
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen sol–gel route for selective growth of 1D rutile N–TiO2: a highly active photocatalyst for H2 generation and environmental remediation under natural sunlight†
Supriya K. Khorea,
Navya Vani Tellabatia,
Sanjay K. Aptea,
Sonali D. Naika,
Prashant Ojhab,
Bharat B. Kale a and
Ravindra S. Sonawane*a
a and
Ravindra S. Sonawane*a
aCentre for Materials for Electronic Technology, Government of India, Panchawati, Off Pashan Road, Pune 411008, India. E-mail: sonawaner@yahoo.com; sonawane@cmet.gov.in
bNaval Materials Research Laboratory (NMRL), Ministry of Defence, Ambernath 421506, India
First published on 29th June 2017
Abstract
We report selective growth of N–TiO2 1D nanorods using a green aqueous sol–gel method followed by hydrothermal treatment. Titanium tetraisopropoxide, diethanolamine, and H2O2 were used as precursors for preparing an aqueous gel. Trifluoroacetic acid (TFA) was used as a growth regulator for selective growth of desired structure and morphology. Effects of TFA on the structure and morphology of N–TiO2 were studied by varying concentration of TFA between 1–10% by volume. Structural characterization using XRD confirmed formation of a specific rutile phase with slight crystal disorder with N-doped TiO2 samples. FESEM and HRTEM analysis showed formation of 1D rice grain shaped N–TiO2 in the presence of 1% TFA solution. One directional growth along the (211) plane was confirmed by both HRTEM and XRD. Optical characterization using UV-visible revealed a red shift in the absorption edge which is marginally extended in the visible region. Subsequent Tauc plot analysis showed decrease in the band gap reflecting a decrease in the energy threshold of TiO2 from N-doping. A FTIR spectrum showed Ti–O and Ti–N vibrations confirming the presence of N in the TiO2 matrix. XPS analysis was used to examine electronic states of nitrogen incorporation; peaks at 397.1 and 399.5 eV were attributed to substitutional (Ti–N–O) and interstitial (Ti–O–N) site doping, respectively. Quantification of N content in 5% N-doped TiO2 showed nitrogen concentration of 4.5% which is very close to the doped concentration, suggesting no loss of N in the hydrothermal reaction. The applicability of both undoped and N–TiO2 was tested for photosplitting of H2O and degradation of methyl orange (MO) under artificial solar light (xenon lamp) and natural solar light. Results of hydrogen production in aqueous solution of methanol under natural solar light were compared with artificial sunlight; N–TiO2 showed excellent photocatalytic activity under natural sunlight for H2O splitting and MO degradation. The rice grain shaped N–TiO2 sample showed a H2 generation rate of 7990 μmol g−1 h−1 under natural sunlight but only 4740 μmol g−1 h−1 under a xenon lamp. The superior photoactivity under natural sunlight may be due to the presence of both UV and visible light.
A. Introduction
Among transition metal oxide photocatalysts, titania (TiO2) has been an intensively investigated candidate within the ambit of various potential applications such as photocatalytic water splitting,1–3 organic pollutant degradation,4–7 supercapacitors,8 dye sensitized solar cells (DSSC),9–12 Li-ion battery photo anodes,13,14 gas-sensors,15,16 and super-hydrophilicity.17 This can be ascribed to its fascinating electronic and optical properties, namely, favourable electronic band structure, non-toxicity, biocompatibility, long term chemical stability towards photo-corrosion, and, importantly, commercial cost-effectiveness.18,19 However, photocatalytic quantum efficiency of TiO2 in solar applications is still limited due to its broad bandgap (3.0 and 3.2 eV for rutile and anatase phases, respectively) responding to UV radiation only (λ < 387 nm).20 But, UV radiation accounts for merely 5% of solar photons. Subsequently, another important drawback with TiO2 is a high recombination rate of photogenerated electron–hole pairs. To compensate for the above challenges, many attempts have been made to decrease the threshold energy of TiO2 or photo-excitation by red-shifting its photo-response to visible light having a wavelength (λ) range between 400–800 nm.21,22Semiconductors possess a void energy region where no energy levels are available to promote electrons and holes by photo-activation.23 Usually, dopants added to a lattice matrix would generate structural defects and thereby generate an impurity energy level in the band gap, thus resulting in narrowing it. Owing to several drawbacks of metal doping such as thermal instability and poor photocatalytic activity, non-metals such as C, S, P, and N doping are more favourable. Among these anion dopants, nitrogen seems to be the most effective and investigated anionic dopant24,25 which tailors electronic properties by providing aspects such as comparable atomic size with oxygen, metastable state formation, and small ionization energy.26 This reflects in narrowing the band gap of TiO2 by a significant overlapping of N2p states over the O2p states which ultimately results in easy promotion of electrons from VB to CB and red-shift in photo-response.27,28
Besides the band gap, another limiting factor is the high degree of recombination of electron–hole pairs which significantly relies on morphology, surface roughness, and microstructure of TiO2. It is, in turn, strongly affected by the synthesis method.29 Several physical and chemical protocols have been used to achieve N-doped titania, such as a wet chemical method,30 sputtering method,31,32 plasma activated chemical vapour deposition (PACVD),26 solvothermal/hydrothermal,33,34 anodization,35,36 spray pyrolysis,37 thermal treatment under NH3 gas flow,38 and the sol–gel method.23,39,40 But, amongst these methods, the low temperature wet chemical sol–gel process is most exploited for nano-titania. Therefore, that has been studied in detail because it is most versatile, eco-friendly, and non-polluting. In this method, size and shape14 of the nanomaterials can be easily monitored depending on hydrolysis rate, reaction time, pH, temperature, solvent used, and growth regulator (i.e., capping agent).41,42
TiO2 occurs in three different crystalline forms viz. anatase (tetragonal), rutile (tetragonal), and brookite (orthorhombic). Among these polymorphs, the anatase phase is extensively investigated for photocatalytic applications whereas the rutile phase is less used.
The novelty of the present investigation is selective growth of one dimensional (1D) rice grain shaped N-doped rutile TiO2 by a greener sol–gel followed by a hydrothermal method. Different approaches are growing day by day to enhance photocatalytic activity by controlling morphology and size using a growth controller. In essence, the morphological regulator usually acts as a physical barrier to restrict the free access of reactants or surfactant surrounded by the nucleated particle. That naturally restricts the particle from growing further and forming larger sized particles.43 In the past, various growth regulators such as acetic acid, oleic acid, and HF have been used. In this work, we studied the common effects of acetic acid and F− ions in the form of trifluoroacetic acid on morphology and crystal phase. TFA has been used to reach a low pH of a solution; Cheng et al. reported the rutile phase of TiO2 selectively synthesized in a low pH solution.34 To the best of our knowledge, H2 production under natural solar light by using rice grain like rutile N–TiO2 has not been attempted yet; hence, it is reported here.
B. Experimental method
B.1. Materials and reagents
Chemicals and raw materials were used as received without further purification; i.e., titanium(IV) tetraisopropoxide (Ti(OCH(CH3)2)4), ACROS organics; 30% w/w hydrogen peroxide (H2O2), Fisher Scientific; trifluoroacetic acid (CF3COOH) and diethanolamine from Qualigen Chemicals Ltd.B.2. Preparation of peroxo-titanate (PT) gel
PT gel was prepared by an aqueous sol–gel method; titanium oxide can be prepared from hydrolysed titanium alkoxide. In a typical synthesis, 7.45 mL of titanium(IV) isopropoxide (TTIP) was hydrolysed by drop-wise addition of TTIP in 100 mL distilled water. White precipitate of titanium hydrous oxide was formed instantly; the suspension was stirred for 10 min to complete the hydrolysis process and then kept undisturbed to settle the precipitate. After that, supernatant liquid was separated by decantation and the precipitate was washed thoroughly with distilled water for complete removal of alcohol formed during hydrolysis. The beaker containing precipitate was kept in an ice-bath and 30 mL aqueous hydrogen peroxide (30%) was added slowly over 3–5 min to get the transparent sol. An ice-bath was used for two purposes: to slow the complex formation rate to synthesize nano-particles as well as to remove heat (this reaction is exothermic; heat is evolved during formation of PT complex as an orange coloured sol). After complete dissolution of precipitate, the sol was diluted to 100 mL using distilled water to avoid immediate formation of dense gel. To prepare nitrogen-doped TiO2 an appropriate quantity of diethanolamine solution was added (1–10% N-doping) into the above orange coloured sol. The sol was stirred for 2 h for homogeneity. After keeping undisturbed for 24 h, the solution transformed into a more viscous sol, and with increasing time a transparent yellow coloured gel was obtained due to internal chemical reactions and polymerization. The final gel volume was reduced to 80 mL due to decomposition of hydrogen peroxide (H2O2) and evaporation of water over 24 h. Then, the gel was used for further treatment i.e., hydrothermal reactions as per requirements.B.3. Hydrothermal synthesis of rice grain TiO2
The product obtained by the sol–gel method is generally amorphous; thus, to induce crystallization, it is essential to anneal the gel at a higher temperature or apply hydrothermal treatment at a moderate temperature. The hydrothermal method has advantages over the calcination method, as the moderate reaction temperature will inhibit grain growth similarly to calcination and the growth of particles/crystallites is generally uniform in size and shape as well. Granular spherical TiO2 particles are obtained after treating wet PT gel hydrothermally at 180 °C due to the lower surface energy of spherical-shaped particles. However, synthesis of other morphologies (nanotubes, nanorods, and nanobelts) is kinetically less favourable due to greater surface energy. Therefore, it is advisable to use certain growth controlling agents for selective growth of a desired morphology and phase.In brief, to adjust to an acidic pH, different percentages (1, 3, 5, and 10% by volume) of TFA is added to the yellow coloured wet gel (80 mL) followed by addition of distilled water to make the final volume of solution to 100 mL. The pH of the as prepared PT gel (without addition of TFA) is measured at around 6 whereas, by addition of 1, 3, 5, and 10% TFA into the gel, the pH changes to 2.0, 1.7, 1.5, and 1.3, respectively. After adding TFA, the reaction mixture was stirred vigorously using a magnetic stirrer for homogenization each time. It is then transferred to a Teflon-coated stainless steel autoclave and kept in an oven at 180 °C for different times (i.e., 12, 24, 36, and 48 h). After completion of the reaction, the reactor was cooled to room temperature naturally and the product was collected by centrifugation. Subsequently, the precipitate was then washed with copious amounts of distilled water and finally with AR grade ethanol. The product was dried at 100 °C for 4 h. The products obtained were named as TiO2–12F, TiO2–24F, TiO2–36F, and TiO2–48F. In our previous study, the 5% N-doped TiO2 showed better activity towards degradation of methylene blue (MB);44 hence, only the 5% N-doped sample was used for further study i.e., selective growth of rutile nanorods. The 5% N-doped TiO2 gel was treated in presence of 1, 3, 5, and 10% TFA solution and the products/samples obtained after this treatment were labelled as 1NTFA, 3NTFA, 5NTFA, and 10NTFA, respectively. TiO2-48 is a sample obtained by hydrothermal treatment of gel under identical conditions but in absence of TFA and diethanolamine. These products were then subjected to characterization using various characterization techniques such as XRD, UV-visible-DRS, FTIR, XPS, FESEM, and HRTEM. After characterization, the application potential of these samples was tested for photodegradation of methyl orange and photo splitting of H2O under artificial and natural solar light.
B.4. Characterization techniques
X-ray diffraction patterns of the powders were recorded on a Bruker X-ray diffractometer (D8 Advanced) using Ni-filtered Cu Kα radiation (λ = 1.5418 Å). The crystallite size was estimated by applying the Scherrer formula to the fwhm (full width at high maximum) of the prominent peak. Optical properties of the undoped and N-doped titania nanoparticles were studied by a UV-visible spectrophotometer in diffuse reflectance mode over a spectral range of 250–800 nm. The measurements were carried out with a PerkinElmer spectrophotometer. IR measurements were performed using a PerkinElmer Spectrum One B spectrophotometer over a range of 500–4000 cm−1. Samples for measurements were prepared by mixing and grinding titania-based sample powders with KBr and pelletized using a pelletizer machine. Surface morphology and microstructure were analysed using a Hitachi FESEM instrument. For these measurements the powder samples were first sonicated in ethanol using an ultrasonic bath and then drop cast onto carbon tape. HRTEM measurements were performed with a JEOL instrument operated at an accelerating voltage of 300 kV. Samples for HRTEM analysis were prepared by vacuum drying the drop cast TiO2 nanoparticle solutions onto carbon-coated copper grids. Image processing and interplanar distance (d) evaluations were performed with the help of micrograph Gatan software. Powder samples were used for XPS measurements. The measurements were performed on a VG Microtech ESCA3000 instrument at a pressure of >1 × 10−9 Torr. General scan C1s, O1s, N1s, and Ti2p core level spectra were recorded with non-monochromatized Mg Kα radiation (photon energy – 1253.6 eV) at a pass energy of 50 eV and an electron take off angle (angle between electron emission direction and surface plane) of 55°. The overall resolution was 0.2 eV for XPS measurements. Core level spectra were background corrected using the Shirley algorithm, and chemically distinct species were resolved using a nonlinear least-squares fitting procedure. Baseline correction and peak fitting for all the samples were done using software package XPS Peak-41. Core level binding energies (BEs) were aligned with respect to the C1s binding energy of 285 eV. Equally weighed powder samples were used for PL measurements. The entrance and exit slit widths were kept the same.C. Photocatalytic study
C.1. Photocatalytic decomposition of methyl orange (MO)
Photocatalytic activity of prepared photocatalysts was tested for degradation of methyl orange using a 150 mL quartz reactor with a flat optical entry window and water circulation to maintain constant temperature. Initial concentration of the model dye solution was 20 mg L−1. A suspension was prepared by mixing 0.3 g N–TiO2 photocatalyst in 100 mL of methyl orange solution and sonicated for 10 min. Before illumination, the suspension was stirred in the dark for 1 h to ensure that adsorption–desorption phenomenon had reached equilibrium. After that, it was irradiated at ambient condition under natural solar light during sunny days (March to May months) between 10 am to 3 pm at Pune, located in Maharashtra state of India, for stipulated times. At regular intervals, samples were collected, centrifuged at 5000 rpm for 20 min to separate the photocatalyst particles, and the supernatant solution was analysed using a UV-visible spectrophotometer (Shimadzu).Changes in absorbance of methyl orange at 464 nm with irradiation time were monitored and compared with a blank i.e., experiment without catalyst under identical experimental conditions. The instrument was calibrated using known concentrations of (10–100 mg L−1) methyl orange solutions and a calibration graph was plotted. A linear relationship was developed between absorbance and concentration of methyl orange. The concentration of a tested solution was estimated from the calibration graph.
Degradation efficiency (DE) in percentage was calculated using following equation,
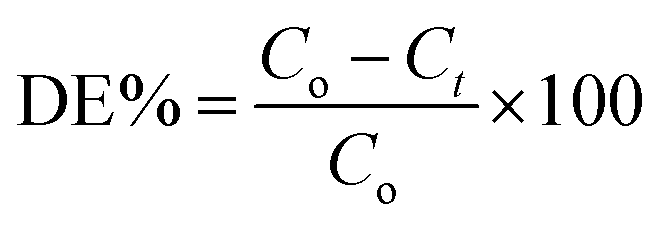
C.2. Photocatalytic H2 generation
Furthermore, the photocatalytic activity of photocatalyst for H2 generation from an aqueous solution containing methanol was performed in an air-tight quartz reactor (total volume of 70 mL) with a cooling jacket for water circulation. The reaction was carried out in double distilled water and aqueous methanol (CH3OH) solution, at ambient conditions under natural solar light on sunny days (March to May months) between 10 am to 3 pm at Pune, located in Maharashtra state of India, as well as under a 300 W xenon lamp for comparison. Intensity of solar light was measured using a digital Lux meter. The observed average intensity of sunlight reaching the surface of earth was 125![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Lux.
000 Lux.
Here, a known quantity (15 mg) of powdered photocatalyst was suspended in 20 mL distilled water and 5 mL methanol. Total volume of the reaction mixture was 25 mL with 45 mL of free space in the reactor. The reactor was made air-tight with a rubber septum, sonicated for 5 min, and then stirred for 30 min to homogenize the suspension. Finally, it was bubbled with high purity nitrogen (N2) gas to remove all dissolved gases and purging was continued further for 30 min to ensure an inert atmosphere in the reactor. After purging, the gas flow of nitrogen was stopped and the reactor was made air tight. Before and after irradiation with light, gas in the free space of the reactor was analysed using gas chromatography (GC). From primary indications, there might be an increase in gas pressure in the photoreactor after irradiation with light due to production of H2 during reaction but it was not measured quantitatively. With the xenon lamp, a UV Schott GG 400 filter with cut-off wavelength of 400 ± 6 nm was used to get rid of UV radiation.
The photo reactor was exposed to natural sunlight/xenon lamp with continuous stirring and cooling. At regular time intervals, samples of hydrogen (H2) gas in the empty reactor space were collected using a gas tight syringe. The samples were analysed immediately using a GC equipped with a thermal conductivity detector (TCD) and Porapak-Q packed column with N2 as a carrier gas.
Cyclic stability of a photocatalyst was tested by repeating the photocatalytic reaction several times under identical conditions. In the 1st cycle experiment, the reaction mixture was irradiated for 10 h in natural sunlight. Purity of the H2 gas generated in the reactor was analysed at regular time intervals by GC. The amount of H2 gas evolved was calculated by using the GC data. Before the 2nd cycle experiment, the air-tight photo-reactor was kept in the dark for 8–10 h and it was de-aerated again by purging with high purity N2 gas. To ensure inertness, specific volume of the gas were analysed by GC and then the reactor was irradiated again using solar light. The same procedure was repeated for the 3rd cycle experiments as well. Effects of Pt loading on the amounts of hydrogen generated was studied by varying the percentage of Pt loading (0.5–2% wt) on a photocatalyst surface.
D. Results
D.1. Structural analysis using X-ray diffraction (XRD)
Phase purity and crystallite size of samples were investigated using powder X-ray diffraction (XRD-D8, Advance, Bruker-AXS) with Cu-Kα radiation (λ = 1.5406 Å) in the 2θ range 20–80 degrees. Phase and particle size are essential parameters which significantly influence activity of photocatalysts.Fig. 1 shows the XRD pattern of PT gel hydrothermally treated in aqueous media at 180 °C for different times: 12, 24, 36, and 48 h. All peaks in Fig. 1 curve A (12 h) and B (24 h) match well with anatase TiO2 with a prominent peak around 25.4° corresponding to the (101) plane as the strongest reflection (JCPDS no. 21-1272). As the time of hydrothermal treatment is increased further, intensities of all peaks increased but, in addition to the anatase phase, a small shoulder from a rutile peak appeared in the XRD pattern (curve C) of a sample obtained by treatment of the gel for 36 h. However, the sample obtained after 48 h shows peaks indexed to the rutile phase as the strongest reflection, and intensity of peaks corresponding to the anatase phase decreased as compared with the 36 h sample. This clearly reveals that, at 36 h crystallization of the rutile phase starts and at 48 h more intense prominent peaks of stable rutile phase of TiO2, along with very weak peaks of anatase, have been observed, signifying increase in percentage of the rutile phase. However, a further increase in reaction time beyond 48 h does not show much change in percentage of the rutile phase; hence, the time of treatment was kept constant for all reactions.
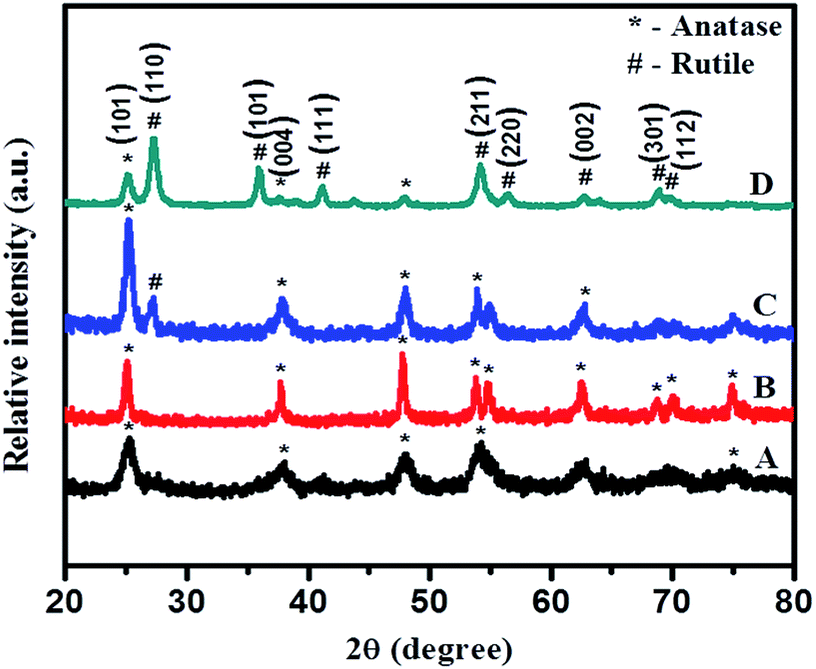 | ||
| Fig. 1 XRD pattern of TiO2 obtained by hydrothermal treatment of PT-gel at 180 °C for (A) 12, (B) 24, (C) 36, and (D) 48 h, respectively. | ||
Rutile is the most stable phase of TiO2 and it is very difficult to synthesize at normal conditions as at lower particle dimensions anatase, the metastable phase of TiO2, is predominant. For selective growth of rutile nanoparticles, TFA was used as a growth regulator. XRD reveals that using TFA, selective growth of rutile phase is quite feasible. Fig. 2 shows the XRD pattern of samples synthesized hydrothermally in the presence of aqueous 3% TFA solution at 180 °C for different times of 12, 24, 36, and 48 h. Characteristic diffraction peaks located at 27.37°, 35.93°, 41.20°, 54.31°, 56.68°, 62.77°, 68.86°, and 69.78° are indexed to (110), (101), (111), (211), (220), (002), (301), and (112) crystallographic planes (JCPDS no. 21-1276), respectively (shown in Fig. 3).
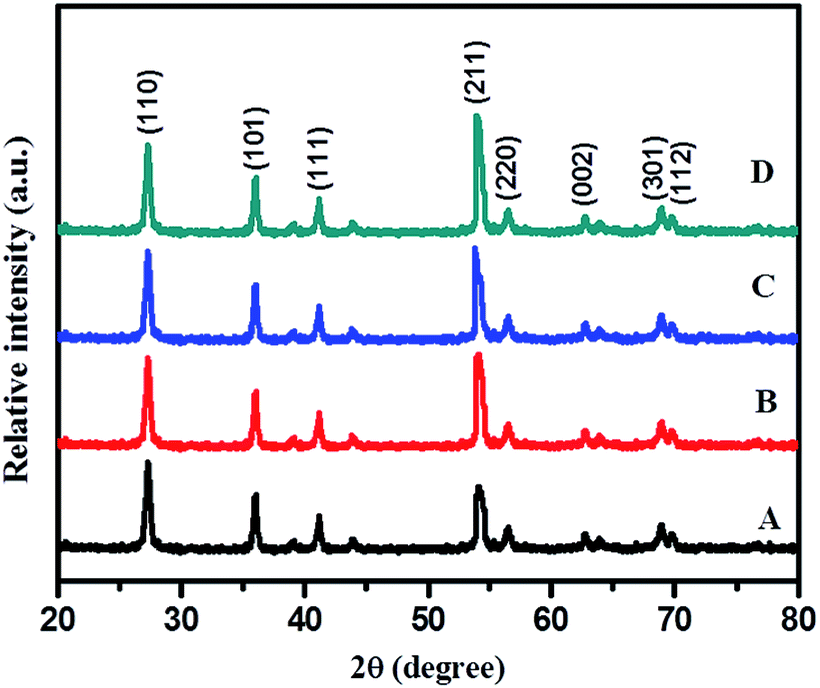 | ||
| Fig. 2 XRD pattern of (A) TiO2–12F, (B) TiO2–24F, (C) TiO2–36F, and (D) TiO2–48F. | ||
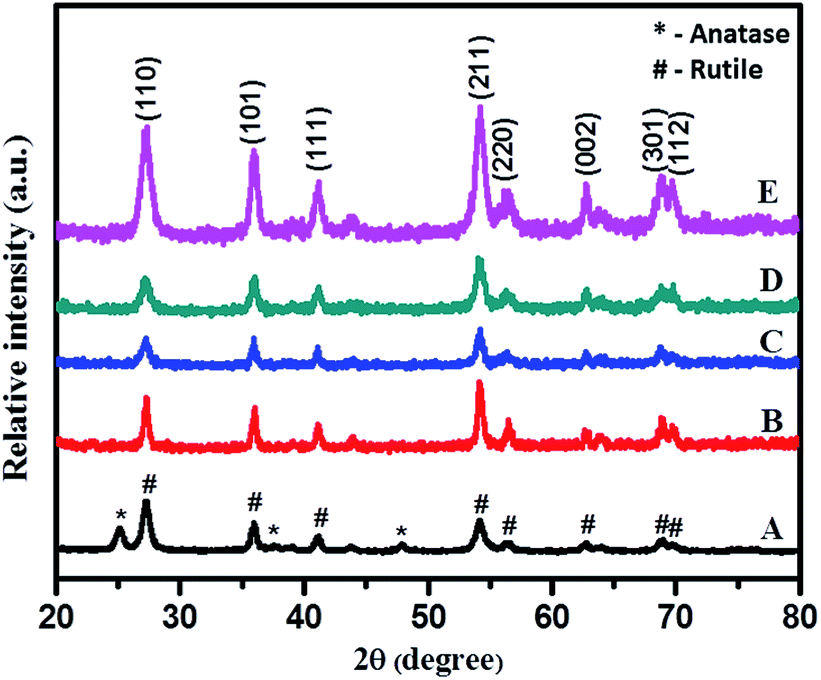 | ||
| Fig. 3 XRD pattern of (A) TiO2-48, (B) 1NTFA, (C) 3NTFA, (D) 5NTFA, and (E) 10NTFA. | ||
This clearly reveals formation of the hexagonal rutile phase. The XRD pattern of the 24 h sample shows less intense broad peaks suggesting the just beginning of crystallization and formation of smaller size particles at these conditions. As time of hydrothermal treatment is increased, the diffraction peaks become sharper and marginally intense, possibly due to growth of nanocrystals into slightly larger sized particles with time via Oswald's ripening process. The highest intensified peaks are observed for TiO2 obtained at 48 h, signifying complete growth of crystals within this duration. No appreciable change was observed in intensity and position of diffraction peaks upon increasing the treatment time beyond 48 h, suggesting complete crystallization of TiO2 within that period. Therefore, a temperature of 180 °C and 48 h time are considered to be optimum; hence, further reactions were carried out only at these optimized conditions. Diffraction patterns of all these samples show (211) as the high intensity peak (100% intensity) instead of (110), a prominent peak in a normal rutile phase which is quite interesting. This may be because of rod-like directional (1D) growth of TiO2 along the (211) plane.
Identical experiments were carried out to explore effects of TFA concentration on the growth of N–TiO2. XRD results are shown in Fig. 3. The XRD pattern of pure TiO2 (a) (without addition of TFA and nitrogen source) show peaks indexed to both anatase (JCPDS 21-1272) and rutile (JCPDS-21-1276) phases with a dominating rutile phase peak around the 2θ (theta) value of 27.37° of the (110) plane. In order to study effects of growth, the regulator, and pH, the wet PT gel was hydrothermally treated at identical conditions by varying concentrations of TFA (1–10% by volume). Surprisingly, all these samples treated at 48 h clearly revealed peaks indexed specifically to the rutile phase with (211) at 2θ value 54.31° as the strongest reflection. The sample obtained with 10% TFA (Fig. 3E) shows sharp peaks with higher intensity as compared with other samples, which may be due to more crystallinity. Furthermore, we observed that the broadening of peaks increased with increasing percentage of TFA, as expected due to increase in surface area of the nanomaterials by decreasing their particle size.45
Fig. 4 illustrates an enlarged version of the (110) peak of rutile TiO2. Here, a marginal shift in the peak position of the (110) plane to a higher angle (lower d value) was noticed with N-doped TiO2 (curve B) sample which may be due to nitrogen incorporation into the TiO2 matrix. A lower d value implies compressive strain; the origin of strain may be the difference in bonding characteristics of nitrogen and oxygen.44 This is vital for the noteworthy modification to the electronic states due to substitution of oxygen sites with nitrogen in case of N-doped TiO2, which is essential for the sizeable shift in the optical absorption spectra in the visible region. This definitely is the case for our samples and it will be discussed in a later part of this paper.
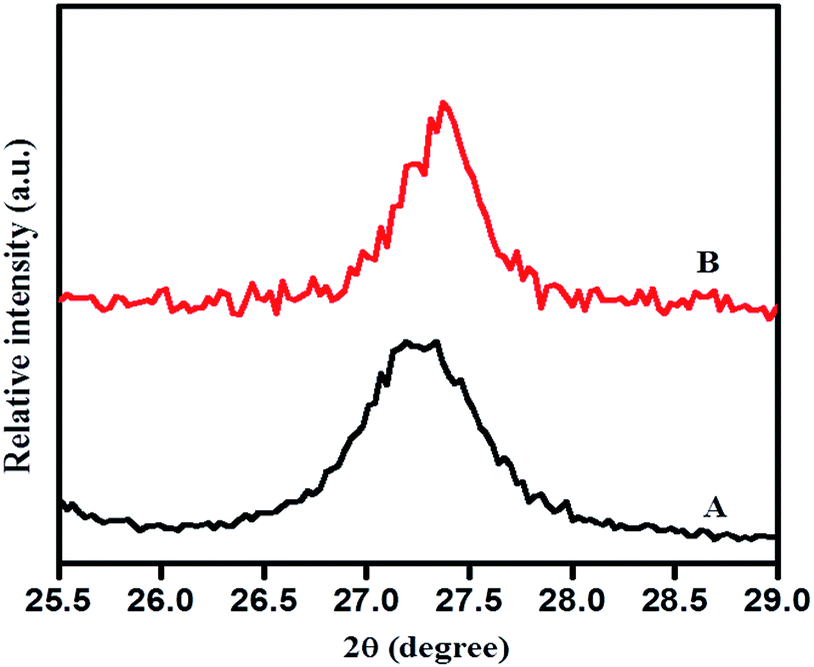 | ||
| Fig. 4 Enlarged version XRD pattern of (110) plane (A) TiO2-48 and (B) 1NTFA. | ||
D.2. UV-visible-DRS spectra
In order to see application potentials of this photo catalyst, and discuss the results of photocatalysis, it is instructive to examine the optical properties of undoped and N–TiO2 nanoparticles since this is a logically transparent way to reveal effects of dopant-induced electronic properties. Photocatalytic activity of a semiconductor catalyst is strongly related to optical absorption ability in the visible/solar region. Fig. 5 shows the ultra violet-visible diffuse reflectance spectra (UV-vis-DRS) of the TiO2-48 and 5% N-doped TiO2 gel treated hydrothermally using different concentrations of TFA.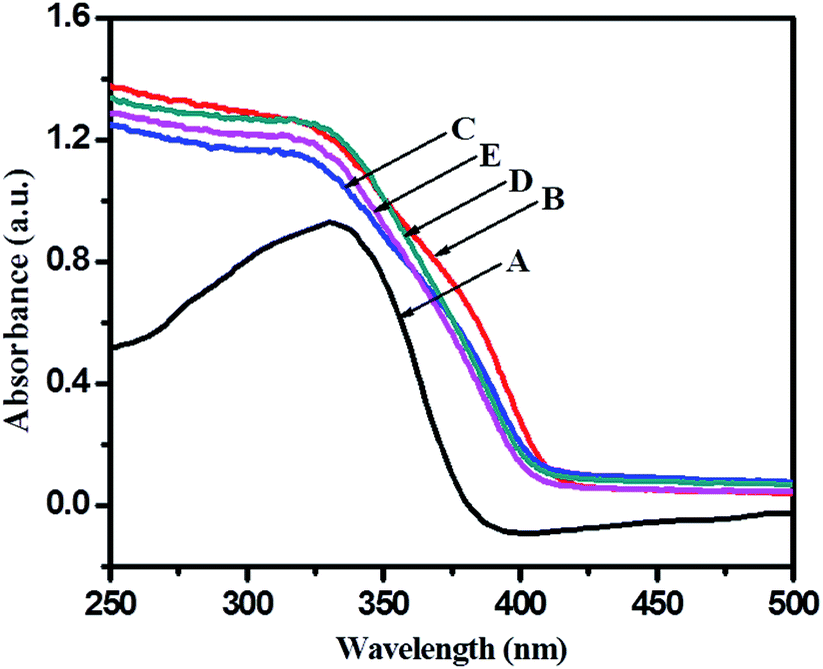 | ||
| Fig. 5 UV-visible diffuse reflectance spectra of (A) TiO2-48, (B) 1NTFA, (C) 3NTFA, (D) 5NTFA, and (E) 10NTFA. | ||
UV-visible spectra of an undoped sample (TiO2-48) shows absorption edge at around 388 nm (3.05 eV) which is very close to the fundamental absorption edge of TiO2. With N-doped samples there is marginal shift in the absorption edge towards the visible side (red shift) of the spectrum. Generally, when doping nitrogen in TiO2, nitrogen may occupy either some of the oxygen sites by substitution or occupy a position at the interstitial sites corresponding to decrease in band gap due to formation of a mid-gap energy level between VB and CB.46 Band gap energy can be calculated from the UV-visible absorption cut-off wavelength or from a Tauc plot. As mentioned earlier, to increase photocatalytic activity of a semiconductor, it is crucial to enhance visible light absorption, which can be realized through band structure engineering.40,47 The absorption edge for TiO2-48 is around 388 nm whereas 1NTFA, 3NTFA, 5NTFA, and 10NTFA samples show absorption edge at 425, 421, 419, and 416 nm, respectively. The 1NTFA sample shows highest absorption edge and, with increasing concentration of TFA in the hydrothermal treatment, the absorption wavelength of NTFA catalysts continuously decreased. The reason may be a decrease in particle size with increasing concentration of TFA. In XRD results also there is broadening of peaks with increasing concentrations of TFA. This peak broadening may be due to smaller particle dimensions. So, the results of UV-visible and XRD match well and are complementary to each other.
A Tauc plot (Fig. 6) shows that there has been a substantial decrease in band gap energy for N-doped samples. The plot of (αhν)1/2 versus band gap energy (hν) in eV between VB and CB (Tauc plot) shows that pristine TiO2 displayed an energy threshold in the UV region corresponding to an estimated energy band gap (Ebg) of 3.05 eV. 1NTFA, 3NTFA, 5NTFA, and 10NTFA show a band gap of 2.81, 2.86, 2.90, and 2.93 eV, respectively. Details of absorption edge (nm) with their corresponding band gap values in eV are tabulated in Table 1. Among all NTFA samples the lowest excitation energy (band gap) was derived for 1NTFA. Therefore, the 1NTFA sample shows good response to visible light which may be useful for photocatalytic activity in sunlight. The presence of nitrogen in TiO2 in NTFA samples was further confirmed by FT-IR analysis.
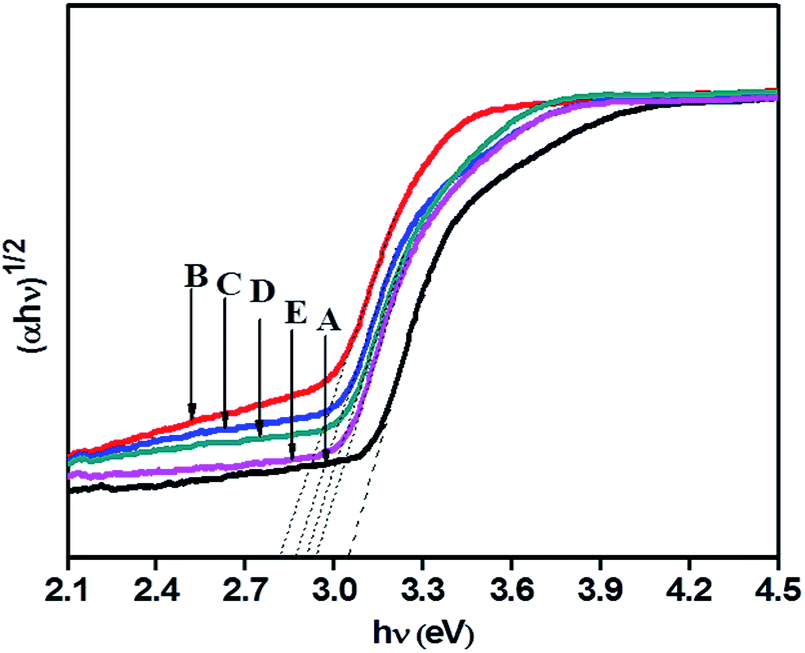 | ||
| Fig. 6 Tauc plot of (A) TiO2-48, (B) 1NTFA, (C) 3NTFA, (D) 5NTFA, and (E) 10NTFA. | ||
| Sr. No. | Sample name | Absorption edge (nm) | Band gap (eV) | Specific surface area (m2 g−1) |
|---|---|---|---|---|
| 1 | TiO2-48 | 388 | 3.05 | 65.2 |
| 2 | 1NTFA | 425 | 2.81 | 57.94 |
| 3 | 3NTFA | 421 | 2.86 | 44.64 |
| 4 | 5NTFA | 419 | 2.90 | 62.1 |
| 5 | 10NTFA | 416 | 2.93 | NA |
D.3. FT-IR
Fig. 7 displays Fourier Transform Infra-Red (FTIR) spectra of hydrothermally treated undoped (TiO2-48) and N-doped TiO2 (1NTFA) analysed between 500–4000 cm−1. In IR spectra, a sharp band centred at 500–750 cm−1 is attributed to a metal oxygen (Ti–O) bond as well as the bridging Ti–O–Ti stretching mode.44,48 The IR signal in the range of 400–1250 cm−1 is characteristic of the formation of the O–Ti–O lattice. Note that in a NTFA curve, the structure in this region is characteristically different as compared to undoped TiO2. The very broad peak at 3300 to 3750 cm−1 is assigned to fundamental stretching vibrations of monodentate O–H (free or bonded) and surface adsorbed water molecules or a hydroxy (-OH) group. The weak band located at 1620–1630 cm−1 corresponds to the bending vibration of O–H. The band at 1076.5 cm−1 is attributed to vibration of the Ti–N bond25,49 and the band at 1041 cm−1 is due to nitrate and hyponitrate groups.50 In the IR spectrum, peaks corresponding to hydrocarbons are absent. Similarly, there is no evidence of any amine complex formation with titanium in the 1NTFA sample. The signal at 1390 cm−1, which is present in N-doped TiO2, corresponds to the symmetric weak IR band (stretching) of nitro compounds and the band at 1076.5 cm−1 to the Ti–N bond indicating presence of nitrogen in the TiO2 matrix.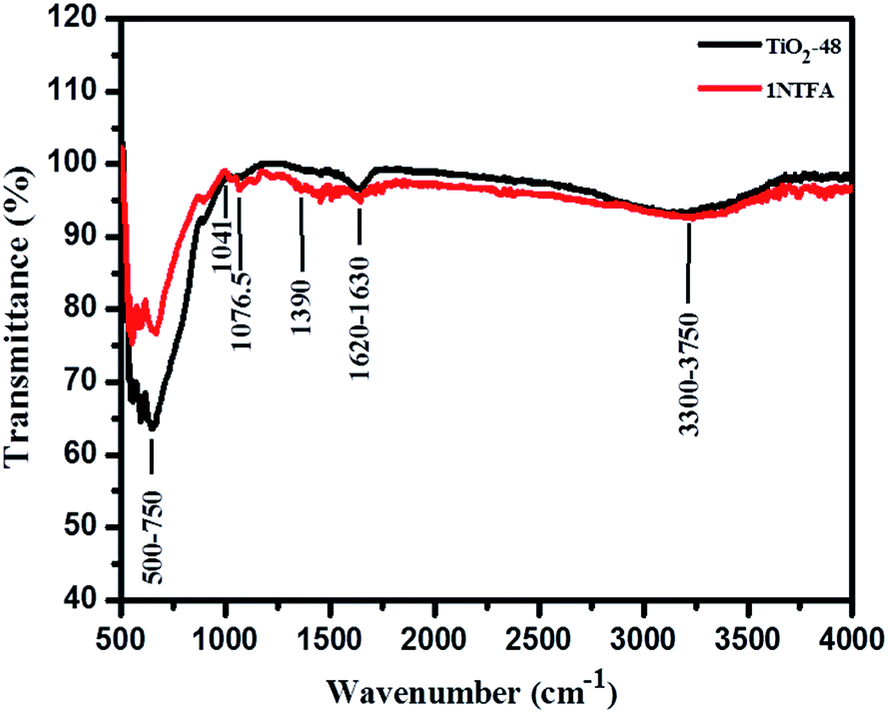 | ||
| Fig. 7 FT-IR spectra of TiO2-48 and 1NTFA samples. | ||
D.4. Surface morphology using FESEM and HRTEM
Particulate surface morphology and microstructure was examined by field emission scanning electron microscopy (FESEM). Influence of TFA concentration and time on morphology was studied by increasing the percentage of TFA in a reaction mixture and by varying reaction time. Fig. 8a and b shows the images of TiO2 obtained after hydrothermal treatment in the presence of 3% TFA for 24 h which provides clues on initialization formation of rods on bulk gel island surfaces. Upon increasing treatment time to 36 h, wire like clusters are formed on the surface of bulk gel (Fig. 8c and d). Further extending the reaction time to 48 h shows growth of different microstructures such as nanorods, cubes, and spheres (Fig. 8e and f) which is quite interesting. However, in XRD as well as FESEM analysis, it was observed that there was no further growth of particles; hence, 48 h was decided as the optimum reaction time for complete growth of TiO2 nanostructures.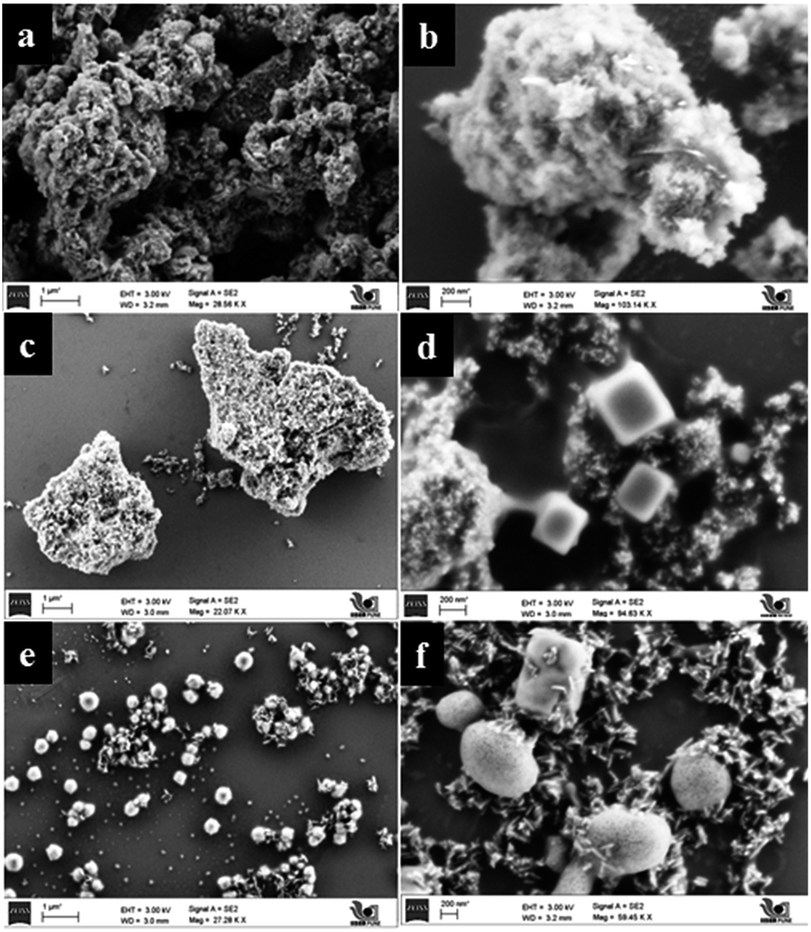 | ||
| Fig. 8 FESEM micrographs of (a and b) TiO2–24F, (c and d) TiO2–36F, and (e and f) TiO2–48F. | ||
In Fig. 9a and b, pure TiO2 (TiO2-48) shows a porous structure with larger size agglomerated lumps without any crystallization. Influence of pH on morphology was studied under similar conditions by only changing the percentage of TFA and adjusting pH to the acidic side. Fig. 9c–f shows FESEM images of samples obtained by treating the gel in presence of TFA (1–3% by volume). The SEM image of a sample obtained by treating gel in presence of 3% TFA (3NTFA) reveals growth of mixed morphological TiO2 including clusters of nanowires/nanorods, cubes, and spheres shown in Fig. 9e and f. To obtain specific morphology of the rutile phase, the same reaction mixture was tried with varying acidic conditions. When NTFA gel was treated using 1% TFA solution, nearly uniform shaped nanorods were obtained for 1NTFA sample (Fig. 9c and d). These morphologies looked like rice grain. Careful observation suggests that all rice shaped nanorods are nearly uniform in size with an aspect ratio (longer to shorter distance morphology) 6–7. Aspect ratio plays a very important role in photocatalytic activity; diameters of the nanorods are predominately in the range of 30–35 nm and length of 170–200 nm.
 | ||
| Fig. 9 FESEM micrographs of (a and b) TiO2-48, (c and d) 1NTFA, and (e and f) 3NTFA. | ||
Furthermore, by increasing percentage of TFA in a reaction mixture to 5% by volume (5NTFA), larger size cubes along with other morphological shapes are observed with an increase in size of TiO2 rods. SEM images of 5NTFA and 10NTFA samples are shown in Fig. S1(a–d) in ESI.† Careful observation of ESI Fig. S1(a and b†) suggests small nanorods start assembling with each other. Whereas, N–TiO2 obtained by treatment in 10% TFA solution i.e., (10NTFA) shows TiO2 particles which are very large and agglomerated (Fig. S1c and d in ESI†).
D.5. HR-TEM
The surface morphological investigation of NTFA samples was confirmed using high resolution transmission electron microscopy (HRTEM). TEM images (Fig. 10a and b) also confirm the formation of rice-like nanorods. It is clearly observed that, though diameters and lengths of nanorods differ, their aspect ratios are nearly the same. Fig. 10c is a lattice image of TiO2 nanorods which provides information on crystal orientation and interplanar distance (d). It also shows the good-crystalline nature of TiO2 nanorods with lattice distance (d) 0.168 nm corresponding to a (211) plane, which supports anisotropic one directional growth along it. The dot-like selected electron diffraction (SAED) pattern (Fig. 10d) confirms the single crystalline nature of rutile titania. The dotted structure corresponds to different planes, (111), (211), and (303) with d spacing of 0.219, 0.168, and 0.083 nm, respectively. HRTEM images of 3NTFA and 5NTFA are shown in ESI Fig. S2 and S3.† In HRTEM of the 3NTFA (ESI Fig. S2†) sample it was observed that there is formation of many nanorods and they are coupled with each other (a and b). The lattice image of these TiO2 nanorods show an interplanar distance (d) of 0.168 nm which corresponds to the (211) plane. Here, surprisingly, the SAED pattern is ring type suggesting a polycrystalline nature with planes (110), (101), and (211) corresponding to interplanar distances (d) 3.24, 2.48, and 1.68 Å, respectively. | ||
| Fig. 10 High resolution transmission electron micrographs (a) low magnification image (b) high magnification image (c) lattice image and (d) ED pattern of 1NTFA. | ||
D.6. XPS
XPS analysis served as a decisive tool to determine nitrogen presence and explore its chemical state in N-doped TiO2 samples. Fig. 11 summarizes XPS analytical results of N-doped TiO2 nanoparticles. It is well-known that, although XPS is a surface technique, a significant portion of the total volume of N-doped TiO2 nanorods still should be sampled because their diameters range between 18–30 nm and the particles are randomly oriented on the substrate. X-ray photoelectron spectroscopy (XPS), which investigates elemental composition, chemical states of the elements, and existence of defects in crystal structure, served to be a critical technique. A high resolution survey spectrum (Fig. 11a) of 1NTFA XPS confirms the presence of predominantly Ti2p, N2p, and O1s elements. The N2p XPS spectrum (Fig. 11c) indicated one broad peak deconvoluted into three peaks at 397.1, 399.5, and 401.4 eV suggesting three different environments of nitrogen within the same sample. A low binding energy peak around 397.4 eV is attributed to substitutional nitrogen doping (Ti–N–O) by replacing oxygen atoms from TiO2.38,51,52 The actual N peak from Ti–N–O is located at 397.1 eV; this slight variation in binding energy is explained by variations in chemical environment and oxidation states of the elements.53 The peak positioned at 399.5 eV indicates presence of interstitial nitrogen in the form of Ti–O–N species25,38,40 while the peak at 401.4 eV can be ascribed to the presence of molecularly chemisorbed dinitrogen (γ-N2) at the surface.54 Thus, we can definitely conclude that N is incorporated in two ways: Ns (substitutionally) and Ni (interstitially) into the TiO2 lattice. However, a mixed phase TiO2–TiN can be ruled out because X-ray diffraction does not show any peaks of TiN. Also, absence of a peak at or near 396 eV for the N1s core level once again confirms that the TiN phase was not formed. In both cases, the O1s core level peak appears at the same place, indicating the nature of oxygen to be similar. However, a small broadening of the O1s core level is clearly visible in the N-doped TiO2 sample. This might be due to the presence of oxygen and nitrogen in the same lattice units of TiO2. Here, in Ti2p XPS spectra, two symmetric peaks located at binding energy 458.3 and 464.4 eV relating to Ti2p(3/2) and Ti2p(1/2) are shown in Fig. 11b. Typically, FWHM of each spin orbit component is the same, but the Ti2p(1/2) orbit is broader than Ti2p(3/2) and therefore Ti2p(1/2) is shorter than Ti2p(3/2). This spin doublet can be assigned due to spin–orbital splitting of Ti4+ in the TiO2 lattice, since splitting between two peaks are 5.7 eV.15,25,55 Moreover, presence of Ti3+ can act as colour centres to actively absorb visible light, which is as shown in the XPS peak at 457.5 eV.56 Furthermore, both peaks of Ti2p contribute to three different oxidation states of titanium i.e., Ti(IV), Ti(III), and Ti(II). The O1s XPS spectra fit into three Gaussian components located at 529.5, 531.9, and 533.4 eV (Fig. 11b). The lowest peak binding energy (529.5 eV) is assigned to oxygen bound to Ti4+ of TiO2 (Ti–O–Ti) i.e., lattice oxygen, whereas the peak around 531.9 eV is ascribed to hydroxy groups adsorbed at the surface of TiO2 (Ti–OH).51,55,57 Further, the peak around 533.4 eV is attributed to presence of carboxyl oxygen (C–O/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). Quantitative estimation of the N/O ratio was also made by XPS measurements with both TiO2 and 1NTFA samples. We observed that the estimated nitrogen concentration in 5% N-doped TiO2 (1NTFA) is 4.5% which closely matched a result between the theoretical and observed values. Absence of a peak at around 292 eV (CF2) and 294 eV (CF3) signifies no chemisorbed TFA molecules present on the surface of 1NTFA.58 Non-existence of peaks at around 687.5–688.0 eV in F1s spectra, which correspond to CF3 groups of chemisorbed TFA, and a peak at 684.8 eV, due to physically adsorbed fluoride ions on the TiO2 surface or ligand exchange from Ti–OH to Ti–F, confirms the absence of chemisorbed or physically adsorbed F in NTFA.59,60 Therefore, the possibility of presence of F in the TiO2 matrix is ruled out. Fig. S4 in ESI† summarizes the XPS spectra of pure TiO2 (TiO2-48). Survey spectra (Fig. S4a in ESI†) just show peaks of Ti2p, and O1s, suggesting presence of both these elements only. Here, absence of peaks related to N2p suggests the formation of pure TiO2 phase without any impurities.
O). Quantitative estimation of the N/O ratio was also made by XPS measurements with both TiO2 and 1NTFA samples. We observed that the estimated nitrogen concentration in 5% N-doped TiO2 (1NTFA) is 4.5% which closely matched a result between the theoretical and observed values. Absence of a peak at around 292 eV (CF2) and 294 eV (CF3) signifies no chemisorbed TFA molecules present on the surface of 1NTFA.58 Non-existence of peaks at around 687.5–688.0 eV in F1s spectra, which correspond to CF3 groups of chemisorbed TFA, and a peak at 684.8 eV, due to physically adsorbed fluoride ions on the TiO2 surface or ligand exchange from Ti–OH to Ti–F, confirms the absence of chemisorbed or physically adsorbed F in NTFA.59,60 Therefore, the possibility of presence of F in the TiO2 matrix is ruled out. Fig. S4 in ESI† summarizes the XPS spectra of pure TiO2 (TiO2-48). Survey spectra (Fig. S4a in ESI†) just show peaks of Ti2p, and O1s, suggesting presence of both these elements only. Here, absence of peaks related to N2p suggests the formation of pure TiO2 phase without any impurities.
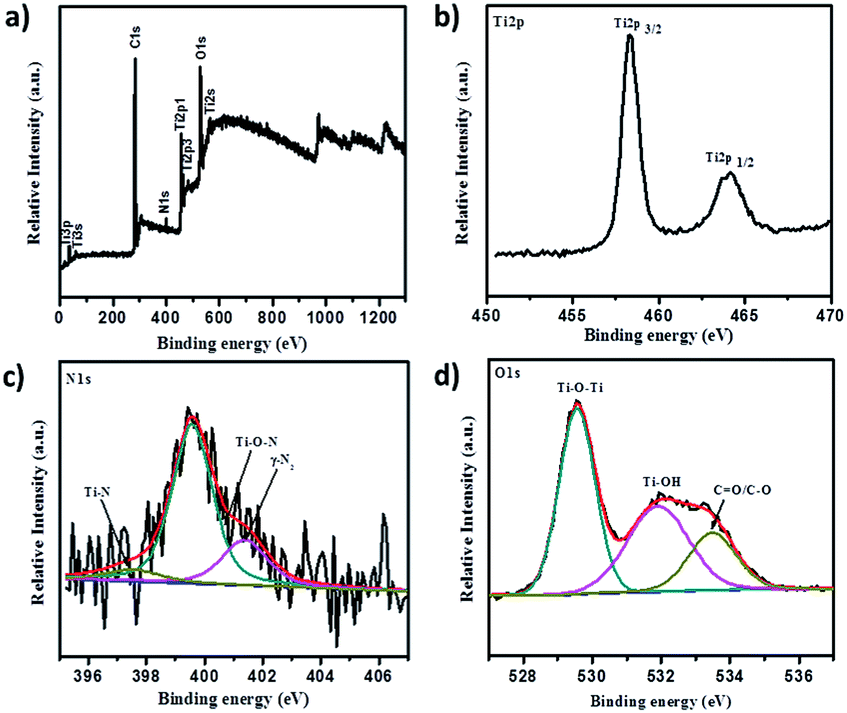 | ||
| Fig. 11 X-ray photoelectron spectra of 1NTFA (a) survey spectra, (b) Ti2p XPS spectra, (c) N1s XPS spectra, and (d) O1s XPS spectra. | ||
D.7. Photoluminescence (PL) spectra
Photoluminescence spectra were investigated for radiative recombination efficiency of electron hole charge carriers and are considered to be a sensitive technique. This depends on the carrier density of upper energy states, density of lower energy states, and transition probability of upper energy states to lower vacant states. Fig. 12 shows room temperature photoluminescence spectra of TiO2-48 and 1NTFA samples treated under identical conditions. Each sample was excited using an excitation source wavelength of 280 nm. The emission peak around 382 nm in the UV range originates from near band edge (NBE) free excitation emission i.e., direct transition from VB to CB.44,55 The latter additional emission signals at 450, 468, 482, and 492 nm originate from the charge transfer transition from oxygen vacancy trapped electrons.61 This effective quenching in PL spectra is observed by two tracks: first, created electrons trapped by oxygen vacancy and holes by doped nitrogen in a TiO2 lattice and, second, excited electron transfer from the valence band to a new existing energy level introduced by nitrogen incorporation.62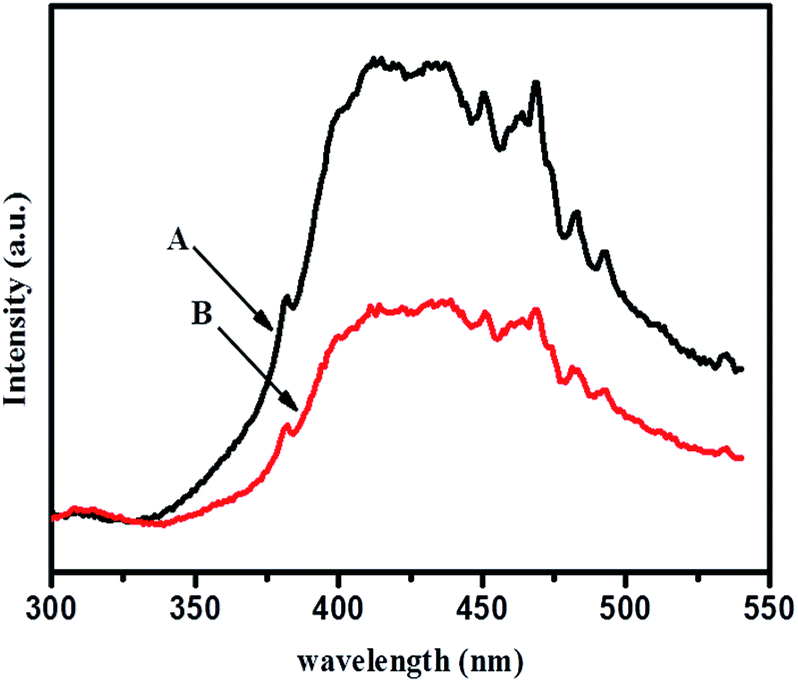 | ||
| Fig. 12 Photoluminescence spectra of (A) TiO2-48 and (B) 1NTFA. | ||
E. Discussion
Titania has been extensively used in photo-catalysis due to its unique semi-conductive properties. It has been already proven that size and shape of the material affects photocatalytic activity. Different shapes of TiO2 have been prepared by phase transformation of titanium peroxo gel in the presence of shape controllers. One directional (1D) morphology is expected to provide a wide variety of properties. TiO2 nanorods accelerate the reaction rate by providing an efficient electron transport pathway, high anisotropy, and a high surface to volume ratio63 as compared to nearly spherical nanoparticles (resulting in large defects, surface states, and grain boundaries). A high surface to volume ratio provides a high density of active sites available for surface reactions and high interfacial charge carrier transfer rate. Moreover, the probability of e−/h+ recombination is reduced by increasing delocalization of charge carriers in rods where they are free to move throughout the length of a crystal.64 The size and shape of a nanomaterial can be controlled by the kind of surfactant used and improved by its quantity. Various methods have been reported for synthesis of TiO2 nanorods e.g., sol–gel,65 hydrothermal,66 template assisted synthesis,51 and a magnetron sputtering method.32 Different surface ligands, which bind selectively to a respective plane, can control nanorod formation.In the present work, we employed a modified sol–gel method followed by hydrothermal treatment at low temperature. Here, a homogenous chemical route is presented to synthesize rutile TiO2 1D nanorods selectively under mild conditions. 1D growth of TiO2 demonstrated two main reactions: first is a condensation–dehydration reaction of the titania precursor to form a Ti–O–Ti complex species, and second is anisotropic growth along a specific crystallographic direction by TFA which acts as a surface ligand. It has been reported that one directional growth can be attenuated by fast hydrolysis of a reaction precursor. The growth of an extensive Ti–O–Ti network proceeds through two steps: hydrolysis to form an unstable titanium hydroxy-alkoxide intermediate and, then poly-condensation by elimination of water molecules.67 Fig. 13 clearly shows that an amorphous Ti–O–Ti gel network is formed by fast hydrolysis and poly-condensation. Under hydrothermal treatment there is destruction of oxo gel into small nanoparticles which then act as nuclei to form large particles by the Ostwald ripening process.64 In absence of TFA, pH of the solution was near 7 (neutral). At this stage, H+ ion concentration at the TiO2 surface is very low and therefore the H+/Ti ratio is also low, which is most favourable for anatase phase formation observed in the XRD (Fig. 3A i.e., TiO2-48). The growth of TiO2 can be controlled by using a growth regulator. In the presence of TFA (acidic condition), partial dissociation of TFA into fluoride ions at the surface of TiO2 can act as a ligand resulting in a high H+/Ti ratio which directly reflects on phase stability (and that is very important according to application viewpoints). These terminated fluorine anions facilitate 1D anisotropic growth rather than inhibiting growth along different crystallographic directions. At optimum TFA, the nuclei result in formation of rutile (a thermodynamically more stable phase) TiO2 nanorods by sharing opposite edges (each share with an adjacent neighbour in a linear chain) by poly-condensation of TiO6 octahedrons.68 It has been proposed that, under acidic conditions, rutile fibrils precipitate out whereas anatase particles are retained in dilute acid. This hypothesis supports the formation of anatase particles and rutile rods derived in our study. FESEM and HRTEM images confirm the dual role of TFA i.e., as a growth regulator and to adjust pH to the acidic side of a reaction mixture. In addition, higher protonation resulted in nucleation of rutile nuclei and further growth of these nuclei results in forming rutile TiO2 nanorods. It is well known that particle size and morphology can be controlled by nucleation and growth procedures.69
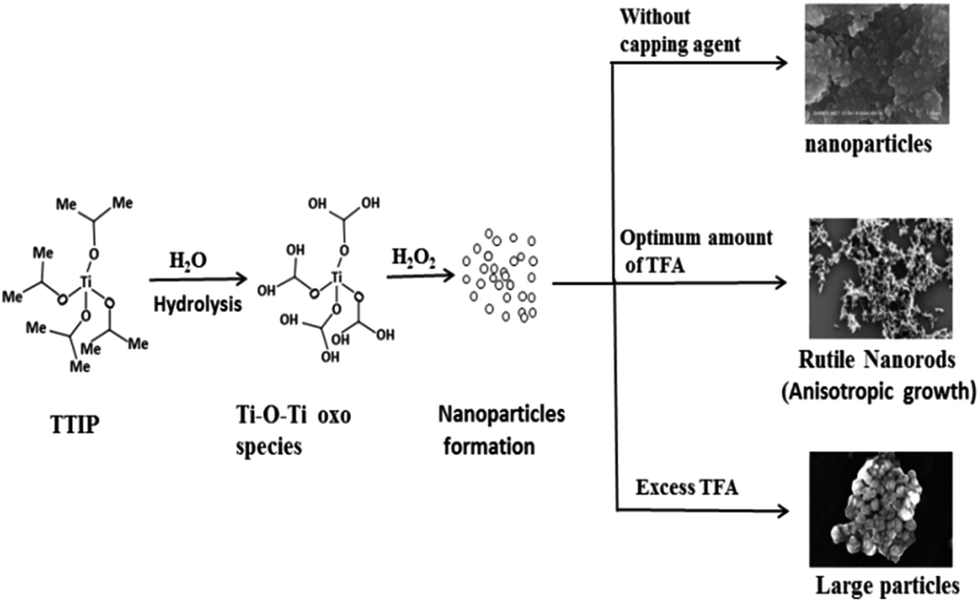 | ||
| Fig. 13 Mechanism for the synthesis of TiO2 nanoparticles and nanorods. | ||
F. Photocatalytic study
TiO2 semiconductor is white coloured pigment used from ancient times. Titania has a wide band gap (∼3.2 eV) which can only absorb UV radiation (λ < 400 nm). To make an environmental friendly society, it is essential to use solar radiation, which is sought to improve optical performance of this semiconductor material in visible light (λ < 400–800 nm). Worldwide per year energy consumption is ∼1023 watts which is mainly obtained by burning fossil fuels e.g., coal etc., while energy reaching earth's surface is 1034 watts, which is far higher than global energy consumption. It is about twice as much as energy obtained from all other of earth's non-renewable resources such as coal, oil, natural gas, and mined uranium. Therefore, different ways have been chosen to utilize this ample amount of solar light. Every day, the total energy hitting the ground is around ∼1120 W m−2. Amongst them, 42–43% visible light (400–800 nm) and 3–5% UV (<400 nm) is available at the surface of earth. Direct sunlight has luminous efficacy of ∼93 lumens (total quantity of visible light) per watt of radiant flux. Luminous efficacy is a measure of how efficiently any light source produces visible energy. Of course, this is much higher efficacy than artificial lighting. Many researchers have used xenon lamps (visible light source) as artificial sunlight for photocatalytic hydrogen production in an aqueous solution of methanol in the presence of Schott GG glass filters to eliminate UV radiation. Although a xenon lamp spectrum produces continuous light in the visible range and is very close to daylight, it is very costly to use because we must supply energy to illuminate the lamp. A xenon arc lamp, having luminous efficacy of about 30–50 lm W−1 (lumen per watt) per watt with a 1000 W bulb, has an overall luminous efficacy around 4.4–7.3% (expressed in dimension form) since all lamps use electric ballasts. By knowing all these, instead of using high watt incandescent bulbs, photocatalytic water splitting in presence of a sacrificial reagent in natural sunlight produces a hydrogen economy having more scalability in terms of system cost, and it is eco-friendly as well.F.1. Photocatalytic degradation of methyl orange
Methyl orange (MO) is toxic, mutagenic, and carcinogenic; therefore, its discharge from colour industries causes many health problems, so it is very essential to decompose it using green technology. It is not readily biodegradable as compared with methylene blue, because MO molecules have a nitrogen bearing a positive charge involved in a double bond. Therefore, in typical photocatalytic decomposition experiments, MO is chosen as a model industrial organic pollutant to evaluate photocatalytic degradation.6 Photocatalytic activity of undoped and N-doped TiO2 photocatalysts in an aqueous solution of MO at neutral pH under natural solar light was carried out, since it is very stable dye. The degradation mechanism is illustrated in detail. When TiO2 is irradiated with light of energy greater than or equal to band gap energy, it promotes electrons from valence band (VB) to the high energy conduction band (CB), with simultaneous generation of a hole at VB. Holes at the VB trapped by water or surface hydroxy groups at the surface of a TiO2 catalyst, results in formation of very reactive hydroxyl radicals (˙OH). Simultaneously, a superoxide radical (O2˙−) generates by reaction of dissolved molecular oxygen with electrons at the CB. Protonation of O2˙− immediately generates a hydroperoxy radical HO2˙ and further protonation generates a very reactive hydrogen peroxide (H2O2) molecule. Degradation of H2O2 rapidly forms O2˙− which serves to be a powerful oxidising agent for organic dye degradation to H2O and CO2 since building units of any dye molecule are carbon, oxygen, and hydrogen. The mechanism of organic dye degradation is as follows:
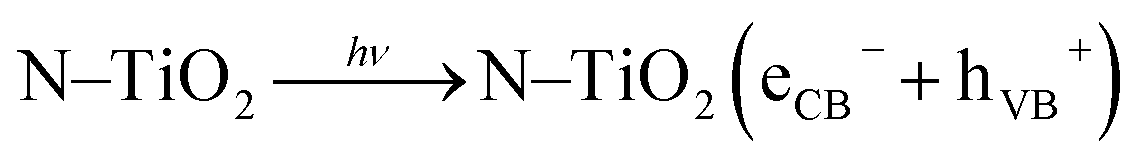 | (1) |
| e− + O2 → O2˙− | (2) |
| h+ + OH− → OH˙ | (3) |
| O2˙− + H2O → HO2˙ + OH− | (4) |
| HO2˙ + H2O → H2O2 + OH˙ | (5) |
| H2O2 → 2OH˙ | (6) |
Photocatalytic activity of undoped and modified N-doped rutile TiO2 nanorods was evaluated by degradation of methyl orange under solar light. Change in concentration of methyl orange with irradiation time was monitored by analysing the samples at regular intervals using a UV-visible spectrophotometer. UV-visible spectra of MO degradation using TiO2-48, 3NTFA, 5NTFA, and 10NTFA photocatalyst are shown in Fig. S5 in ESI.† The instrument first was calibrated using methyl orange solutions of known concentrations. The plot of change in concentration with irradiation time is as shown in Fig. 14. Direct decomposition of MO without catalyst in visible light was not observed. As well as to exclude the masking effect of surface adsorption, the reaction mixture was kept in the dark to establish the adsorption–desorption equilibrium between MO molecules and catalyst.49 In Fig. 14a, it is observed that the intensity of the characteristic absorption peak of MO (464 nm) was significantly decreased with increasing irradiation time. Amongst TiO2 and N–TiO2, 1NTFA shows superior degradation activity with 86.3% degradation under natural solar light in 180 min; this was expected due to rice shaped morphology and nitrogen doping, simultaneously. The photo-degradation efficiency of MO was observed in order of 1NTFA > 3NTFA >10NTFA > 5NTFA as shown in Fig. 14b. It was concluded that, though visible light performance was strongly enhanced by N doping (which has obvious influence on degradation of MO), degradation is also dependent on surface microstructure and particle size.
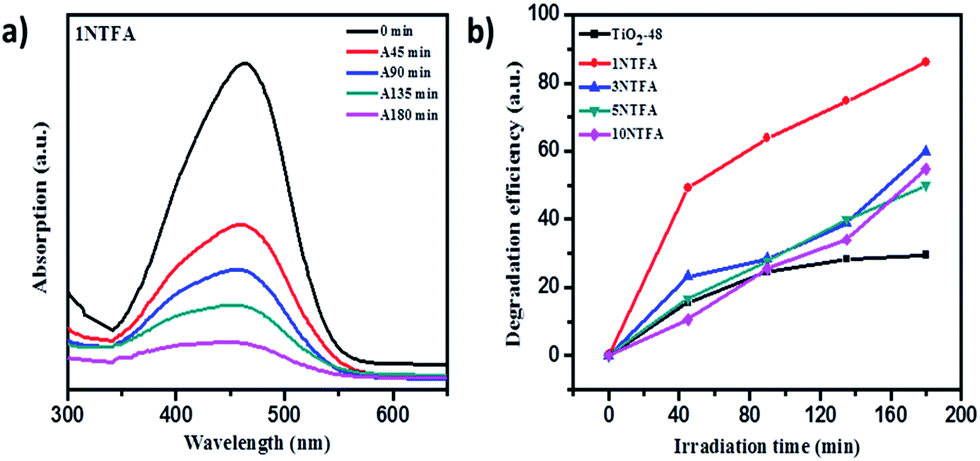 | ||
| Fig. 14 (a) UV-visible absorption spectra of MO irradiated for different time intervals using 1NTFA photocatalyst. (b) Plot of degradation efficiency versus irradiation time in min using TiO2 and N-doped TiO2 (1NTFA). | ||
In contrast, as expected, undoped TiO2 (TiO2-48) shows less decomposition of MO (around 28.9%) as compared to other N-doped samples i.e., 3NTFA, 5NTFA, and 10NTFA which show 59.9, 50.2, and 55.1% degradation under the same exposure times.
F.2. Photocatalytic H2 production
In order to evaluate photo activity, the prepared photocatalysts were further tested for hydrogen generation by water (H2O) splitting under artificial and natural solar light. This study was carried in the presence of a sacrificial agent and co-catalyst. One of the main disadvantages associated with TiO2 is fast recombination of photo-generated electrons at CB and holes at VB by releasing energy in the form of photons.69 In order to achieve higher quantum efficiency, organic compounds such as alcohols can be used as electron donors.72 An electron donor irreversibly scavenges the photo-generated holes at VB and converts itself to an oxidative product, resulting in a reduced recombination rate. Simultaneously, electrons at CB are involved in a reduction process to produce H2. Therefore, it is important to use a hole scavenger.Loading a noble metal on the surface of a photocatalyst can act as a co-catalyst which plays an important role in the charge separation process. Pt, Pd, Au, Rh, Cu, and Ni could be used as a co-catalyst. Amongst these, Pt is considered to be one of the most efficient as the Fermi level of this metal is located just below the CB of TiO2, therefore photo-excited electrons can be easily transferred to the metal co-catalyst resulting in efficient separation of the charge carrier. These accumulated electrons at the metal surface also are involved in the reduction reaction, and consequently on H2 generation, due to longer lifetimes for interfacial charge transfer.69,72
The amount of H2 produced with time was monitored by analysing the samples using a gas chromatograph (GC). The samples were collected at regular intervals and analysed for H2 content. Fig. 15 illustrates the resulting amounts of H2 produced with time using TiO2-48 and NTFA photocatalysts. Pure TiO2 (TiO2-48) and 10NTFA samples show the lowest H2 generation rate; note the curve also is not linear. Among all these photocatalysts, 1NTFA shows the highest H2 generation rate and the curve obtained is also linear suggesting no saturation in rate of H2 generation even after 5 h irradiation. The 3NTFA curve is linear until 3 h of irradiation and saturation starts after that. These results suggest that 1NTFA is a good photocatalyst for H2 generation from an aqueous solution of methanol; hence, effects of Pt loading and cyclic stability were performed using this catalyst only.
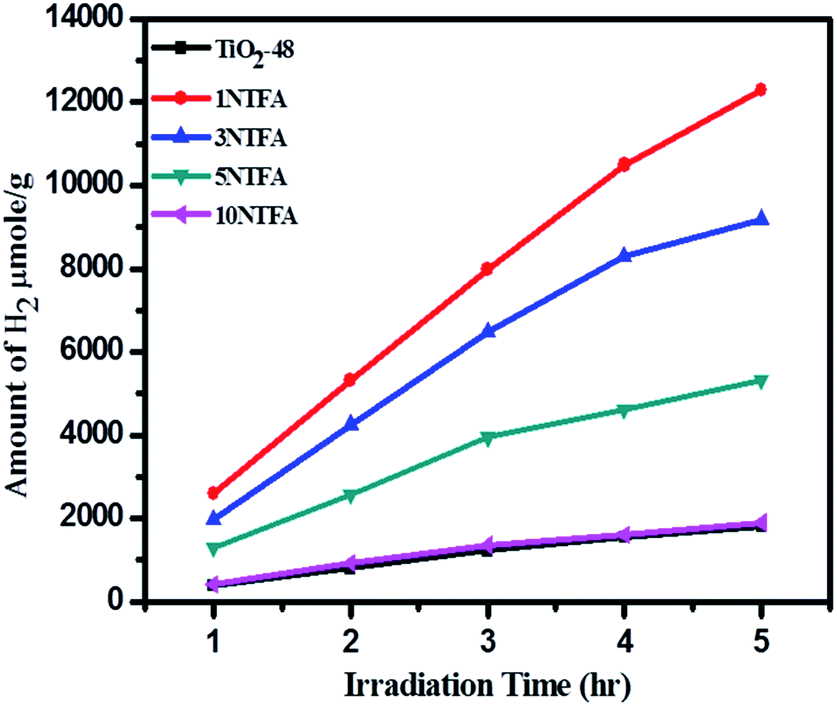 | ||
| Fig. 15 Change in amounts of H2 in μmol g−1 with irradiation time. | ||
Effects of Pt loading on catalyst surface and photocatalytic activities were studied over 1NTFA (Fig. 16) under both xenon and natural sunlight. Pt loading was varied between 0.5–2% by weight of TiO2 and used as photocatalysts for H2 generation. Amongst 0.5, 1, 1.5, and 2 (w/w) Pt loading, 1% Pt loading shows highest H2 production i.e., 7990 μmol g−1 h−1 under natural sunlight and 4740 μmol g−1 h−1 in artificial sunlight under identical conditions. High concentrations of co-catalyst might have acted as a recombination centre and ultimately increased rates of recombination of electrons and holes exponentially because the average distance between trapping sites is decrease by increases in noble metal concentration.73 Therefore, eventually, hydrogen production is monotonically stimulated with sufficient amounts of co-catalyst because accumulation of electrons becomes suppressed. In fact, a Pt co-catalyst shows higher activities for oxidation reactions.5 Therefore, a 1% loading was found to be optimum for better activity towards H2O splitting.
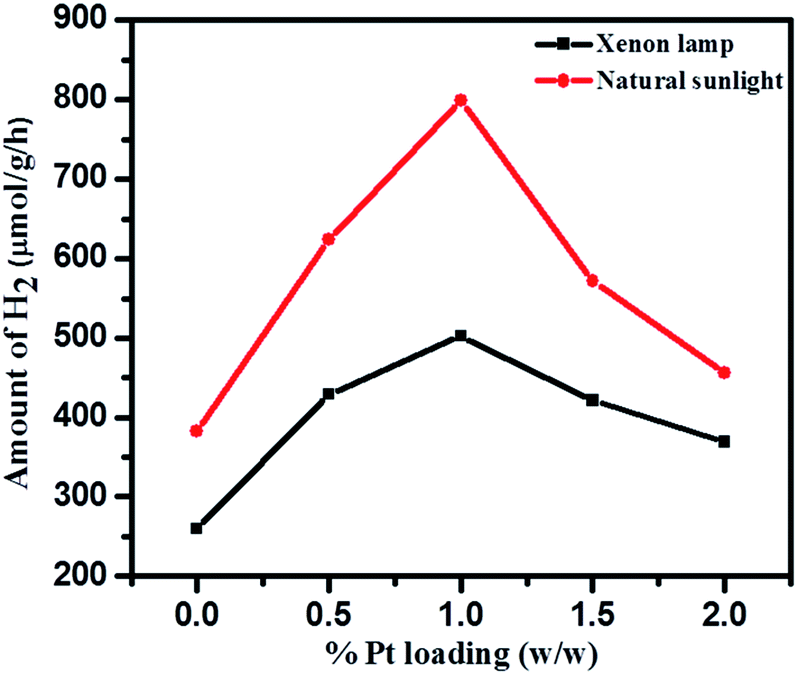 | ||
| Fig. 16 Effect of Pt loading on H2 evolution under xenon lamp and natural solar light. | ||
Cumulative H2 production over different photocatalysts under artificial and natural light is as shown in Fig. 17. The blank experiment was conducted by irradiating only a reaction mixture (water, methanol, & Pt) to confirm the possibility of H2O splitting without a photocatalyst. It was confirmed that very negligible amounts of H2 (2 and 20 μmol g−1 h−1) are produced under xenon and natural sunlight, respectively in the absence of photocatalyst. Pure TiO2-48 (without N doping) also shows less activity under a xenon lamp since UV radiation is eliminated. In contrast, TiO2-48 shows 1230 μmol g−1 h−1 hydrogen production under natural solar light because of availability of both UV and visible light. Amongst all catalysts, 1NTFA showed the highest rate for H2 generation under xenon and natural solar light i.e., 4740 and 7990 μmol g−1 h−1, respectively. 3NTFA, 5NTFA, and 10NTFA show H2 generation of 6480, 3950, and 1360 μmol g−1 h−1 under natural solar light and 3850, 2010, and 740 μmol g−1 h−1, under the xenon lamp, respectively. Table 2 lists results of H2 generation in μmol g−1 h−1 under xenon and natural solar light.
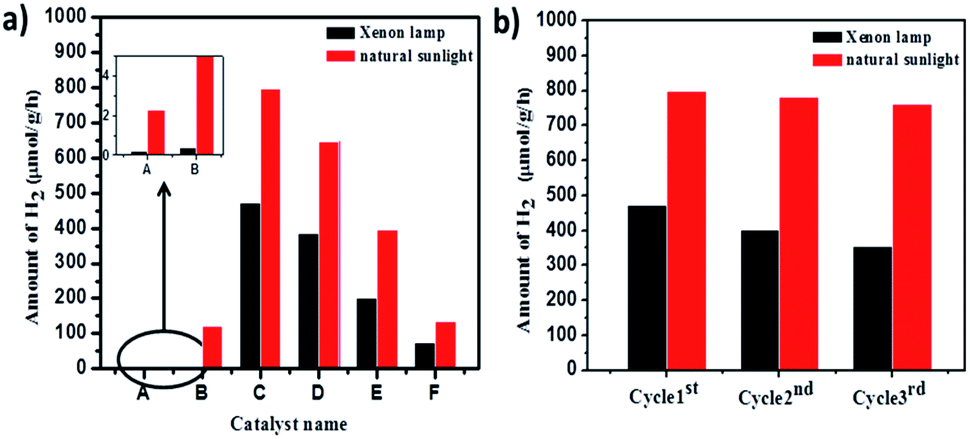 | ||
| Fig. 17 (a) Amount of hydrogen produced with time using (A) blank, (B) TiO2-48, (C) 1NTFA, (D) 3NTFA, (E) 5NTFA, and (F) 10NTFA photocatalysts. (b) Shows hydrogen in μmol g−1 h−1 after 3 cycles under xenon and natural light. | ||
| Sr. No. | Sample name | Under xenon lamp H2 (μmol g−1 h−1) | Under natural sunlight H2 (μmol g−1 h−1) |
|---|---|---|---|
| 1 | Blank | 2 | 20 |
| 2 | TiO2-48 | 4 | 1230 |
| 3 | 1NTFA | 4740 | 7990 |
| 4 | 3NTFA | 3850 | 6480 |
| 5 | 5NTFA | 2010 | 3950 |
| 6 | 10NTFA | 740 | 1360 |
In catalysis reactions, stability of the catalyst is an important criterion for long-term use of the catalyst and commercial viability of the process. Therefore, stability of the 1NTFA photocatalyst was tested under artificial and natural sunlight by recycling the same catalyst 3 times in the hydrogen generation reactions. A suspension of the photocatalyst was irradiated for 10 h in each cycle. Hydrogen generation was then converted into μmol g−1 h−1. After each cycle, the catalyst was reused for the next experiment. Recyclability was checked 3 times for the most active 1NTFA photocatalyst. Fig. 17b shows sustained and consistent activity up to the 3rd cycle. With photocatalytic activity performed under natural solar light, we observed that there was little decrease in activity of the 1NTFA photocatalyst after 3 cycles. Note results of 7990, 7830, and 7620 μmol g−1 h−1 after the 1st, 2nd, and 3rd cycles, respectively. However, under a xenon lamp activity was decreased; here, 4740, 4030, and 3540 μmol g−1 h−1 H2 was observed after the 1st, 2nd, and 3rd cycles, respectively.
G. Conclusion
A simple and green sol–gel method followed by hydrothermal treatment was developed for preparing rice-like rutile N–TiO2 nanorods. Hydrothermal treatment of the gel at 180 °C for 48 h showed highest crystallinity; hence, 180 °C temperature and 48 h exposure time is optimum for crystallizing the desired morphology and phase. Hydrothermal treatment of aqueous PT gel produces anatase TiO2 aggregates. Therefore, trifluoroacetic acid was used as growth regulator for selective growth of the rutile phase with different morphologies. A 5% N-doped TiO2 gel treated in 1% TFA solution selectively produces TiO2 nanorods; however, the gel, when treated with different concentrations of TFA, converts it into a mixed morphological N–TiO2. TFA plays a binary role i.e., adjusting acidic pH of the solution mixture to control the morphology of TiO2 and for selective growth of the rutile phase. N-doping also creates an energy level in between VB and CB of TiO2, which results in narrowing the band gap to shift the absorption to visible light. Application of N-doped TiO2 for degrading methyl orange and H2 generation shows excellent photocatalytic activity. Among all doped samples, the 1NTFA sample shows the highest activity over undoped TiO2 and other N–TiO2 samples. The N–TiO2 samples prepared are very useful for converting natural solar energy into green fuel H2.Acknowledgements
The authors would like to thank ER & IPR, DRDO, Ministry of Defence, Government of India, New Delhi, for sanctioning the grant for this work. Also authors are grateful to Director General C-MET, Pune, for the encouragement. Ms. Supriya K. Khore would like to thank Centre for Material for Electronics Technology (C-MET) for providing research facilities. The authors are very grateful to the nanocrystalline materials group C-MET, Pune for their support.References
- M. K. Kumar, K. Bhavani, B. Srinivas, S. N. Kumar, M. Sudhakar, G. Naresh and A. Venugopal, Appl. Catal., A, 2016, 515, 91–100 CrossRef CAS.
- M. N. Chong, B. Jin, C. W. K. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- S. R. Kadam, V. R. Mate, R. P. Panmand, L. K. Nikam, M. V. Kulkarni, R. S. Sonawane and B. B. Kale, RSC Adv., 2014, 4, 60626–60635 RSC.
- C. C. Wang, J. R. Li, X. L. Lv, Y. Q. Zhang and G. Guo, Energy Environ. Sci., 2014, 7, 2831–2867 CAS.
- C. Zhang, H. He and K. i. Tanaka, Appl. Catal., B, 2006, 65, 37–43 CrossRef CAS.
- J. Kaur, S. Bansal and S. Singhal, Phys. B, 2013, 416, 33–38 CrossRef CAS.
- M. U. D. Sheikh, G. A. Naikoo, M. Thomas, M. Bano and F. Khan, New J. Chem., 2016, 40, 5483–5494 RSC.
- S. Liu, Z. Cai, J. Zhou, A. Pan and S. Liang, J. Mater. Chem. A, 2016, 4, 18278–18283 CAS.
- H. Sun, J. Deng, L. Qiu, X. Fang and H. Peng, Energy Environ. Sci., 2015, 8, 1139–1159 CAS.
- B. Roose, S. Pathak and U. Steiner, Chem. Soc. Rev., 2015, 44, 8326–8349 RSC.
- Z. S. Wang, K. Sayama and H. Sugihara, J. Phys. Chem. B, 2005, 109, 22449 CrossRef CAS PubMed.
- J. Song, H. B. Yang, X. Wang, S. Y. Khoo, C. C. Wong, X. W. Liu and C. M. Li, ACS Appl. Mater. Interfaces, 2014, 4, 3712–3717 Search PubMed.
- M. H. Ryu, K. N. Jung, K. H. Shin, K. S. Han and S. Yoon, J. Phys. Chem. C, 2013, 117, 8092–8098 CAS.
- M. Ge, C. Cao, J. Huang, S. Li, Z. Chen, K. Q. Zhang, S. S. AlDeyab and Y. Lai, J. Mater. Chem. A, 2016, 4, 6772–6801 CAS.
- Y. Li, Z. Wang and X. J. Lv, J. Mater. Chem. A, 2014, 2, 15473–15479 CAS.
- F. Chekin, S. Bagheri and S. B. Abd Hamid, Sens. Actuators, B, 2013, 177, 898–903 CrossRef CAS.
- C. W. Dunnill, Z. Ansari, A. Kafizas, S. Perni, D. J. Morgan, M. Wilson and I. P. Parkin, J. Mater. Chem., 2011, 21, 11854–11861 RSC.
- G. Liu, L. Wang, H. G. Yang, H. M. Cheng and G. Q. Lu, J. Mater. Chem., 2009, 20, 831–843 RSC.
- A. Sadeghzadeh Attar, M. Sasani Ghamsari, F. Hajiesmaeilbaigi, S. Mirdamadi, K. Katagiri and K. Koumoto, J. Mater. Sci., 2008, 43, 5924–5929 CrossRef.
- H. Zeng, J. Xie, H. Xie, B. L. Su, M. Wang, H. Ping, W. Wang, H. Wang and Z. Fu, J. Mater. Chem. A, 2015, 3, 19588–19596 CAS.
- J. Zhang, Y. Wu, M. Xing, S. A. K. Leghari and S. Sajjad, Energy Environ. Sci., 2010, 3, 715–726 CAS.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- J. Li and N. Wu, Catal. Sci. Technol., 2014, 5, 1360–1384 Search PubMed.
- S. A. Ansari, M. M. Khan, M. O. Ansari and M. H. Cho, New J. Chem., 2016, 40, 3000–3009 RSC.
- G. Yang, Z. Jiang, H. Shi, T. Xiao and Z. Yan, J. Mater. Chem., 2010, 20, 5301–5309 RSC.
- S. A. Bakar and C. Ribeiro, J. Photochem. Photobiol., C, 2016, 27, 1–29 CrossRef.
- C. Di Valentin and G. Pacchioni, Catal. Today, 2011, 206, 12–18 CrossRef.
- L. Sun, J. Cai, Q. Wu, P. Huang, Y. Su and C. Lin, Electrochim. Acta, 2013, 108, 525–531 CrossRef CAS.
- H. Xu, S. Ouyang, L. Liu, P. Reunchan, N. Umezawa and J. Ye, J. Mater. Chem. A, 2014, 2, 12642–12661 CAS.
- K. J. Hwang, J. W. Lee, S. J. Yoo, S. Jeong, D. H. Jeong, W. G. Shim and D. W. Cho, New J. Chem., 2013, 37, 1378–1384 RSC.
- W. Zhang, Y. Liu, D. Zhou, J. Wen, W. Liang and F. Yang, RSC Adv., 2015, 5, 57155–57163 RSC.
- L. Meng, H. Chen, C. Li and M. P. dos Santos, Thin Solid Films, 2015, 577, 103–108 CrossRef CAS.
- S. Wang, J. Xu, H. Ding, S. Pan, Y. Zhang and G. Li, CrystEngComm, 2012, 14, 7672–7679 RSC.
- H. Cheng, J. Ma, Z. Zhao and L. Qi, Chem. Mater., 1995, 7, 663–671 CrossRef CAS.
- R. S. Hyam and D. Choi, RSC Adv., 2013, 3, 7057–7063 RSC.
- Y. Jun, J. H. Park and M. G. Kang, Chem. Commun., 2012, 48, 6456–6471 RSC.
- N. C. Raut, T. Mathews, P. K. Ajikumar, R. P. George, S. Dash and A. K. Tyagi, RSC Adv., 2012, 2, 10639–10647 RSC.
- J. Wang, D. N. Tafen, J. P. Lewis, Z. Hong, A. Manivannan, M. Zhi, M. Li and N. Wu, J. Am. Chem. Soc., 2009, 131, 12290–12297 CrossRef CAS PubMed.
- M. Li, S. Zhang, Y. Peng, L. Lv and B. Pan, RSC Adv., 2014, 5, 7363–7369 RSC.
- Z. Jiang, F. Yang, N. Luo, B. T. T. Chu, D. Sun, H. Shi, T. Xiao and P. P. Edwards, Chem. Commun., 2008, 6372–6374 RSC.
- J. Gu, Y. W. Zhang and F. Tao, Chem. Soc. Rev., 2012, 41, 8050–8065 RSC.
- T. D. Nguyen, Nanoscale, 2013, 5, 9455–9482 RSC.
- J. Yu and L. Shi, J. Mol. Catal. A: Chem., 2010, 326, 8–14 CrossRef CAS.
- T. C. Jagadale, S. P. Takale, R. S. Sonawane, H. M. Joshi, S. I. Patil, B. B. Kale and S. B. Ogale, J. Phys. Chem. C, 2008, 112, 14595–14602 CAS.
- L. Jing, W. Zhou, G. Tian and H. Fu, Chem. Soc. Rev., 2013, 42, 9509–9549 RSC.
- R. Asahi, T. Morikawa, H. Irie and T. Ohwaki, Chem. Rev., 2014, 114, 9824–9852 CrossRef CAS PubMed.
- K. Yang, Y. Dai, B. Huang and S. Han, J. Phys. Chem. B, 2006, 110, 24011–24014 CrossRef CAS PubMed.
- B. Erdem, R. A. Hunsicker, G. W. Simmons, E. D. Sudol, V. L. Dimonie and M. S. ElAasser, Langmuir, 2001, 17, 2664–2669 CrossRef CAS.
- W. Wang, Y. Liu, J. Qu, Y. Chen and Z. Shao, RSC Adv., 2016, 6, 40923–40931 RSC.
- S. Amreetha, S. Dhanuskodi, A. Nithya and K. Jothivenkatachalam, RSC Adv., 2016, 6, 7854–7863 RSC.
- T. Sreethawong, S. Laehsalee and S. Chavadej, Catal. Commun., 2009, 10, 538–543 CrossRef CAS.
- J. Wang, C. Fan, Z. Ren, X. Fu, G. Qian and Z. Wang, Dalton Trans., 2014, 43, 13783–13791 RSC.
- J. Lynch, C. Giannini, J. K. Cooper, A. Loiudice, I. D. Sharp and R. Buonsanti, J. Phys. Chem. C, 2015, 119, 7443–7452 CAS.
- B. Viswanathan and K. R. Krishanmurthy, Int. J. Photoenergy, 2012, 10 Search PubMed.
- A. Esfandiar, S. Ghasemi, A. Irajizad, O. Akhavan and M. R. Gholami, Int. J. Hydrogen Energy, 2012, 37, 15423–15432 CrossRef CAS.
- J. Yu, X. Zhao and Q. Zhao, Mater. Chem. Phys., 2001, 69, 25–29 CrossRef CAS.
- S. Sun, P. Gao, Y. Yang, P. Yang, Y. Chen and Y. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 18126–18131 CAS.
- T. Giannakopoulou, N. Todorova, T. Vaimakis, S. Ladas and C. Trapalis, J. Sol. Energy Eng., 2008, 130, 041007 CrossRef.
- E. M. Samsudin, S. B. A. Hamid, J. C. Juan, W. J. Basirun, A. E. Kandjani and S. K. Bhargava, Appl. Surf. Sci., 2016, 365, 57–68 CrossRef CAS.
- K. Lv, J. Yu, L. Cui, S. Chen and M. Li, J. Alloys Compd., 2011, 509, 4557–4562 CrossRef CAS.
- H. Tang, H. Berger, P. E. Schmid, F. Levy and G. Burri, Solid State Commun., 1993, 87, 847–850 CrossRef CAS.
- Y. Cong, J. Zhang, F. Chen and M. Anpo, J. Phys. Chem. C, 2007, 111, 6976–6982 CAS.
- B. F. Cottam, S. Krishnadasan, A. J. deMello, J. C. deMello and M. S. P. Shaffer, Lab Chip, 2007, 7, 167–169 RSC.
- P. D. Cozzoli, A. Kornowski and H. Weller, J. Am. Chem. Soc., 2003, 125, 14539–14548 CrossRef CAS PubMed.
- B. Koo, J. Park, Y. Kim, S. H. Choi, Y. E. Sung and T. Hyeon, J. Phys. Chem. B, 2006, 110, 24318–24323 CrossRef CAS PubMed.
- J. Archana, M. Navaneethan and Y. Hayakawa, Mater. Lett., 2013, 98, 38–41 CrossRef CAS.
- T. A. Kandiel, R. Dillert, A. Feldhoff and D. W. Bahnemann, J. Phys. Chem. C, 2010, 114, 4909–4915 CAS.
- T. Y. Ke, P. C. Chen, M. H. Yang, H. T. Chiu and C. Y. Lee, CrystEngComm, 2011, 13, 5292–5295 RSC.
- C. H. Liao, C. W. Huang and J. C. S. Wu, Catalysts, 2012, 2, 490 CrossRef CAS.
- Z. He, W. Que, J. Chen, X. Yin, Y. He and J. Ren, ACS Appl. Mater. Interfaces, 2012, 4, 6816–6826 CAS.
- L. Gomathi Devi and R. Kavitha, RSC Adv., 2014, 4, 28265–28299 RSC.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- W. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669–13679 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: FESEM images of 5NTFA and 10NTFA samples, HRTEM of 5NTFA and 10NTFA, degradation of methyl orange by TiO2-48, 3NTFA, 5NTFA, and 10NTFA. See DOI: 10.1039/c7ra01648d |
| This journal is © The Royal Society of Chemistry 2017 |