catalysts with advanced low-temperature catalytic activity remains a great challenge.
Extensive studies have revealed that the low-temperature catalytic performances of Ni based catalysts are greatly dependent on various influencing parameters, such as the catalytic support, promotor, and preparation method.2,7,28 Specifically, the catalytic support has a significant effect on the high dispersion of the metallic active sites, which greatly contributes to the activation and dissociation of the H2 molecule.18,29 Thus, the materials with large surface areas, big pore volumes, and unblocked pore channels, such as mesoporous zeolite and mesoporous metal oxides, have been investigated as the supports of the CO2 methanation catalysts.23,30–32 As for the CO2, it is an extremely stable molecule with eight out-layer electron structure. In order to decrease its activation energy, the alkaline promotors, such as MgO, CaO, La2O3, and so on, are often used to intensify the surface basicity, which will intensify the chemisorption and activation of the CO2.33–35 As a result, the catalytic activity at low reaction temperature will be promoted.
For Ni based catalyst, another disadvantage is the thermal sintering of the metallic active sites during the processes of reduction and CO2 methanation reaction due to its low Tammann temperature (590 °C), above which the thermal sintering of metallic Ni will easily take place.36 Besides, there are possible hot spots existing among the catalyst bed due to exothermic feature of the CO2 methanation reaction.37,38 Thus, the temperature of the catalyst bed is usually higher than the reactor temperature. This will aggravate the thermal agglomeration of the metallic Ni active sites and decrease the stability of the catalysts. In order to address this challenge, the Ni catalytic sites are often stabilized by well-defined crystalline structures (e.g. solid solution, spinel, perovskite, etc.),39–41 rigid mesoporous frameworks (e.g. Ni-MCM-41, NiO–Al2O3, etc.),30,31 and core–shell structures (e.g. Ni@SiO2),42 especially for high temperature reactions (such as dry reforming and steam reforming). With the aid of these strategies, the metallic Ni active sites can be firmly confined and its thermal sintering can be effectively controlled. Among these materials, the catalysts with mesoporous structures can be considered as promising candidates due to their outstanding structural properties, which can expose sufficient active sites for the reactants. For example, Haller et al. reported that the Ni-MCM-41 displayed outstanding catalytic performance comparable to the best results of Ru/SiO2 noble catalysts toward CO2 methanation owing to its excellent sintering-resistance and textual properties.30 Following this guideline, we had successfully synthesized ordered mesoporous NiO–Al2O3 composite oxide for CO2 methanation, which displayed much better catalytic performance than both Ni/Al2O3 supported and non-porous NiO–Al2O3 catalysts.31 However, the low-temperature catalytic activity over ordered mesoporous NiO–Al2O3 catalysts still requires further improvement.
Herein, a series of ordered mesoporous NiO–MgO–Al2O3 materials with different Mg contents were fabricated by one-pot evaporation-induced self-assembly (EISA) method and directly employed as the catalysts for CO2 methanation reaction. Similar to the ordered mesoporous NiO–Al2O3 catalyst, the present materials with wide Mg/Al molar ratio (0–10%) also possessed outstanding textural properties and the Ni active sites were firmly confined by the mesoporous matrix, promising excellent catalytic activities and stabilities. Besides, the incorporation of the Mg greatly promoted the surface basicity, which could further intensify the chemisorption of the CO2. The influence the Mg promotor on the catalytic activity had been carefully investigated. It was found that the presence of the Mg greatly favored the promotion of low-temperature catalytic activity and only appropriate Mg content could maximally promote the activity. The relationship between the structure and performance over these catalysts had been elaborated with the aid of the various analyzing techniques.
2. Experimental
2.1 The fabrication of the ordered mesoporous NiO–MgO–Al2O3 composite metal oxide catalysts
The ordered mesoporous NiO–MgO–Al2O3 composite metal oxides with different Mg contents were synthesized by one-pot evaporation induced self-assembly (EISA) strategy, which had been reported elsewhere.43–45 In these materials, the Ni/Al molar ratio was controlled at 10% and the Mg/Al molar ratios were controlled at x% (x = 0, 1, 3, 5, 8, and 10), respectively. Specifically, 1.0 g (EO)20(PO)70(EO)20 (Pluronic P123, Mn = 5800, Sigma-Aldrich) structure directing agent was dissolved in 20.0 mL absolute ethanol with vigorous agitation. 1.6 mL 67.0 wt% nitric acid, 10 mmol aluminum isopropoxide (>98%, Sigma-Aldrich), 1 mmol Ni(NO3)2·6H2O (Sigma-Aldrich), and quantitative Mg(NO3)2·6H2O (Sigma-Aldrich) were added to above solution in sequence. After 5 h vigorous agitation, the final transparent light green solution was transferred into a Petri dish, which was put into a convection oven with appropriate temperature (60 °C) and relative humidity (<50%) to carry out the EISA process for 48 h. The final light green xerogel was calcined at 700 °C for 5 h with a ramping heat rate of 1 °C min−1. For convenience, these ordered mesoporous NiO–MgO–Al2O3 materials with different Mg contents were abbreviated as OMA-10NixMg, where “OMA”, “10”, an “x” referred to the “ordered mesoporous alumina matrix”, “Ni/Al molar ratio”, and “Mg/Al molar ratio”, respectively.The Ni/γ-Al2O3 supported catalyst was prepared via incipient wetness impregnation method using Ni(NO3)2·6H2O (Sigma-Aldrich) as the precursor and investigated as the reference catalyst, which had identical Ni/Al molar ratio (10%) to the OMA-10NixMg materials. The catalyst calcined at 700 °C was denoted as 10Ni/Al2O3, where “10” stood for the “Ni/Al molar ratio”.
2.2 Catalyst characterizations
Small-angle and wide-angle X-ray diffraction (XRD) patterns were recorded on the X'pert Pro multipurpose diffractometer (PANalytical, Inc.) with Ni-filtered Cu Kα radiation (0.15046). The N2 adsorption and desorption analyses were conducted by using a NOVA 2200e (Quantachrome) at liquid nitrogen temperature (−196 °C). Prior to the regular analysis, the sample should be degassed at 200 °C under vacuum condition for 4 h to remove physisorbed water and surface impurities. The inductive coupled plasma atomic emission spectrometer (ICP-AES) was determined on the Optima 7300DV (Perkin Elmer) to obtain the actual Ni/Al and Mg/Al ratios. Transmission electron microscopy (TEM), selected area electron diffraction (SAED), and energy-dispersive spectroscopy (EDS) characterization were carried out on a JEOL 2010F microscope using 200 kV as the working voltage. The powder of the sample ought to be highly dispersed in ethanol with the aid of ultrasonic apparatus before dropping onto the carbon-coated copper grid. X-ray photoelectron spectroscopy (XPS) analyses of the catalysts were recorded over a VG ESCALAB 210 (Thermo Scientific) spectrometer. The binding energy of the C 1s (284.8 eV) was used as the standard reference of the XPS data. For XPS analysis, the sample should be loaded on a sample holder coated with conductive adhesive tape and pretreated in high vacuum environment. The H2 temperature programmed reduction (H2-TPR) measurements of the catalysts were performed on a Quantachrome ChemBET Pulsar TPR/TPD apparatus. Specifically, 0.1 g sample was loaded on the U-type quartz tube connected with the equipment. Prior to the formal test, the sample should be purged with He stream (30 mL min−1) at 300 °C for 1 h to remove the physical-adsorption water and other surface impurities. Then, the H2-TPR measurement was carried out in 5 vol% H2–95 vol% He stream (80 mL min−1) with a 10 °C min heating rate up to 1100 °C and the amount of hydrogen consumption was recorded by TCD detector. The CO2 temperature programmed desorption (CO2-TPD) measurements of the catalysts were carried out on the Micromeritics AutoChem II 2920 Chemisorption analyzer. In particular, 0.1 g sample loaded in the U-type tube was pretreated at 300 °C in He stream for 1 h to remove the surface impurities. After cooled down to the 25 °C, the sample was exposed to pure CO2 atmosphere (50 mL min−1) for 1 h to make the sample surface saturated with chemisorbed CO2. Then, the gas was changed into the He stream (50 mL min−1) to purge the physically absorbed CO2 until the baseline was stable. Finally, the CO2-TPD was conducted with a heating rate of 20 °C min−1 to 900 °C and the desorbed CO2 signal was also recorded by the TCD detector.2.3 Catalyst evaluation
The CO2 methanation reaction was carried out in a continuous-flow fixed-bed tubular quartz reactor (i.d. = 10 mm). The reaction temperature was determined and controlled by a coaxial thermocouple located at the center of the catalyst bed. The flow rates of the reactant gases were regulated by calibrated mass flow controllers. Before the catalytic reaction, the catalyst ought to be pre-reduced under H2/N2 (10/20 mL min−1) mixed flow at 800 °C for 2 h with the ramping heating rate of 1.5 °C min−1. After purging the reactor with N2 to remove chemisorbed H2, the mixture of the H2 and CO2 (molar ratio H2/CO2 = 4) without any dilution was introduced into the reactor at a gas hourly space velocity (GHSV) of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL (g−1 h−1). For each catalyst, the catalytic performance was investigated from 200 to 450 °C at the interval of 50 °C. Lab-scale stability tests were conducted at 400 °C with the GHSV of 15000 mL (g−1 h−1) for 50 h under atmospheric pressure. Before flowing into the on-line GC system, the product gases effluent from the reactor was mixed with the N2 (5 mL min−1) as an internal standard gas. Finally, the mixture of product gases were injected on line via the automatic six-port valve injectors and subsequently analyzed by the GC machine (Agilent 7890B) equipped with a packed column (TDX-01) for TCD and an alumina capillary column for FID. However, there was no C2+ hydrocarbon detected by the FID detector. Therefore, the CO2 conversion (abbreviated as CCO2) and CH4 selectivity (denoted as SCH4) were calculated based on the following formulas.
000 mL (g−1 h−1). For each catalyst, the catalytic performance was investigated from 200 to 450 °C at the interval of 50 °C. Lab-scale stability tests were conducted at 400 °C with the GHSV of 15000 mL (g−1 h−1) for 50 h under atmospheric pressure. Before flowing into the on-line GC system, the product gases effluent from the reactor was mixed with the N2 (5 mL min−1) as an internal standard gas. Finally, the mixture of product gases were injected on line via the automatic six-port valve injectors and subsequently analyzed by the GC machine (Agilent 7890B) equipped with a packed column (TDX-01) for TCD and an alumina capillary column for FID. However, there was no C2+ hydrocarbon detected by the FID detector. Therefore, the CO2 conversion (abbreviated as CCO2) and CH4 selectivity (denoted as SCH4) were calculated based on the following formulas.| CCO2 = (FCO2,inlet − FCO2,outlet)/FCO2,inlet × 100% | (1) |
| SCH4 = FCH4,outlet/(FCH4,outlet − FCO,outlet) × 100% | (2) |
In all formulas, the Fx,inlet and Fx,outlet represented the flow rate of the x species, which flowed into and out of the reactor, respectively.
3. Results and discussion
3.1 Physical and chemical properties of the fresh catalysts
The small-angle (0.5–5°) and wide-angle (20–80°) XRD patterns of the fresh OMA-10NixMg catalysts were displayed in Fig. 1(1) and (2), respectively. It could be observed in Fig. 1(1) that all the OMA-10NixMg catalysts with different Mg contents exhibited two groups of peaks in the range of 0.76–0.78° and 1.30–1.50°, which could be attributed to the (1 0 0) and (1 1 0) directed inflections, respectively. This suggested the presence of two dimensional p6mm hexagonal ordered mesoporous channel with long-range orderliness. Besides, the incorporation of the Mg species almost had no obviously adverse effect on the ordered mesostructure of the OMA-10NixMg. The d-space(1 0 0) values of these samples were calculated based on the peak position of the (1 0 0) directed reflection according to Bragg's equation. As observed in the Table 1, their values were in the range of 11.3–11.9 nm, which belonged to mesopore (2–50 nm) based on the IUPAC definition.46 As for the wide-angle XRD patterns in Fig. 1(2), all the OMA-10NixMg samples with identical Ni loading amount did not display any diffraction peaks related with Ni species, such as NiO, NiAl2O4, and so on, suggesting the high dispersion of Ni species among the ordered mesoporous framework. However, the diffraction peak of MgAl2O4 spinel phase (PDF-#-21-1152) gradually appeared and became stronger and stronger with the increase of Mg/Al molar ratio from 0% to 10%. In contrast, the XRD pattern of the conventional 10Ni/Al2O3 supported catalyst in Fig. S1 (ESI†) displayed apparent NiO (PDF-#-44-1159) diffraction peak, suggesting its relatively poorer Ni dispersion than OMA-10NixMg counterparts.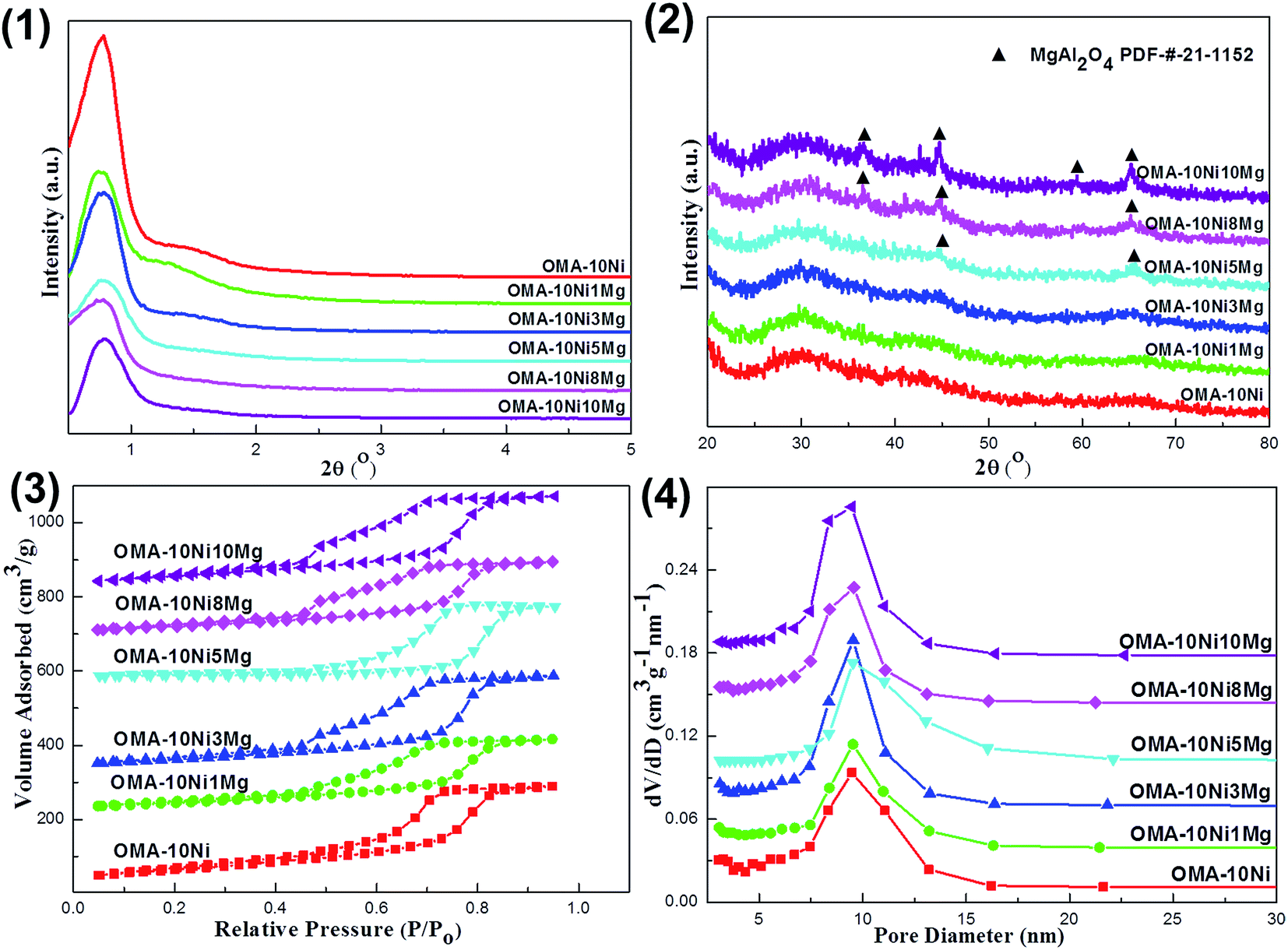 | ||
| Fig. 1 (1) Small-angle and (2) wide-angle X-ray diffraction patterns of the fresh OMA-10NixMg catalysts; (3) nitrogen adsorption–desorption isotherms and (4) pore size distribution curves of the fresh OMA-10NixMg catalysts. | ||
| Samples | SBETa (m2 g−1) | VBJHb (cm3 g−1) | APDc (nm) | d(100)e (nm) | WTf (nm) | Ni/Alg | Mg/Alg | Eah (kJ mol−1) | Isotherm type |
|---|---|---|---|---|---|---|---|---|---|
a SBET stands for the specific area calculated based on Brunauer–Emmett–Teller theory.b VBJH stands for the pore volume calculated based on Barrett–Joyner–Halenda theory.c APD stands for average pore diameter.d SP stands for 100 h spent catalyst.e d(100) stands for the d-spacing of the (1 0 0) direction calculated by the Bragg's law: d = nλ/2sinθ, where λ is the wavelength of the X-ray wave (0.15406 nm).f WT stands for the wall thickness (calculated by 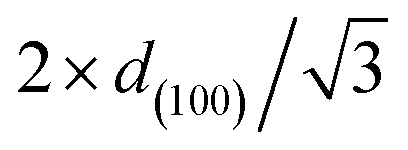 − pore diameter).g Ni/Al and Mg/Al molar ratios were obtained by ICP-AES analyses.h Ea stood for the apparent activation energy of CO2. − pore diameter).g Ni/Al and Mg/Al molar ratios were obtained by ICP-AES analyses.h Ea stood for the apparent activation energy of CO2. |
|||||||||
| OMA-10Ni | 232.8 | 0.42 | 9.5 | 11.3 | 3.5 | 0.097 | — | 75.2 | IV H1 |
| OMA-10Ni1Mg | 182.1 | 0.40 | 9.5 | 11.9 | 4.2 | 0.098 | 0.011 | 68.6 | IV H1 |
| OMA-10Ni3Mg | 180.8 | 0.43 | 9.5 | 11.3 | 3.4 | 0.095 | 0.032 | 63.6 | IV H1 |
| OMA-10Ni5Mg | 188.7 | 0.37 | 9.5 | 11.6 | 3.9 | 0.097 | 0.053 | 57.6 | IV H1 |
| OMA-10Ni8Mg | 187.6 | 0.39 | 9.6 | 11.3 | 3.4 | 0.094 | 0.078 | 65.5 | IV H1 |
| OMA-10Ni10Mg | 218.8 | 0.41 | 9.5 | 11.3 | 3.5 | 0.096 | 0.104 | 65.1 | IV H1 |
| 10Ni/Al2O3 | 158.0 | 0.17 | 5.5 | — | — | 0.095 | — | 77.1 | IV H2 |
| SP-OMA-10Ni | 139.0 | 0.27 | 8.5 | — | — | 0.098 | — | — | IV H1 |
| SPd-OMA-10Ni3Mg | 138.4 | 0.31 | 8.3 | — | — | 0.093 | 0.034 | — | IV H1 |
| SP-OMA-10Ni5Mg | 135.9 | 0.32 | 8.4 | — | — | 0.095 | 0.049 | — | IV H1 |
| SP-10Ni/Al2O3 | 107.0 | 0.11 | 5.0 | 0.096 | — | — | IV H2 | ||
The N2 adsorption–desorption isotherms and corresponding pore size distribution curves of the OMA-10NixMg materials were presented in Fig. 1(3) and (4). As shown in Fig. 1(3), all the materials possessed IV type isotherms with steep H1 shaped hysteresis loops in the range of 0.5–0.9 P/P0 owing to the capillary condensation of N2 among the mesoporous channels, suggesting the presence of mesoporous channels with cylindrical shapes.45 Besides, the Fig. 1(4) described the corresponding pore size distribution curves of these materials, which were calculated according to the Barrett–Joyner–Halenda (BJH) method using the adsorption branch. It could be observed that all the OMA-10NixMg materials regardless of the Mg contents displayed greatly narrow pore size distributions around 9.5 nm. As a comparison, the fresh 10Ni/Al2O3 catalyst in Fig. S2 (ESI†) displayed IV H2 shaped isotherm and narrow pore size distribution around 6.0 nm. This suggested the presence of the mesoporous structure, which was mainly originated from the γ-Al2O3 support, not from the assembly process. Furthermore, the textural properties, such as Brunauer–Emmett–Teller (BET) specific surface areas, pore volumes, BJH pore diameters, and wall thickness of the mesopores, were summarized in Table 1. It was noticeable that all the OMA-10NixMg materials were still provided with large surface areas up to 232.8 m2 g−1 and pore volumes up to 0.43 cm3 g−1 after calcination at 700 °C, which were somewhat better than 10Ni/Al2O3 supported catalyst. The wall thicknesses of these samples were in the range of 3.4–4.2 nm. The influence of the Mg modification on the textural properties of OMA-10NixMg was not obvious. Besides, the values of actual Ni/Al and Mg/Al molar ratios for the fresh OMA-10NixMg and 10Ni/Al2O3 catalysts based on the ICP-AES measurements were also summarized in Table 1. It was noticeable that the actual values of the Ni/Al and Mg/Al molar ratios did not deviate too much from their corresponding theoretical values.
The TEM images of the fresh OMA-10NixMg materials were shown in Fig. 2. It could be observed that all the samples exhibited the alignments of cylindrical shaped mesopores along [1 1 0] direction (Fig. 2(a–c) and (e–g)) and [0 0 1] direction (Fig. 2(d)), which confirmed the presence of 2D p6mm hexagonally structured mesoporous channels. This was well consistent with the deduction based on the small-angle XRD and N2 adsorption–desorption analyses. Similar to wide-angle XRD, the selected area electron diffraction (SAED) could also provide information related with the crystallinity of the materials. As displayed in the insets of Fig. 2(a–c) and (e–g), the diffraction rings of these samples were not very distinguishable, implying the poor crystallinity of the mesoporous framework. Therefore, the results of XRD and SAED were in good agreement with each other. Furthermore, the EDS characterization was conducted over OMA-10Ni10Mg and its profile was displayed in Fig. 2(h). The characteristic peaks of Ni, Mg, Al, O, C, and Cu elements could be identified in the EDS profile. For the C and Cu peaks, they ought to be derived from the copper grid coated with carbon film. The co-presence of Ni, Mg, Al, O peaks suggested that these elements had been successfully introduced into ordered mesoporous OMA-10Ni10Mg material.
 | ||
| Fig. 2 TEM, SAED, and EDS measurements of the fresh OMA-10NixMg catalysts: (a) OMA-10Ni, (b) OMA-10Ni1Mg, (c and d) OMA-10Ni3Mg; (e) OMA-10Ni5Mg; (f) OMA-10Ni8Mg; (g and h) OMA-10Ni10Mg. | ||
The interaction strength between the Ni species and the ordered mesoporous Al2O3 matrix was investigated by the H2-TPR over the OMA-10NixMg catalysts with different Mg contents. Their profiles were displayed in Fig. 3(1). It was worth noting that each sample only possessed one pronounced H2 reduction peak around 820.0 °C in the whole temperature range investigated (100–1070 °C). This suggested that the strong metal-framework interaction had been formed. The absence of the reduction peak in low temperature region (300–500 °C) demonstrated that there was no disassociated NiO species weakly bonded with mesoporous framework.47 The reason for this phenomenon could be attributed to the unique “one-pot” fabrication strategy. Different with the conventional supported catalysts, the Ni active sites were in situ introduced into the ordered mesoporous framework during the one-pot EISA process. It was supposed that the location of the Ni species was homogenously embedded among the ordered mesoporous Al2O3 matrix owing to the advantage of the “one-pot” synthesis strategy. As a result, there was almost no big difference between the “surface” and “bulk” Ni species in their chemical coordination environment, accounting for the only one evident reduction peak for each OMA-10NixMg sample. However, the H2-TPR profile of Ni/Al2O3 supported catalyst in Fig. S3(1) (ESI†) exhibited two pronounced peaks centered at 517.6 °C and 891.9 °C, which could be identified as the NiO species with weak and strong interactions with the γ-Al2O3 support, respectively. This result suggested that the Ni species was heterogeneously distributed over the surface of Al2O3 support after impregnation process. Therefore, compared with OMA-10NixMg, it was relatively easier to reduce the weakly interacted Ni species over Ni/Al2O3 catalyst, which was well consistent with the previous reports.31,47,48 Thus, the metallic Ni active sites in OMA-10NixMg would be endowed with enhanced anti-sintering performance.
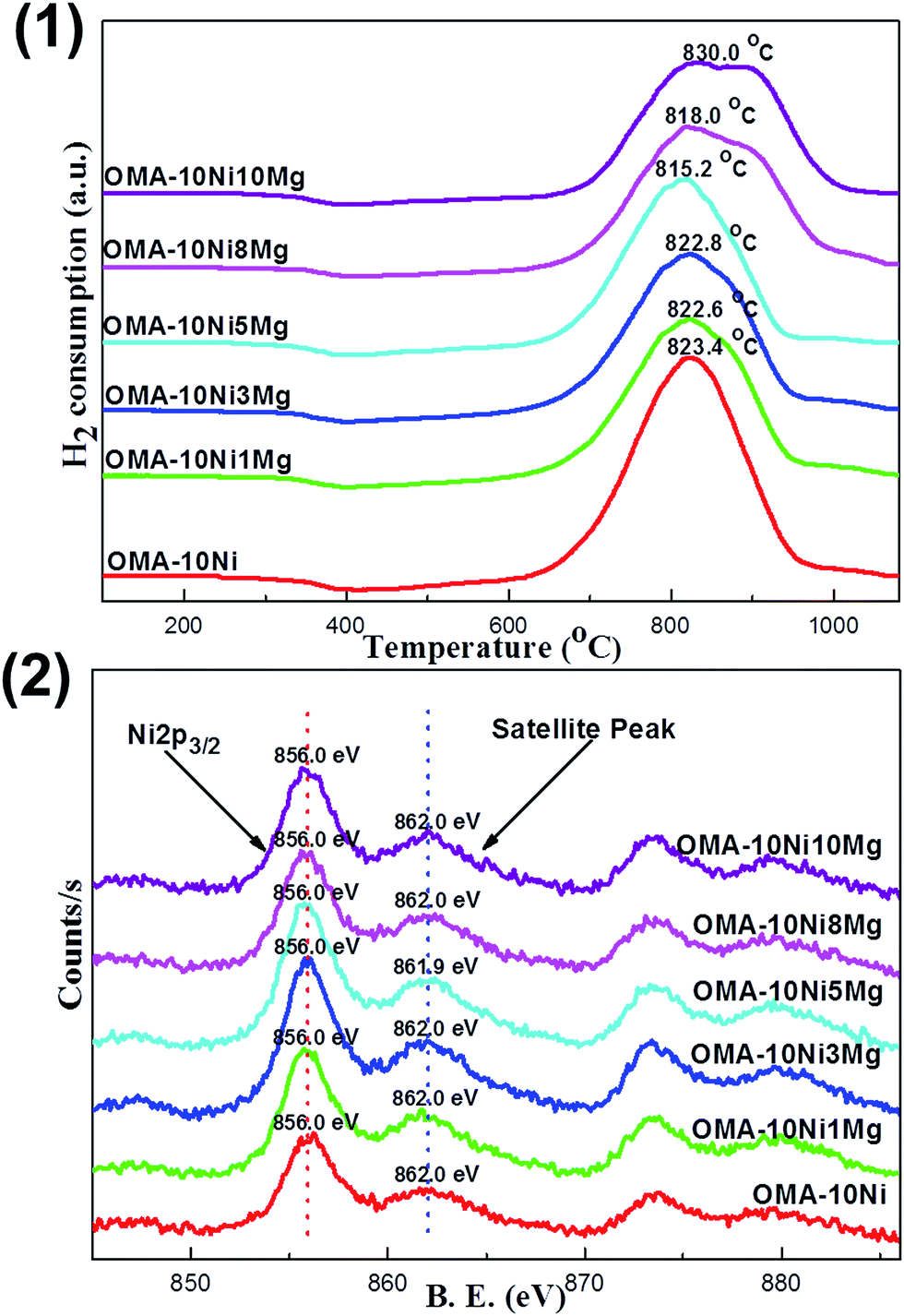 | ||
| Fig. 3 (1) H2-TPR and (2) Ni 2p XPS profiles of the fresh OMA-10NixMg catalysts. | ||
Fig. 3(2) showed the XPS profiles of Ni 2p for the fresh OMA-10NixMg materials with different Mg contents. It was of great interest to find that all the samples irrespective of the amount Mg contents displayed identical Ni 2p profiles in the shape. Specifically, each sample exhibited one main Ni 2p3/2 peak around 856.0 eV and another satellite peak around 862.0 eV, which were the featured peaks Ni2+ in the form of NiAl2O4 spinel species.49 This indicated that the NiAl2O4 species existed over the surface of the OMA-10NixMg catalysts and the incorporation of the Mg alkaline promotor into the mesoporous framework did not influence the chemical coordination environment of the Ni elements. For OMA-10NixMg catalysts, the Ni species were homogenously distributed among the whole materials due to the advantage of “one-pot” strategy. There ought to be no difference between the bulk and surface Ni species. Thus, it can be supposed that the Ni species embedded in the inner part of the mesoporous framework was also in the form of NiAl2O4 phase. Actually, the single-reduction peak over these OMA-10NixMg materials in Fig. 3(1) had already demonstrated the presence of the NiAl2O4 species, which only could be reduced at high temperature above 800 °C. Therefore, the absence of NiAl2O4 diffraction peak in Fig. 1(2) was because of the high dispersion of the NiAl2O4 among the amorphous Al2O3 framework. As for Ni/Al2O3 supported catalyst, its Ni 2p XPS profile in Fig. S3(2) (ESI†) displayed the 2p3/2 peak at 855.8 eV, which was a bit lower than peak position of Ni 2p3/2 in OMA-10NixMg. The shift toward to the lower binding energy region ought to be attributable to the surface NiO species, which could be reduced at low temperature according to H2-TPR analysis in Fig. S3(1).†
The CO2-TPD measurements had been carried out over the fresh OMA-10NixMg materials to investigate the surface basicity, which would greatly affect the chemisorption and activation of the CO2 during the catalytic reaction. Their CO2-TPD profiles were displayed in the Fig. 4. For all the samples, their desorption peaks of CO2 were very similar in the shapes, which were mainly centered at 83.2 and 460.5 °C. The first peak at 83.2 °C might be attributed to the weakly chemisorbed CO2 molecules, which possibly interacted with the surface basic sites in the form of monodentate carbonate species.50,51 The second peak at 460.5 °C ought to be derived from the decomposition of bidentate and/or polydentate carbonates, which strongly interacted with the basic sites.50,51 Besides, it could be noticeable that the intensity of the CO2 desorption peak was greatly related with the Mg contents, which was in agreement with previous reports.52,53 Specifically, with the increase of Mg/Al molar ratio from 0% to 10%, the surface basicity progressively increased and the formation of MgAl2O4 phase did not influence the total surface basicity. In summary, the incorporation Mg species into the mesoporous framework could simultaneously promote the weak basic site and strong basic site, which would be favorable in subsequent processes of CO2 chemisorption and activation during CO2 methanation reaction.
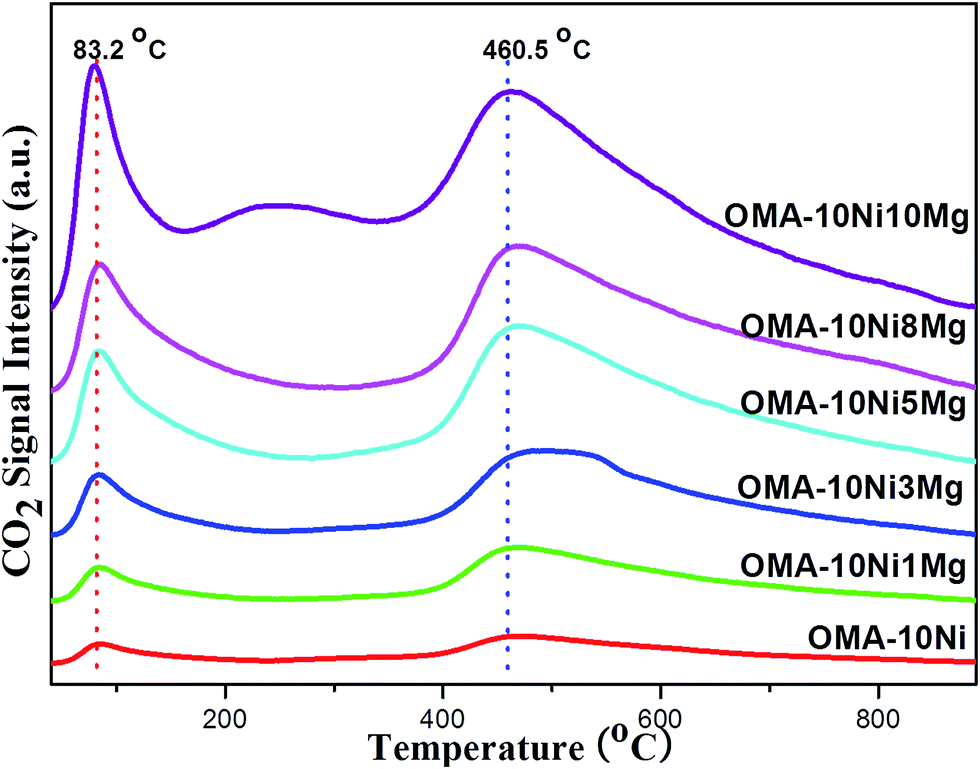 | ||
| Fig. 4 CO2-TPD profiles of the fresh OMA-10NixMg catalysts. | ||
3.2 Catalytic performances for CO2 methanation
Prior to the regular catalyst evaluation, the blank test was conducted without loading catalysts. Almost no catalytic activity was detected in the temperature range of 200–450 °C, demonstrating the critical role of catalyst in promoting catalytic activity. For the catalytic activity tests, the data related with CO2 conversion and CH4 selectivity was the average values of three times.The effect of the Mg basic modifier on the catalytic activities at different reaction temperatures had been investigated over OMA-10NixMg catalysts under specified conditions (H2/CH4 = 4, GHSV = 15000 mL (g−1 h−1), 1 atm). The curves of the CO2 conversion versus Mg/Al molar ratio at different reaction temperatures were described in Fig. 5. It was noticeable that these curves at different temperatures were similar to “volcano-shape curve” in the shape, which was very universal in the catalysis field.54,55 Specifically, with the increase of Mg/Al molar ratio from 0% to 5%, the CO2 conversion continuously increased until reaching the maximum value; however, further increasing the Mg/Al molar ratio up to 10% would cause the decline of the CO2 conversion. This suggested that only appropriate amount of Mg basic modifier could maximally promote the catalytic activity, which was well consistent with pioneer report.34 According to the above CO2-TPD analysis, the presence of Mg evidently increased the surface basicity, which was proportional to the amount of Mg species. This could greatly intensify the chemisorption and subsequent activation of CO2 during the reaction, which could finally decrease the activation energy of CO2. As a result, the catalytic activity of the alkaline modified catalyst, especially at low reaction temperature could be promoted. However, excessive Mg basic promotor would firmly absorb the CO2 and the subsequent desorption step of CO2 would become difficult, which might cause the coverage of neighboring Ni active sites and the blockage of following activation of H2. Consequently, the CO2 conversion suffered some decline because of the excessive surface basicity. Therefore, only Mg/Al molar ratio (5%) could evidently promote the catalytic activity to the utmost.
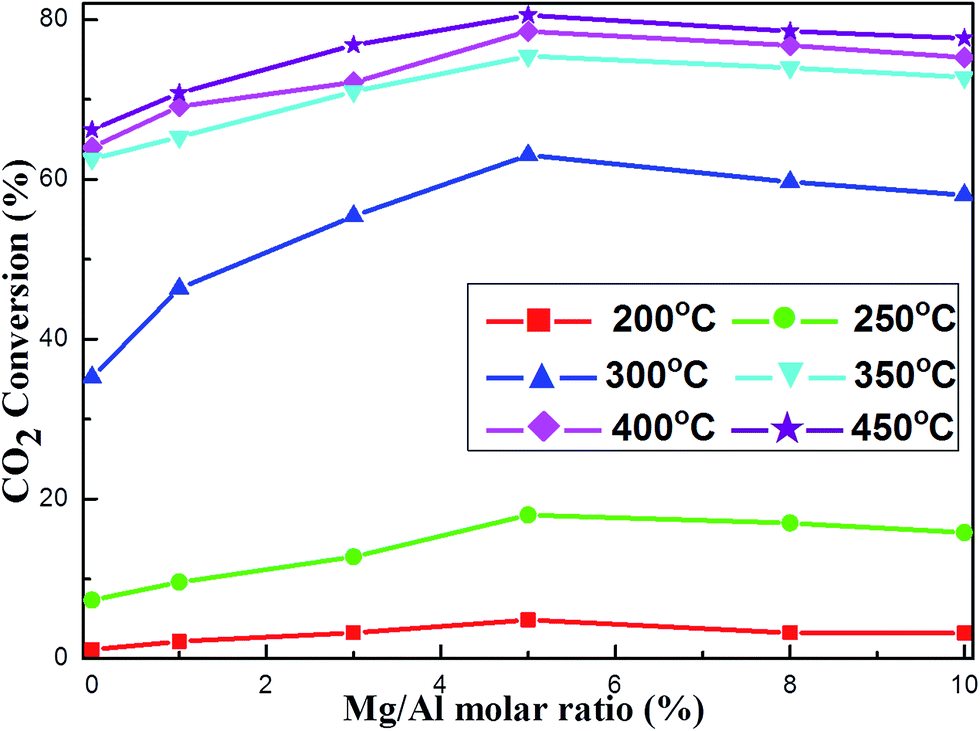 | ||
| Fig. 5 The curve of the CO2 conversion versus Mg/Al molar ratio at different reaction temperatures over the OMA-10NixMg catalysts; reaction condition: H2/CO2 = 4, GHSV = 15000 mL g−1 h−1, 1 atm. | ||
The role of Mg alkaline promotor in enhancing the catalytic activity over OMA-10NixMg catalysts toward CO2 methanation, especially at low temperature, was further confirmed by the kinetic study. The Arrhennius plots of the OMA-10NixMg catalysts with different Mg/Al molar ratios were displayed in Fig. S4 (ESI†) and their corresponding apparent activation energies of CO2 were summarized in Table 1. It was of great interest to find that the catalysts with basic modification displayed much lower apparent activation energies of CO2 than pristine OMA-10Ni catalyst without any modification. The results suggested that the incorporation of the Mg alkaline promotor possessed positive roles in CO2 activation. Specifically, with the increase of Mg/Al molar ratio from 0% to 5%, the CO2 activation energy gradually decreased from 75.2 kJ mol−1 to 57.6 kJ mol−1. However, further increasing the Mg/Al molar ratio up to 10% caused the increase of the CO2 activation energy. This indicated that only appropriate Mg/Al molar ratio (5%) could achieve the lowest activation energy of the CO2 because of “volcano-shape curve” phenomenon.
The influence of reaction temperature on the catalytic activity and selectivity was studied over OMA-10Ni, OMA-10Ni5Mg, and 10Ni/Al2O3 representative catalysts in the region of 200–450 °C. As observed in Fig. 6(1), the CO2 conversions over most of these catalysts gradually increased with the increase of the reaction temperature from 200 to 400 °C and achieved the maximum values at 400 °C; however, the further increase of the temperature to 450 °C caused the decline of the CO2 conversion. Actually, the trends of these three curves were completely different with that of the theoretical equilibrium CO2 conversion curve, which gradually decreased with the increase of the reaction temperature.28 Although the low reaction temperature was favorable for the high CO2 conversion, the kinetic limitation for CO2 activation was a great barrier for the achievement of outstanding low-temperature activity.7 In order to overcome this difficulty, the alkaline promotor was used for intensifying the processes of chemisorption and activation. Therefore, compared with the OMA-10Ni, the OMA-10Ni5Mg behaved much higher CO2 conversion, which was more close to the theoretical equilibrium conversion. Furthermore, it was also noticeable to find that the CO2 conversions over both OMA-10Ni and OMA-10Ni5Mg catalysts were also much higher than that over 10Ni/Al2O3 catalyst, especially in the low temperature region. This could be attributed to the better textural properties of the ordered mesoporous catalysts, such as larger surface area and unimpeded mesoporous channel, which made the mass diffusion of the gaseous reactants toward metallic Ni active sites become easier. This accounted for the lower CO2 activation energies over OMA-10NixMg catalysts than 10Ni/Al2O3 catalyst (77.1 kJ mol−1, see Table 1). Besides, the relationship between the CH4 selectivity and the reaction temperature was reflected in the Fig. 6(2). It could be observed that the equilibrium CH4 selectivity gradually decreased with the increase of the temperature due to the reverse water–gas shift (RWGS) side reaction, which could generate the CO and decrease the CH4 selectivity.56 For the CH4 selectivity over these three real catalysts, their values at different temperatures were lower than the corresponding theoretical equilibrium selectivity. Specifically, the OMA-10Ni and OMA-10Ni5Mg displayed their highest CH4 selectivity at 350 °C and 10Ni/Al2O3 showed its highest CH4 selectivity at 200 °C. Besides, the OMA-10Ni5Mg with basic modification displayed a bit higher CH4 selectivity than the pristine OMA-10Ni. This suggested that the presence of Mg basic site was beneficial to the enhancement of the CH4 selectivity. For the reaction mechanism of the CO2 methanation, though they had been intensively investigated over different kinds of active sites, the arguments related with the reaction intermediates had not reached a consensus. For the Ni based catalysts, it was reported that the CO2 methanation reaction was initiated by the dissociation of CO2 into a carbon species (COads) and an oxygen species (Oads) over the catalyst surface and the COads subsequently reacted with H atom to generate CH4 on Ni active site, which was responsible for dissociating the H2 into H atom.27,57,58 The presence of the Mg basic modifier might intensify the formation of the COads species and suppress the desorption process of COads into CO, which accounted for the relatively higher CH4 selectivity over OMA-10Ni5Mg catalyst. Besides, the CH4 selectivity was also closely related with the state of the metallic Ni active site. If the thermal sintering of the Ni active sites took place, the H2 dissociation into Hads would be blocked, which would also cause the decrease of the CH4 selectivity due to lack of Hads source. Therefore, the lower CH4 selectivity over 10Ni/Al2O3 catalyst than the other mesoporous catalysts at high temperature above 300 °C might be caused by the thermal sintering of the metallic Ni active sites.
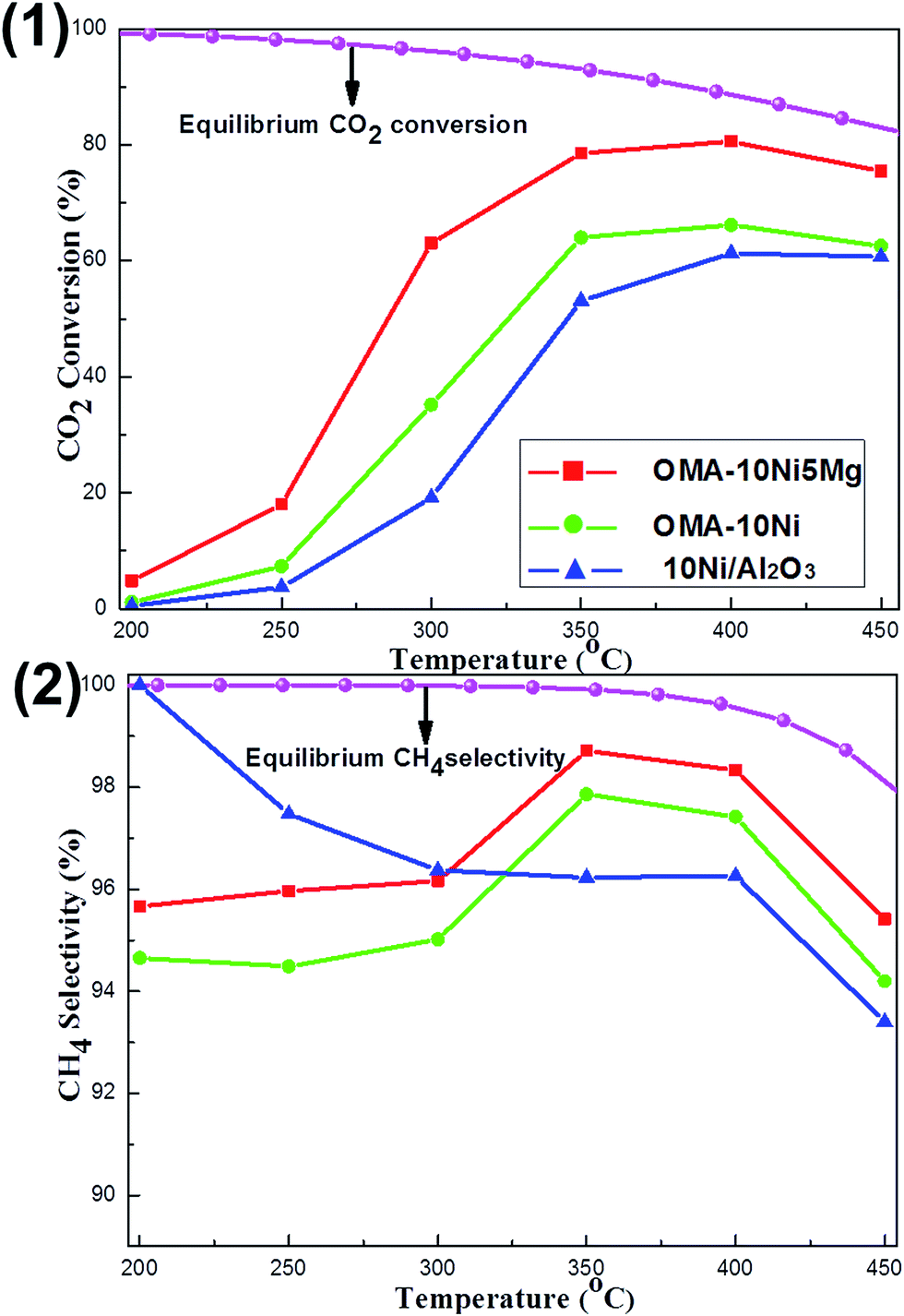 | ||
| Fig. 6 The curves of the (1) CO2 conversion and (2) CH4 selectivity versus reaction temperature over OMA-10NixMg and 10Ni/Al2O3 catalysts; reaction condition: H2/CO2 = 4, GHSV = 15000 mL g−1 h−1, 1 atm. | ||
The development of the Ni based catalyst with long-term catalytic stability was also an important concern because the thermal sintering of the Ni active sites easily took place. Therefore, the 50 h lab-scale long term stability tests were carried out under given conditions (H2/CO2 = 4, 400 °C, GHSV = 15000 mL (g−1 h−1), 1 atm). The OMA-10Ni, OMA-10Ni3Mg, OMA-10Ni5Mg, and 10Ni/Al2O3 catalysts were chosen as the representatives for the stability tests and their catalytic performances were summarized in Fig. 7. As shown in the Fig. 7(1), there was no obvious deactivation observed over OMA-10Ni, OMA-10Ni3Mg, and OMA-10Ni5Mg mesoporous catalysts after 50 h stability tests, displaying greatly excellent catalytic stabilities. This suggested that the deactivation caused by the thermal sintering of the Ni active sites had been successfully avoided owing to the confinement effect of the mesoporous framework, which could stabilize the metallic Ni nanoparticles by the mesoporous framework. In contrast, the CO2 conversion over traditional 10Ni/Al2O3 supported catalyst gradually decreased from 61.2% to 55.3% during the whole 50 h stability test, which ought to be caused by the thermal sintering of the metallic Ni active sites due to weak metal-support interaction. Furthermore, it was noticeable that the CO2 conversions over OMA-10Ni3Mg (76.3%) and OMA-10Ni5Mg (80.3%) were still higher than that over OMA-10Ni (67.8%), once more demonstrating the positive roles of alkaline promotors in enhancing the catalytic activity. Besides, the catalytic performances related with CH4 selectivity were depicted in Fig. 7(2). Similar to the CO2 conversions, the corresponding CH4 selectivity over OMA-10Ni, OMA-10Ni3Mg, and OMA-10Ni5Mg catalysts kept stable during the whole 50 h time on stream. However, the general trend of the CH4 selectivity over 10Ni/Al2O3 supported catalyst was descendent because of thermal sintering of the metallic Ni active sites. As for the CH4 selectivity, it was also very sensitive to the thermal sintering of the metallic active sites based on the previous reports, which were responsible for the dissociation of the H2 into Hads.18,31,57 The thermal agglomeration of the metallic active sites would cause the deceleration of the H2 dissociation, finally leading to the decomposition of the COads into CO due to the lack of Hads.18,31 For the current OMA-10NixNi catalysts, the steadiness of their CH4 selectivity during the long-term stability tests was attributed to the enhanced thermal stability of the metallic Ni active sites, which were stabilized by the ordered mesoporous Al2O3 matrix. Therefore, compared with the 10Ni/Al2O3 supported catalyst, the OMA-10NixMg catalysts exhibited more excellent catalytic stabilities.
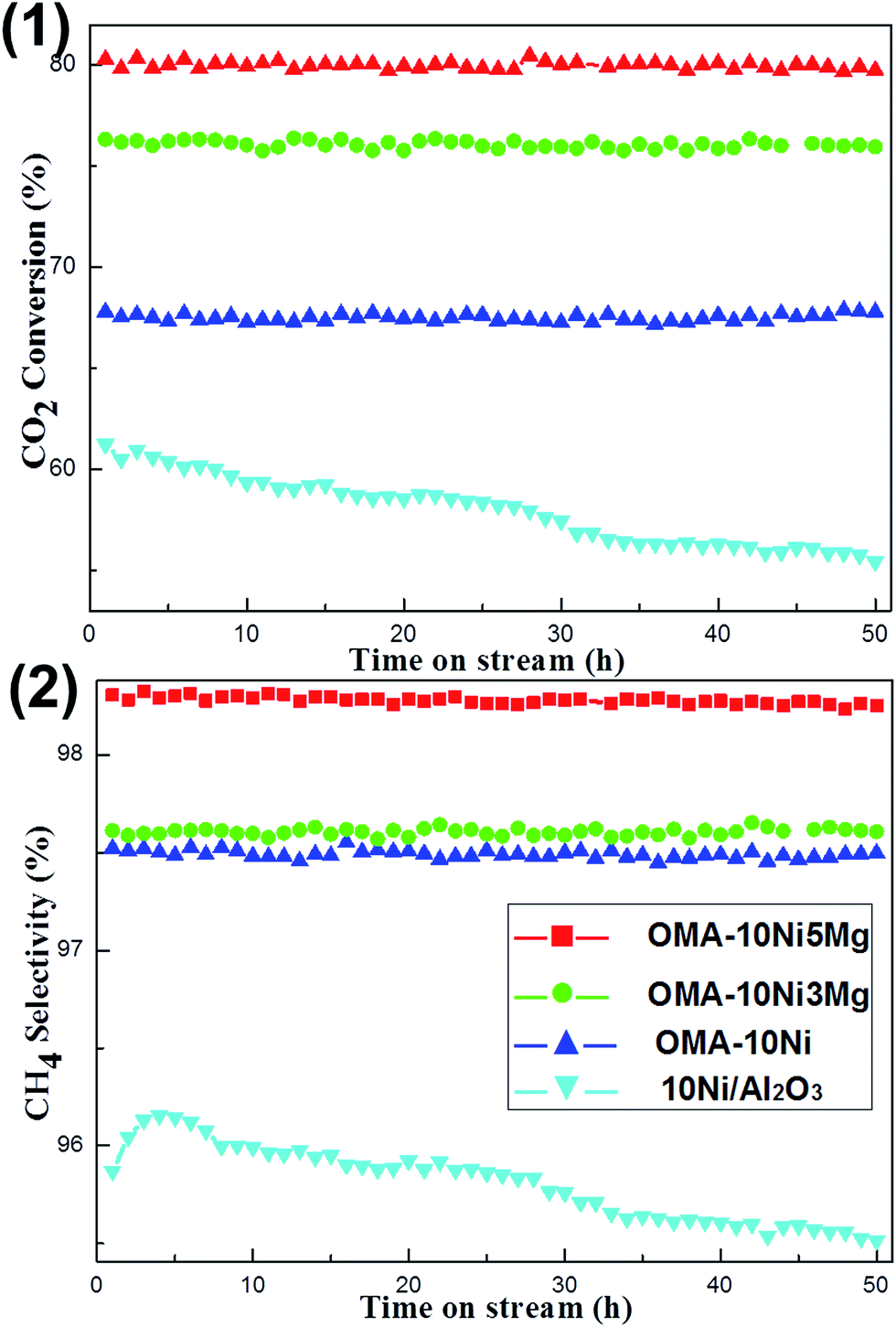 | ||
| Fig. 7 50 h long-term stability tests over OMA-10NixMg and 10Ni/Al2O3 catalysts: (1) CO2 conversion and (2) H2 selectivity; reaction conditions: H2/CO2 = 4, GHSV = 15000 mL g−1 h−1, 400 °C, 1 atm. | ||
Overall, the low-temperature catalytic activity over OMA-10NixMg catalysts had been obviously enhanced by the incorporation of Mg alkaline promotor by intensifying the surface basicity. Compared with conventional 10Ni/Al2O3 supported catalyst, these ordered mesoporous catalysts were provided with better structural properties, higher metallic Ni dispersion, and stronger metal-support interaction, which endowed them with more excellent catalytic performances. However, the obtained CO2 conversions over these OMA-10NixMg catalysts were still much lower than the chemical equilibrium value and had enough room to be improved. As for the Ni–Al based catalysts, except for the current mesoporous catalysts, the Ni–Al hydrotalcite catalysts recently had been extensively investigated for CO2 methanation due to their unique structural properties, where the Ni active sites could be highly dispersed among the layers of hydrotalcite crystalline structure.58–60 Abate et al. reported that the Ni–Al hydrotalcite catalysts (75–80 wt% NiO) could approximately approach the equilibrium CO2 conversion at the temperature above 300 °C with highly diluted feed gases (H2/CO2/N2 = 10/2.5/87.5, GHSV = 20000 h−1, 5 bar).59 Batte et al. reported a series (Ni, Mg, Al)-hydrotalcite-like catalysts (59 wt% Ni) for CO2 methanation with diluted feed gases (H2/CO2/Ar/N2 = 18.5/4.6/12.8/64.1, WHSV = 1100 mL (gcat min)−1).60 The maximum CO2 conversion of (74 ± 2)% could be achieved at the temperatures between 330 and 350 °C. Wierzbicki et al. fabricated (Ni, La, Mg, Al)-hydrotalcite derived catalysts (15 wt% Ni) for CO2 methanation with diluted feed gases (H2/CO2/Ar = 12/3/5, GHSV = 12000 h−1).58 The CO2 conversion of ca. 46.0% could be obtained over the catalysts with 2 and 4 wt% La at 250 °C owing to the enhancement of medium strength basic sites by La. Apparently, the Ni–Al hydrotalcite and (Ni, La, Mg, Al) catalysts displayed higher CO2 conversions at low temperature than the OMA-10NixMg catalysts (about 10.0 wt% Ni). However, these catalysts with different metallic Ni loading amounts were investigated under different reaction conditions. Therefore, these results could not be directly comparable with each other. The common advantages of Ni–Al hydrotalcite and OMA-10NixMg catalysts were that the Ni active sites were effectively confined and the sintering of the metallic Ni could be inhibited, which accounted for their good catalytic stabilities. Based on this concept, if the Ni–Al hydrotalcite based ordered mesoporous catalyst could be fabricated, its low-temperature catalytic activity and stability would be further improved because of their own unique advantages.
3.3 Characterization of the used catalysts
The XRD characterizations of the 50 h spent (denoted as SP) OMA-10Ni, OMA-10Ni3Mg, OMA-10Ni5Mg, and 10Ni/Al2O3 catalysts were carried out and their XRD patterns were exhibited in Fig. 8. As observed, the SP-10Ni/Al2O3 supported catalyst displayed much stronger metallic Ni diffraction peaks than the other SP-OMA-10NixMg catalysts. This illustrated that the OMA-10NixMg catalysts possessed much stronger sintering-resistant abilities than the 10Ni/Al2O3 supported catalyst owing to the confinement effect of the mesoporous framework. Thus, the metallic Ni active sites among the OMA-10NixMg catalysts could be effectively stabilized by the mesoporous Al2O3 matrix, accounting for no deactivation after 50 h long-term stability tests. Furthermore, the spent OMA-10NixMg catalysts also displayed characteristic diffraction peaks of metallic γ-Al2O3 and/or MgAl2O4 phases, which were different with their corresponding fresh samples. The appearance of the γ-Al2O3 and/or MgAl2O4 diffraction peaks over SP-OMA-10NixMg catalysts might be attributed to the pre-reduction procedure at high temperature (800 °C), which promoted the phase transformation from the amorphous to crystalline γ-Al2O3 and/or MgAl2O4 phases.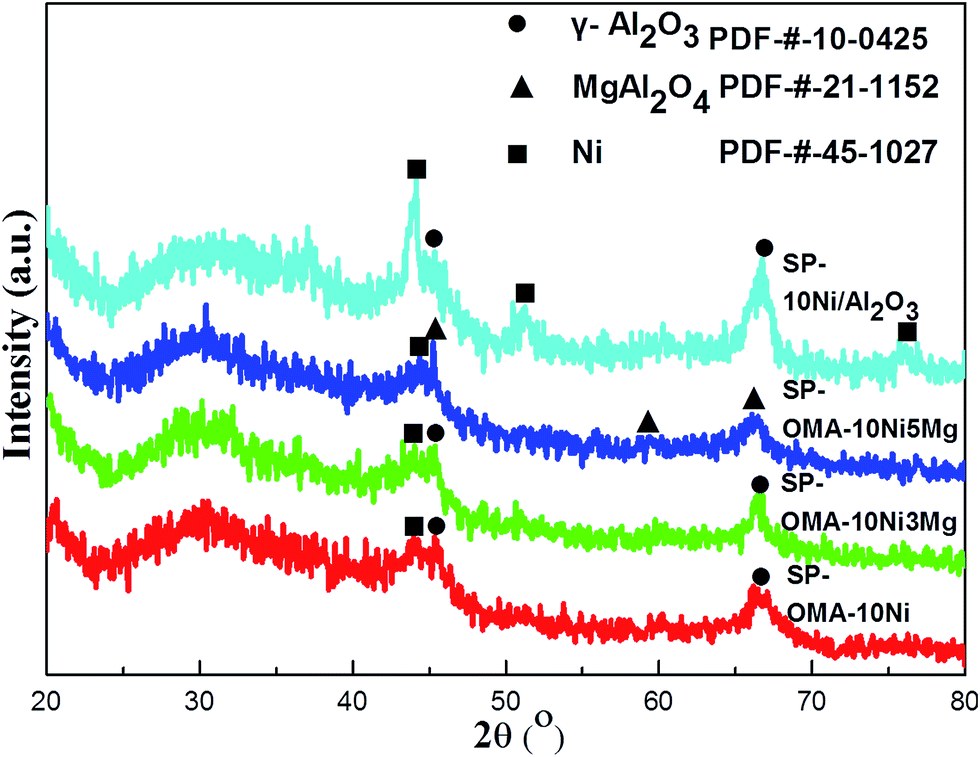 | ||
| Fig. 8 X-ray diffraction patterns of the 50 h spent OMA-10NixMg and 10Ni/Al2O3 catalysts. | ||
The nitrogen adsorption and desorption analyses were also carried out over the 50 h spent catalysts and their results were displayed in Fig. 9 and S2 (ESI†). As can be seen in Fig. 9(1), all the spent OMA-10NixMg catalysts still showed IV isotherms with steep H1-shaped hysteresis loops in the range of 0.5–0.9 P/P0. This indicated that the ordered cylindrical mesoporous channels had been successfully retained after experiencing the 2 h high temperature reduction at 800 °C and 50 h long-term stability tests at 400 °C. Furthermore, as can be seen in Fig. 9(2), their corresponding pore size distribution curves centered at 8.3–8.5 nm were very narrow. As for the SP-10Ni/Al2O3 catalyst, similar to its fresh precursor, the spent sample also displayed IV-H2 type isotherm and narrow pore size distribution curve in Fig. S2 (ESI†). Besides, as shown in the Table 1, the values of the specific surface areas and pore volumes of all the spent catalysts also suffered some decline. As for the SP-OMA-10NixMg catalysts, the decline was mainly derived from the thermal shrinkage of the mesoporous framework during the processes of reduction and reaction for these catalysts. But the thermal shrinkage had not cause the deformation and collapse of the ordered mesoporous skeletons according to their IV H1-shaped isotherms and narrow pore size distributions, demonstrating outstanding thermal stability. For the SP-10Ni/Al2O3 catalyst, the decrease in its surface area and pore volume might be mainly because of the thermal sintering of the metallic Ni, which would cause the blockage of the pore channel by large clusters. In addition, the Ni/Al and Mg/Al molar ratios (see Table 1) of the spent catalysts were also determined by ICP-AES. The values were comparable to their corresponding fresh catalysts, suggesting that the serious loss of the metallic Ni active sites did not occur.
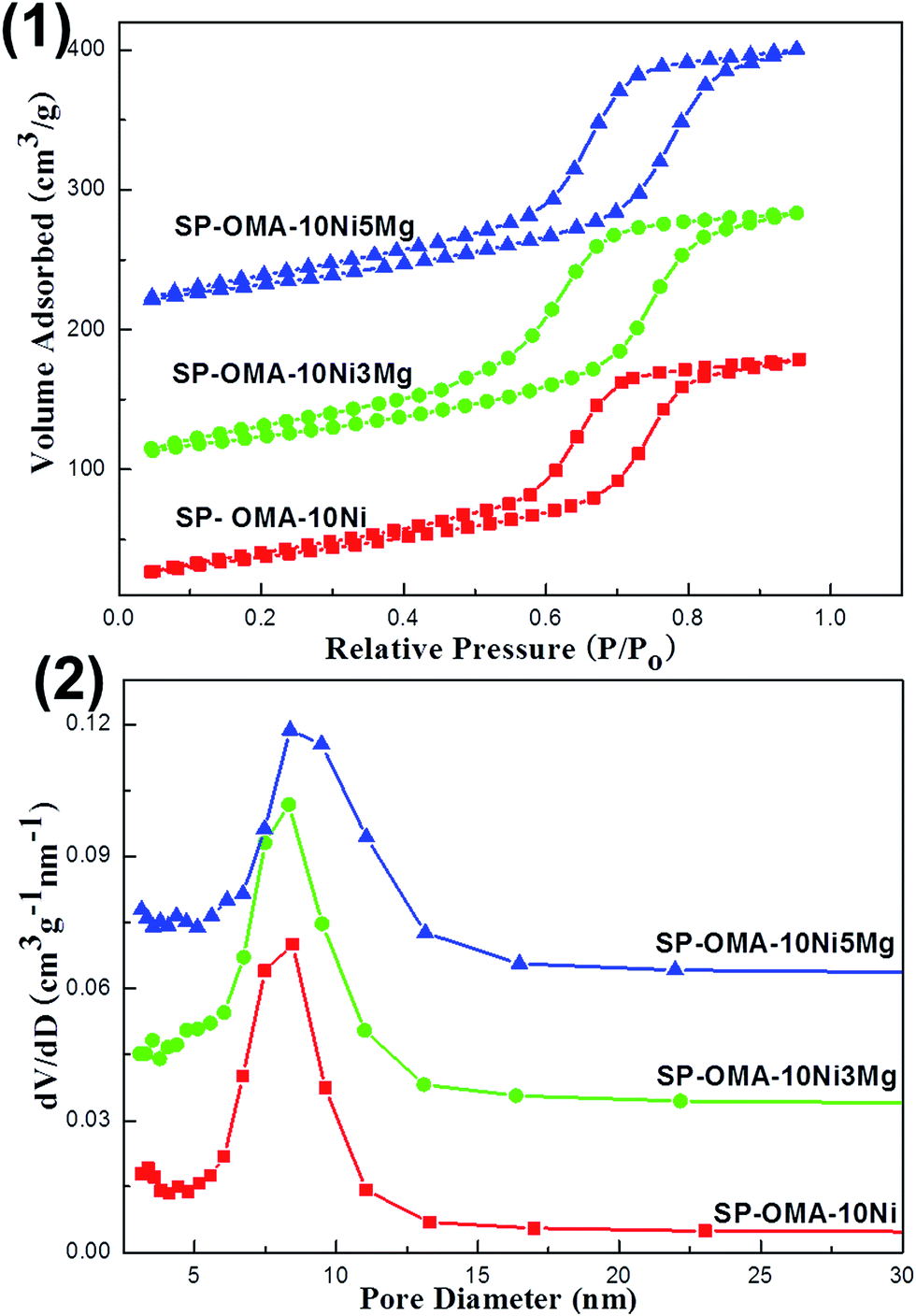 | ||
| Fig. 9 (1) Nitrogen adsorption–desorption isotherms and (2) pore size distribution curves of the 50 h spent OMA-10NixMg catalysts. | ||
TEM analyses of the 50 h spent catalysts were conducted to further investigate the thermal sintering property of the metallic Ni active sites and/or the morphology of the ordered mesoporous channels. As shown in the Fig. 10, the uniformly cylindrical mesopore channels viewed along the [1 1 0] direction could be observed over these 50 h SP-OMA-10NixMg catalysts, suggesting that the ordered mesoporous structures had been successfully retained after 50 h stability tests. Besides, it was worth noting that no obvious metallic Ni cluster could be observed over all the SP-OMA-10NixMg catalysts, demonstrating excellent thermal sintering-resistance property. In sharp contrast, as can be seen in the Fig. S5 (ESI†), the large metallic Ni clusters appeared over SP-10Ni/Al2O3 catalyst. This proved that serious thermal sintering of the metallic Ni active sites had taken place, which had been already confirmed by the XRD characterization in Fig. 8. Therefore, the current OMA-10NixMg catalysts displayed much better anti-sintering property than 10Ni/Al2O3 supported catalyst.
 | ||
| Fig. 10 TEM and SAED images of the 50 h spent OMA-10NixMg catalysts: (a and b) SP-OMA-10Ni, (c and d) SP-OMA-10Ni3Mg, (e and f) SP-OMA-10Ni5Mg. | ||
4. Conclusions
In summary, a series of ordered mesoporous NiO–MgO–Al2O3 composite oxides with different Mg contents had been facilely fabricated by one-pot EISA strategy. The obtained materials with outstanding textural properties were directly employed as the catalysts for CO2 methanation. The incorporation of the Mg alkaline promotor greatly enhanced the surface basicity, which was in favor of the chemisorption and activation of CO2. As a result, their low-temperature catalytic activity and CH4 selectivity toward CO2 methanation were promoted. The “volcano-shape curve” relationship between the catalytic activity and Mg contents was observed. It was found that only the appropriate Mg/Al molar ratio (5%) could maximally promote the catalytic activity. Besides, these alkaline promoted catalysts were also provided with enhanced catalytic stability owing to the confinement effect of the mesoporous framework. The Ni active site and Mg basic promotor were in situ embedded among the alumina matrix by the one-pot fabrication strategy, which could effectively prevent the metallic active sites from sintering. Owing to these favorable advantages, the ordered mesoporous NiO–MgO–Al2O3 composite oxides could be considered as a series of potential catalysts for CO2 methanation reaction.Acknowledgements
The authors sincerely acknowledge the financial support from National Natural Science Foundation of China (Grant No. 21503113, 21577065, 21503142, and 91543115), International ST Cooperation Program of China (2014DFA90780), Key Projects in the National Science & Technology Pillar Program of Jiangsu Province (BE2014602), Natural Science Foundation of Jiangsu Province (BZ201306) and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.References
- M. R. Raupach, G. Marland, P. Ciais, C. Le Quéré, J. G. Canadell, G. Klepper and C. B. Field, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10288–10293 CrossRef CAS PubMed.
- M. Aziz, A. Jalil, S. Triwahyono and A. Ahmad, Green Chem., 2015, 17, 2647–2663 RSC.
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- P. Nema, S. Nema and P. Roy, Renewable Sustainable Energy Rev., 2012, 16, 2329–2336 CrossRef.
- G. A. Meehl, W. M. Washington, W. D. Collins, J. M. Arblaster, A. Hu, L. E. Buja, W. G. Strand and H. Teng, Science, 2005, 307, 1769–1772 CrossRef CAS PubMed.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- W. Wei and G. Jinlong, Front. Chem. Sci. Eng., 2011, 5, 2–10 CrossRef.
- I. Omae, Catal. Today, 2006, 115, 33–52 CrossRef CAS.
- Q. Zhang, Y. Z. Zuo, M.-H. Han, J.-F. Wang, Y. Jin and F. Wei, Catal. Today, 2010, 150, 55–60 CrossRef CAS.
- G. Bonura, M. Cordaro, C. Cannilla, F. Arena and F. Frusteri, Appl. Catal., B, 2014, 152, 152–161 CrossRef.
- R. Zhou, N. Rui, Z. Fan and C. J. Liu, Int. J. Hydrogen Energy, 2016, 41, 22017–22025 CrossRef CAS.
- S. Schiebahn, T. Grube, M. Robinius, V. Tietze, B. Kumar and D. Stolten, Int. J. Hydrogen Energy, 2015, 40, 4285–4294 CrossRef CAS.
- Y. H. P. Zhang, Int. J. Hydrogen Energy, 2010, 35, 10334–10342 CrossRef CAS.
- S. Rönsch, J. Schneider, S. Matthischke, M. Schlüter, M. Götz, J. Lefebvre, P. Prabhakaran and S. Bajohr, Fuel, 2016, 166, 276–296 CrossRef.
- P. J. Lunde and F. L. Kester, Ind. Eng. Chem. Process Des. Dev., 1974, 13, 27–33 CAS.
- J. Xu, X. Su, H. Duan, B. Hou, Q. Lin, X. Liu, X. Pan, G. Pei, H. Geng, Y. Huang and T. Zhang, J. Catal., 2016, 333, 227–237 CrossRef CAS.
- S. Tada, O. J. Ochieng, R. Kikuchi, T. Haneda and H. Kameyama, Int. J. Hydrogen Energy, 2014, 39, 10090–10100 CrossRef CAS.
- C. Swalus, M. Jacquemin, C. Poleunis, P. Bertrand and P. Ruiz, Appl. Catal., B, 2012, 125, 41–50 CrossRef CAS.
- J. N. Park and E. W. McFarland, J. Catal., 2009, 266, 92–97 CrossRef CAS.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono, R. R. Mukti, Y. H. Taufiq-Yap and M. R. Sazegar, Appl. Catal., B, 2014, 147, 359–368 CrossRef CAS.
- H. H. Shin, L. Lu, Z. Yang, C. J. Kiely and S. McIntosh, ACS Catal., 2016, 6, 2811–2818 CrossRef CAS.
- M. Guo and G. Lu, React. Kinet., Mech. Catal., 2014, 113, 101–113 CrossRef CAS.
- G. Zhou, T. Wu, H. Xie and X. Zheng, Int. J. Hydrogen Energy, 2013, 38, 10012–10018 CrossRef CAS.
- J. Liu, C. Li, F. Wang, S. He, H. Chen, Y. Zhao, M. Wei, D. G. Evans and X. Duan, Catal. Sci. Technol., 2013, 3, 2627–2633 CAS.
- Q. Pan, J. Peng, S. Wang and S. Wang, Catal. Sci. Technol., 2014, 4, 502–509 CAS.
- S. Rahmani, M. Rezaei and F. Meshkani, J. Ind. Eng. Chem., 2014, 20, 1346–1352 CrossRef CAS.
- W. Zhen, B. Li, G. Lu and J. Ma, Chem. Commun., 2015, 51, 1728–1731 RSC.
- J. Gao, Q. Liu, F. Gu, B. Liu, Z. Zhong and F. Su, RSC Adv., 2015, 5, 22759–22776 RSC.
- C. K. Vance and C. H. Bartholomew, Appl. Catal., 1983, 7, 169–177 CrossRef CAS.
- G. Du, S. Lim, Y. Yang, C. Wang, L. Pfefferle and G. L. Haller, J. Catal., 2007, 249, 370–379 CrossRef CAS.
- L. Xu, F. Wang, M. Chen, J. Zhang, K. Yuan, L. Wang, K. Wu, G. Xu and W. Chen, RSC Adv., 2016, 6, 28489–28499 RSC.
- N. Perkas, G. Amirian, Z. Zhong, J. Teo, Y. Gofer and A. Gedanken, Catal. Lett., 2009, 130, 455–462 CrossRef CAS.
- M. Guo and G. Lu, Catal. Commun., 2014, 54, 55–60 CrossRef CAS.
- M. Guo and G. Lu, RSC Adv., 2014, 4, 58171–58177 RSC.
- G. Zhi, X. Guo, Y. Wang, G. Jin and X. Guo, Catal. Commun., 2011, 16, 56–59 CrossRef CAS.
- D. L. Trimm, Catal. Today, 1999, 49, 3–10 CrossRef CAS.
- M. S. Duyar, A. Ramachandran, C. Wang and R. J. Farrauto, J. CO2 Util., 2015, 12, 27–33 CrossRef CAS.
- O. Görke, P. Pfeifer and K. Schubert, Catal. Today, 2005, 110, 132–139 CrossRef.
- K. Tomishige, Y.-g. Chen and K. Fujimoto, J. Catal., 1999, 181, 91–103 CrossRef CAS.
- B. Liu and C. Au, Catal. Lett., 2003, 85, 165–170 CrossRef CAS.
- C. J. Liu, J. Ye, J. Jiang and Y. Pan, ChemCatChem, 2011, 3, 529–541 CrossRef CAS.
- F. Wang, L. Xu and W. Shi, J. CO2 Util., 2016, 16, 318–327 CrossRef CAS.
- L. Xu, H. Song and L. Chou, Appl. Catal., B, 2011, 108, 177–190 CrossRef.
- W. Shen, K. Komatsubara, T. Hagiyama, A. Yoshida and S. Naito, Chem. Commun., 2009, 6490–6492 RSC.
- Q. Yuan, A. X. Yin, C. Luo, L. D. Sun, Y. W. Zhang, W. T. Duan, H. C. Liu and C. H. Yan, J. Am. Chem. Soc., 2008, 130, 3465–3472 CrossRef CAS PubMed.
- K. S. Sing, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- C. Li and Y. W. Chen, Thermochim. Acta, 1995, 256, 457–465 CrossRef CAS.
- D. Hu, J. Gao, Y. Ping, L. Jia, P. Gunawan, Z. Zhong, G. Xu, F. Gu and F. Su, Ind. Eng. Chem. Res., 2012, 51, 4875–4886 CrossRef CAS.
- M. Lenglet, A. D'Huysser, J. P. Bonelle, J. Dürr and C. K. Jørgensen, Chem. Phys. Lett., 1987, 136, 478–482 CrossRef CAS.
- L. Li, X. Wen, X. Fu, F. Wang, N. Zhao, F. Xiao, W. Wei and Y. Sun, Energy Fuels, 2010, 24, 5773–5780 CrossRef CAS.
- F. Wang, L. Xu, W. Shi, J. Zhang, K. Wu, Y. Zhao, H. Li, H. X. Li, G. Q. Xu and W. Chen, Nano Res., 2017, 10, 364–380 CrossRef CAS.
- V. García, J. J. Fernández, W. Ruíz, F. Mondragón and A. Moreno, Catal. Commun., 2009, 11, 240–246 CrossRef.
- K. Y. Koo, H. S. Roh, Y. T. Seo, D. J. Seo, W. L. Yoon and S. B. Park, Appl. Catal., A, 2008, 340, 183–190 CrossRef CAS.
- R. Espinal, E. Taboada, E. Molins, R. J. Chimentao, F. Medina and J. Llorca, Appl. Catal., B, 2012, 127, 59–67 CrossRef CAS.
- D. Wang, J. Jiang, H. F. Wang and P. Hu, ACS Catal., 2015, 6, 733–741 CrossRef.
- W. Luhui, S. Zhang and L. Yuan, J. Rare Earths, 2008, 26, 66–70 CrossRef.
- S. J. Choe, H. J. Kang, S. J. Kim, S. B. Park, D. H. Park and D. S. Huh, Bull. Korean Chem. Soc., 2005, 26, 1682–1688 CrossRef CAS.
- D. Wierzbicki, R. Debek, M. Motak, T. Grzybek, M. E. Gálvez and P. Da Costa, Catal. Commun., 2016, 83, 5–8 CrossRef CAS.
- S. Abate, K. Barbera, E. Giglio, F. Deorsola, S. Bensaid, S. Perathoner, R. Pirone and G. Centi, Ind. Eng. Chem. Res., 2016, 55, 8299–8308 CrossRef CAS.
- N. Bette, J. Thielemann, M. Schreiner and F. Mertens, ChemCatChem, 2016, 8, 2903–2906 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01673e |
| This journal is © The Royal Society of Chemistry 2017 |