
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvolvement of P-glycoprotein and multidrug and toxin extrusion protein 1 in hepatic and renal berberine efflux in mice
Guofeng Wang†
a,
Jingyi Jin†a,
Jiakai Zenga,
Rong Shia,
Yan Daia,
Jiasheng Wua,
Yuanyuan Lia,
Tianming Wanga and
Yueming Ma *ab
*ab
aDepartment of Pharmacology, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China. E-mail: mayueming@shutcm.edu.cn; Fax: +86-21-51322200; Tel: +86-21-51322200
bShanghai Key Laboratory of Compound Chinese Medicines, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China
First published on 11th July 2017
Abstract
Berberine (BBR) is a natural alkaloid and commonly used drug with extensive pharmacological effects. The blood exposure of BBR is known to be extremely low while its tissue exposure is extremely high, especially in the liver and kidney where its uptake is mediated by organic cation transporters and organic anion-transporting polypeptides. However, the efflux mechanism in the liver and kidney is not clear. Therefore, the present study was designed to investigate the mechanism of hepatic and renal efflux of BBR. A transport assay of BBR using rat Madin–Darby canine kidney (MDCK)-multidrug and toxin extrusion protein 1 (rMate1) cells and a transcellular transport assay using MDCK-multidrug resistance protein 1 (MDR1) cells were conducted to evaluate the efflux mechanism. Tissue distribution and excretion of BBR were measured using liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of specimens from C57BL/6J mice co-administered BBR and itraconazole (ITZ, a P-glycoprotein, P-gp inhibitor) or pyrimethamine (PYR, a MATE1 inhibitor). The inhibitory effect of ITZ and PYR on the combined effects of BBR with P-gp and MATE1 was verified using Glide docking within the Schrodinger program. BBR uptake was observed to be pH-, time-, temperature- and concentration-dependent and was inhibited by PYR in MDCK-rMate1 cells. ITZ significantly decreased the Pappb→a values of BBR in MDCK-MDR1 cells. Co-administration with PYR or ITZ increased the concentration of BBR in the liver and kidney and decreased its excretion in urine and bile in mice. In addition, co-administration with ITZ increased the plasma concentration of BBR. Both ITZ and PYR had higher docking scores than BBR did to P-gp and rMate1, respectively and separately prevented the combination of BBR with rMate1 and P-gp. This study indicated that both MATE1 and P-gp mediated hepatic and renal efflux of BBR. These results provide salient information that enhances the understanding of the pharmacokinetics properties of BBR and its interaction with other drugs.
1. Introduction
Berberine (BBR), a benzylisoquinoline alkaloid, is mainly isolated from Rhizoma coptidis, which is a widely used traditional Chinese medicine (TCM). It has extensive pharmacological effects including cholesterol-lowering,1 antimicrobial,2 antiprotozoal,3 antidiabetic,4 anti-inflammatory,5 and antitumor.6 The multiple pharmacological properties of BBR contribute to its broad clinical application. Clinical studies have reported the effectiveness of BBR in treating conditions such as chronic congestive heart failure, hypercholesterolemia, type 2 diabetes, polycystic ovary syndrome, and irritable bowel syndrome.7 However, oral BBR has been reported to cause extrapyramidal system reactions, arrhythmia, liver dysfunction, and even death in clinics in China,8 as well as acute toxicity in animals.9 To guide rational clinical drug therapy, it is necessary to elucidate the pharmacokinetic properties of BBR.There are numerous reports on the pharmacokinetics of berberine, which is mainly absorbed in the small intestine with a bioavailability of <1% after oral administration.10 Although its plasma concentration was shown to be extremely low, the tissue concentration of BBR was considerably higher than that in the plasma.11 For example, the BBR concentration in the liver was 70-fold higher than that in the plasma.10 Berberine undergoes oxidative demethylation metabolism mediated mainly mediated by cytochrome P450 (CYP) 2D6 in mice and humans.12 The phase I metabolites further undergo sulfation and glucuronidation metabolism through uridine diphosphate glucuronosyltransferase family 1 member A (1UGT1A1) and UGT2B1 in rats.13 In the final process, 19% of BBR is excreted as a prototype after oral administration.14
It has been reported that some active transport mediated by transporters could partially explain why the tissue distribution of BBR is concentrated in the liver. For example, the organic cation transporter 1 (OCT1) and organic anion-transporting polypeptides (OATPs) were reported to contribute to the hepatic uptake of BBR in rats.15 Moreover, BBR is a substrate of OCT2 and OCT3.16 However, current knowledge of the efflux of BBR indicates that it is a substrate of P-glycoprotein (P-gp, also known as multidrug resistance protein 1, MDR1) in Caco-2 cells as well as OCT1 and P-gp in double-transfected Madin–Darby canine kidney (MDCK) cells;17,18 however the effect of P-gp on BBR efflux from the liver and kidney is unknown in vivo. In addition, it is not clear whether BBR is a substrate of multidrug and toxin extrusion 1 (MATE1), which is expressed in the liver and especially, the kidney19 and mediates the biliary clearance and renal tubular secretion of organic cations.20 Furthermore, whether MATE1 effects the efflux of BBR from the liver and kidney in vivo is unclear. Therefore, in the present study, we investigated whether P-gp and MATE1 are involved in BBR efflux from the liver and kidney using transfected cells, mice, and molecular docking analysis.
2. Material and methods
2.1 Chemicals and reagents
BBR was purchased from Sigma-Aldrich Chemie GmbH (Buchs, Switzerland) while PYR was obtained from Dr Ehrenstorfer GmbH (Augsburg, Germany). ITZ was supplied by CNM Technologies GmbH Company (Düsseldorf, Germany) and acetonitrile and methanol were of high-performance liquid chromatography (HPLC) grade from Burdick & Jackson Company (Ulsan, Korea). Formic acid and ammonium sulfate (both HPLC grade) were purchased from CNW Technologies GmbH Company (Düsseldorf, Germany) and Sigma-Aldrich Chemie GmbH (Buchs, Switzerland), respectively. Ultra-pure water was purified using a Milli-Q system (Millipore, Bedford, MA, USA) and all other reagents were analytical grade.2.2 Animals
Six-week-old male C57BL/6J mice (Certificate No. SYXK 2014-0008) were purchased from Shanghai B&K Universal Group Limited. The animals were housed and acclimatized for 2 weeks prior to the experiment in cage racks with free access to food and water on a 12 h light/dark cycle (lights on from 7:00 am to 7:00 pm) at a temperature of 22–24 °C and a relative humidity of 50 ± 10%. Animals were fasted overnight before all experiments with free access to water. The animal studies were conducted according to the Institutional Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Committee of Shanghai University of Traditional Chinese Medicine (TCM).2.3 Cell culture
MDCK cells stably expressing rat MATE1 (MDCK-rMate1) and green fluorescent protein (GFP) as an indicator protein (MDCK-GFP, mock cells obtained via the virus vector method were purchased from Shanghai Gene Chemistry Co., Ltd., (Shanghai, China) with qualified results from western blot analysis and fluorescent microscopy and verified by specific substrates. The MDCKII-MDR1 cells were provided by Professor Min Huang (School of Pharmaceutical Science, Sun Yat-sen University, Guangzhou, China). The MDCK-rMate1, MDCK-MDR1 and MDCK-GFP cells were maintained at 37 °C exposed to an atmosphere of 5% CO2 in a 100 × 20 mm tissue culture dish in 10 mL Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% penicillin, and streptomycin.2.4 Transport assay of BBR using MDCK-rMate1 cells
As an organic cation/H+ antiporter, rMate1 could play an uptake role by changing the intracellular and extracellular pH.21 To assess the optimum pH on BBR transport in cells, the following experiment was conducted. The MDCK-rMate1 and mock cells were grown on 24-well plates at a density of 2 × 105 cells per well initially for 48 h. The cells were washed twice with incubation medium (pH 7.4, in mM: 125 sodium chloride [NaCl], 4.2 sodium bicarbonate [NaHCO3], 4.8 potassium chloride [KCl], 1.2 calcium [CaCl2], 5.6C6H12O6·H2O, magnesium chloride hexahydrate [MgCl2·6H2O], 1.2 magnesium sulfate [MgSO4·7H2O], 1.2 potassium dihydrogen phosphate [KH2PO4], 0.36 sodium dihydrogen phosphate dodecahydrate [Na2HPO4·12H2O], and 25 HEPES). After preincubation with 0.5 mL incubation medium (pH 6.0, acidifying) containing ammonium chloride (NH4Cl, 30 mM) for 30 min at 37 °C, the transport assay was initiated by adding 0.5 mL incubation medium (pH 6.5–9.0) containing BBR (50 μM) at 37 °C. After 30 min, the medium was aspirated, the 24-well plates were rapidly washed thrice with 0.5 mL ice-cold incubation medium (pH 7.4), and then the cells were dissolved in 0.2 mL pure water. The 24-well plates were placed at −80 °C for three 24 h freeze–thaw cycles.Uptake experiments were performed under the optimum pH conditions for BBR transport using the same method described above. Briefly, cells were incubated in medium containing BBR at 37 and 4 °C to assess the influence of temperature. Furthermore, BBR uptake was measured at regular intervals (5, 10, 20, and 30 min) and concentrations of 0.1–10 μM to assess the time- and concentration-dependence. PYR, an inhibitor of rMate1,22 was used to determine the effect of rMate1 on BBR uptake. The protein content of cells was measured using a bicinchoninic acid (BCA) protein assay kit.
2.5 Transcellular transport of BBR in MDCK-MDR1 cells
Before culturing the MDCK-MDR1 cells, polyester membrane clear transwell plates (No. 3450, Corning, NY) were preincubated with DMEM, 10% FBS, and 1% penicillin and streptomycin for 1 h. Then, the MDCK-MDR1 cells were seeded in transwell filter inserts at an initial density of 5.6 × 105 cells per well. The plates were incubated with DMEM (1.5 and 2.6 mL in the transwell insert and lower compartment, respectively) at 37 °C, exposed to an atmosphere of 5% CO2 for 96 h. The culture medium was refreshed every other day.To measure the integrity of the cell monolayers, the permeability of fluorescein disodium salt was determined before conducting the transport experiment. The apparent permeability coefficient (Papp) values of fluorescein disodium salt were <10−6 cm s−1 after culturing at 37 °C in an atmosphere of 5% CO2 for 96 h, which confirmed the compactness and function of the monolayers. For the transcellular inhibition assay, the cells were prewashed twice with incubation medium, preincubated with or without the P-gp inhibitor at 37 °C 1 h, followed by the addition of BBR with or without the inhibitor to the apical (1.5 mL) or basolateral (2.6 mL) side, and then the opposite compartments were filled with or without the inhibitor. Samples (50 μL) were collected at 1, 2, 3, and 4 h from the receiver compartment (which was then supplemented with the blank medium with or without the inhibitor) to determine the drug transmembrane concentration.
2.6 Distribution of BBR in mouse liver and kidney
Sixty healthy male mice were randomly divided into three groups, which were intraperitoneally injected with ITZ (20 mg kg−1), PYR (10 mg kg−1), or normal saline separately, followed by intraperitoneal administration of BBR (8 mg kg−1) 30 min later. Five mice per group were euthanized at each per time point, and blood samples, as well as the livers and kidneys, were collected at 0.25, 1, 2, and 6 h after BBR administration. The collected samples were stored at −80 °C for subsequent analysis.2.7 Urinary excretion of BBR in mice
Fifteen male mice were randomly divided into three groups and then individually transferred to metabolic cages 12 h before the commenced experiment for adaptation and collection of blank urine. The administration route and dosage were the same as those used in the tissue distribution experiment. After administration of BBR, urine samples were collected overnight for 12 h and subsequently stored at −80 °C after the volume was measured.2.8 Biliary excretion of BBR in mice
Fifteen male mice were randomly divided into three groups, which were intraperitoneally injected with the inhibitors or normal saline separately, and then anesthetized 10 min after injection. After opening the abdominal cavity of each mouse, the common bile duct was ligated, a polythene catheter with a 0.28 mm inner diameter was cannulated to the gallbladder, fixed with a string, and then the surgical incision was finally sutured. BBR was also intraperitoneally injected into mice 30 min after administration of the inhibitors and normal saline. Centrifuge tubes were used to collect bile samples for 90 min after BBR injection. The bile samples were stored at −80 °C for subsequent analytical procedures.2.9 Sample preparation
To prepare the cellular samples, 50 μL acetonitrile was added to 50 μL of samples (collected from the 24-well plates and the receiver compartment of the transwell membrane) and vortexed for 1 min, followed by the addition of 100 μL pure water containing carbamazepine (internal standard, 10 ng mL−1). After the samples had been vortexed for another 1 min, they were centrifuged at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 10 min, and 150 μL of the supernatant was transferred into a vial for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. For plasma samples, 90 μL acetonitrile was added to 30 μL plasma sample, and the mixture was vortexed for 1 min, followed by the addition of 120 μL ultrapure water containing the IS (10 ng mL−1). After vortexing and centrifuging the mixture at 16000 × g for 10 min, a 2 μL aliquot was injected into the LC-MS/MS system for analysis.
000 × g for 10 min, and 150 μL of the supernatant was transferred into a vial for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. For plasma samples, 90 μL acetonitrile was added to 30 μL plasma sample, and the mixture was vortexed for 1 min, followed by the addition of 120 μL ultrapure water containing the IS (10 ng mL−1). After vortexing and centrifuging the mixture at 16000 × g for 10 min, a 2 μL aliquot was injected into the LC-MS/MS system for analysis.
The liver, kidney, and bile samples were similarly treated except for the dilution ratio. The liver and kidney were weighed and homogenized in 2 volumes of ultra-pure water and diluted to degrees varying from twice to fifteen times according to different time points while the bile samples were diluted 50 times. Acetonitrile (150 μL) was added to 50 μL each of the liver and kidney homogenates and diluted bile sample, which were vortexed for 1 min, diluted with 200 μL ultrapure water containing the IS (50 ng mL−1), vortexed again, and then centrifuged at 16000 × g for 10 min. Then, a 10 μL aliquot was injected into the LC-MS/MS system for analysis. For the urine sample, 180 μL acetonitrile and 20 μL urine sample were mixed and vortexed for 3 min, 200 μL ultrapure water containing IS (10 ng mL−1) was added to the mixture, which was vortexed and centrifuged at 16000 × g for 10 min, and then a 10 μL aliquot was injected into the LC-MS/MS system for analysis.
2.10 Determination of BBR using LC-MS/MS
The BBR content of the samples was determined using LC-MS/MS using a slightly modified version of the previous method.23 The plasma samples were analyzed using a Waters ACQUITY™ system (Milford, MA, USA) and a triple-quadrupole mass spectrometer (API 5500, Applied Biosystems, CA, USA) equipped with an electrospray ionization (ESI) source. Chromatographic separation was performed using an ACQUITY ultra-pure liquid chromatography (UPLC) BEH (2.1 × 100 mm, 1.7 μm) column under a binary gradient consisting of 0.0625% formic acid/5 mM ammonium formate/water (A) and 0.0625% formic acid/5 mM ammonium formate/acetonitrile (B). The schedule was run as follows, 50 to 90% from 0.01 to 3.90 min, 90 to 50% from 3.9 to 4.0 min, and 50% from 4 to 6 min at a flow rate and temperature of 0.2 mL min−1 and 40 °C, respectively. The mass spectrometer was operated in the positive ESI mode, and quantification was performed using multiple reaction monitoring (MRM) of the transitions from 336.1 to 320.1 for BBR and 237.2 to 194.3 for carbamazepine (IS).The tissue, urine, and bile samples were analyzed using an LC-MS/MS system consisting of a Shimadzu Prominence UFLCXR system (Shimadzu, Japan) coupled to a Thermo Scientific TSQ Quantum Ultra triple-quadrupole mass spectrometer controlled using the Xcalibur software (version 1.0.2.65 SP2, Thermo Fisher Scientific, San Jose, CA, USA). LC separations were achieved using a Hypersil Gold C18 column (100 mm × 2.1 mm, 5 μm) under a binary gradient. The mobile phase was the same as described before using the following different schedule: B, 0.01 → 7.00 min, 27 → 70%; 7.00 → 7.01 min, 70 → 27%; and 7.01 → 9.00 min, 27 → 27% at a flow rate of 0.3 mL min−1 and temperature of 40 °C. The mass spectrometer was operated in the positive ion mode with selected reaction monitoring (SRM). The iron pairs monitored were the same as described above and the analytical method met the requirements for the quantitation of biological samples.
2.11 Molecular docking
The rMate1 docking model was constructed using the sequence obtained from the Universal Protein Resource Knowledgebase (UniProtKB) with accession number Q5I0E9. The complex crystal structures of NorM and R6G with PDB code 4HUN were selected as templates. The rMatel sequence was aligned to NorM using Clustalx. The three-dimensional (3D) model of rMate1 was generated according to the standard protocol of MODELLER and the model with the best score was then minimized using the Nanoscale Molecular Dynamics (NAMD) program. Altogether, 5000 steps of the steepest descent and 2000 steps of the conjugate gradient optimization were performed. Finally, a Ramachandran plot was obtained using the PROCHECK program to verify the quality of the model.The ligand structures of BBR, ITZ, and PYR (Fig. 1) were optimized using the Optimized Potentials for Liquid Simulations (OPLS) 2011 force field within Maestro environment. The previously constructed structure of rMate1 and reported the crystal structure of P-gp (PDB code: 3G60) were used for the docking template.24 The proteins were prepared using the protein preparation wizard in Maestro using default options. The ligands were then docked into rMate1 or P-gp using the Glide XP method with default settings.
 | ||
| Fig. 1 Structures of berberine (BBR) (A), pyrimethamine (PYR) (B), and itraconazole (ITZ) (C). | ||
2.12 Data analysis
| Papp = (V × ΔC)/Δt/(A × C0) |
3. Results
3.1 Transport assay of BBR in MDCK cells
The uptake of BBR in MDCK-rMate1 cells increased significantly with decreasing pH and reached the maximum at pH 6.5 while that of the mock cells varied slightly (Fig. 2A). The uptake in MDCK-rMate1 cells was also temperature- (Fig. 2B) and time-dependent (Fig. 2C). As shown in Fig. 2D, a concentration-dependent uptake of BBR by MATE1 was observed. The Km and Vmax values were 9.84 ± 2.43 μM and 55.98 ± 4.00 pmol per mg per protein per min, respectively. PYR significantly inhibited BBR uptake at concentrations of 0.1–10 μM (Fig. 2E). The half-maximal inhibitory concentration (IC50) of PYR was 388.00 nM. In the MDCK-MDR1 cell monolayers, the Pappb→a value of BBR was obviously higher than the Pappa→b values. In the presence of the P-gp inhibitor ITZ (10 μM), the Pappb→a values significantly decreased (p < 0.01, Fig. 2F).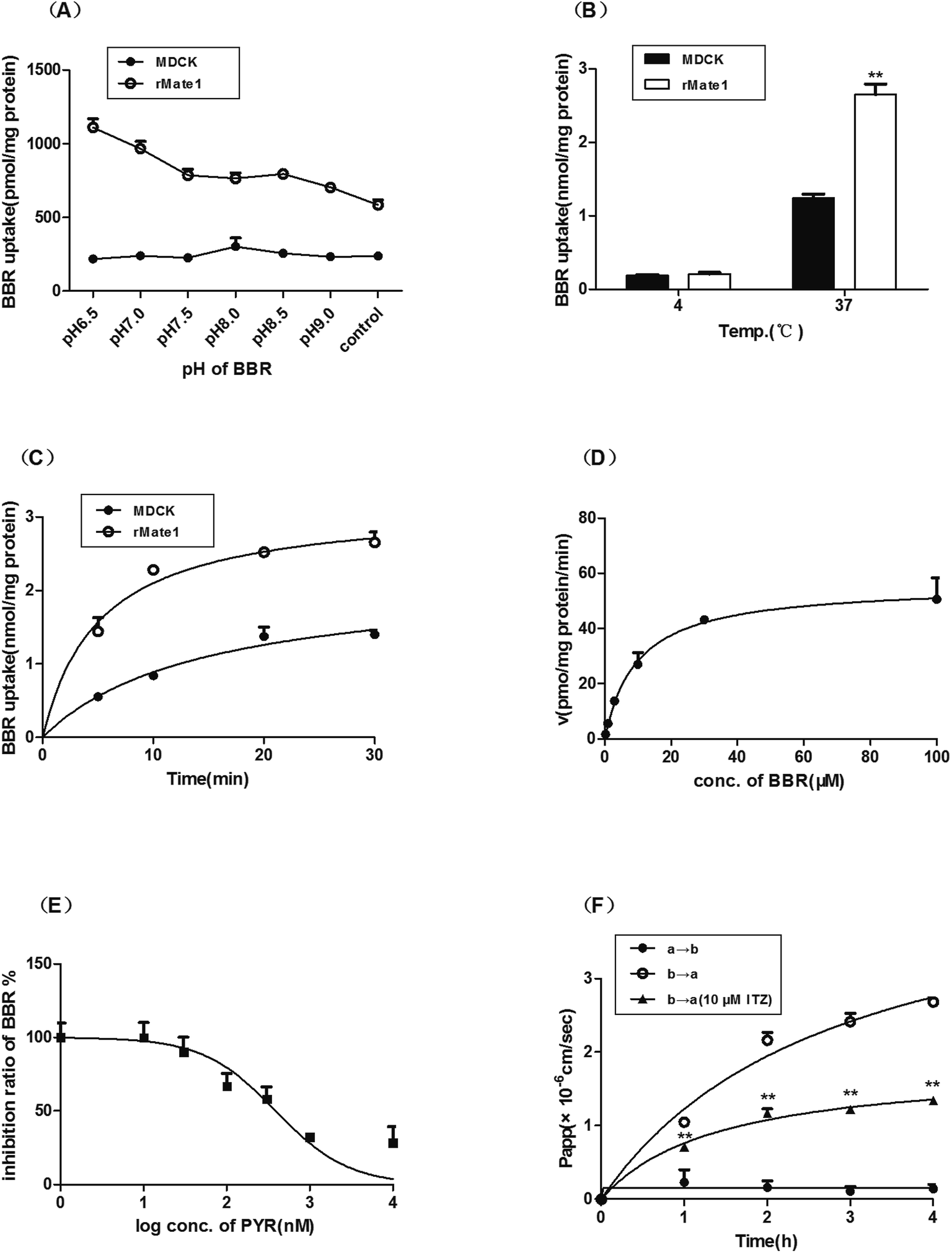 | ||
| Fig. 2 Transport of berberine (BBR) in (A–E) Madin–Darby canine kidney (MDCK)-rat multidrug and toxin extrusion protein 1 (rMate1) and (F) MDCK-multidrug resistance protein 1 (MDR1) cells (A) Effect of pH on uptake of BBR (50 μM). (B) Temperature-dependence of BBR uptake. (C) Time course of BBR uptake. (D) Concentration-dependence of BBR uptake. (E) Effect of pyrimethamine (PYR) on BBR uptake. (F) Inhibition of itraconazole (ITZ) on transcellular transport of BBR in MDCK-MDR1 cells. Results are three independent experiments and data are means ± standard error of the mean (SEM, n = 3). | ||
3.2 Effect of ITZ and PYR on tissue distribution of BBR in mice
The concentration of BBR in the plasma, liver, and kidney after administration to mice is shown in Fig. 3. The time- (0–6 h) and group-related two-way ANOVA showed that the plasma concentration of BBR significantly increased when co-administered with ITZ (p < 0.01). The liver concentration of BBR significantly increased when co-administered with ITZ or PYR (p < 0.01). In the kidney tissue, the increase in BBR concentration induced by PYR (p < 0.01) was more significant than that induced by ITZ (p < 0.05). The area under the concentration–time curve (AUC0–6 h) values of BBR in the plasma, liver, and kidney are summarized in Table 1. The AUC0–6 h values of BBR in the liver and kidney were >70 times higher than the values in the plasma.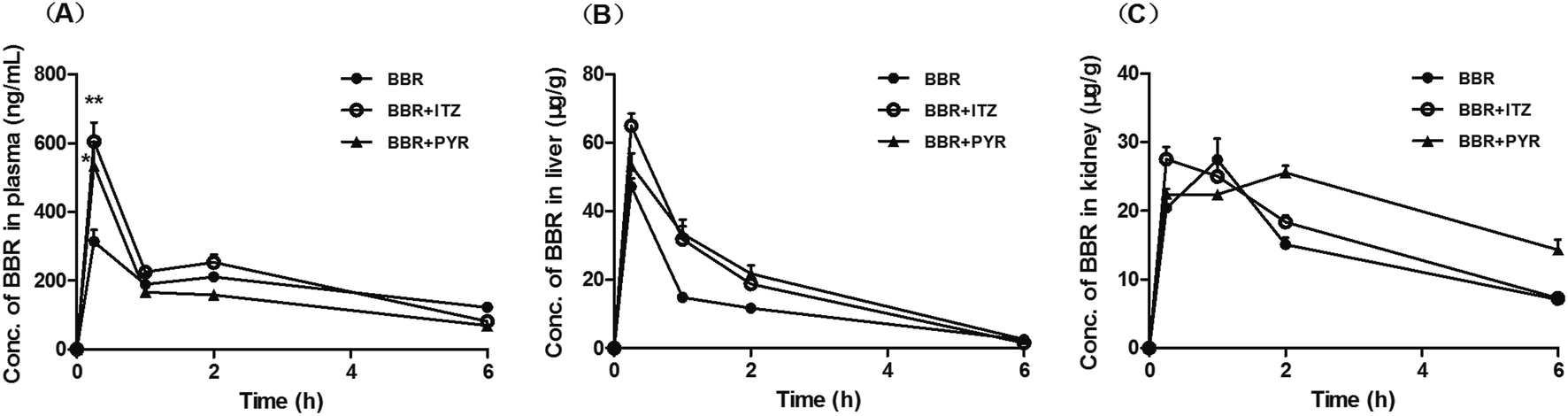 | ||
| Fig. 3 Effect of itraconazole (ITZ) and pyrimethamine (PYR) on berberine (BBR) concentration in mouse plasma (A), liver (B), and kidney (C) data are mean ± standard error of the mean (SEM, n = 5). *p < 0.05 and **p < 0.01 compared with control group (one-way analysis of variance [ANOVA] and least significant difference [LSD]). | ||
| Groups | AUC0–6 h (ng h mL−1) | ||
|---|---|---|---|
| Plasma | Liver | Kidney | |
| BBR | 1095 | 70884 |
86066 |
| BBR + ITZ | 1295 | 110081 |
96004 |
| BBR + PYR | 946 | 115459 |
123370 |
3.3 Effect of ITZ and PYR on urinary and biliary excretion of BBR in mice
The urinary excretion of BBR (Fig. 4) significantly decreased in the ITZ- and PYR-treated groups (p < 0.05 and < 0.01, respectively) compared to that in the control group. In the biliary excretion experiment, the excretion of BBR also decreased in both the ITZ and PYR groups compared to that in the control group (p < 0.01 and < 0.05, respectively).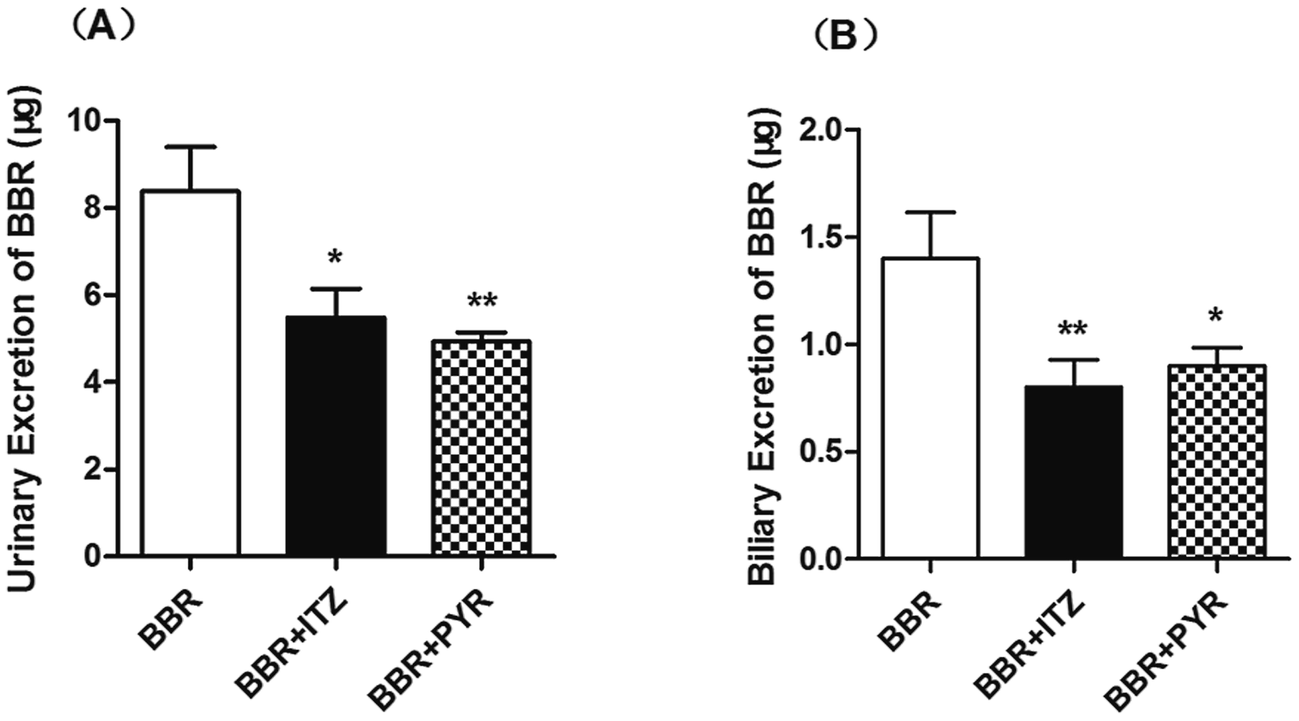 | ||
| Fig. 4 Effect of itraconazole (ITZ) and pyrimethamine (PYR) on cumulative excretion of berberine (BBR) in urine (A) and bile (B) data are mean ± standard error of the mean (SEM, n = 5). *p < 0.05 and **p < 0.01 compared with control group. | ||
3.4 Molecular docking of BBR to rMate1 and P-gp
The rMate1 structure was used as a receptor to perform molecular docking to evaluate the docking of BBR and PYR. As shown in Fig. 5A and B, the binding sites of BBR and PYR were located at similar positions in the rMate1 structure; however, the docking score of PYR (−6.417) was lower than that of BBR (−4.977). In addition, Fig. 5D shows a strong pi–pi interaction between PYR and the benzene ring on SER 299. These results revealed that PYR had a stronger binding capacity than BBR did and, therefore, PYR prevented the combination of BBR with rMate1 to some extent.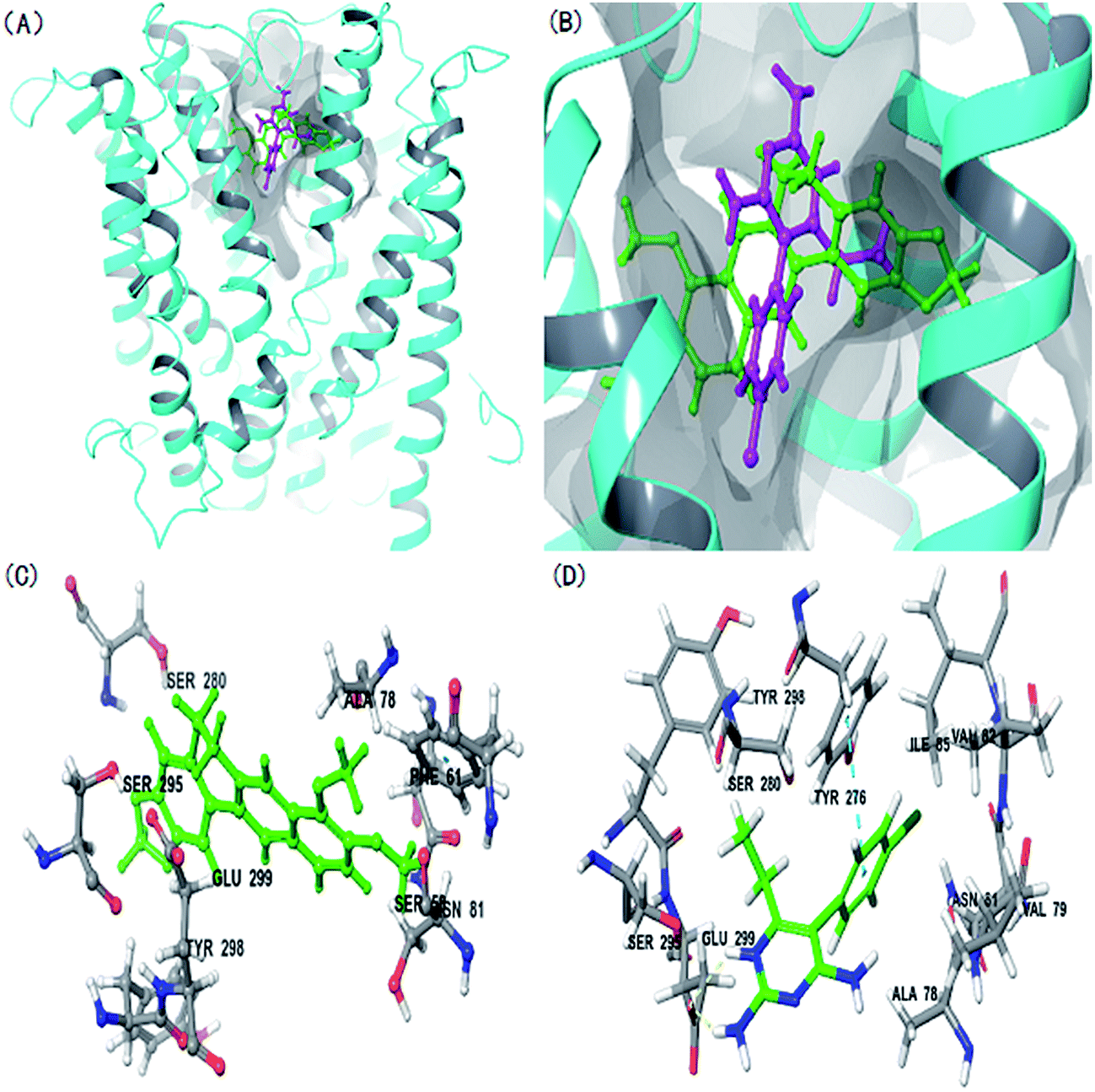 | ||
| Fig. 5 Molecular docking of berberine (BBR) and pyrimethamine (PYR) to rat multidrug and toxin extrusion protein 1 (rMate1). (A), (B) Whole and partial view of BBR and PYR binding model of rMate1. Ribbons, sticks, surface represents protein, BBR (green) and PYR (purple), binding pocket, respectively. Detailed binding model of BBR (C) and PYR (D) with rMate1. White and green sticks represent amino acid residues and ligands, respectively. Yellow and blue dash lines represent hydrogen bond and pi–pi interactions, respectively. | ||
The P-gp structure was used as a receptor to perform molecular docking while evaluating the mechanisms of action of BBR and ITZ. As indicated in Fig. 6, ITZ had a large molecular structure. When ITZ combined with P-gp, it occupied some amino acids that were also sites for BBR such as LEU 971, PHE 724, and PHE 332. In addition, the docking score of ITZ (−10.234) was lower than that of BBR (−8.960). These results also showed that ITZ prevented the combination of BBR with P-gp.
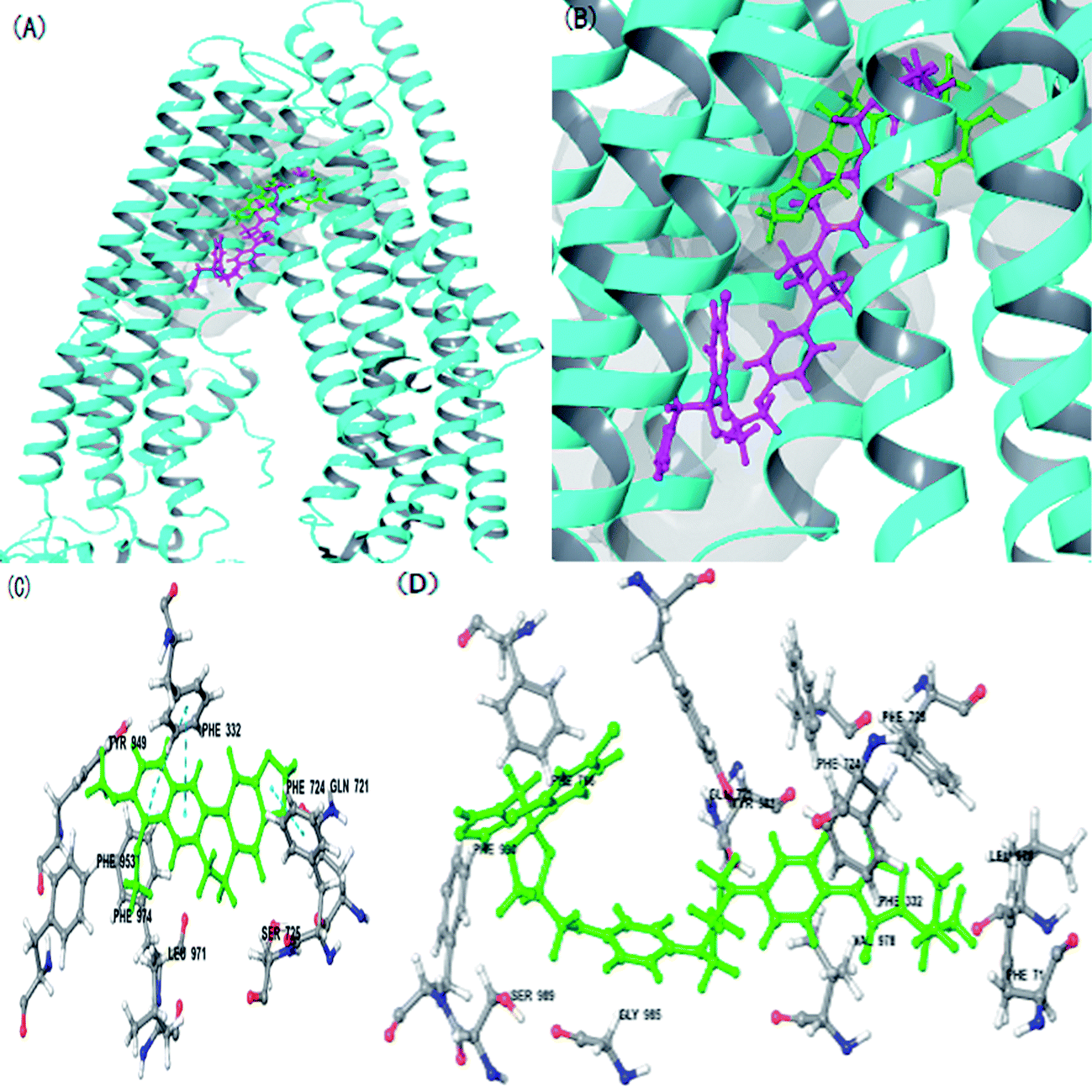 | ||
| Fig. 6 Molecular docking of berberine (BBR) and itraconazole (ITZ) to P-glycoprotein (P-gp). (A), (B) Whole and partial view of BBR and ITZ binding model of P-gp. Ribbons, sticks, surface represent protein, BBR (green) and ITZ (purple), binding pocket, respectively. Detailed binding model of BBR (C) and ITZ (D) with P-gp. White and green sticks represent amino acid residues and ligands, respectively. Blue dash line presents pi–pi interaction. | ||
4. Discussion
BBR is a commonly used drug in China, and although there are numerous reports of its pharmacokinetics, its efflux mechanism from the liver and kidney was not well known before the present study. The main efflux transporters expressed in the luminal membranes of the bile canaliculi of the liver and urinary tubules in the kidney include P-gp and MATE1, which mediate the final excretion step for organic cations. In the present study, we chose MATE1 and P-gp to investigate the efflux mechanism of BBR, an organic cation, in the liver and kidney.Two main types of experimental methods are used to study the transport function of MATE1 in cells: one uses double-transfected MDCK cells stably expressing OCT1/2 and MATE1 to investigate the transcellular translocation of organic cations25 while the other transforms the efflux transporter MATE into an uptake transporter under acidic conditions.26 Compared to the first method, the second one is more convenient and economical. In our study, we transformed rMate1 into an uptake transporter according to a previous method.26 Because MATE1 is an H+-dependent efflux transporter, the uptake of BBR in MDCK-rMate1 cells may be affected by different intracellular and extracellular pH levels. We choose the optimal pH condition for the following assay. The results showed that BBR is a substrate of rMate1. Further, in mouse experiments co-administration with PYR increased the concentration of BBR in the liver and kidney and decreased its excretion in the urine and bile. This result showed that the inhibition of MATE1 reduced the excretion of BBR from the liver and kidney, thereby inducing its accumulation in the liver and kidney. Furthermore, this result indicates that MATE1 mediated the hepatic and renal efflux of BBR. According to the related research,19 mMate1 was mainly expressed in kidney, secondly in liver in mice and few in the intestine. In present study, the increased plasma Cmax of BBR + PYR may be caused by reduced efflux because of the inhibitory effect on MATE1.
Studies such as in vitro experiments in double-transfected MDCK expressing OCT1 and P-gp showed BBR to be a substrate of P-gp.27 However, the effect of P-gp on BBR excretion from the liver and kidney in vivo has not been elucidated before this study. ITZ, a relatively specific inhibitor of P-gp,28 was selected to study the P-gp-mediated transport of BBR. Firstly, the efflux of BBR across MDCK-MDR1 cell monolayers was inhibited by ITZ. Further, in mice, co-administration with ITZ increased the concentration of BBR in the plasma, liver, and kidney, and decreased its excretion in bile and urine, which showed that the P-gp inhibition reduced the excretion of BBR from the liver and kidney, leading to its accumulation in the liver and kidney. These results indicate that P-gp mediated the hepatic and renal efflux of BBR, as well.
The structure of BBR is very different from those of ITZ and PYR. To illustrate the individual inhibitory effects of ITZ and PYR on BBR transport mediated by P-gp and MATE1, the molecular docking of BBR to rMate1 and P-gp was studied. The results showed that PYR and ITZ each individually prevented the combination of BBR with rMate1 and P-gp. It is becoming increasing evident that herb–drug interactions should be taken seriously. In addition to ITZ and PYR, numerous clinical drugs such as verapamil, ritonavir, and quinidine are inhibitors of both P-gp and MATE1. Therefore, the combination of BBR with these drugs could increase the concentration in the liver or kidney, which may enhance the effects or toxic reaction of BBR, leading to potential adverse such as liver dysfunction. However, the interactions between BBR and these drugs require further investigation.
Because BBR is usually administered orally in clinic, the evaluation that the effect of P-gp and MATE1 inhibitors on the pharmacokinetics behavior of oral BBR would match the clinical application. However there was larger individual differences in absorption of BBR. In this article, to avoid the influence of oral absorption on the results, BBR was given to mice by intraperitoneal administration. As block P-gp in the intestine could increase the absorption of berberine,29 it is expected that ITZ may increase the plasma concentration of berberine after orally administration, but we have not observed the phenomenon (data not shown) after oral administration of BBR. The reason call further investigation.
5. Conclusion
In this study, we investigated the potential involvement of P-gp and MATE1 in the efflux of BBR from the liver and kidney using transfected cells, a mouse model, and molecular docking. We discovered that BBR is a substrate of MATE1, which along with P-gp, mediate the hepatic and renal efflux of BBR in mice. These results provide pertinent information that enhances the understanding of the pharmacokinetic properties of BBR and its potential interaction of with other drugs.Acknowledgements
The authors are grateful for the financial support from National Natural Science Foundation of China (81273658 and 81303296) and Professor Min Huang (School of Pharmaceutical Science, Sun Yat-sen University, Guangzhou, China) for providing human MDCKII-MDR1 cells.References
- W. Kong, J. Wei, P. Abidi, M. Lin, S. Inaba, C. Li, Y. Wang, Z. Wang, S. Si, H. Pan, S. Wang, J. Wu, Y. Wang, Z. Li, J. Liu and J. D. Jiang, Nat. Med., 2004, 10, 1344–1351 CrossRef CAS PubMed.
- K. Karaosmanoglu, N. A. Sayar, I. A. Kurnaz and B. S. Akbulut, OMICS, 2014, 18, 42–53 CrossRef CAS PubMed.
- M. A. Dkhil, M. S. Metwaly, S. Al-Quraishy, N. E. Sherif, D. Delic, S. Y. Al Omar and F. Wunderlich, Parasitol. Res., 2015, 114, 1581–1593 CrossRef PubMed.
- Y. Dong, Y. T. Chen, Y. X. Yang, X. J. Zhou, S. J. Dai, J. F. Tong, D. Shou and C. Li, Phytother. Res., 2016, 30, 823–828 CrossRef CAS PubMed.
- F. Li, Y. B. Zhao, D. K. Wang, X. Zou, K. Fang and K. F. Wang, J. Huazhong Univ. Sci. Technol., Med. Sci., 2016, 36, 64–69 CrossRef CAS PubMed.
- J. M. Hur and D. Kim, Toxicol. Res., 2010, 26, 109–115 CrossRef CAS PubMed.
- C. Caliceti, P. Franco, S. Spinozzi, A. Roda and A. F. Cicero, Curr. Med. Chem., 2016, 23, 1460–1476 CrossRef CAS PubMed.
- H. Li, X. Cai and L. XF, Med. J. Chin. Peop. Armed Poli. Forc., 2008, 7, 661–663 Search PubMed.
- M. M. Kheir, Y. Wang, L. Hua, J. Hu, L. Li, F. Lei and L. Du, Food Chem. Toxicol., 2010, 48, 1105–1110 CrossRef CAS PubMed.
- Y. T. Liu, H. P. Hao, H. G. Xie, L. Lai, Q. Wang, C. X. Liu and G. J. Wang, Drug metabolism and disposition: the biological fate of chemicals, 2010, 38, 1779–1784 CrossRef CAS PubMed.
- X. S. Tan, J. Y. Ma, R. Feng, C. Ma, W. J. Chen, Y. P. Sun, J. Fu, M. Huang, C. Y. He, J. W. Shou, W. Y. He, Y. Wang and J. D. Jiang, PLoS One, 2013, 8, e77969 CAS.
- Y. Guo, F. Li, X. Ma, X. Cheng, H. Zhou and C. D. Klaassen, Xenobiotica, 2011, 41, 996–1005 CrossRef CAS PubMed.
- Y. Liu, H. Hao, H. Xie, H. Lv, C. Liu and G. Wang, J. Pharm. Sci., 2009, 98, 4391–4401 CrossRef CAS PubMed.
- J. Y. Ma, R. Feng, X. S. Tan, C. Ma, J. W. Shou, J. Fu, M. Huang, C. Y. He, S. N. Chen, Z. X. Zhao, W. Y. He, Y. Wang and J. D. Jiang, J. Pharm. Sci., 2013, 102, 4181–4192 CrossRef CAS PubMed.
- C. Chen, Z. T. Wu, L. L. Ma, X. Ni, Y. F. Lin, L. Wang, K. P. Chen, C. G. Huang and G. Pan, Xenobiotica, 2015, 45, 1138–1146 CrossRef CAS PubMed.
- S. Sun, K. Wang, H. Lei, L. Li, M. Tu, S. Zeng, H. Zhou and H. Jiang, Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 2014, 49, 1–6 CrossRef CAS PubMed.
- H. J. Maeng, H. J. Yoo, I. W. Kim, I. S. Song, S. J. Chung and C. K. Shim, J. Pharm. Sci., 2002, 91, 2614–2621 CrossRef CAS PubMed.
- A. T. Nies, E. Herrmann, M. Brom and D. Keppler, Naunyn-Schmiedeberg's Arch. Pharmacol., 2008, 376, 449–461 CrossRef CAS PubMed.
- A. J. Lickteig, X. Cheng, L. M. Augustine, C. D. Klaassen and N. J. Cherrington, Life Sci., 2008, 83, 59–64 CrossRef CAS PubMed.
- T. Terada, S. Masuda, J. Asaka, M. Tsuda, T. Katsura and K. Inui, Pharm. Res., 2006, 23, 1696–1701 CrossRef CAS PubMed.
- K. Y. Ohta, K. Inoue, Y. Hayashi and H. Yuasa, Drug metabolism and disposition: the biological fate of chemicals, 2006, 34, 1868–1874 CrossRef CAS PubMed.
- S. Ito, H. Kusuhara, Y. Kuroiwa, C. Wu, Y. Moriyama, K. Inoue, T. Kondo, H. Yuasa, H. Nakayama, S. Horita and Y. Sugiyama, J. Pharmacol. Exp. Ther., 2010, 333, 341–350 CrossRef CAS PubMed.
- B. Zan, R. Shi, T. Wang, J. Wu, Y. Ma and N. Cheng, B. Chromatogr, 2011, 25, 816–826 CAS.
- S. G. Aller, J. Yu, A. Ward, Y. Weng, S. Chittaboina, R. Zhuo, P. M. Harrell, Y. T. Trinh, Q. Zhang, I. L. Urbatsch and G. Chang, Science, 2009, 323, 1718–1722 CrossRef CAS PubMed.
- J. Konig, O. Zolk, K. Singer, C. Hoffmann and M. F. Fromm, Br. J. Pharmacol., 2011, 163, 546–555 CrossRef CAS PubMed.
- K. Y. Ohta, Y. Imamura, N. Okudaira, R. Atsumi, K. Inoue and H. Yuasa, J. Pharmacol. Exp. Ther., 2009, 328, 628–634 CrossRef CAS PubMed.
- A. T. Nies, E. Herrmann, M. Brom and D. Keppler, Naunyn-Schmiedeberg's Arch. Pharmacol., 2008, 376, 449–461 CrossRef CAS PubMed.
- A. Lam, J. D. Hoang, A. Singleton, X. Han and B. S. Bleier, Int. Forum. Allergy Rhinol., 2015, 5, 477–480 CrossRef PubMed.
- Y. Q. Shan, G. Ren, Y. X. Wang, J. Pang., Z. Y. Zhao, J. Yao, X. F. You, S. Y. Si, D. Q. Song, W. J. Kong and J. D. Jiang, Metabolism, 2013, 62, 446–456 CrossRef CAS PubMed.
Footnote |
| † Co-first authors. |
| This journal is © The Royal Society of Chemistry 2017 |