
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of the acceptor pattern and substitution position on the properties of N-phenyl-carbazolyl based donor–acceptor–donor molecules†
Xuejing Songa,
Min Wangb,
Lingqian Kongc and
Jinsheng Zhao *d
*d
aDongguan University of Technology, Dongguan, 523808, P. R. China
bLiaocheng Pepole's Hospital, Liaocheng, 252000, P. R. China
cDongchang College, Liaocheng University, 252059, P. R. China
dDepartment of Chemistry, Liaocheng University, Liaocheng, 252059, P. R. China. E-mail: zhaojinsheng@lcu.edu.cn; Fax: +86-635-8539607; Tel: +86-635-8539607
First published on 24th March 2017
Abstract
Four optoelectronic compounds based on the same donor (N-phenylcarbazole) and different acceptors of benzothiadiazole or benzoselenadiazole were designed and synthesized to investigate the effects of the substitution position and effects of the acceptor heteroatom on the properties of N-phenyl-carbazolyl based organic optoelectronic compounds. The results demonstrated that these four compounds have band gaps (2.25 to 2.59 eV) that are lower than that of carbazole (3.20 eV). For the acceptor moiety, the replacement of benzothiadiazole with benzoselenadiazole was beneficial for the polarization of the D–A compound. Different acceptors substituted at the 3-position of N-phenylcarbazole led to molecules with a higher degree of planar conjugation than molecules derived from acceptors substituted at the 2-position. The increase in polarity and conjugation degree of the D–A compounds gave rise to lower band gaps and redshifts in the UV-vis absorption spectra and fluorescence emission spectra compared to the corresponding compounds. Solvatochromism effects were observed for these four compounds in different polar solvents, accompanied by redshifts in the fluorescence emission spectra, which ranged from 509 nm to 596 nm. Using thermogravimetric analysis, the decomposition temperatures of all four materials were found to be above 400 °C.
Introduction
Carbazole is an industrial raw material that is inexpensive and available in large quantities. Carbazole and its derivatives can be fused with many aromatic units to generate novel compounds,1 which have applications in a variety of fields, such as in photoelectric materials,2,3 dyes,4,5 medicines6 and biosensors.7 For carbazole, facile substituted modification on the 9′-nitrogen atom and optional link of the 2′- or 3′-sites enable the precise regulation of the band gaps of the resultant molecule.4 The important merits of carbazole derivatives include their high hole mobilities, perfect luminous performance, high molar extinction coefficients and capacity to undergo polymerization.8Donor (D)–acceptor (A)-type compounds usually have low band gaps due to the push–pull effect along the molecular skeleton; these molecules have become an increasingly important class of conjugated materials.9–15 Carbazole and its derivatives are frequently used as end-capping donor units due to their strong electron-donating abilities and the obtained molecules often have fluorescence properties.16–18 Przemyslaw Ledwon et al. synthesized a compound with a D–π–A–π–D structure employing carbazole as the D unit, benzothiadiazole (BTD) as the A unit, and 2-vinyl-3-hexyl-thiophene as the π linker; the molecule has a unique deep red color and is infrared emitting.8
As a strongly electron deficient unit, BTD is frequently used for the construction of D–A type conjugated molecules. Some of the resultant molecules have light-emitting properties and can be used as active materials in many fields, including heavy metal ion assay,14,15 biosensors and florescence imaging probes.18–20 Pei jian group prepared a series of D–A–D type oligomers taking BTD as the acceptor and oligo(thienylene vinylene)s with distinct lengths as the donor unit, which showed the broad absorption bands between 300–800 nm, and their emission bands were centered at approximately 650 nm.21 Sanjio S. Zade et al. reported three BTD derivatives with the D–A–D configuration and phenyl, thiophene and selenophene as the donor units. Respectively, the emissive colours of the three molecules were green (phenyl), orange (thiophene) and red (selenophene).22 In brief, the properties of D–A type materials depend on the proper combination of the A unit and D unit, as well as the substitution sites.23
Benzoselenadiazole (BSe) is an analogue of BTD, that is obtained by replacing an S atom with an Se.24 The selenium atom has a larger size and is less electronegative than sulphur.25 Therefore, BSe-containing compounds usually have red shifts in both absorption spectra and fluorescence-emitting spectra relative to the corresponding BTD-containing compounds.24,25 A series of dyes were designed and synthesized using 5-thienyl-BTD or 5-thienyl-BDS as the electron-deficient cores the fluorene derivatives as the D units. The resultant compounds had valuable NIR emission properties with high quantum yields.20 The replacement of the BTD unit with a BDS unit also shifted the absorption of the molecules toward longer wavelengths.20
There are no reports concerning the direct combination of N-phenylcarbazole with BTD for the construction of D–A–D-type materials; novel fluorescent dyes with a high performance could be anticipated for such a combination. The effects of the replacement of BTD by BSe on the fluorescence properties of the obtained D–A–D-type molecules also deserve study. Changes in the substitution positions on the carbazole derivative units would also have a direct influence on the properties of the resultant compounds and still need to be studied. Based on these considerations, we present in this study the synthesis of four D–A–D-type analogous molecules employing N-phenylcarbazole and benzothiadiazole (or benzoselenadiazole) as the D and A units, respectively, with the aim of exploring the effects of the substitution positions and heteroatom replacement within the acceptor units on the properties of the obtained substance.26 Their specific properties, including optical, redox and thermal stabilities, were studied in detail. Interestingly, the fluorescent dyes obtained emitted yellow, green and red colours upon irradiation, that is, the primary colours. Any colour can be made from these primary colours, which makes them important and leads them to have wide application prospects in the organic electroluminescent field. All of the compounds showed the solvatochromism effect in different polar solvents and had simultaneous high thermal stability and redox activities. As anticipated, the replacement of BTD by BSe as the acceptor unit could also reduce the band gap of the resultant molecules.
Experimental
Materials
2-Boric acid-N-phenylcarbazole (98%), 3-boric acid-N-phenylcarbazole (98%), 4,7-bibromine-2,1,3-benzothiadiazole (98%), sodium borohydride (NaBH4, 98%), sodium carbonate (Na2CO3, 98%), selenium dioxide (SeO2, 99%), tetra (triphenylphosphine)palladium (Pd(PPh3)4), tetra-n-butylammonium fluoride (TBAF), ethanol (EtOH, 99.9%), methylbenzene (AR), n-hexane (AR), trichloromethane (AR), N,N-dimethylformamide (DMF, AR), methanol (AR), dimethyl sulfoxide (AR) and anhydrous magnesium sulphate (AR) were all purchased from the Aldrich Chemical Reagent Company and used directly without further treatment. Tetrabutylammonium hexafluorophosphate (TBAPF6, 98%) was purchased from Alfa Aesar. Thin layer chromatography (TLC) silica gel (GF254) and column layer chromatography silica gel (200–300 meshes) were provided by Qingdao Marine Chemical Factory.Characterization
1H and 13C NMR were recorded using a Varian AMX 400 nuclear magnetic resonance spectrometer (CDCl3, 400 MHz), and tetramethylsilane was used as the internal standard substance. Elemental analysis was performed on a Thermo Finnigan Flash EA 1112 analyser. MALDI-TOF MS spectra were recorded using Bruker Daltonics Flexanalysis.UV-vis absorption spectra were collected by Varian Cary 5000 spectrometer. A serials of solvents including n-hexane, dichloromethane, trichloromethane, methylbenzene, tetrahydrofuran, acetonitrile, DMF, methanol and dimethyl sulfoxide were used as the solvent to prepare solutions. The D–A–D compounds, including 4,7-bis(9-phenyl-9H-carbazole-2-yl)benzo[c][1,2,5] thiadiazole (BPC-2BTD), 4,7-bis(9-phenyl-9H-carbazole-2-yl)benzo[c][1,2,5]selenadiazole (BPC-2BSe), 4,7-bis(9-phenyl-9H-carbazole-3-yl)thiadiazole (BPC-3BTD) and 4,7-bis (9-phenyl-9H-carbazole-3-yl)selenadiazole (BPC-3BSe), were dissolved in the above solvents to a respectively, with the determined concentration of 1 × 10−5 mol L−1. The prepared solutions were undertaken for UV-vis measurement with a scan range of 200–800 nm.
Fluorescence spectra were measured on a LS55 fluorescence photometer (Perkin Elmer). The fluorescence emission spectra of the compounds were measured using a 1 cm standard quartz cuvette with a slit width of 6.0 nm, appropriate excitation wavelength, a voltage of 700 V and a scanning speed of 300 nm min−1. The redox property of the compounds was studied using cyclic voltammetry method in a dry DCM solution containing 0.2 M TBAPF6. A three-electrode system was employed with a Pt wire (Φ 0.5 mm) as the working electrode, a Pt ring (Φ 0.5 mm) as the counter electrode, and a Ag/AgCl electrode as the reference electrode. The thermal stabilities of compounds were tested using a NETZSCH STA 499C TG-DSC tester under an N2 atmosphere, temperature range of 40–800 °C, and heating rate of 10 °C min−1. A Canon Power Shot A3000 IS was used to take pictures of the four compounds and its solutions and their solutions.
Synthesis of acceptor unit
The acceptor unit 4,7-dibromo-2,1,3-benzoselenadiazole was synthesized using the methods reported previously in the literature24–28 (Scheme 1(a)): 5.75 g (19.56 mM) of 4,7-dibromo-2,1,3-benzothiadiazole and 300 mL of ethanol were added into a 500 mL three-necked flask in a thermostat water bath. Under electromagnetic stirring at 30 °C, an excessive of 16 g (422 mM) NaBH4 was added to the above suspension in three portions over an interval of 8 h. Forty-eight hours later, the reaction was terminated by pouring the mixture directly into 500 mL of distilled water; abundant white floccules precipitated. The mixture was stirred mechanically for 5 min and filtered by decompression suction to afford white solid 3,6-dibromo-1,2-phenylenediamine. The solid was dried in a vacuum oven for 20 hours and reserved for further application. 1H NMR (CDCl3, 400 MHz, ppm): δ = 6.84 (s, 2H, Ar H), 3.89 (s, 4H). 13C NMR (CDCl3, δ, ppm): δ = 133.70, 123.24, 109.68.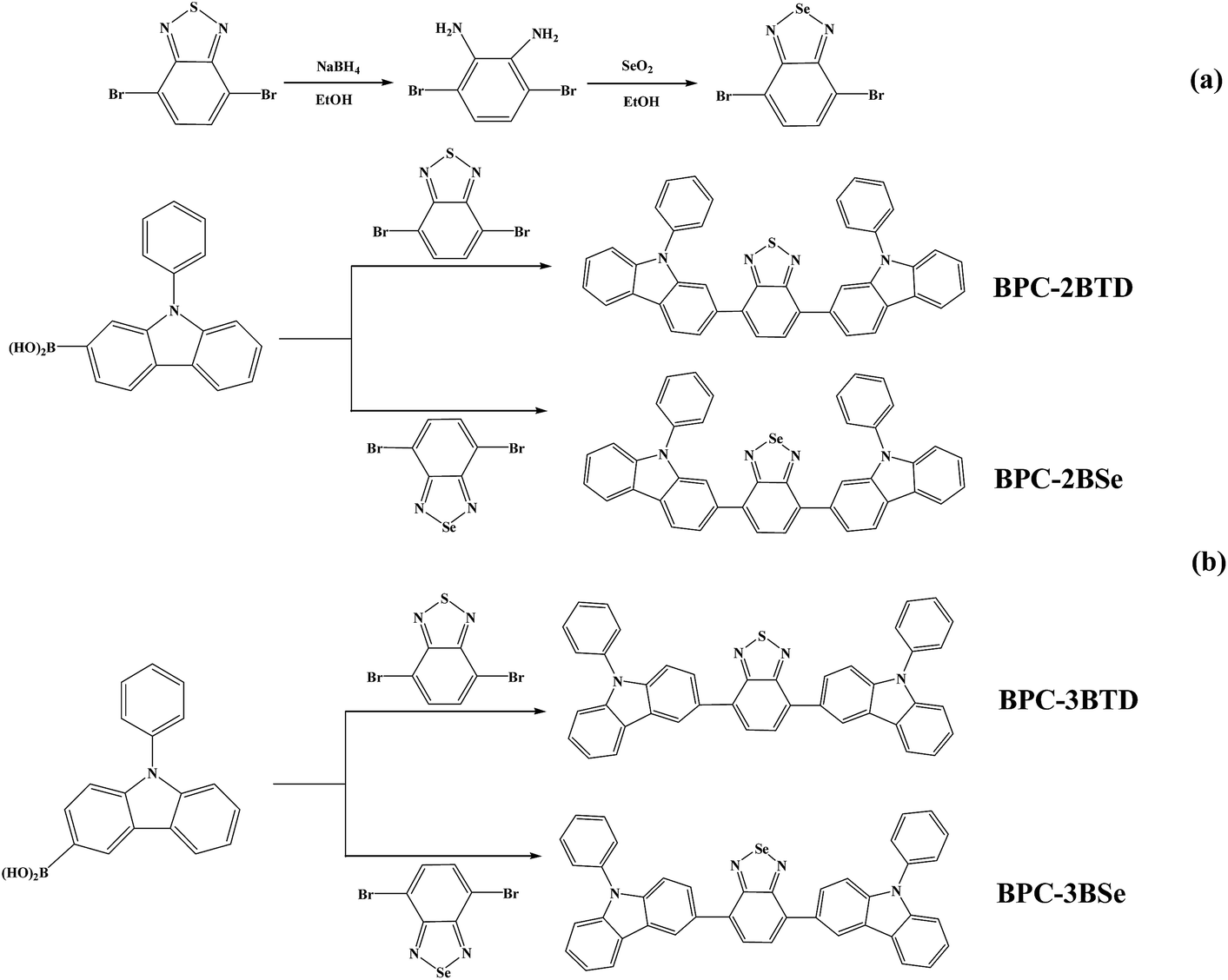 | ||
| Scheme 1 Synthetic routes of acceptor and four D–A–D compounds. | ||
To prepare 4,7-dibromo-2,1,3-benzoselenadiazole, 9.47 g (36.91 mM) 3,6-dibromo-1,2-phenylenediamine was dissolved in 200 mL ethyl alcohol in a flask, and the solution was heated to 90 °C in a water bath. At the same time, 4.2 g of SeO2 (37.85 mM) was dissolved into 50 mL hot water, and the solution was added to the reaction flask slowly. Yellow insoluble substances were produced. The mixture was refluxed for 2 h, and 4,7-dibromo-2,1,3-benzoselenadiazole was afforded from vacuum filtration.28 The yellow solid was then washed with ethanol three times, with water three times and dried under vacuum at 50 °C. 1H NMR (CDCl3, 400 M Hz, ppm): δ = 7.64 (s, 2H, Ar H). 13C NMR (CDCl3, δ, ppm): δ = 158.16, 144.00, 139.30.
Synthesis of D–A–D compounds
The synthetic routes are outlined in Scheme 1(b). Four molecules including BPC-2BTD, BPC-2BSe, BPC-3BTD and BPC-3BSe were synthesized using Suzuki coupling reactions.29 The specific synthetic process for BPC-2BTD was as follows: 3 g of (10.45 mM) 2-boric acid-N-phenylcarbazole, 1.53 g of (5.22 mM) 4,7-dibromo-2,1,3-benzoselenadiazole, 40 mL of (2 mol L−1) NaCO3 solution and 60 mL of methylbenzene (volume ratio of methylbenzene and NaCO3 was 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) were added into a round-bottom flask. Then, 0.6 g of Pd(PPh3)4 (molar mass of reactant was 5%) and 0.1 g of phase transfer agent TBAF were added to the flask. The flask was subjected to gas displacement until the atmosphere in the flask was replaced by argon gas. The flask was stirred magnetically under argon, heated to 120 °C and maintained for 48 h. After the reaction was completed, the solvent was removed under vacuum. The residue was dissolved in DCM, washed three times with water and dried over magnesium sulphate. The resulting crude product was obtained by distillation of the solvent, and purified using column chromatography (3:1, n-hexane, dichloromethane). Finally, 2.2 g of yellow powder was collected (yield of 68%), and termed BPC-2BTD. 1H NMR (CDCl3, 400 MHz, ppm): δ = 8.27 (d, 2H, ArH), 8.19 (d, 2H), 7.97 (d, 2H), 7.88 (d, 2H), 7.78 (s, 2H), 7.60 (m, 8H), 7.44 (m, 6H), 7.31 (m, 2H) (see Fig. S1a in ESI†). 13C NMR (CDCl3, δ, ppm): δ = 158.90, 146.13, 145.69, 142.16, 139.97, 138.42, 134.54, 132.96, 132.12, 131.77, 130.84, 128.00, 127.74, 126.07, 125.15, 124.96, 124.75, 115.27, 114.48 (see Fig. S1b in ESI†). MS (MALDI-TOF): m/z: calcd for 618.19; found 618.75. Elemental analysis calcd (%): C 81.53, H 4.24, N 9.05, S 5.18; found: C 81.35, H 4.27, N 9.13, S 5.25.
:2) were added into a round-bottom flask. Then, 0.6 g of Pd(PPh3)4 (molar mass of reactant was 5%) and 0.1 g of phase transfer agent TBAF were added to the flask. The flask was subjected to gas displacement until the atmosphere in the flask was replaced by argon gas. The flask was stirred magnetically under argon, heated to 120 °C and maintained for 48 h. After the reaction was completed, the solvent was removed under vacuum. The residue was dissolved in DCM, washed three times with water and dried over magnesium sulphate. The resulting crude product was obtained by distillation of the solvent, and purified using column chromatography (3:1, n-hexane, dichloromethane). Finally, 2.2 g of yellow powder was collected (yield of 68%), and termed BPC-2BTD. 1H NMR (CDCl3, 400 MHz, ppm): δ = 8.27 (d, 2H, ArH), 8.19 (d, 2H), 7.97 (d, 2H), 7.88 (d, 2H), 7.78 (s, 2H), 7.60 (m, 8H), 7.44 (m, 6H), 7.31 (m, 2H) (see Fig. S1a in ESI†). 13C NMR (CDCl3, δ, ppm): δ = 158.90, 146.13, 145.69, 142.16, 139.97, 138.42, 134.54, 132.96, 132.12, 131.77, 130.84, 128.00, 127.74, 126.07, 125.15, 124.96, 124.75, 115.27, 114.48 (see Fig. S1b in ESI†). MS (MALDI-TOF): m/z: calcd for 618.19; found 618.75. Elemental analysis calcd (%): C 81.53, H 4.24, N 9.05, S 5.18; found: C 81.35, H 4.27, N 9.13, S 5.25.
The synthesis of BPC-2BSe was the same as for BPC-2BTD, which was obtained as a fluffy yellow solid with a yield of 71%. 1H NMR (CDCl3, 400 MHz, ppm): δ = 8.26 (d, 2H, ArH), 8.18 (d, 2H), 7.86 (d, 2H), 7.80 (d, 2H), 7.62 (s, 2H), 7.58 (m, 8H), 7.43 (m, 6H), 7.30 (m, 2H) (see Fig. S2a in ESI†). 13C NMR (CDCl3, δ, ppm): δ = 159.88, 141.43, 140.96, 137.53, 135.95, 135.60, 129.86, 128.68, 127.43, 127.12, 126.11, 123.26, 123.12, 121.73, 120.47, 120.12, 120.06, 110.76, 109.80 (see Fig. S2b in ESI†). MS (MALDI-TOF): m/z: calcd for 666.13; found 666.17. Elemental analysis calcd (%): C 75.78, H 3.94, N 8.42, Se 11.86; found: C 75.56, H 4.00, N 8.48, Se 11.96.
BPC-3BTD was obtained as a the yellow solid with a yield of 72%, and the procedure used was the same as the above two compounds. 1H NMR (CDCl3, 400 MHz, ppm): δ = 8.75 (d, 2H, ArH), 8.25 (d, 2H), 8.04 (d, 2H), 7.85 (s, 2H), 7.61 (m, 8H), 7.55 (d, 2H), 7.46 (m, 6H), 7.33 (m, 2H) (see Fig. S3a in ESI†). 13C NMR (CDCl3, δ, ppm): δ = 154.52, 141.31, 140.75, 137.51, 133.21, 129.91, 129.58, 127.96, 127.55, 127.43, 127.06, 126.17, 123.70, 123.53, 121.20, 120.56, 120.16, 109.94, 109.83 (see Fig. S3b in ESI†). MS (MALDI-TOF): m/z: calcd for 618.19; found 618.33. Elemental analysis calcd (%): C 81.53, H 4.24, N 9.05, S 5.18; found: C 81.41, H 4.24, N 9.15, S 5.20.
Similarly, the fourth compound BPC-3BSe was produced as a reddish-orange solid in a yield of 68.7%. 1H NMR (CDCl3, 400 MHz, ppm): δ = 8.68 (d, 2H, ArH), 8.25 (d, 2H), 8.00 (q, 2H), 7.76 (s, 2H), 7.64 (m, 8H), 7.57 (d, 2H), 7.48 (m, 6H), 7.34 (m, 2H) (see Fig. S4a in ESI†). 13C NMR (CDCl3, δ, ppm): δ = 160.26, 141.30, 140.71, 137.53, 135.14, 130.19, 129.90, 128.40, 127.71, 127.55, 127.09, 126.11, 123.58, 123.50, 121.38, 120.53, 120.09, 109.90, 109.67 (see Fig. S4b in ESI†). MS (MALDI-TOF): m/z: calcd for 666.13; found 666.25. Elemental analysis calcd (%): C 75.78, H 3.94, N 8.42, Se 11.86; found: C 75.59, H 3.98, N 8.53, Se 11.90.
Results and discussion
Synthesis and characterization
The general synthetic method of acceptor unit 4,7-dibromo-2,1,3-benzoselenadiazole and four D–A–D compounds is shown in Scheme 1. In the ethanolic solution, 4,7-dibromo-2,1,3-benzothiadiazole reacted with NaBH4 to produce 3,6-dibromo-1,2-phenylenediamine at a yield of 75%. SeO2 was dissolved in hot water and added to an ethanol solution of 3,6-dibromo-1,2-phenylenediamine to produce the acceptor unit 4,7-dibromo-2,1,3-benzoselenadiazole (yield of 62%). Four analogous D–A–D compounds (BPC-2BTD, BPC-2BSe, BPC-3BTD and BPC-3BSe) were synthesized using Suzuki coupling reactions between 2-boric acid-N-phenylcarbazole (as electron donor) and 4,7-bibromine-2,1,3-benzothiadiazole or 4,7-dibromo-2,1,3-benzoselenadiazole (as electron acceptor) at a high yield. The four compounds exhibited high solubility in common solvents, including hexane, dichloromethane, chloroform, toluene and tetrahydrofuran, etc. The obtained compounds were confirmed by 1H NMR, 13C NMR spectroscopy, mass spectroscopy and elemental analysis.UV-vis absorption spectra
The UV-vis absorption spectra of four compounds in various solvents were analyzed; the results are shown in Fig. 1.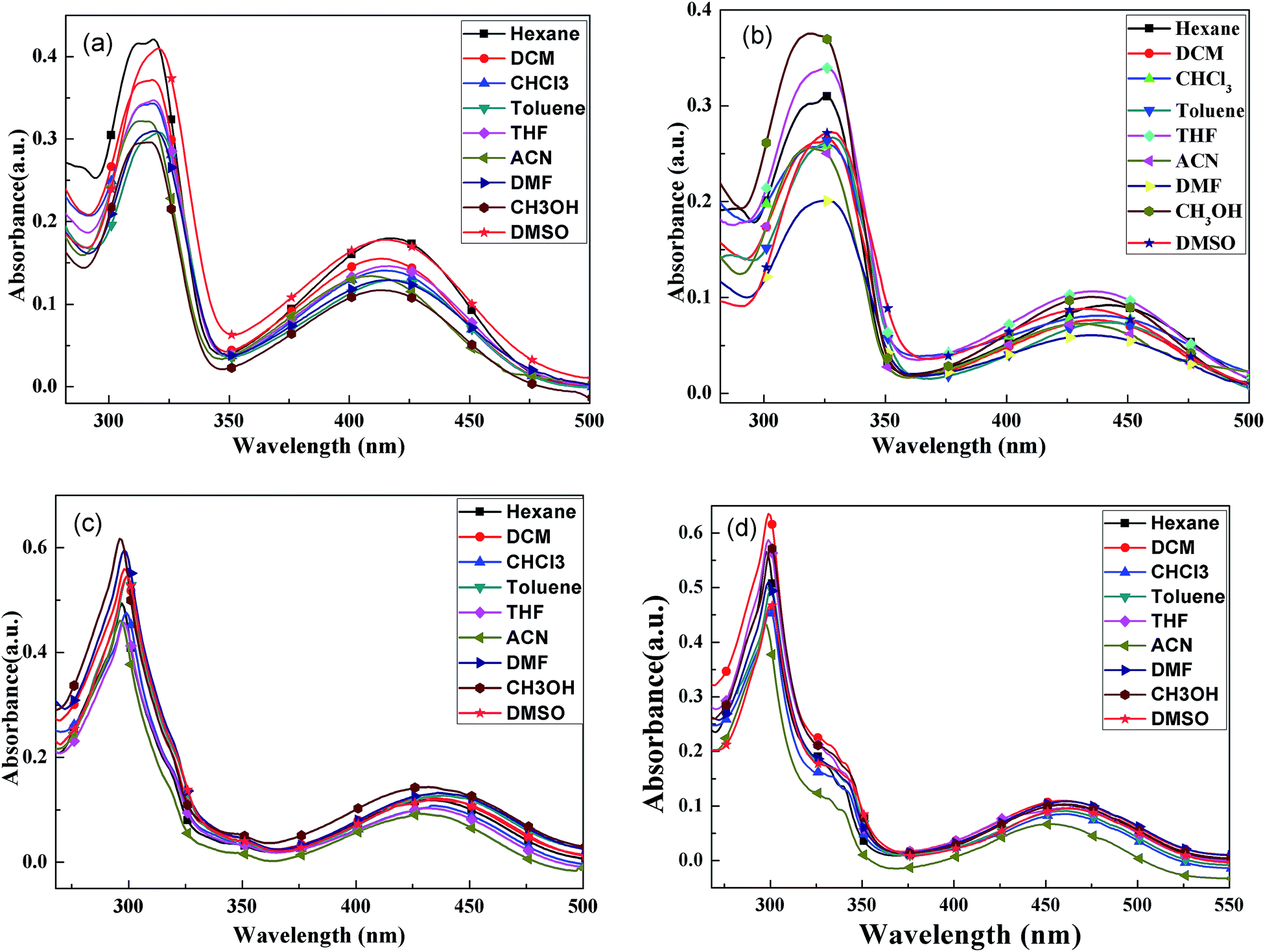 | ||
| Fig. 1 UV-vis absorption spectra of BPC-2BTD (a), BPC-2BSe (b), BPC-3BTD (c) and BPC-3BSe (d) in different solvents. | ||
In Fig. 1, all of the compounds showed two characteristic absorption bands in all of the solvents. There was one intense absorption at short wavelength and a second weak absorption at long wavelengths. The short wavelength absorption bands were attributed to π–π* transitions in the N-phenylcarbazole moieties, and the long wavelength absorption bands were assigned to the intramolecular charge-transfer (ICT) from the electron-donating N-phenylcarbazole moiety to the central electron-withdrawing group, that is BTD or BSe.24,25 The UV-vis absorption spectra of each compound in different solutions are similar, but slightly solvent-dependent. With increased solvent polarity, the absorption bands of four compounds showed slight redshifting (less than 10 nm), indicating a slight solvatochromic effect in the ground state of four compounds. The positions of the characteristic absorption peaks in Fig. 1 are given in Table 1.
| Solvent λmax/nm | BPC-2BTD | BPC-2BSe | BPC-3BTD | BPC-3BSe |
|---|---|---|---|---|
| Hexane | 317, 414 | 326, 438 | 297, 432 | 298, 459 |
| Dichloromethane | 318, 414 | 327, 438 | 298, 433 | 299, 460 |
| Chloroform | 318, 415 | 327, 437 | 299, 434 | 300, 461 |
| Toluene | 321, 417 | 329, 442 | 299, 439 | 300, 462 |
| Tetrahydrofuran | 318, 416 | 326, 436 | 298, 431 | 299, 460 |
| Acetonitrile | 317, 409 | 319, 432 | 296, 429 | 297, 453 |
| Dimethyl formamide | 319, 416 | 325, 436 | 298, 436 | 299, 462 |
| Methanol | 317, 413 | 319, 435 | 297, 432 | 300, 460 |
| Dimethyl sulfoxide | 321, 416 | 328, 441 | 300, 436 | 300, 463 |
The UV-vis absorption spectra of four compounds in dichloromethane are discussed as an example. The absorption maxima of BPC-2BTD were located at 318 nm and 414 nm, while those of BPC-2BSe were located at 327 nm and 438 nm. The two characteristic absorption bands of BPC-2BSe were redshifted by 9 nm and 24 nm compared with those of BPC-2BTD, respectively. The same phenomenon was observed between BPC-3BTD and BPC-3BSe. The Se atom is larger in size than the S atom and less electronegative than the S atom. Thus, the Se atom exerts a weaker binding force on external electrons.24–26 Therefore, the molecular polarization was intensified when the heteroatom S in the acceptor unit was replaced by Se. The excited state energies of BPC-2BSe and BPC-3BSe in a polar solvent were lower than those of BPC-2BTD and BPC-3BTD, respectively, leading to a redshift in the both high and low energy absorption bands.
The changes of the substitution positions may also have an effect on the absorption properties of the compounds. It was observed that the short wavelength absorption maximum of BPC-3BTD was blueshifted (20 nm) compared to that of BPC-2BTD, while the long wavelength absorption maximum was simultaneously redshifted (19 nm). The same phenomenon was observed between BPC-2BSe and BPC-3BSe. Two characteristic absorption bands moved in different directions, caused by their different transition mechanisms. The short wavelength absorption bands were mainly attributed to the π–π* transition of the N-phenylcarbazole unit in the D–A–D type molecule, whereas the low energy absorption bands were caused by the ICT between the donor and acceptor units.26
The 3-position substitution molecules including BPC-3BTD and BPC-3BSe, appear to have better coplanarity than their corresponding analogues BPC-2BTD and BPC-2BSe, as further confirmed by the quantum chemistry calculation method in the following section. The less twisted conjugationed structure of BPC-3BTD or BPC-3BSe as compared to their respective isomers BPC-2BTD or BPC-2BSe, resulted in more effective conjugation and stronger ICT interactions. Consequently, the electron cloud densities of the N-phenylcarbazole units substituted at the 3-position were reduced moderately more than the N-phenylcarbazole unit substituted at the 2-position. This led to a blue shift of the high energy absorption bands of BPC-3BTD or BPC-3BSe compared with their respective analogues. As mentioned above, the low-energy absorption bands were caused by ICT, and the stronger ICT within the 3-position-substituted molecules than their respective analogues (2-position) caused a redshift of low-energy absorption bands. Based on the low energy absorption edges (λonset) in the UV-vis absorption spectra, the optical band gaps (Eg, Eg = 1241/λonset) of BPC-2BTD, BPC-2BSe, BPC-3BTD and BPC-3BSe were calculated to be 2.59 eV, 2.45 eV, 2.48 eV and 2.33 eV, respectively. Furthermore, it was observed that the band gaps of the 3-position substitution products were slightly lower than those of the 2-position substitution products. According to the literature, the band gap of carbazole is 3.20 eV,30 and the D–A–D compounds reported in the present study achieved far lower band gaps than this value. This proves that building a D–A–D conjugation system by introducing acceptor units between Bi–N-phenylcarbazole is beneficial for reducing the band gap of carbazole derivatives.
Fluorescence emission spectra
The fluorescence emission spectra of BPC-2BTD, the fluorescence emission spectra of BPC-2BTD, BPC-2BSe, BPC-3BTD and BPC-3BSe in different solvents are shown in Fig. 2.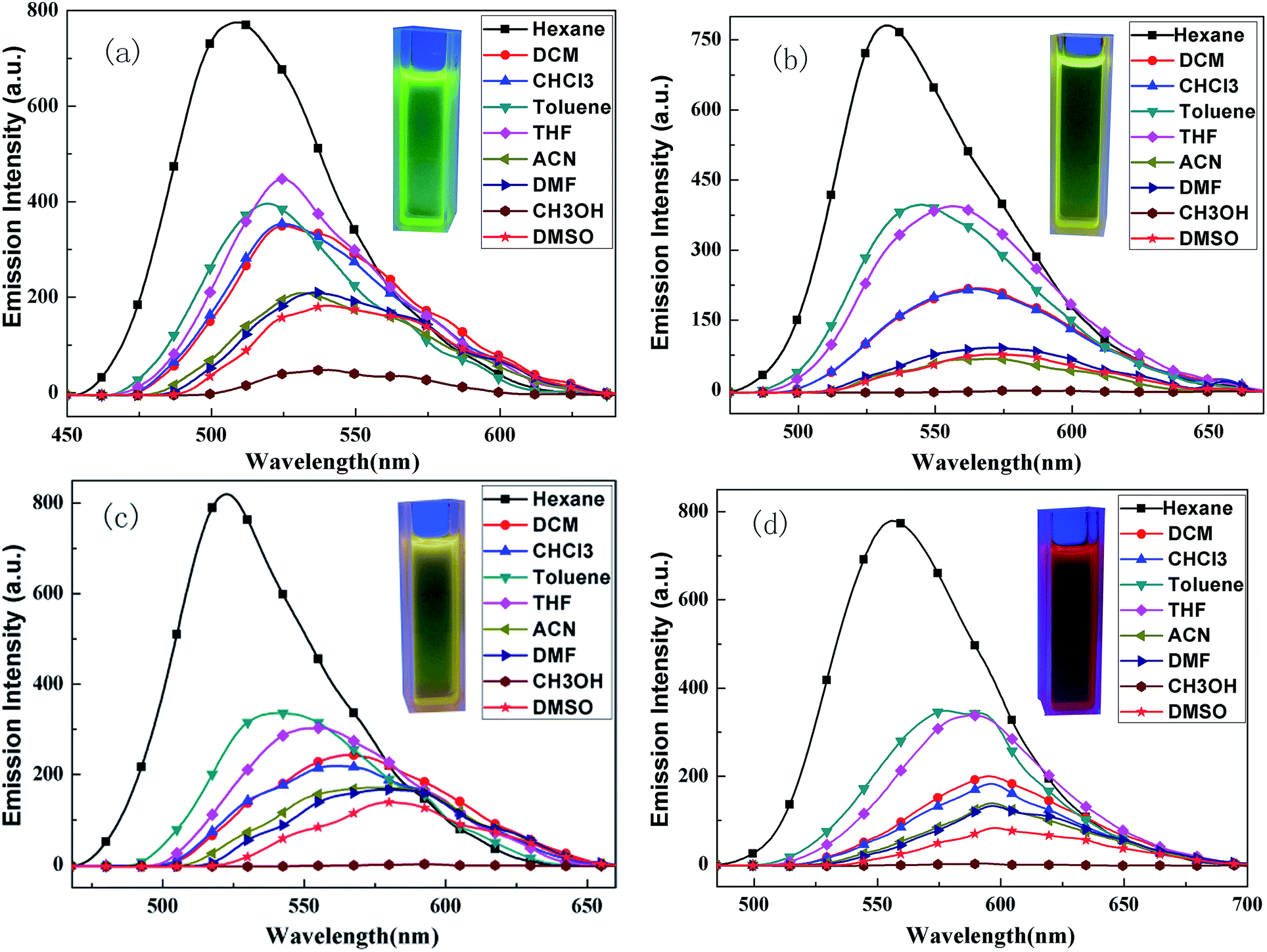 | ||
| Fig. 2 Fluorescence spectra of BPC-2BTD (a), BPC-2BSe (b), BPC-3BTD (c) and BPC-3BSe (d) in different solvents. Photos show DCM solutions of four D–A–D compounds illuminated by ultraviolet light at 360 nm. | ||
The fluorescence emission spectra of four compounds are similar with each other in different solvents. Unlike the UV-vis absorption spectra, all of the fluorescence emission curves of the D–A–D compounds demonstrate only one resolved ICT emission and showed a strong solvent dependence. With the increase in solvent polarity, the emission peaks exhibited large redshifts (ranging from 31 to 58 nm), displaying a significant solvatochromic effect.31–34 This phenomenon might be related to the inherent ICT in the D–A molecules. Many studies have shown that ICT was strengthened upon excitation, which increases the molecular dipole moment.30,31 The dipole moment in the excited state is greater than that of the molecule in the ground state. The increase in solvent polarity is beneficial for electron delocalization and the stabilization of the molecule in the excited state. The stability of the molecule in the excited state is enhanced by the increase in solvent polarity, thereby reducing the energy level of the excited state and making the energy gap between the excited state and ground state smaller. Thus, the fluorescence emission wavelength has a large redshift.35
Additionally, the fluorescence emission spectra of four compounds in methanol solvent were approximately straight lines without distinct emission peaks. This is because the hydrogen-bond interaction between the solvent and molecules quenched the fluorescence completely.36 The maxima emission wavelength data of the fluorescence emission spectra are listed in Table 2.
| Solvent λmax/nm | BPC-2BTD | BPC-2BSe | BPC-3BTD | BPC-3BSe |
|---|---|---|---|---|
| Hexane | 509 | 532 | 522 | 556 |
| Dichloromethane | 525 | 565 | 565 | 595 |
| Chloroform | 525 | 565 | 562 | 596 |
| Toluene | 520 | 545 | 540 | 577 |
| Tetrahydrofuran | 524 | 556 | 555 | 588 |
| Acetonitrile | 532 | 572 | 575 | 597 |
| Dimethyl formamide | 536 | 571 | 578 | 597 |
| Methanol | — | — | — | — |
| Dimethyl sulfoxide | 540 | 570 | 580 | 598 |
The fluorescence emission spectra of four compounds in dichloromethane are discussed in detail for the sake of contrast. The emission maxima of BPC-2BTD and BPC-2BSe were located at 525 nm and 565 nm, respectively. An apparent redshift of 40 nm was observed for BPC-2BSe relative to that of BPC-2BTD. Similarly, the emission band of BPC-3BSe had a redshift of 30 nm compared to that of BPC-3BTD. The cause of this phenomenon was similar to the redshift of the UV-vis absorption spectra. Replacing the heteroatom S with Se was beneficial for molecular polarization and resulted in a lower excited state energy in the polar solvent, leading to a lower band gap and a redshift of the emission wavelength. With the same acceptor unit but a different position of the substituent, the emission peak of BPC-3BTD was 565 nm, which had a redshift by 40 nm compared to BPC-2BTD. The same tendency was observed between BPC-3BSe and BPC-2BSe. This is consistent with the trend in their absorption spectra and originates from the much stronger ICT interactions in compounds BPC-3BTD and BPC-3BSe compared to their isomers, BPC-2BTD and BPC-2BSe.
Fluorescence yield
The compound 4-methylamino-7-nitro-2,1,3-benzodioxazole (NBD) was used as a reference and has fluorescence quantum yield (FQE) of 0.38 in acetonitrile.37,38 Based on this criterion, the FQE values of the four molecules were measured in dichloromethane. Under the same conditions (fluorescence scanning range: 300–700 nm; slit width: 6.0 nm; voltage: 700 V; scanning speed: 300 nm min−1), the fluorescence emission spectra of the test compounds and the reference were scanned and their integral fluorescence intensities were calculated. Then, the FQE values of different compounds were calculated from eqn (1). The results are shown in Table 3.
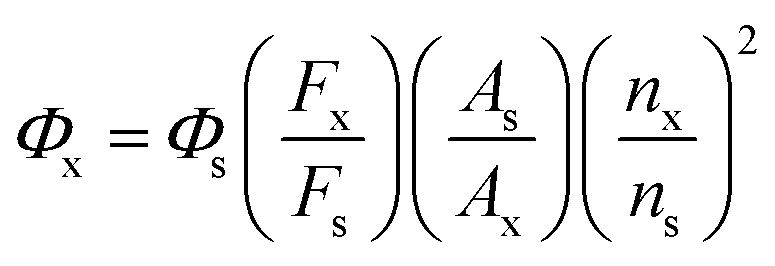 | (1) |
| Compounds | Φs | Fx | Fs | Ax | As | nx | ns | Φx |
|---|---|---|---|---|---|---|---|---|
| BPC-2BTD | 0.38 | 50893 |
38253 |
0.038 | 0.038 | 1.42 | 1.34 | 0.57 |
| BPC-2BSe | 0.38 | 23196 |
51861 |
0.040 | 0.040 | 1.42 | 1.34 | 0.19 |
| BPC-3BTD | 0.38 | 33083 |
51861 |
0.039 | 0.039 | 1.42 | 1.34 | 0.27 |
| BPC-3BSe | 0.38 | 8263 | 50541 |
0.037 | 0.037 | 1.42 | 1.34 | 0.07 |
Table 3 gives the FQE values of four compounds. BPC-2BSe had a lower fluorescence efficiency than BPC-2BTD. Similarly, the fluorescence efficiency of BPC-3BSe was lower than that of BPC-3BTD. The Se atom has a larger molecular weight than the S atom, which might exert a heavy atom effect on the energy level transitions.24,26 For example, when the singlet state in the excited state transforms into the triplet state, it cannot expediently return to the ground state through intersystem crossing due to the presence of the large Se atom. Therefore, the fluorescence efficiencies of the compounds using BSe as the acceptor were lower than for compounds using BTD as acceptor. By comparing the same acceptor pattern (BPC-3BTD and BPC-2BTD or BPC-3Be and BPC-2Be), substitution at the 2-position of N-phenylcarbazole had higher fluorescence luminous efficiencies than the molecules with a 3-position substitution pattern. This was caused by the large steric hindrance of substitution at the 2-position. Higher molecular rigidity is beneficial for increasing fluorescence efficiency.23–27
Electrochemical properties
The electrochemical properties of the compounds were tested by Cyclic Voltammetry (CV). CV curves of four compounds are presented in Fig. 3.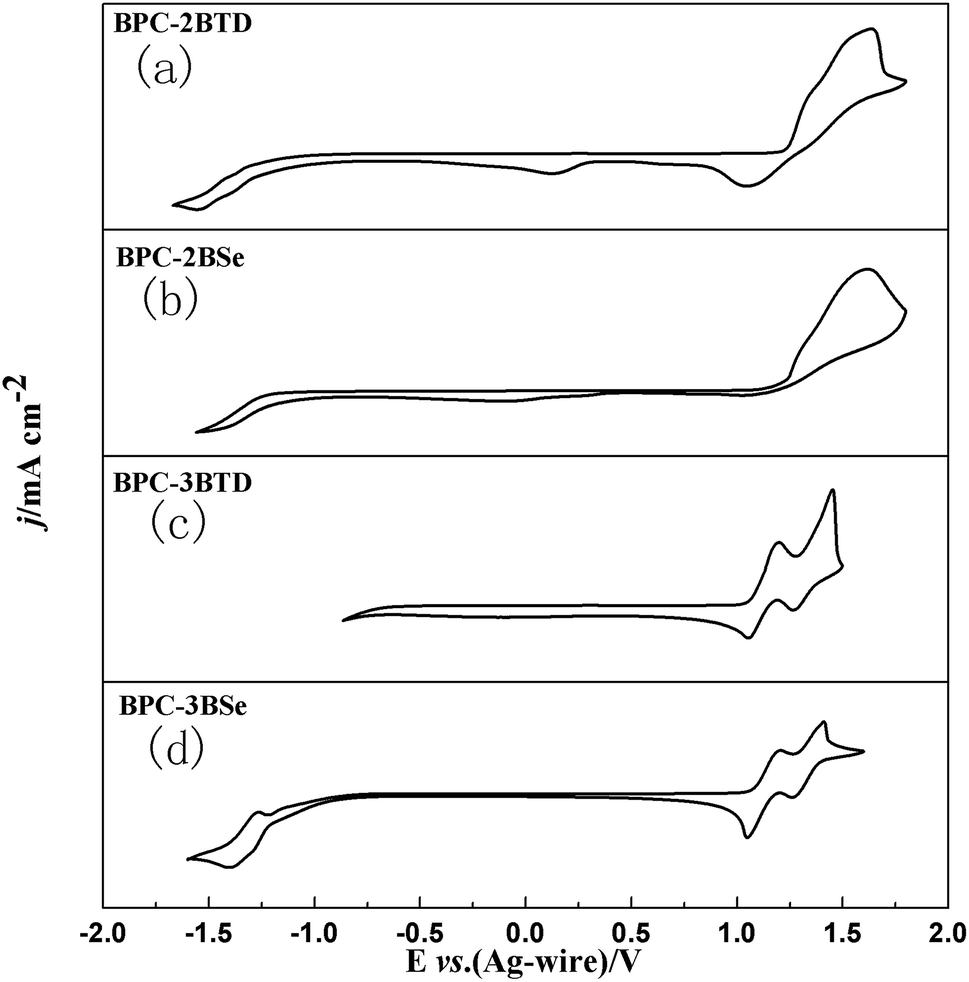 | ||
| Fig. 3 CVcurves of BPC-2BTD (a), BPC-2BSe (b), BPC-3BTD (c) and BPC-3BSe (d). | ||
For BPC-2BTD, there are two irreversible redox peaks located at 1.63 V/1.04 V and an additional weak reduction peak was observed at 0.12 V. There are no any apparent redox peaks observed in the negative scan up to −1.60 V. For BPC-2BSe, only one oxidation peak was observed at a potential of 1.62 V, and no obvious redox peaks were found in the negative scan. Interestingly, there are two groups of redox peaks for both BPC-3BTD and BPC-3BSe, located at 1.44 V/1.25 V, 1.18 V/1.02 V and 1.41 V/1.26 V and 1.18 V/1.01 V, respectively. It is worth mentioning that two redox peaks at −1.26 V/−1.39 V were found in the negative scan of BPC-3BSe. The different redox behaviours of four compounds suggested that the substitution position and the type of acceptor have greatly influence on the properties of the compounds. The 3-position substitution has much lower oxidation potentials than that of the 2-position analogues, suggesting a higher degree of conjugation in the former compounds than the latter. The onset oxidation potentials of the compounds were gained from the corresponding tangent lines of the CV curves shown in Fig. 3. The values were 1.22 V, 1.20 V, 1.06 V and 1.05 V for BPC-2BTD, BPC-2BSe, BPC-3BTD and BPC-3BSe, respectively. BPC-2BSe had a lower onset oxidation potential than that of BPC-2BTD. The same phenomenon was observed between BPC-3BTD and BPC-3BSe. BPC-3BSe had a lower onset oxidation potential than that of BPC-3BTD, which suggested a higher HOMO level for the former compound than the latter. The HOMO levels of the compounds could be drawn from the following formula, HOMO = −e(Eonset + 0.02 + 4.4),38 and the HOMO energy levels of BPC-2BTD, BPC-2BSe, BPC-3BTD, and BPC-3BSe were calculated as −5.64 eV, −5.62 eV, −5.48 eV and −5.47 eV, respectively. It was suggested that the HOMO levels of the D–A type molecules could be reflected by the HOMO levels of the donor unit, as confirmed by the comparable HOMO values of the four compounds, since they have the same donor unit. According to the formula: LUMO = HOMO + Eg,39,40 the LUMO energy levels of the four compounds were −3.05 eV, −3.17 eV, −3.00 eV and −3.14 eV, respectively, for BPC-2BTD, BPC-2BSe, BPC-3BTD and BPC-3BSe. Comparing the HOMO and LUMO values of four compounds, it was found that using BSe as the acceptor increased the energy levels of HOMO and decrease the energy levels of LUMO synchronously. Thus, the band gaps of the BSe-containing molecules are smaller than the BDT-containing analogues. Moreover, substitution of the acceptor at the 3-position of the donor increased HOMO values and decrease LUMO values synchronously, also leading to the reduction of the band gap of the accordingly obtained molecule. The data for the electrochemical and optical properties of four compounds were summarized in Table 4.
| Compounds | Eonset, vs. (Ag-wire) (V) | λmax (nm)/λonset (nm) | Ega (eV) | HOMOb (eV) | LUMOc (eV) | Egd (eV) | HOMOd (eV) | LUMOd (eV) |
|---|---|---|---|---|---|---|---|---|
| a Band gap calculated from λonset, Eg = 1241/λonset.b Calculated from onset oxidation potentials of the compounds, HOMO = −e(Eonset + 0.02 + 4.4).c Estimated using empirical equations LUMO = HOMO + Eg.d Eg, HOMO and LUMO are calculated based on density functional theory (DFT). | ||||||||
| BPC-2BTD | 1.22 | 318, 414/480 | 2.59 | −5.64 | −3.05 | 2.63 | −5.51 | −2.88 |
| BPC-2BSe | 1.20 | 327, 438/507 | 2.45 | −5.62 | −3.17 | 2.63 | −5.46 | −2.83 |
| BPC-3BTD | 1.06 | 298, 433/500 | 2.48 | −5.48 | −3.00 | 2.42 | −5.21 | −2.79 |
| BPC-3BSe | 1.05 | 299, 460/533 | 2.33 | −5.47 | −3.14 | 2.44 | −5.18 | −2.74 |
Theoretical calculation
To obtain more accurate structural information for the molecules, quantum chemical computations of the compounds were undertaken. The theoretical values of Egd, HOMOd and LUMOd of four compounds are given in Table 4, which were obtained at the B3LYP/6-31G level employing the Gaussian 03 program.23,26,35–38 The quantum chemical calculations were used to compare the theoretical values with the experimental results and to further understanding of the relationship between the molecule structure and its properties, as well as to gain a more intuitive display of the frontier orbital state of molecules. The HOMO and LUMO of the optimized molecular structures are shown in Fig. 4 (cartesian coordinates in ESI†). For optimized molecular structures, different substitution positions led to different planarities of the compounds with different HOMO and LUMO energy levels. Dihedral angles between the donor and acceptor are 31.78 and 32.95 for BPC-2BTD, 28.57 and 28.02 for BPC-3BTD, 34.92 and 33.87 for BPC-2BSe, and 31.23 and 30.65 for BPC-2BSe (Fig. 4). The introduction of the acceptor units (BTD or BSe) between the 3-position of two N-phenylcarbazole molecules generates a molecular system that has better coplanarity than the isomer obtained at the 2-position substitution. The distributions of π-electrons in the HOMO orbit of each of four molecules are mainly distributed on the N-phenylcarbazole donor unit, while the excited electron in the LUMO orbit of the four molecules was mainly distributed on the acceptor moieties, including benzothiadiazole and benzoselenadiazole. This indicates that ICT occurred from the ground state to the excited state.22,26 The substitution position influenced the band gap significantly, but the changes from BTD to BSe as the acceptor unit had only a slight influence on the band gaps. Additionally, the 3-position substitution increased the HOMO value of the obtained compound. The replacement of BTD by BSe increased the HOMO and LUMO levels simultaneously. The HOMO and LUMO values obtained from the theoretical calculation are comparable with the data from experiments (Table 1), which further validated the correctness of the predicted molecular configuration as shown in Fig. 4.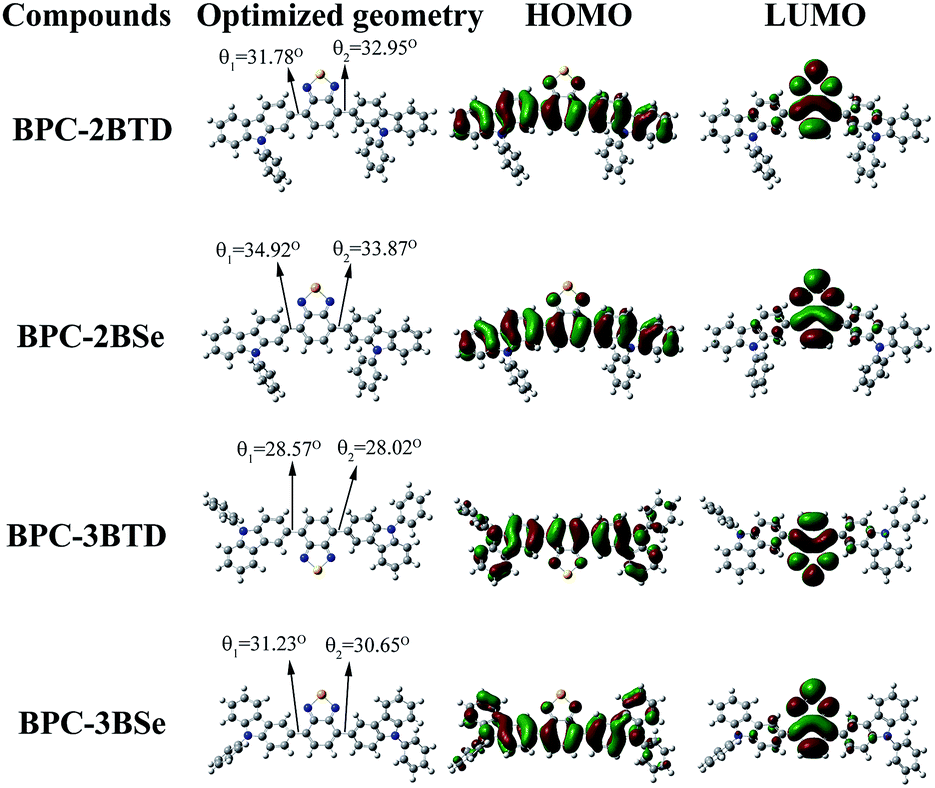 | ||
| Fig. 4 Molecular orbital diagrams of HOMO and LUMO of four compounds. | ||
Thermostability analysis
The thermal stabilities of four compounds were evaluated by thermogravimetric analysis (TGA). The TGA curves of BPC-2BTD is shown in Fig. 5, and the TGA curves of BPC-2BSe, BPC-3BTD and BPC-3BSe are presented in Fig. S5a–c in ESI.† These four compounds showed only a one-stage decomposition pattern, and the corresponding on-set degradation temperatures (Td) reached 428 °C, 415 °C, 438 °C, and 418 °C, respectively. Differential thermal gravity (DTG) curves are also shown in Fig. 5 and S5a–c,† from which the maximum degradation rates of four compounds occurred at 525 °C, 469 °C, 511 °C and 464 °C, respectively. The data revealed the good thermal stability and high temperature resistance of these four compounds. Weight loss in the case of BPC-2BTD and BPC-3BTD was nearly 90%, whereas in the case of BPC-2BSe and BPC-3BSe, the weight losses were approximately 70% at 800 °C. The residues of BPC-2BSe and BPC-3BSe remained at approximately 30% of their original weight, higher than that of BPC-2BTD and BPC-3BTD. This might be due to the formation of SeO2 during the decomposition of BPC-2BSe and BPC-3BSe. SeO2 does not evaporate, resulting in the increased observed residue in the thermograms.41–44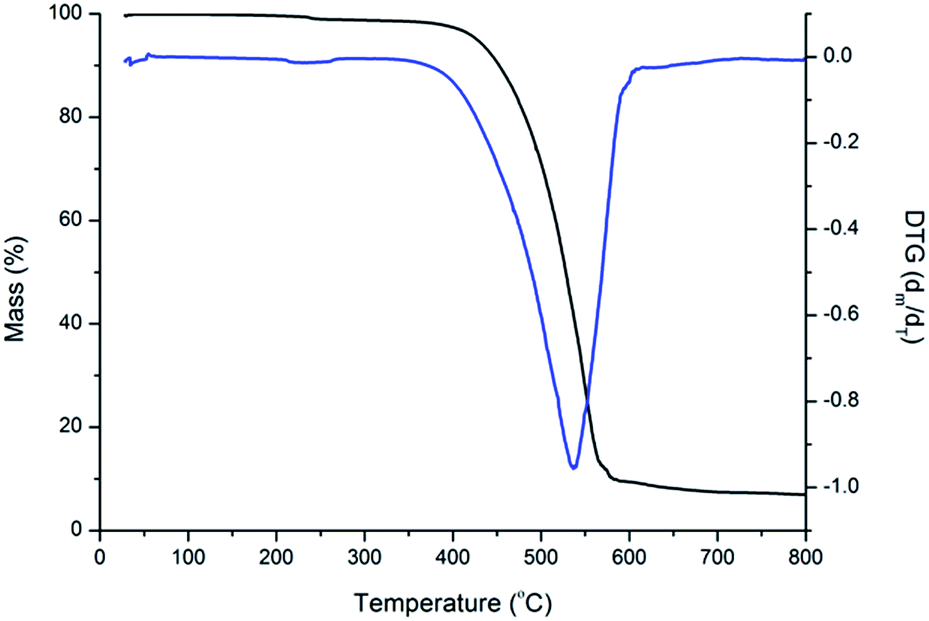 | ||
| Fig. 5 TG and DTA curve of BPC-2BTD. | ||
Conclusion
Four compounds (BPC-2BTD, BPC-2BSe, BPC-3BTD and BPC-3BSe) were synthesized based on Suzuki coupling reactions using benzocarbarzole as the donor and benzothiadiazole or benzoselenadiazole as the acceptor. The effects of the acceptor pattern and substitution position at the donor on the photoelectric properties of these compounds are discussed. The results demonstrate that substitution of the acceptor at the 3-position of N-phenylcarbazole is beneficial to the co-planarity of the obtained molecule reduced the band gaps of the molecules and caused a redshift in the emission spectra. At the same time, replacement of BTD by BSe as the acceptor unit could also reduce the band gap of the resultant molecule. All four compounds showed solvatochromism in different polar solvents. All four compounds had a high thermal stability and redox activities. In chloroform solution, BPC-2BTD, BPC-2BSe, BPC-3BTD and BPC-3BSe emitted green, yellowgreen, yellow and red lights upon irradiation, which are valuable primary colors. In summary, these four compounds are excellent luminescent materials and possess promising application prospects in the organic electroluminescence material fields.Acknowledgements
The work was financially supported by the National Natural Science Foundation of China (51473074, 31400044), the Special Funds for Outstanding Young Innovative Talent Training plan of Guangdong Province (Seedling Project, 2013LYM_0105), and the Training program for Outstanding Young Teachers in Higher Education Institutions of Guangdong Province.Notes and references
- J. Peng, K. Q. Ye, J. B. Sun, Y. Zhan, J. H. Jia, P. C. Xue, G. H. Zhang, Z. Q. Zhang and R. Lu, Dyes Pigm., 2015, 116, 36–45 CrossRef CAS.
- D. Jiang, S. H. Chen, Z. Xue, Y. J. Li, H. B. Liu, W. S. Yang and Y. L. Li, Dyes Pigm., 2016, 125, 100–105 CrossRef CAS.
- L. Han, X. Y. Zu, Y. H. Cui, H. B. Wu, Q. Ye and J. R. Gao, Org. Electron., 2014, 15, 1536–1544 CrossRef CAS.
- R. D. Telore, A. G. Jadhav and N. Sekar, J. Lumin., 2016, 179, 420–428 CrossRef CAS.
- B. Huang, Z. H. Yin, X. X. Ban, W. Jiang, Y. Dai, J. Y. Zhang, Y. Y. Liu, Y. P. Yang and Y. M. Sun, Dyes Pigm., 2015, 117, 141–148 CrossRef CAS.
- E. Wincent, A. Kubota, A. T. Laragy, M. E. Jönsson, M. E. Hahn and J. Stegeman, Biochem. Pharmacol., 2016, 110, 117–129 CrossRef PubMed.
- L. Fan, S. Q. Gao, Z. B. Li, W. F. Niu, W. J. Zhang, S. M. Shuang and C. Dong, Sens. Actuators, B, 2015, 221, 1069–1076 CrossRef CAS.
- P. Ledwon, P. Zassowski, T. Jarosz, M. Lapkowski, P. Wagner, V. Cherpak and P. Stakhira, J. Mater. Chem. C, 2016, 4, 2219–2227 RSC.
- X. Wu, Z. Q. Guo, Y. Z. Wu, S. Q. Zhu, T. D. James and W. H. Zhu, ACS Appl. Mater. Interfaces, 2013, 5, 12215–12220 CAS.
- B. A. D. Neto, P. H. P. R. Carvalho and J. R. Correa, Acc. Chem. Res., 2015, 48, 1560–1569 CrossRef CAS PubMed.
- P. C. Li, H. Tong, J. Q. Ding, Z. Y. Xie and L. Wang, J. Mater. Chem. A, 2013, 1, 8805–8812 CAS.
- M. Paramasivam, A. Gupta, A. M. Raynor and S. V. Bhosale, RSC Adv., 2014, 4, 35318–35331 RSC.
- P. C. Rodrigues, L. S. Berlim, D. Azevedo, N. C. S. Aavedra, P. N. Prasad, W. H. Schreiner, T. D. Z. Atvars and L. Akcelrud, J. Phys. Chem. A, 2012, 116, 3681–3690 CrossRef CAS PubMed.
- C. Saravanan, S. Easwaramoorthi, C. Y. Hsiow, K. Wang, M. Hayashi and L. Wang, Org. Lett., 2014, 16, 354–357 CrossRef CAS PubMed.
- J. F. Wu, G. Q. Lai, Z. F. Li, Y. X. Lu, T. H. Leng, Y. J. Shen and C. Y. Wang, Dyes Pigm., 2016, 124, 268–276 CrossRef CAS.
- P. Thongkasee, A. Thangthong, N. Janthasing, T. Sudyoadsuk, S. Namuangruk, T. Keawin, S. Jungsuttiwong and V. Promarak, ACS Appl. Mater. Interfaces, 2014, 6, 8212–8222 CAS.
- S. S. Palayangoda, X. Cai, R. M. Adhikari and D. C. Neckers, Org. Lett., 2008, 10, 281–282 CrossRef CAS PubMed.
- A. Kimoto and Y. Tajima, Org. Lett., 2012, 14, 2282–2285 CrossRef CAS PubMed.
- O. Çimen, H. Dinçalp and C. Varlikli, Sens. Actuators, B, 2015, 209, 853–863 CrossRef.
- S. Yao, B. Kim, X. L. Yue, M. Y. C. Gomez and M. V. Bondar, ACS Omega, 2016, 1, 1149–1156 CrossRef CAS.
- J. L. Wang, Q. Xiao and J. Pei, Org. Lett., 2010, 12(18), 4164–4167 CrossRef CAS PubMed.
- P. B. Pati and S. S. Zade, Cryst. Growth Des., 2014, 14, 1695–1700 CAS.
- S. Mondal, M. Konda, B. Kauffmann, M. K. Manna and A. K. Das, Cryst. Growth Des., 2015, 15, 5548–5554 CAS.
- S. Toksabay, S. O. Hacioglu, N. A. Unlu, A. Cirpan and L. Toppare, Polymer, 2014, 55, 3093–3099 CrossRef CAS.
- F. B. Koyuncu, E. Sefer, S. Koyuncu and E. Ozdemir, Polymer, 2011, 52, 5772–5779 CrossRef CAS.
- M. Akbayrak and A. M. Önal, Polym. Chem., 2016, 7, 6110–6119 RSC.
- Z. H. Guo, T. Lei, Z. X. Jin, J. Y. Wang and J. Pei, Org. Lett., 2013, 15, 3530–3533 CrossRef CAS PubMed.
- D. Baran, G. Oktem, S. Celebi and L. Toppare, Macromol. Chem. Phys., 2011, 212, 799–805 CrossRef CAS.
- E. J. Zhou, J. Z. Cong, K. Hashimoto and K. Tajima, Macromolecules, 2013, 46, 763–768 CrossRef CAS.
- F. F. Zhang, C. H. Zhou and J. P. Yan, Chin. J. Org. Chem., 2010, 30(6), 783–796 CAS.
- X. F. Lu, S. H. Fan, J. H. Wu, X. W. Jia, Z. S. Wang and G. Zhou, J. Org. Chem., 2014, 79, 6480–6489 CrossRef CAS PubMed.
- T. Ishi-i, M. Sakai and C. Shinoda, Tetrahedron, 2013, 69, 9475–9480 CrossRef CAS.
- J. L. Westrup, L. W. Oenning, M. M. S. Paula, R. C. Duarte, F. S. Rodembusch, T. E. A. Frizon, L. Silva and A. G. Dal-Bó, Dyes Pigm., 2016, 126, 209–217 CrossRef CAS.
- M. D. Damaceanu, C. P. Constantin and L. Marin, Dyes Pigm., 2016, 134, 382–396 CrossRef CAS.
- T. E. A. Frizon, J. C. V. Martínez, J. L. Westrup, R. C. Duarte, E. Zapp, K. G. Domiciano, F. S. Rodembusch and A. G. Dal-Bó, Dyes Pigm., 2016, 135, 26–35 CrossRef CAS.
- G. J. Huang, J. H. Ho, C. Prabhakar, Y. H. Liu, S. M. Peng and J. S. Yang, Org. Lett., 2012, 14, 5034–5037 CrossRef CAS PubMed.
- M. Onoda, S. Uchiyama, T. Santa and K. Imai, Anal. Chem., 2002, 74, 4089–4096 CrossRef CAS PubMed.
- S. Uchiyama, Y. Matsumura, A. P. de Silva and K. Iwai, Anal. Chem., 2003, 75, 5926–5935 CrossRef CAS PubMed.
- E. Sefer, H. Bilgili, B. Gultekin, M. Tonga and S. Koyuncu, Dyes Pigm., 2015, 113, 121–128 CrossRef CAS.
- J. K. Xu, H. T. Liu, S. Z. Pu, F. Y. Li and M. B. Luo, Macromolecules, 2006, 39, 5611–5616 CrossRef CAS.
- L. Benati, P. C. Montevecchi and G. Zanardi, J. Org. Chem., 1977, 42, 575–577 CrossRef CAS.
- S. Lu, M. Yang, J. Luo, Y. Cao and F. Bai, Macromol. Chem. Phys., 2005, 206, 664–671 CrossRef CAS.
- A. M. Alsalme, A. A. B. Alghamdi, A. A. G. Q. Alhamdani and A. Iraqi, Int. J. Electrochem. Sci., 2014, 9, 1920–1941 Search PubMed.
- S. Wen, J. Pei, Y. Zhou, P. Li, L. Xue and Y. Li, Macromolecules, 2009, 42, 4977–4984 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01449j |
| This journal is © The Royal Society of Chemistry 2017 |