
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficiency of hydrophobic phosphonium ionic liquids and DMSO as recyclable cellulose dissolution and regeneration media†
Ashley J. Holdingab,
Arno Parviainena,
Ilkka Kilpeläinena,
Ana Sotob,
Alistair W. T. King *a and
Héctor Rodríguez*b
*a and
Héctor Rodríguez*b
aDepartment of Chemistry, University of Helsinki, A.I. Virtasen Aukio 1, 00014 Helsinki, Finland. E-mail: alistair.king@helsinki.fi
bDepartamento de Enxeñería Química, Universidade de Santiago de Compostela, E-15782, Santiago de Compsotela, Spain. E-mail: hector.rodriguez@usc.es
First published on 20th March 2017
Abstract
Hydrophobic, long-chain tetraalkylphosphonium acetate salts (ionic liquids) were combined with a dipolar aprotic co-solvent, dimethylsulfoxide (DMSO), and the feasibility of these solvent systems for cellulose dissolution and regeneration was studied. A 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 w/w mixture of the ionic liquid tetraoctylphosphonium acetate ([P8888][OAc]) and DMSO was found to dissolve up to 8 wt% cellulose, whilst trioctyl(tetradecyl)phosphonium acetate ([P14888][OAc]) dissolved up to 3 wt% cellulose. Water (an anti-solvent for cellulose) was found to give rise to biphasic liquid–liquid systems when combined with these mixtures, yielding an upper phase rich in ionic liquid and a lower aqueous phase. The liquid–liquid equilibria of the ternary systems were experimentally determined, finding that DMSO strongly partitioned towards the aqueous phase. Thus, a process scheme involving simultaneous regeneration of cellulose and recycling of the solvent system was envisioned, and demonstrated on a large scale using [P8888][OAc]. A large portion of the ionic liquid (ca. 60 wt%) was directly recovered via phase separation, with a further 37 wt% being recovered from the swollen cellulose phase and residual materials, bringing recovery to 97%. XRD analysis of the recovered cellulose materials showed a loss of crystallinity and conversion from Cellulose I to Cellulose II. Non-dissolving compositions of ionic liquid and DMSO did not affect cellulose crystallinity after cellulose pulp treatment.
:40 w/w mixture of the ionic liquid tetraoctylphosphonium acetate ([P8888][OAc]) and DMSO was found to dissolve up to 8 wt% cellulose, whilst trioctyl(tetradecyl)phosphonium acetate ([P14888][OAc]) dissolved up to 3 wt% cellulose. Water (an anti-solvent for cellulose) was found to give rise to biphasic liquid–liquid systems when combined with these mixtures, yielding an upper phase rich in ionic liquid and a lower aqueous phase. The liquid–liquid equilibria of the ternary systems were experimentally determined, finding that DMSO strongly partitioned towards the aqueous phase. Thus, a process scheme involving simultaneous regeneration of cellulose and recycling of the solvent system was envisioned, and demonstrated on a large scale using [P8888][OAc]. A large portion of the ionic liquid (ca. 60 wt%) was directly recovered via phase separation, with a further 37 wt% being recovered from the swollen cellulose phase and residual materials, bringing recovery to 97%. XRD analysis of the recovered cellulose materials showed a loss of crystallinity and conversion from Cellulose I to Cellulose II. Non-dissolving compositions of ionic liquid and DMSO did not affect cellulose crystallinity after cellulose pulp treatment.
Introduction
Ionic liquids (ILs) are widely studied for their ability to dissolve cellulose and other lignocellulosic biopolymers,1–4 and have been applied as media for homogenous derivatization reactions,5–7 biomass pre-treatment,8–11 dissolution of wood and other biomass,12,13 biomass fractionation via extraction or selective precipitation,3,14–19 catalytic processing of lignocellulose,20,21 and spinning of regenerated cellulose fibres.22–25 Ionic liquids have been arbitrarily defined26 as salts which are liquid below 100 °C and most are composed of a large, organic cation and an inorganic or organic counter anion. The main basis of their interaction with cellulose and other biopolymers is the potential for hydrogen-bonding networks to be broken by hydrogen-bond basic27 anions, such as acetate ([OAc]−) and chloride (Cl−) anions. However, other inter-molecular forces including coulombic and dispersion interactions should also be considered, depending on the structures of the solvent and solute. Imidazolium-based ionic liquids are arguably the most studied both in general and with respect to their application towards cellulose and biomass, with ionic liquids such as 1-butyl-3-methylimidazolium chloride ([bmim]Cl),28 1-ethyl-3-methylimidazolium acetate ([emim][OAc]),29 and 1-ethyl-3-methylimidazolium dimethylphosphate ([emim][Me2PO4])27 being some examples.Organic electrolyte solutions30 or mixtures of ionic and molecular solvents for cellulose dissolution have been explored recently due to their ease of dissolution at low temperature, lowered viscosity, and cellulose dissolution efficiency.30–34 Typical combinations include imidazolium30,31,34–38 or onium cation39–42 ionic liquids paired with basic anions, mixed with DMSO (dimethylsulfoxide), DMI (1,3-dimethyl-2-imidazolidinone), DMA (N,N-dimethylacetamide), DMF (N,N-dimethylformamide), TMU (1,1,3,3-tetramethylurea),34 as well as γ-butyrolactone and γ-valerolactone.43
Recently, researchers have demonstrated the possibility of spinning regenerated cellulose fibres from ionic liquid solutions, such as in the ‘IONCELL-F’ process,24 based on Lyocell-type process, whereby strong fibres (having a higher tenacity than both the N-methylmorpholine-N-oxide (NMMO) based Lyocell and the sulphite based viscose fibres) can be produced by dry-jet wet (air-gap) spinning from an ionic liquid dope into a water bath. In this case, the cellulose dissolving ionic liquid 1,5-diazabicyclo[4.3.0]non-5-enium acetate ([DBNH][OAc]) dissolves around 13–14 wt% of cellulose pulp to form the spinning dope. Other ionic liquids have also been studied with respect to regenerated fibre spinning, such as [bmim]Cl23 and [emim][OAc].22 The latter showed favourable physical properties when compared to NMMO Lyocell. After the ionic liquid and cellulose solution is “spun” into water, the ionic liquid remains and must be removed from the spinning bath after a period of time. Water acts as an anti-solvent for cellulose by disrupting the hydrogen-bond network between the cellulose hydroxyl groups and the bound ionic liquid anion.
One issue in particular that has received a lot of attention in recent years has been the recovery and purification of ionic liquids. Ionic liquids are often expensive, sometimes toxic and as such, need to have high recovery rates in order to become economically and environmentally feasible. In addition, non-volatile contaminants including oligomeric or monomeric sugars, inorganics and others are likely to build up and need to be removed by a low cost purification method. Distillation of the ionic liquid is one method, which has been applied to [emim][OAc],44 a well-known ionic liquid for dissolving cellulose, but which requires rather high temperatures and reduced pressures. This ionic liquid has also been shown to react with the cellulose substrate45–47 via a reaction of the imidazolium carbene at the C2 position and the cellulose reducing end, forming a 2-(hydroxymethyl)-substituted imidazolium adduct45,46 thus removing solvent and cellulose from the system. Newer generations of ionic liquids are able to be distilled at much lower vacuum and temperatures, including 1,1,3,3-tetramethylguanidinium acetate ([TMGH][OAc])48 and [DBNH][OAc]49 which are also able to dissolve cellulose. Since water is often used as a precipitating anti-solvent, another possible recycling or purification method is phase-separation of the ionic liquid from water, if it is present.
In a previous study,40 we identified a set of ionic liquids, based on tetraalkylphosphonium cations paired with acetate anions which were effective for dissolving lignin and which dissolved cellulose upon the addition of a dipolar aprotic solvent, such as dimethylsulfoxide (DMSO). As such, these solvents for cellulose should be considered as mixed molecular and ionic solvents or “organic electrolyte” solutions.30 Some of the structures studied were phase-separable from water. One ionic liquid, methyltrioctylphosphonium acetate ([P8881][OAc]) was studied in more detail. When paired with the dipolar, aprotic molecular solvent DMSO it was shown to be a highly effective cellulose solvent, dissolving large amount of cellulose when compared to other types of cellulose dissolving ionic liquids and electrolytes.
A ternary phase system consisting of [P8881][OAc], DMSO and water was studied as a model of a possible solvent and recovery system for a cellulose fibre spinning process. However, the resulting ternary phase diagram showed a limited two-phase region with a large amount of ionic liquid undesirably getting into the aqueous phase. Addition of kosmotropic salts to the aqueous phase, including sodium or potassium acetate, improved the phase-separation but complicated recovery even further. Others have recently investigated hydrophilic and hydrophobic phosphonium ionic liquids as cellulose solvents,50 including the hydrophilic tetrabutylphosphonium acetate ([P4444][OAc]) and the hydrophobic tetraoctylphosphonium acetate ([P8888][OAc]), claiming that they dissolved 0.1 wt% cellulose. Based on our previous investigations and from this study, it is unlikely that the neat ionic liquids (without dipolar aprotic co-solvents) dissolve much more cellulose.
Two future pathways are envisaged for this class of cellulose solvents: the first, is to use smaller-chain ionic liquids such as [P4444][OAc] (less hydrophobic)40 in combination with dipolar aprotic solvents, with recovery taking place by the addition of “salting out” (kosmotropic) salts; the second, is to use longer-chain ionic liquids, such as [P8888][OAc] (which are more hydrophobic)40 with dipolar aprotic solvents and phase-separate them from water without the use of any additives, hopefully simplifying the recycling process and reducing energy demands (compared to distillation of water). However, we showed recently51 how the toxicity of this class of phosphonium ionic liquids increases with chain-length. A trade-off between cellulose dissolution capability, recovery by phase-separation, and toxicity is likely. The best way forward should be to investigate both routes thoroughly and determine exactly which routes are feasible and sustainable. Doing so should give valuable information for optimising cellulose fibre spinning process24 based on these solvents.
In this paper, we will concentrate on the latter route, where hydrophobic ionic liquids (part of an organic electrolyte solution with DMSO) are recovered from water without further additives. Following from our previous study,40 we desired to investigate ionic liquid-based electrolytes which were predicted to be much more hydrophobic than any previously studied ionic liquids or electrolytes, but still dissolved cellulose. The aim was to investigate the dissolution capability of two specific ionic liquids based on large tetraalkylphosphonium cations (tetraoctylphosphonium acetate, [P8888][OAc] and trioctyl(tetradecyl)phosphonium acetate, [P14888][OAc]), in combination with DMSO, whilst using water as a precipitating anti-solvent for cellulose. We aimed to study the feasibility of the phase-separation recycling method by measuring phase diagrams for these ternary systems.
Results and discussion
The two long-chain, hydrophobic phosphonium ionic liquids were synthesized: trioctyl(tetradecyl)phosphonium acetate [P14888][OAc] and tetraoctylphosphonium acetate [P8888][OAc] (Fig. 1). Both ionic liquids were found to be immiscible with water, a property typical of long chain phosphonium ionic liquids.50,52–54 | ||
| Fig. 1 Structure of ionic liquids used in this study. | ||
However, the pure ionic liquids prepared in this study did not dissolve cellulose to any appreciable degree, below a lower tested limit of 0.5 wt%, which fits to our previous understanding40 of some phosphonium acetate ionic liquids (including the ionic liquid [P8881][OAc]) and cellulose dissolution. Thus, we set out to use a molecular co-solvent in conjunction with these ionic liquids to enable cellulose dissolution, as we previously showed this combination to enable some non cellulose-dissolving ionic liquids to dissolve cellulose.40 In general, any molecular organic solvents in combination with an ionic liquid must be both polar and aprotic to act as a so called “co-solvent” for cellulose dissolution. Firstly, the solvent must not disrupt hydrogen bonding between the hydrogen-bond basic anion and cellulose, and thus must not be a hydrogen-bond donator (aprotic) and must be highly polar in order to effectively solvate the anions and cations. From our previous study, it appeared that DMSO was the most effective co-solvent, and has been the most widely studied so far.33,55–58 Its high boiling point (i.e. tolerance of the high cellulose dissolution temperatures), low hydrogen bond acidity, and very high polarity make it an ideal co-solvent. The use of DMSO is not without its disadvantages – although it is non-toxic, its biological solvent action is a particular concern, particularly in combination with ionic liquids. Overall, DMSO plays a synergistic role enabling previously non-dissolving ionic liquids (such as tetraalkylphosphonium acetates), to dissolve cellulose to an extremely high degree when compared to other ionic liquid based solvents.40 The most posited mechanism of action of this “co-solvent effect” is the strong ability of the highly dipolar DMSO (or other dipolar aprotic solvent) to solvate both the cation and anion, increasing ion pair separation and thus ‘freeing’ the hydrogen-bond basic anion to bond to cellulose hydroxyl groups.35,36,56,58 The role of preferential solvation is debated, with Xu et al. suggesting the preferential solvation of the cation plays an important part in the co-solvent effect in the [bmim][OAc]/DMSO solvent system.59
The cation is also speculated to play a major role which differs according to its structure. Imidazolium-based ionic liquids, for example, are able to donate hydrogen bonds from the acidic proton on the C2 position of the imidazolium ring, to the hydroxyl oxygens and hemiacetal oxygen. Phosphonium ionic liquids, on the other hand, lack such an acidic proton but have typically long and non-polar alkyl chains, which we have previously suggested40 to contribute to the dissolution of cellulose by increasing the strength of the non-polar interactions between the cation and the non-polar, hydrophobic surface of cellulose. Recent computational work has shown the importance of the contribution of these dispersion forces60 on cellulose dissolution and there has been much discussion on the significance of cellulose amphiphilicity61,62 on its solubility behaviour.
Two views of the cellulose dissolution in these systems are presented: a weight (weight percentage, wt% – Fig. 2) based view and a molar based view (Fig. 3), where we use the metric “AGU:IL” ratio, or moles of anhydroglucose units “AGU” to moles of IL. Such a metric allows us to compare easily the amount of cellulose dissolved per ionic liquid ion between ILs of different molecular weights and sizes. We compare both of the ionic liquid electrolytes from this study to [P8881][OAc], which was studied previously.40
 | ||
| Fig. 2 Cellulose (MCC) saturation point as wt% in [P8888][OAc]/DMSO and [P14888][OAc]/DMSO solutions, as a function of the DMSO concentration, in wt%. | ||
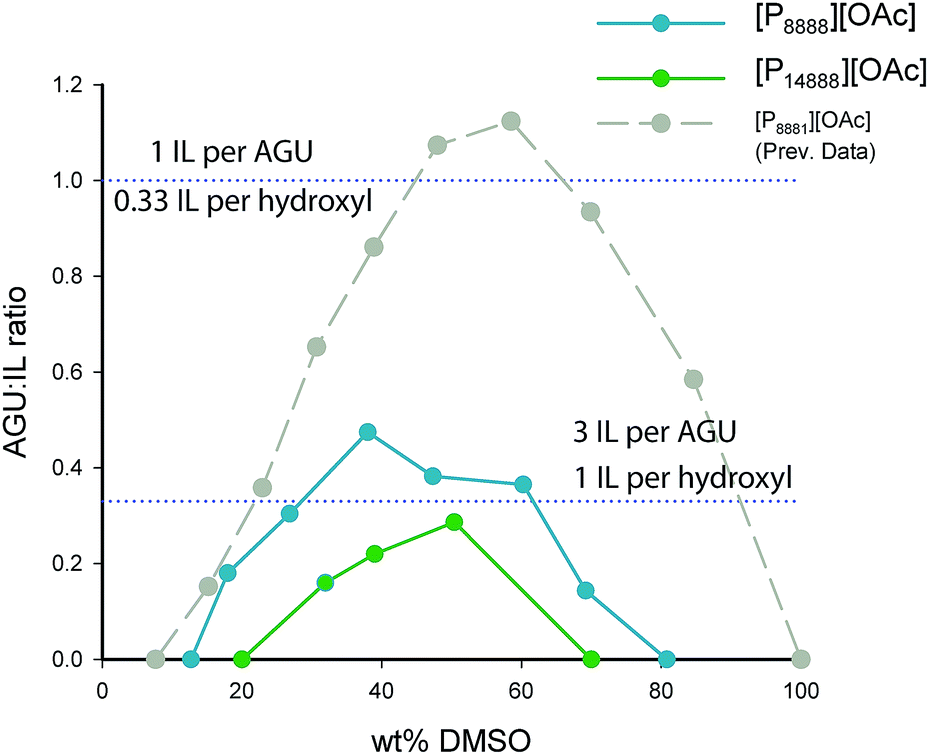 | ||
| Fig. 3 Molar dissolution ratio (AGU:IL ratio) versus wt% of DMSO. Displayed are lines showing the points at which each cellulose hydroxyl is bound by one ionic liquid ion (or 3 mol of IL per AGU) and by 1/3 ionic liquid ion (or 1 mol of IL per AGU). | ||
The cellulose dissolution capabilities in these organic electrolyte solvent systems are typically affected by concentration of the co-solvent component, usually rising and falling around a maximum (Fig. 2, 40–50 wt%). Both ionic liquids have this similar response to DMSO concentration as the previously studied ionic liquid, [P8881][OAc]. [P14888][OAc], with an extremely large cation, dissolves less cellulose than [P8888][OAc] at all DMSO concentration ranges, with maxima of ca. 3 wt% and ca. 8 wt% cellulose dissolve in solution, respectively. Ideal levels of dissolution of cellulose depend heavily on the application – for analytical purposes or small scale cellulose derivatization reactions, smaller amounts will be enough, but for Lyocell-like fibre spinning, larger amounts will be necessary. In the latter case, the rheological parameters would need to be adjusted by tuning the cellulose, DMSO and ionic liquid concentration. Thus, it is likely that only [P8888][OAc] would be able to be applied to Lyocell-like fibre spinning, although [P14888][OAc] may still be useful in niche applications.
When considering the molar dissolution metrics (Fig. 3), at the maximum point, [P8888][OAc]/DMSO can dissolve up to 0.44 mol of AGU per mol of IL (or 2.2 mol of IL per mol of AGU) whilst [P14888][OAc]/DMSO can dissolve up to 0.30 mol of AGU per mol of IL (or 3.3 mol of IL per mol of AGU). What is quite surprising is that cations of such size and bulkiness, in combination with dipolar aprotic co-solvents such as DMSO, compare favourably to ionic liquids with much smaller cations. However, it is clear that solutions containing cations larger than [P14888]+ are approaching the point of insolubility, as there is a clear negative trend in the molar dissolution ratio (AGU:IL) with increase in cation size from [P8881]+ through to [P14888]+. In comparison, [emim][OAc] without any dipolar aprotic co-solvents is typically described as being able to dissolve up to 0.33 mol of AGU per mol of IL, or 3 mol of IL per mol of AGU (ca. 25 wt%). Essentially, it has been speculated57 that this is the dissolution limit, where each hydroxyl is bound by 1 ionic liquid ion, specifically the hydrogen-bond basic anion.
However, from our and other past studies,40,58 it is apparent that there is a clear “co-solvent” effect on the dissolution of cellulose, even upon imidazolium ionic liquids like [emim][OAc]. Although it dissolves cellulose to a high degree, the amount of cellulose able to be dissolved with the addition of DMSO is dramatically increased when compared to the neat ionic liquid, up to 0.79 mol AGU:IL, or 1.7 mol IL:AGU – suggesting that not every hydroxyl is being bound by one anion.40 Equally, in this study we see that [P8888][OAc]/DMSO at its AGU:IL dissolution maximum displays similar characteristics, with less moles of ionic liquid than the amount of cellulose hydroxyls. However, it is a great deal less powerful as a solvent than the previously studied ionic liquid solvent system [P8881][OAc]/DMSO, which dissolved up to 1 mol AGU:IL (Fig. 3).40 Taking all the data into account, the change in the alkyl chain length clearly has a huge impact on the cellulose dissolution capacity. In comparison, the ionic liquids used in this study rank just below some other classes of cellulose dissolving ionic liquids in terms of ‘wt%’ cellulose dissolution capacity. Acid–base conjugate ionic liquids including [TMGH][OAc] (1,1,3,3-tetramethylguanidinum acetate) and [DBNH][OAc] (1,5-diazabicyclo[4.3.0]non-5-enium acetate) have dissolved as much as 15–16 wt% of cellulose.49 However, other onium cation ionic liquids have been studied which were comparable to the solvents in this study, such as [N4444][OAc] (tetrabutylammonium acetate) which was found to dissolve up to 8 wt% cellulose in the presence of DMSO.41 In this respect, it is quite remarkable that the much larger cation present in the solvent [P8888][OAc]/DMSO leads to the same level of cellulose dissolution.
The dependence of the molar or weight ratio of ionic liquid to co-solvent is reflected in other literature regarding mixed ionic and molecular cellulose solvents or organic electrolyte solutions with cellulose dissolution capacity often rising and falling around a maximum and the addition of the co-solvent increasing the amount of cellulose able to be dissolved in some instances.34,58,59
As a general consideration of the mechanism of dissolution in these mixed ionic-molecular solvent systems, it is likely that the increased size of the cation forces hydrogen-bonds from the acetate anion to become shared, as there are now less anions per unit of cellulose in the first solvation shell. This is a view that was recently proposed in Rabideau et al.'s computational studies of cellulose dissolution60,63 where the authors found that increases in alkyl chain length of imidazolium based ionic liquids led to a decrease in the amount of single, non-bridging hydrogen bonds and an increase in shared, bridging hydrogen bonds. In the phosphonium ionic liquids, this effect must in some way be mediated or offset just enough by the increase in the alkyl chain length and the strength of the dispersion forces between the non-polar cation and the non-polar surface of cellulose (in addition to the increased ion pair separation and solvation afforded by the dipolar aprotic component) to allow for dissolution. In solutions with cations significantly larger than [P14888]+, it is unlikely that much dissolution will occur at all, with the cations being simply too large (thus unable to stabilise the non-polar surface of cellulose) with the effective concentration of hydrogen-bond accepting anions in the first solvation shell being too low.
Liquid–liquid equilibria
The liquid–liquid equilibria of ternary systems composed of a mixture of a tetraalkylphosphonium acetate ionic liquid, DMSO, and water was studied to evaluate the recyclability of the solvent systems via phase-separation. Such ternary system is a representation of the state of the mixture after dissolution of cellulose and its subsequent regeneration with water, the anti-solvent. The experimental liquid–liquid equilibrium data for the ternary systems [P8888][OAc] + DMSO + water and [P14888][OAc] + DMSO + water, at 25.0 °C and atmospheric pressure, are reported in ESI Tables 1 and 2† and are presented as triangular phase diagrams in Fig. 4 and 5. Both systems were found to be of Type I according to the classification by Sørensen et al.,64 with water and the ionic liquid showing a large mutual immiscibility and the other two binary combinations (DMSO + water, and DMSO + ionic liquid) being totally miscible in any proportion. | ||
| Fig. 4 Experimental tie-lines for the liquid–liquid equilibrium of the ternary system [P8888][OAc] + DMSO + water at 25.0 °C and atmospheric pressure, in weight fractions. | ||
 | ||
| Fig. 5 Experimental tie-lines for the liquid–liquid equilibrium of the ternary system [P14888][OAc] + DMSO + water at 25.0 °C and atmospheric pressure, in weight fractions. | ||
A large 2-phase region was identified in both systems, consisting of an upper ionic liquid-rich phase, and a lower aqueous phase, with the area of the two-phase region being larger with the ionic liquid [P14888][OAc]. There is a clear effect of the alkyl chain length, or size (molar volume) between the two ionic liquids on the distribution ratio of the DMSO, on the water content of the ionic liquid phase, and on the ionic liquid content of the aqueous phase. As the cation size increases, the ionic liquid content in the aqueous phase drops, as does the water content of the ionic liquid phase. These effects would be consistent with the increase in “hydrophobicity” as afforded by the longer alkyl chains, by increasing the size and non-polar character of the cations. An interesting feature is the very low or even negligible concentration of ionic liquid in the aqueous phase for both ionic liquids, particularly [P14888][OAc]. The recovery of ionic liquid is the limiting factor for scale-up of cellulose dissolving and biomass fractionation processes with ionic liquids, and thus this is a very important consideration. From the liquid–liquid equilibrium data, we can see that the ionic liquid has very low solubility in the aqueous phase in the ternary systems studied; thus minimising the loss of the expensive ionic liquid component into the aqueous waste stream.
The distribution ratio of DMSO in the phases in equilibrium, βDMSO, was calculated by means of eqn (1):
 | (1) |
 | (2) |
 | ||
| Fig. 6 DMSO distribution ratio (βDMSO) for the ternary systems [P8888][OAc] + DMSO + water (blue) and [P14888][OAc] + DMSO + water (red), at 25.0 °C and atmospheric pressure, as a function of the mass fraction of DMSO in the ionic liquid-rich phase. | ||
Cellulose regeneration and ionic liquid recovery
We conducted a larger-scale demonstration of the cellulose dissolution and regeneration capabilities of media based on [P8888][OAc] and DMSO, since this was the ionic liquid with the greatest cellulose dissolution capability. The overall process is illustrated in Fig. 7. Taking a 5 wt% solution of cellulose in a solution of 40 wt% IL and 60 wt% DMSO, water was used as an anti-solvent to precipitate cellulose. The recovery global composition was 80 wt% water, 12 wt% ionic liquid, and 8 wt% DMSO. Even though water and [P8888][OAc], at this concentration, are immiscible, cellulose precipitation still occurs. Initially, the cellulose precipitates as a gel, and requires strong overhead stirring in warm water to break up the gel into a solid suspension, with the ionic liquid and aqueous phases appearing as an emulsion. The amount of water was determined based on the ternary phase diagram and information from the equilibrium experiments, in order to maximize potential recovery and minimize excessively strong emulsion formation. The ionic liquid, DMSO and water mixture is separated from cellulose via centrifugation, with two liquid phases and the solid cellulose phase separating evenly, enabling the ionic liquid-rich phase to be recovered with relative ease from the top phase. However, since the solid cellulose floats at the interface of the two phases, a portion of the ionic liquid is absorbed into the top of the swollen cellulose material, and must be washed with a miscible solvent to be fully recovered. | ||
| Fig. 7 Illustration of larger (lab)-scale dissolution and regeneration experiment showing the recycling scheme. | ||
Thus, the aqueous phase and the solid cellulose was separated via filtration with the cellulose being washed with ethanol. After phase-separation and removal of the cellulose phase, a small amount of ionic liquid remained on the top of the aqueous phase. This portion was carefully decanted and extracted and the side of the flask washed with ethanol to recover more residual ionic liquid. The ethanol wash filtrate was evaporated combined with the ionic liquid-rich phase for the final recovery mass balance. Typically, in a real fibre spinning process, only a small amount of ‘dope’ is spun into a spinning bath at one time, with the fibres being continually removed, after which they are taken through multiple stages of washing baths to remove residual solvent. This is in contrast to our lab scale demonstration, where the whole mass of cellulose is precipitated at once (appearing between the phases) necessitating such recovery techniques. Doing these experiments, however, enables us to gain a reasonable mass balance as an evaluation of the potential of this recovery technique on a lab scale.
The yield of recovered cellulose was high, with 99 wt% being recovered. The yield of ionic liquid recovered from the ionic liquid-rich organic phase in the liquid–liquid separation was 60 wt%, with the bulk of the rest being recovered from the cellulose ethanol wash, around 30 wt%, whilst a small amount of ionic liquid, around 7 wt%, was recovered from the top of the aqueous phase after removal of the ionic liquid and cellulose phases (bringing the total recovery of ionic liquid to 97 wt%), and the rest, around 3 wt% was unaccounted for, with a portion (<0.1 wt%, from the phase diagram) to be in the aqueous phase and the rest due to experimental losses, likely during the filtration stage. From the phase diagram, the recovered ionic liquid-rich phase should have a small amount of DMSO and water (around 10 wt% respectively). The residual water and DMSO can be removed under high-vacuum, leaving a pure and dry ionic liquid. However, from the phase equilibrium data, it should also be possible to remove the residual DMSO by multi-stage or counter-current extraction with water due to the extremely favourably distribution ratio and selectivity for DMSO extraction in the aqueous phase. However, a small amount of water would still remain, which may need to be removed with some sort of appropriate drying method. The recovered cellulose showed no signs of contamination by the ionic liquid, when comparing the infra-red spectra to that of pure cellulose (ESI†).
The crystallinity of the recovered sample from the large-scale dissolution and regeneration was analysed via XRD (X-Ray Diffraction) measurements (Fig. 8). As found with the previous studies on phosphonium ionic-liquid and DMSO-based electrolytes,40 the regenerated cellulose material was found to convert from the Cellulose I to the Cellulose II polymorph with a corresponding relative loss in crystallinity and increase in amorphous content.
 | ||
| Fig. 8 XRD interferograms of two samples: MCC cellulose dissolved and regenerated from [P8888][OAc]:DMSO 40:60 wt% (with water, and washed with ethanol) and untreated MCC. | ||
We also explored the use of the phosphonium ionic liquids as cellulose decrystallisation agents – that is, the ability to remove cellulose crystallinity (Cellulose I for native cellulose) and recover amorphous cellulose from high molecular weight dissolving pulp (Enocell PHK Pulp). This was tested by simple heating (at 100 °C) to 4 hours, followed by removal of the ionic liquid with ethanol. It was found that heating in the neat ionic liquid (0 wt% DMSO) and “non-dissolving” compositions of [P8888][OAc] and DMSO (i.e. 5 and 90 wt% DMSO, based on Fig. 2) lead to very little change in the XRD interferogram, with Cellulose I remaining as the dominant cellulose crystalline form. When viewed under a microscope, the fibrous structure of the pulp remained (ESI Fig. 11 and 13†). This adds further evidence to the non-dissolving ability of these kinds of hydrophobic phosphonium ionic liquids in the absence (or excess) of a dipolar-aprotic co-solvent (such as DMSO) as described by Fig. 2. However, a “dissolving” composition (40 wt% DMSO) with an excess of cellulose (10 wt%, being just over the measured saturation point of ca. 8 wt% cellulose, meaning a portion of the cellulose is swollen and not dissolved) leads to an intermediate mixture of Cellulose I and Cellulose II polymorphs (Fig. 9), which would be consistent with undissolved fibre fragments being present along with dissolved and regenerated Cellulose II (likely with increased amorphous content) (ESI Fig. 12†). The XRD interferograms of cellulose (pre-hydrolysis kraft pulp) treated with pure [P8888][OAc] and varying amounts of DMSO are displayed in Fig. 9.
 | ||
| Fig. 9 XRD interferograms of Enocell PHK Pulp (10 wt% in solution) swollen (not fully dissolved) in [P8888][OAc] and varying compositions of IL and DMSO. | ||
Tracking dissolution, regeneration and recovery with NMR spectroscopy
We have shown previously the excellent ability of this class of cellulose solvents in observing lignocellulosic biopolymers, including high molecular weight cellulose, by NMR spectroscopy.65 Proton NMR spectra of the ionic liquid before dissolution, after dissolution of 5 wt% cellulose in 60:40 wt% [P8888][OAc]/DMSO-d6, after phase separation to an ionic liquid-rich phase, and after drying and full recovery shows the process of dissolution and recovery in these solvents (Fig. 10). After dissolution of cellulose in the solvent, it is clearly visible in the 1H spectra. The spectral area for 1H signals arising from carbohydrates is around 5–3 ppm, with no major conflicting signals from the ionic liquid solvent. However, minor solvent impurities are also visible with low intensity in the cellulose region, but these are easily distinguished from the cellulose backbone peaks. These are visible with very low intensity in the spectrum of the ionic liquid prior to dissolution (ESI Fig. 5†). No residual carbohydrates are visible in this region in the 1H spectrum of the recovered ionic liquid, indicating minimal contamination with oligosaccharides, monosaccharides or residual cellulose in the ionic liquid, such as from degradation processes. After addition of water, and extraction of an NMR sample from the ionic liquid-rich phase, we can see the disappearance of the cellulose peaks, along with a new water peak and a DMSO peak of increased intensity. The recovered ionic liquid is free from DMSO, water, and shows no significant changes to the starting material.
 | ||
| Fig. 10 1H NMR spectra illustrating dissolution and regeneration processes: (a) ionic liquid prior to dissolution in CDCl3; (b) 5 wt% MCC cellulose in [P8888][OAc]/DMSO-d6, (CN refers to the cellulose carbon number, NRE = non reducing end); (c) [P8888][OAc]/DMSO-d6 solution after addition of water and regeneration of cellulose; (d) [P8888][OAc], after phase-separation, recovery and drying, in CDCl3. Note: in DMSO-d6, some chemical shifts, notably the peak arising from the acetate CH3, shift upfield. | ||
Conclusions
Two hydrophobic, long chain phosphonium ionic liquids were found to dissolve cellulose in the presence of DMSO. However, the ionic liquid [P14888][OAc] dissolved significantly less cellulose at the most optimum DMSO concentration than [P8888][OAc] – so a limit on the effective chain length of phosphonium ionic liquids, and ionic liquids in general for cellulose dissolution, is apparent. Ternary systems including basic and hydrophobic phosphonium acetate ionic liquids of a series [PR888][OAc] with DMSO and water were shown to be phase-separable from water, showing a Type I phase diagram. The high DMSO distribution ratios and high selectivity values demonstrate that the ionic liquid and DMSO may be efficiently separated with a minimum of the separating solvent, water. The low solubility of the ionic liquid in the aqueous phase allows quantitative recovery of the ionic liquid when water (as a precipitating anti-solvent for dissolved cellulose) is added to an ionic-liquid and DMSO mixture. Thus, the increased recovery by simple phase-separation compared to previous generations of this solvent class (and other ionic liquids) is a significant advance in the search for a phase-separable solvent medium for cellulose dissolution and regeneration, which could lead to lowered energy demands and thus more sustainable processes. Potential roadblocks for larger scale applications of these solvent systems include the separation of DMSO from waste water streams and toxicity of the component ionic liquids (due to the long alkyl chain length of the cation). However, a decrease in cation alkyl chain length (and its associated lowered toxicity) comes at the expense of the recovery ability by phase-separation – so a balance between these two factors is needed. Future work, including engineering simulations and LCA analyses may be able to elucidate exactly which pathway is more sustainable.Experimental section
Chemicals and raw materials
Trioctylphosphine (CYTOP 380) was a gift from Cytec Ltd. Octyl chloride, tetradecyl chloride, potassium acetate (99.8%, anhydrous crystalline powder), methyl acetate (HPLC grade, >99.8%) and 2-propanol (HPLC grade >99.5%) were purchased from Sigma Aldrich Ltd. Microcrystalline cellulose (MCC) was also purchased from Sigma Aldrich Ltd. Enocell PHK pulp was kindly supplied by Storä Enso Oyj.Ionic liquid synthesis
For both synthesized ionic liquids, trioctylphosphine was charged under argon into a purged 3 necked round bottom flask, with an argon balloon capped condenser and septum on the side neck. The corresponding alkyl chloride, 1.01 equivalent was introduced in small portions through the septum whilst stirring, with the temperature of the mixture being monitored with a thermometer (Scheme 1i). The temperature of the reaction mixture was slowly increased to 145 °C and stirred for 16 hours. Excess reactants and any small-molecular weight by-products were removed under high vacuum at ca. 90 °C, producing a light yellow, highly viscous liquid. | ||
| Scheme 1 Synthesis of phosphonium ionic liquids. (i) Menshutkin quaternization, (ii) anion metathesis. | ||
The tetraalkylphosphonium chloride ionic liquid intermediate was dissolved in isopropanol, in a 1:5 volume ratio and heated to 80 °C with stirring. Potassium acetate, 1.01 molar equivalent, was added in small portions (Scheme 1ii) and stirred to ensure complete dissolution and homogenization in the mixture. After stirring at 80 °C for one hour, the mixture was stirred at room temperature for 16 hours, and refrigerated for a further 16 hours to ensure complete precipitation of KCl. The KCl by-product was vacuum filtered over a glass sinter filter and celite and the isopropanol was removed under reduced pressure. Cold acetone was added to the crude product to precipitate any remaining salts, including KOAc and KCl, and again filtered with a glass sinter filter and celite. Acetone was evaporated under reduced pressure, and the product was dried under high vacuum and high temperature (ca. 90 °C) to produce a highly viscous, dark amber liquid.
Trioctyl(tetradecyl)phosphonium acetate [P14888][OAc]
Trioctylphosphine (83.1 g, 2.24 × 10−1 mol), tetradecylchloride (52.62 g, 2.27 × 10−1 mol), potassium acetate (21.28 g, 2.17 × 10−1 mol). Final product: 135.90 g, 2.17 × 10−1 mol, 96.7% yield.1H: (600 MHz, CDCl3) δ 2.41 (m, P–CH2), 1.94 (s, −OOC–CH3), 1.63 (m, –CH2–), 1.48 (m, –CH2–), 1.36 (m, –CH2–) 1.24 (m, –CH2–), 0.86 (t, J = 6.9 Hz, CH3).
13C: (600 MHz, CDCl3) δ 179.21 (s, CH3CO2−), 33.52 (s, CH2), 33.42 (s, CH2), 32.29 (s, CH2), 32.26 (s, CH2), 32.22 (s, CH2), 32.14 (s, CH2), 31.96 (s, CH2), 31.94 (s, CH2), 31.58 (s, CH2), 27.86 (s, CH3CO2−), 25.29 (s, CH2), 25.20 (s, CH2), 24.61 (d, J = 4.78 Hz, CH2), 21.46 (d, J = 46.7 Hz, P–CH2), 16.72 (s, CH3), 16.65 (s, CH3).
Tetraoctylphosphonium acetate [P8888][OAc]
Trioctylphosphine (83 g, 2.24 × 10−1 mol), octylchloride (33.56 g, 2.27 × 10−1 mol), potassium acetate (22.02 g, 2.24 × 10−1 mol). Final product: 117.6 g, 2.17 × 10−1 mol, 96.7% yield.1H NMR (600 MHz, CDCl3) δ 2.38 (m, P–CH2), 1.90 (s, CH3CO2−), 1.60 (m, –CH2–), 1.46 (m, –CH2–), 1.33 (m, –CH2–), 1.21 (m, –CH2–), 0.82 (t, J = 7 Hz, CH3)
13C: (600 MHz, CDCl3) δ 179.24 (s, CH3CO2−), 34.27 (s, CH2), 33.48 (s, CH2), 33.38 (s, CH2), 31.53 (s, CH2), 27.95 (s, CH3CO2−), 25.15 (s, CH2), 24.56 (d, J = 4.59 Hz, CH2) 21.6 (d, J = 46.4 Hz, P–CH2), 16.60 (s, CH3).
Cellulose dissolution experiments
Dissolution experiments were conducted in a similar manner to our previous work,40 with cellulose being dissolved in a pre-mixed ionic liquid–DMSO solution at a constant temperature, 120 °C and cellulose being added in small increments until a cloud point is reached. The upper boundary is taken as the midpoint between the cloud point and the last addition of cellulose, where each addition is ca. 0.5% of the total mass. Cloudy solutions can be confirmed by optical microscopy to contain fibre fragments.Liquid–liquid equilibria
Mixtures of DMSO, water and ionic liquid ([P8888][OAc] or [P14888][OAc]), with global composition lying in the immiscibility domain of the corresponding ternary system, were prepared and introduced into jacketed glass cells especially designed for liquid–liquid equilibrium experiments. The temperature of the cells was kept at 298.2 K by means of an Ultratherm-200P Selecta water-circulating thermostatic bath, with an uncertainty of 0.1 K. All mixtures were vigorously stirred (magnetic stirring) for a minimum of 2 h, and then allowed to settle until complete separation of the phases in equilibrium (a minimum of 12 h, and typically several days). It was found that at lower DMSO concentrations, the heterogeneous mixtures formed strong emulsions and phase separation times were unreasonably large. To overcome this problem, such mixtures were placed in centrifuge tubes, then centrifuged twice for 15 minutes each at 4000 rpm, and the centrifuge tubes were placed into the thermostatic water bath at 298.2 K. In all cases, after equilibration and separation, both phases were sampled with syringes attached to needles, with avoidance or minimisation of the disturbance of the liquid–liquid interface. Thereafter, these samples were placed in glass vials, weighed, and mixed with known amounts of an external standard and a solvent, as described in the following paragraph, for compositional analysis by gas chromatography.The compositions of the samples were analysed using a HP 6890 series gas chromatograph, equipped with a thermal conductivity detector and a HP-FFAP capillary column (25 m × 0.2 mm × 0.33 μm), coupled with an empty pre-column to collect the ionic liquid that was not retained by the liner. To allow calculation of the ionic liquid concentration by difference, methyl acetate was used as an external standard. An organic solvent, namely 2-propanol, was used as solvent for the mixtures of sample and external standard. Helium was used as the mobile phase, and the injection volume was 1 μL with a split ratio of 50:1 and a pressure of 20.42 psi. Injection temperature was 250 °C. The initial oven temperature was 50 °C held for 2.7 minutes. There were two heating ramps, the first from 50–175 °C at 12 °C per minute, and the second from 175–240 °C at 120 °C per minute, leading to a total run time of 18.16 minutes. Under these conditions, an adequate separation between the signals of the external standard, solvent, water, and DMSO was observed. Calibration curves were constructed with samples prepared by weight and with compositions in the (homogeneous) vicinity of the solubility curve, previously determined via the “cloud point” method. The water content of the ionic liquids was measured via Karl-Fischer titration in a MetrOhm 737 KF coulometer, and was factored into the mass balance of ionic liquid and water. The peak areas ratio for the analytes DMSO and H2O over the external standard were correlated with the mass ratios of the analytes over the internal standard. The standard mass fraction errors associated with the compositional analysis procedure described were found to be: 0.006 and 0.007 for the ionic liquid-rich phase and water-rich phase respectively in the system [P8888][OAc] + DMSO + water; and 0.006 and 0.005 for the ionic liquid-rich phase and water-rich phase respectively in the system [P14888][OAc] + DMSO + water.
Large scale dissolution-regeneration experiment
Dry [P8888][OAc] (8.7 g) was mixed with DMSO-d6 (5.8 g), to which cellulose (0.77 g) was added, and the solution stirred until a homogenous dispersion was achieved. The solution was heated at 100 °C for ca. 1 hour and 120 °C for ca. 30 minutes, until a clear solution was observed. To the cellulose/ionic liquid/DMSO solution water was added (10 mL), initially inducing cellulose regeneration to a homogenous gel state. Overhead mechanical stirring was used to stir the mixture vigorously, whilst the remaining portion of water (13 mL) was added, forming the precipitated cellulose as a suspension. The mixture was stirred for ca. 2 hours with mechanical stirring at room temperature. The mixture was transferred to a centrifugation tube and centrifuged for 45 minutes at 40 °C to separate the ionic liquid, aqueous and solid cellulose phases. From the resulting 3-phase system, the ionic liquid layer was extracted from the top with a needle and syringe leaving the cellulose phase, floating at the top of the aqueous phase, swollen with the ionic liquid phase. The mixture was centrifuged once again, for 1 hour at 40 °C and a smaller amount of the ionic liquid phase was removed. The remaining mixture was filtered to remove the swollen cellulose phase, leaving the filtrate, the aqueous phase, with a small amount of ionic liquid “scum” or film on the surface. The cellulose was washed multiple times with ethanol, and stirred with warm (65 °C) ethanol for ca. 12 hours to remove all traces of ionic liquid, and subsequently dried (0.76 g, 99% recovery by mass). Ethanol was used to wash the remaining glassware and remove final traces of ionic liquid. The solid cellulose was filtered and dried, and the combined ethanol wash and filtrate evaporated to produce a recovered fraction of ionic liquid (3.2 g). The fraction from the phase-separation was dried under high-vacuum conditions at 80 °C for ca. 16 hours to remove traces of water and DMSO for the main recovery fraction (5.1 g). The recovered cellulose was analysed by FT-IR, Optical Microscopy, and XRD.Pulp swelling and decrystalisation experiments
The pulp (Enocell PHK pulp) fibres were dispersed in the ionic liquid, or ionic liquid–DMSO solutions with the aid of a vortexer. The mixtures were stirred at 100 °C for 4 hours, after which ethanol was added (5× volume), thoroughly mixed, filtered and this repeated twice. To remove traces of ionic liquids from the cellulose precipitate, the solid materials were heated for ca. 4 hours in ethanol (ca. 65 °C), filtered and this repeated twice. The precipitate was finally filtered over vacuum, and dried under vacuum at 60 °C for 1 hour.XRD, FTIR, microscopy & NMR measurements
Cellulose samples were pressed into a tablet-form using a hydraulic press designed for preparing FT-IR tablets, to ensure consistency between samples. XRD measurements were performed by using a PANalytical X'Pert Pro MPD in Bragg–Brentano geometry. The measurement range was 5–50° 2θ, which was scanned at 0.03° s−1 with 0.08° 2θ steps by using a PIXcel detector in the 1D mode. Cellulose samples were studied with a Bruker Alpha ATR-FT-IR spectrometer and Olympus Optical Microscope, under various magnification amounts (×5 displayed in ESI†). All NMR spectra were obtained at 27 °C on a Varian UNITY INOVA 600 MHz spectrometer and 5 mm triple resonance (1H, 13C, 15N) pulsed field gradient probe-head.
Acknowledgements
We would like to thank COST for the Short Term Scientific Mission funding under the EXIL (Exchange on Ionic Liquids) action and CLIC Innovation Oy for the continued funding under the ACel program. We would also like to thank CYTEC (Cytec Solvay Group) and Stora Enso Oyj for the provision of phosphines/phosphonium salts and Enocell pre-hydrolysis kraft pulp, respectively.References
- S. Zhu, Y. Wu, Q. Chen, Z. Yu, C. Wang, S. Jin, Y. Ding and G. Wu, Green Chem., 2006, 8, 325–327 RSC.
- H. Wang, G. Gurau and R. D. Rogers, Chem. Soc. Rev., 2012, 41, 1519–1537 RSC.
- P. Mäki-Arvela, I. Anugwom, P. Virtanen, R. Sjöholm and J. P. Mikkola, Ind. Crops Prod., 2010, 32, 175–201 CrossRef.
- Z. Z. Chowdhury, S. M. Zain, S. B. Abd Hamid and K. Khalid, BioResources, 2014, 9, 1787–1823 CrossRef.
- J. Wu, J. Zhang, H. Zhang, J. He, Q. Ren and M. Guo, Biomacromolecules, 2004, 5, 266–268 CrossRef CAS PubMed.
- Y. Cao, J. Wu, J. Zhang, H. Li, Y. Zhang and J. He, Chem. Eng. J., 2009, 147, 13–21 CrossRef CAS.
- L. Feng and Z. Chen, J. Mol. Liq., 2008, 142, 1–5 CrossRef.
- M. Mora-Pale, L. Meli, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2011, 108, 1229–1245 CrossRef CAS PubMed.
- A. Brandt, J. P. Hallett, D. J. Leak, R. J. Murphy and T. Welton, Green Chem., 2010, 12, 672–679 RSC.
- G. Bokinsky, P. P. Peralta-Yahya, A. George, B. M. Holmes, E. J. Steen, J. Dietrich, T. S. Lee, D. Tullman-Ercek, C. A. Voigt, B. A. Simmons and J. D. Keasling, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 19949–19954 CrossRef CAS PubMed.
- C. Li, B. Knierim, C. Manisseri, R. Arora, H. V. Scheller, M. Auer, K. P. Vogel, B. A. Simmons and S. Singh, Bioresour. Technol., 2010, 101, 4900–4906 CrossRef CAS PubMed.
- I. Kilpeläinen, H. Xie, A. King, M. Granstrom, S. Heikkinen and D. S. Argyropoulos, J. Agric. Food Chem., 2007, 55, 9142–9148 CrossRef PubMed.
- D. A. Fort, R. C. Remsing, R. P. Swatloski, P. Moyna, G. Moyna and R. D. Rogers, Green Chem., 2007, 9, 63–69 RSC.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- A. Stark, Energy Environ. Sci., 2011, 4, 19–32 CAS.
- T. Leskinen, A. W. T. King, I. Kilpeläinen and D. S. Argyropoulos, Ind. Eng. Chem. Res., 2011, 50, 12349–12357 CrossRef CAS.
- A. Pinkert, D. F. Goeke, K. N. Marsh and S. Pang, Green Chem., 2011, 13, 3124–3136 RSC.
- N. Sun, M. Rahman, Y. Qin, M. L. Maxim, H. Rodríguez and R. D. Rogers, Green Chem., 2009, 11, 646–655 RSC.
- M. C. Castro, H. Rodríguez, A. Arce and A. Soto, Ind. Eng. Chem. Res., 2014, 53, 11850–11861 CrossRef CAS.
- R. Rinaldi, R. Palkovits and F. Schüth, Angew. Chem., Int. Ed., 2008, 47, 8047–8050 CrossRef CAS PubMed.
- R. Rinaldi, N. Meine, J. vom Stein, R. Palkovits and F. Schüth, ChemSusChem, 2010, 3, 266–276 CrossRef CAS PubMed.
- D. Ingildeev, F. Effenberger, K. Bredereck and F. Hermanutz, J. Appl. Polym. Sci., 2013, 128, 4141–4150 CrossRef CAS.
- G. Jiang, Y. Yuan, B. Wang, X. Yin, K. S. Mukuze, W. Huang, Y. Zhang and H. Wang, Cellulose, 2012, 19, 1075–1083 CrossRef CAS.
- H. Sixta, A. Michud, L. Hauru, S. Asaadi, Y. Ma, A. W. T. King, I. Kilpeläinen and M. Hummel, Nord. Pulp Pap. Res. J., 2015, 30, 043–057 CAS.
- M. Hummel, A. Michud, M. Tanttu, S. Asaadi, Y. Ma, L. K. J. Hauru, A. Parviainen, A. W. T. King, I. Kilpeläinen and H. Sixta, Ionic Liquids for the Production of Man-Made Cellulosic Fibers: Opportunities and Challenges, Springer, Berlin Heidelberg, 2015, pp. 1–36 Search PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- Y. Fukaya, K. Hayashi, M. Wada and H. Ohno, Green Chem., 2008, 10, 44–46 RSC.
- J. S. Moulthrop, R. P. Swatloski, G. Moyna and R. D. Rogers, Chem. Commun., 2005, 1557–1559 RSC.
- R. P. Swatloski, R. D. Rodgers and J. D. Holbrey, WO2003029329, 2003.
- R. Rinaldi, Chem. Commun., 2011, 47, 511–513 RSC.
- M. Gericke, T. Liebert, O. A. El Seoud and T. Heinze, Macromol. Mater. Eng., 2011, 296, 483–493 CrossRef CAS.
- Y. Zhao, X. Liu, J. Wang and S. Zhang, J. Phys. Chem. B, 2013, 117, 9042–9049 CrossRef CAS PubMed.
- J.-M. Andanson, E. Bordes, J. Devémy, F. Leroux, A. A. H. Pádua and M. F. C. Gomes, Green Chem., 2014, 16, 2528 RSC.
- D. L. Minnick, R. A. Flores, M. R. DeStefano and A. M. Scurto, J. Phys. Chem. B, 2016, 120, 7906–7919 CrossRef CAS PubMed.
- A. Xu, X. Guo and R. Xu, Int. J. Biol. Macromol., 2015, 81, 1000–1004 CrossRef CAS PubMed.
- A. Xu and Y. Zhang, J. Mol. Struct., 2015, 1088, 101–104 CrossRef CAS.
- A. Xu, Y. Zhang, Y. Zhao and J. Wang, Carbohydr. Polym., 2013, 92, 540–544 CrossRef CAS PubMed.
- A.-R. Xu, X. Guo and J. Ma, J. Macromol. Sci., Part B: Phys., 2016, 55, 559–565 CrossRef CAS.
- Y.-B. Huang, P.-P. Xin, J.-X. Li, Y.-Y. Shao, C.-B. Huang and H. Pan, ACS Sustainable Chem. Eng., 2016, 4, 2286–2294 CrossRef CAS.
- A. J. Holding, M. Heikkilä, I. Kilpeläinen and A. W. T. King, ChemSusChem, 2014, 7, 1422–1434 CrossRef CAS PubMed.
- J. Miao, H. Sun, Y. Yu, L. Zhang and X. Song, RSC Adv., 2014, 4, 36721–36724 RSC.
- X. Q. Meng, J. Devemy, V. Verney, A. Gautier, P. Husson and J.-M. Andanson, ChemSusChem, 2017 DOI:10.1002/cssc.201601830.
- E. Gale, R. H. Wirawan, R. L. Silveira, C. S. Pereira, M. A. Johns, M. S. Skaf and J. L. Scott, ACS Sustainable Chem. Eng., 2016, 4, 6200–6207 CrossRef CAS.
- A. W. T. King, A. Parviainen, P. Karhunen, J. Matikainen, L. K. J. Hauru, H. Sixta and I. Kilpeläinen, RSC Adv., 2012, 2, 8020–8026 RSC.
- M. T. Clough, K. Geyer, P. A. Hunt, S. Son, U. Vagt and T. Welton, Green Chem., 2015, 17, 231–243 RSC.
- M. T. Clough, J. A. Griffith, O. Kuzmina and T. Welton, Green Chem., 2016, 18, 3758–3766 RSC.
- G. Ebner, S. Schiehser, A. Potthast and T. Rosenau, Tetrahedron Lett., 2008, 49, 7322–7324 CrossRef CAS.
- A. W. T. King, J. Asikkala, I. Mutikainen, P. Järvi and I. Kilpeläinen, Angew. Chem., Int. Ed., 2011, 50, 6301–6305 CrossRef CAS PubMed.
- A. Parviainen, A. W. T. King, I. Mutikainen, M. Hummel, C. Selg, L. K. J. Hauru, H. Sixta and I. Kilpeläinen, ChemSusChem, 2013, 6, 2161–2169 CrossRef CAS PubMed.
- M. Abe, K. Kuroda, D. Sato, H. Kunimura and H. Ohno, Phys. Chem. Chem. Phys., 2015, 17, 32276–32282 RSC.
- S.-K. Mikkola, A. Robciuc, J. Lokajová, A. J. Holding, M. Lämmerhofer, I. Kilpeläinen, J. M. Holopainen, A. W. T. King and S. K. Wiedmer, Environ. Sci. Technol., 2015, 49, 1870–1878 CrossRef CAS PubMed.
- Y. Fukaya and H. Ohno, Phys. Chem. Chem. Phys., 2013, 15, 4066 RSC.
- J. Kagimoto, S. Taguchi, K. Fukumoto and H. Ohno, J. Mol. Liq., 2010, 153, 133–138 CrossRef CAS.
- M. G. Freire, P. J. Carvalho, R. L. Gardas, L. M. N. B. F. Santos, I. M. Marrucho and J. A. P. Coutinho, J. Chem. Eng. Data, 2008, 53, 2378–2382 CrossRef CAS.
- Y. Lv, J. Wu, J. J. Zhang, Y. Niu, C.-Y. Liu, J. He and J. J. Zhang, Polymer., 2012, 53, 2524–2531 CrossRef CAS.
- F. Huo, Z. Liu and W. Wang, J. Phys. Chem. B, 2013, 117, 11780–11792 CrossRef CAS PubMed.
- K. A. Le, C. Rudaz and T. Budtova, Carbohydr. Polym., 2014, 105, 237–243 CrossRef CAS PubMed.
- Y. Zhao, X. Liu, J. Wang and S. Zhang, J. Phys. Chem. B, 2013, 117, 9042–9049 CrossRef CAS PubMed.
- A. Xu, Y. Zhang, Y. Zhao and J. Wang, Carbohydr. Polym., 2013, 92, 540–544 CrossRef CAS PubMed.
- B. D. Rabideau, A. Agarwal and A. E. Ismail, J. Phys. Chem. B, 2014, 118, 1621–1629 CrossRef CAS PubMed.
- B. Medronho, A. Romano, M. G. Miguel, L. Stigsson and B. Lindman, Cellulose, 2012, 19, 581–587 CrossRef CAS.
- W. G. Glasser, R. H. Atalla, J. Blackwell, R. Malcolm Brown, W. Burchard, A. D. French, D. O. Klemm and Y. Nishiyama, Cellulose, 2012, 19, 589–598 CrossRef CAS.
- B. D. Rabideau and A. E. Ismail, Phys. Chem. Chem. Phys., 2015, 17, 5767–5775 RSC.
- J. M. Sørensen, T. Magnussen, P. Rasmussen and A. Fredenslund, Fluid Phase Equilib., 1979, 2, 297–309 CrossRef.
- A. J. Holding, V. Mäkelä, L. Tolonen, H. Sixta, I. Kilpeläinen and A. W. T. King, ChemSusChem, 2016, 9, 880–892 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01662j |
| This journal is © The Royal Society of Chemistry 2017 |