
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIncorporating a silicon unit into a polyether backbone—an effective approach to enhance polyether solubility in CO2†
Yongfei Zhang,
Zewen Zhu,
Zhenguo Bai,
Wei Jiang,
Fengqi Liu and
Jun Tang *
*
Department of Polymer Science, College of Chemistry, Jilin University, Changchun 130012, China. E-mail: chemjtang@jlu.edu.cn; Fax: +86-431-88498179
First published on 15th March 2017
Abstract
A series of poly(silyl ether)s were prepared by condensation polymerization and hydrosilation polymerization through incorporating a silicon unit into a polyether backbone. The phase behavior of poly(silyl ether)s in CO2 was measured in terms of concentration, molecular weight and temperature. Through incorporating the silicon unit, the poly(silyl ether)s exhibited high solubility in CO2 compared to the precursors of polyether. For example, the cloud point pressure decreased from 24.6 MPa for poly(1,2-propene glycol) (PPG) to 16.5 MPa for poly(dimethylsiloxane-alt-propene glycol) (PSPG) with a concentration of 0.6 wt% at 30 °C. Moreover, the molecular weight dependence of solubility for PSPG and PSDPG in CO2 compared with PPG was weakened. The key factor to enhance the solubility of poly(silyl ether)s in CO2 was systematically researched via surface tension and glass transition temperature. The results demonstrated that higher solubility of synthesized poly(silyl ether)s in CO2 compared to PPG was mainly attributed to lower polymer–polymer interactions.
1. Introduction
Due to the characteristics of green, safety and controllable solvent power, liquid and supercritical CO2 is considered to be an attractive substitute for hazardous organic solvents in polymer syntheses.1,2 However, its feeble solvation ability for polar and high molecular weight materials bring several difficulties for its wide practical applications.3–6 To address this limitation, the use of “CO2-philes” that can optimize the solvent character of CO2 has been suggested.7–9 Unfortunately, only few “CO2-philic” polymers exhibit high solubility in CO2, such as fluorinated poly(meth)acrylates,10,11 perfluoropolyethers12 and poly(dimethylsiloxane) (PDMS).13,14 These existing polymers cannot meet the growing demand of polymer progress and the pursuit of material cost reduction.Polyether is CO2-philic due to the nature of the high chain flexibility (glass transition temperature (Tg) of poly(1,2-propylene glycol) (PPG) is about −68 °C (ref. 15)) and special Lewis acid–Lewis base interactions between the ether oxygen of polyether and the carbon atom of CO2.16 As “CO2-philic” chain, polyether has already gained considerable attention and was designed for surfactants,17–20 and stabilizers21–24 in the field of CO2 as solvent and membranes and absorbents for CO2 capture.25–30 In order to obtain advanced polyether materials with high CO2-philicity or solubility, efficient polyether modification approaches and insight into the thermodynamic parameters affecting the solubility of polyether in CO2 had been studied. Beckman and co-workers31 masterly designed ether-carbonate copolymers through incorporating CO2-philic carbonyl group32 into a polyether backbone by aluminum-catalyzed copolymerization of cyclic ethers with CO2; the resulting statistical copolymers exhibited extremely high solubility in CO2. Subsequently, Cooper et al.33 developed a step growth polymerization route to synthesize the ether-carbonate copolymers and Wick34 further certified the major role of ether-carbonate copolymers–CO2 interactions on the increased solubility of the polymer in CO2 through Gibbs ensemble Monte Carlo simulations. In addition, the introduction of fluorine into the polyether backbone remarkably enhanced the solubility of polyether in CO2.12 Chen and colleagues35 further evaluated the key role of intermolecular interactions between (fluorinated) polyether and CO2 by in situ ATR FTIR monitoring system. Polyether solubility can also be enhanced by modifying CO2-phobic hydroxyl end-group into CO2-philic ether,36,37 acetate moieties31 and trimethylsilyl group.38 It is necessary to further develop a versatile polyether modification method for enhancing polyether solubility, and the factors affecting polyether solubility in CO2 need to be further explored.
In this study, we demonstrate an effective approach to enhance polyether solubility in CO2 by incorporating a silicon unit into a polyether backbone and further explored the key factors to increase the solubility of the polymer in CO2. After incorporating silicon unit of Si–CH3 with low intermolecular forces39 into the polyether, the polymer–polymer interactions of the modified polyether should be weaker than that of polyether. PPG was chosen as our model polyether due to its relatively high solubility in CO2 among polyethers.36 The structures of designed poly(silyl ether)s are presented in Scheme 1. Condensation polymerization and hydrosilation polymerization routes were taken to prepare the target polymers and then the phase behavior of target polymers and PPG was studied by cloud point measurements. Surface tension and glass transition temperature tests were implemented to systematically evaluate the key factor to enhance the solubility of polyether in CO2.
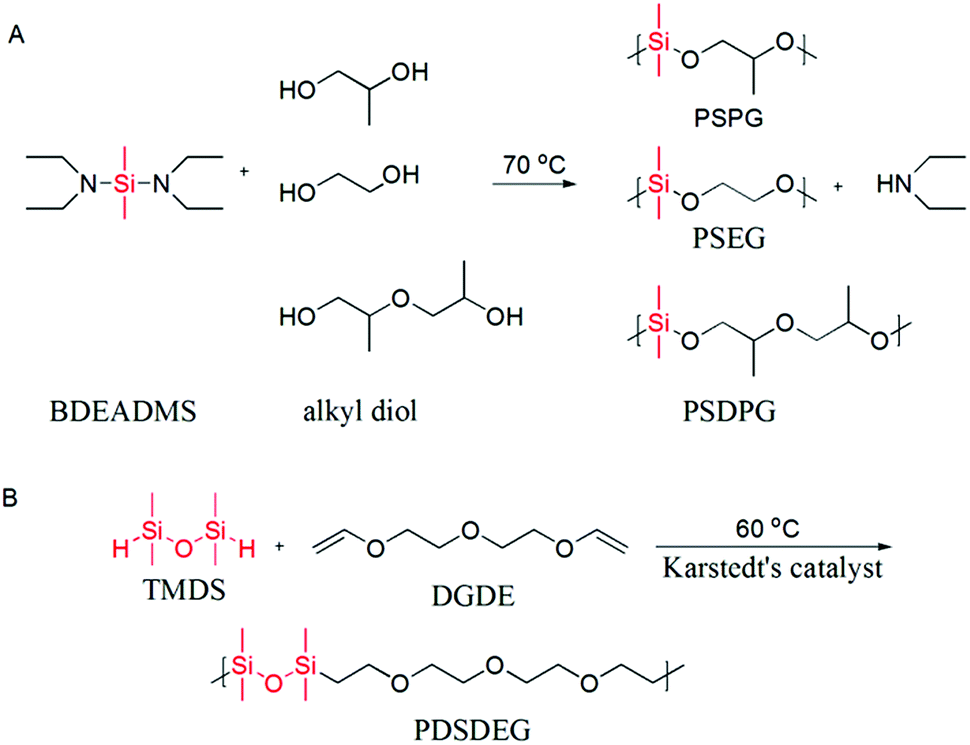 | ||
| Scheme 1 (A) Condensation polymerization of alkyl diols and BDEADMS. (B) Hydrosilation polymerization of TMDS and DGDE. | ||
2. Experimental
2.1. Materials
Dichlorodimethylsilane (DCDMS, 99%), diethylamine (DEA, 99.5%), propylene oxide (PO, 99.5%) and poly(ethylene glycol) (PEG, Mw = 2000 g mol−1) were purchased from J&K Scientific. Boron fluoride·ethyl ether complex (47–48% boron fluoride) was obtained from Strem, and 1,2-propanediol (PG, 99.5%), dipropylene glycol (DPG, 99%, mixture of isomers), ethylene glycol (EG, 99.8%), 1,1,3,3-teramethyldisiloxane (TMDS) and platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex solution in xylene, Pt ∼ 2% (Karstedt's catalyst) were purchased from Sigma-Aldrich. PG, DPG and EG were thoroughly dried by applying vacuum for 3 hours at 45 °C before use. Diethylene glycol divinyl ether (DGDE, stabilized with KOH, ≥98%) was obtained from TCI. Toluene was purchased from Beijing Chemical Works and freshly distilled before use from a purple Na/benzophenone ketyl solution. Carbon dioxide (CO2, 99.95%) was purchased from Changchun Juyang gas industry.2.2. Structure characterization
The nuclear magnetic resonance (NMR) spectra were conducted on a 400 MHz Bruker Avance III NMR spectrometer or a 300 MHz VARIAN NMR spectrometer. Chemical shifts (in ppm) were recorded in CDCl3 list as “residual internal CHCl3 (δ = 7.26)”. The molar masses of the poly(silyl ether)s were detected by gel permeation chromatography (GPC) using a Malvern apparatus equipped with a VE 3580 refractive index detector thermostat-controlled at 35 °C, a 100 μL injection loop and a set of three Malvern columns (T5000, T3000, T1000) thermostat-controlled at 35 °C. Tetrahydrofuran (THF) was used as the eluent at a flow rate of 1.0 mL min−1 and the detection was calibrated with polystyrene standards of narrow molecular weight distribution. Differential scanning calorimetric measurement (DSC) was performed using a TA instruments Q20 under a nitrogen atmosphere. The samples were heated and cooled at a rate of 10 °C min−1.2.3. Surface tension measurement
It had been reported that surface tension (γ) is approximately a positive correlation to the cohesive energy density of polymer.5,40 In this study, surface tension measurements (Dataphysics OCA20, pendant drop) were used to evaluate the polymer–polymer interactions.2.4. Solubility measurements of polymer in CO2
The solubility was based on cloud point pressure (Pcloud) that was observed using a high pressure phase equilibrium apparatus composed of a variable-volume view cell (from 40.0 mL to 85.0 mL) with two quartz glass windows and a magnetic stirrer. The temperature of the cell was controlled by a thermostat-controlled bath and measured by a thermocouple (T type, precision of ±0.1 °C) inside the cell. The pressure was measured via a pressure transducer (precision of ±0.01 MPa) equipped with a pressure numerical display. In a typical test, an accurate amount of sample was weighed and placed at the bottom platform of the cell. The cell was sealed and heated to the desired temperature and then flushed four times with CO2 at 0.5 MPa to remove the air. Subsequently CO2 was injected into the cell until the desired pressure was reached using a positive displacement pump (Model SFC-24, SSI, USA). The mass of CO2 was calculated based on the displaced volume, temperature and pressure of the cell with the data of NIST webbook (http://webbook.nist.gov/chemistry/fluid/). The polymer–CO2 solution was stirred and equilibrated for 30–40 min. The Pcloud was examined by carefully and gradually depressurizing the autoclave and the entire phase transition process was observed and recorded by a camera connected to a computer. The Pcloud was defined as the point at which it was no longer possible to see the syringe needle through the CO2/polymer mixture, which was because the syringe was behind the view cell; this was the reference. The process was repeated at least three times for each measurement, and an average of the results was taken as the cloud point. For a given polymer, deviations were approximately ±0.5 MPa. All the measurements were conducted at 30 °C unless otherwise noted.2.5. Synthesis of poly(propylene glycol) (PPG)
In order to compare the solubility, poly(propylene glycol) was synthesized as follows. In a typical experiment, the mixture of 15 mL toluene, EG (1.24 g, 20.0 mmol) and boron fluoride ethyl ether complex (0.52 mL) was stirred for 30 min at room temperature, and then the solution of 28 mL (0.4 mol) of PO in 10 mL of toluene was added dropwise into the reaction solution over 30 min in an ice-water bath. The reaction was allowed to proceed for 4 h and was terminated with aqueous NaOH. The polymer solution was washed three times with pure water, and the organic phase was dried with magnesium sulphate. The solvent was removed under vacuum at 50 °C.2.6. Synthesis of bis(diethylamino)dimethylsilane (BDEADMS)
The synthesis process of bis(diethylamino)dimethylsilane was similar to the previous report.41 A solution of dichlorodimethylsilane (28.40 g, 0.22 mol) in toluene (20 mL) was slowly added dropwise over 1 hour to diethylamine (69.48 g, 0.95 mol) dissolved in toluene (400 mL) with ice-water bath cooling and then continued to react for 12 hours at room temperature. The resulting white precipitate was removed via vacuum filtration under N2 atmosphere. Most of the solvent was removed by rotary evaporation at 30 °C, and the crude production was further purified by vacuum distillation twice to yield BDEADMS (31 g, yield 70%). The product was identified and confirmed by 1H NMR. 1H NMR (400 MHz, CDCl3): δ 0.03 ppm (s, 6H), 0.95 ppm (t, 12H), 2.79 (q, 8H).2.7. Synthesis of poly(silyl ether)s by condensation polymerization
Poly(dimethylsiloxane-alt-propene glycol) (PSPG), poly(dimethylsiloxane-alt-dipropene glycol) (PSDPG) and poly(dimethylsiloxane-alt-ethylene glycol) (PSEG) were prepared by condensation polymerization of BDEADMS with PG, DPG and EG, respectively. The synthesis route is presented in Scheme 1A. In a typical experiment, PG (0.761 g, 10.00 mmol) and BDEADMS (1.928 g, 9.52 mmol) were charged into a 25 mL round-bottomed flask equipped with a magnetic stirring bar, a reflux condenser and a vacuum source. The reaction was performed at 70 °C under reduced pressure 35 kPa for 23 h and then 5 kPa for 1 h for the removal of diethylamine, and a colourless or yellowish oily product (1.103 g, yield 85%) was thus collected.2.8. Synthesis of poly(silyl ether) by hydrosilation polymerization
Poly(teramethyldisiloxane-alt-diethylene glycol divinyl ether) (PDSDEG) was prepared via hydrosilation polymerization of TMDS and DGDE (Scheme 1B). A typical polymerization reaction was performed in a 25 mL round reaction flask, which was equipped with a magnetic stirring bar and a reflux condenser. 1.5854 g (10 mmol) DGDE, 1.3433 g (10 mmol) TMDS and 10 mL anhydrous toluene were charged into the reactor and stirred for 10 min, followed by adding three drops of Karstedt's catalyst. Subsequently, the reactant mixture was gradually heated to 60 °C and stirred for 48 h. The color of the mixture turned from slightly yellow to yellow-brown. After removing toluene by evaporation under reduced pressure, the crude product was redissolved in chloroform for the removal of Karstedt's catalyst by treating the solution with activated charcoal. The product was transferred into Spectra/Por 6 regenerated cellulose dialysis tubing (molecular weight cut-off: 1 kDa) and dialyzed against ethanol for one day. The finally purified polymer was a colourless or slightly yellowish, viscous fluid (2.1963 g, yield 75%), and it was characterized by GPC and 1H NMR.3. Results and discussion
3.1. Preparation of poly(silyl ether)s
In this study, condensation polymerization and hydrosilation polymerization were used to incorporate silicon unit into the polyether backbone and synthesize a series of poly(silyl ether)s with different molecular weight and repeat unit structures (see Table 1). Microstructure of poly(silyl ether)s can be precisely designed via altering specific comonomers on account of the step growth mechanism. Based on the structure of PPG, PSPG with 50 mol% silicon compositions and PSDPG with 33 mol% silicon contents were synthesized by BDEADMS with PG and DPG, respectively. Moreover, based on PEG, PSEG and PDSDEG with the same silicon compositions as that of PSPG were synthesized (shown in Scheme 1). The detailed experimental results of synthesized poly(silyl ether)s with different molecular weight and repeat unit structures are listed in Table 1. The evolution of condensation polymerization of poly(silyl ether)s was monitored by GPC. The kinetic plots of molecular weight versus reaction time of PSPG, PSDPG and PSEG are shown in Fig. 1. The molecular weight of PSPG, PSDPG and PSEG increased until about 1300 min, after which the molecular weight almost no longer increased, and the polydispersity index (PDI, Mw/Mn) reached the typical value of about 2 for polycondensation. With the same reaction conditions, the final product of PSPG showed a higher molecular weight than PSDPG, which may be attributed to the higher reaction activity of primary hydroxyl group in PG (DPG contains less primary hydroxyl groups due to the type of isomer mixture). The molecular weight of the poly(silyl ether)s could be controlled by adjusting the feed ratio of alkyl diols and BDEADMS. The structure of products was confirmed by 1H NMR spectroscopy (Fig. S1–S3†). PDSDEG was synthesized by hydrosilation polymerization; GPC and 1H NMR spectra (Fig. S4 and S5†) of the final purified PDSDEG showed that hydrosilation polymerization is also a good method to modify polyether.| Run | Sample | Mn (g mol−1) | Mw (g mol−1) | PDI | Tg (°C) | γ ± 0.5 (mN m−1) |
|---|---|---|---|---|---|---|
| a n.d. = not determined.b Data from ref. 15. | ||||||
| 1 | PSPG-2500 | 2470 | 3570 | 1.40 | −75 | 22.0 |
| 2 | PSPG-3300 | 3260 | 6460 | 1.98 | −75 | 22.3 |
| 3 | PSPG-5000 | 5030 | 9250 | 1.84 | −74 | 22.5 |
| 4 | PSDPG-2000 | 1960 | 2890 | 1.50 | −73 | 21.9 |
| 5 | PSDPG-3000 | 3020 | 5350 | 1.77 | n.d.a | 22.7 |
| 6 | PSDPG-5100 | 5060 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 030 030 |
1.98 | −72 | 22.8 |
| 7 | PSEG-4300 | 4260 | 8840 | 1.98 | −88 | 22.4 |
| 8 | PDSDEG-2900 | 2860 | 4840 | 1.69 | −87 | 24.5 |
| 9 | PDSDEG-1900 | 1940 | 2794 | 1.44 | n.d.a | 24.0 |
| 10 | PPG-2000 | 2040 | 2600 | 1.27 | −68b | 30.1 |
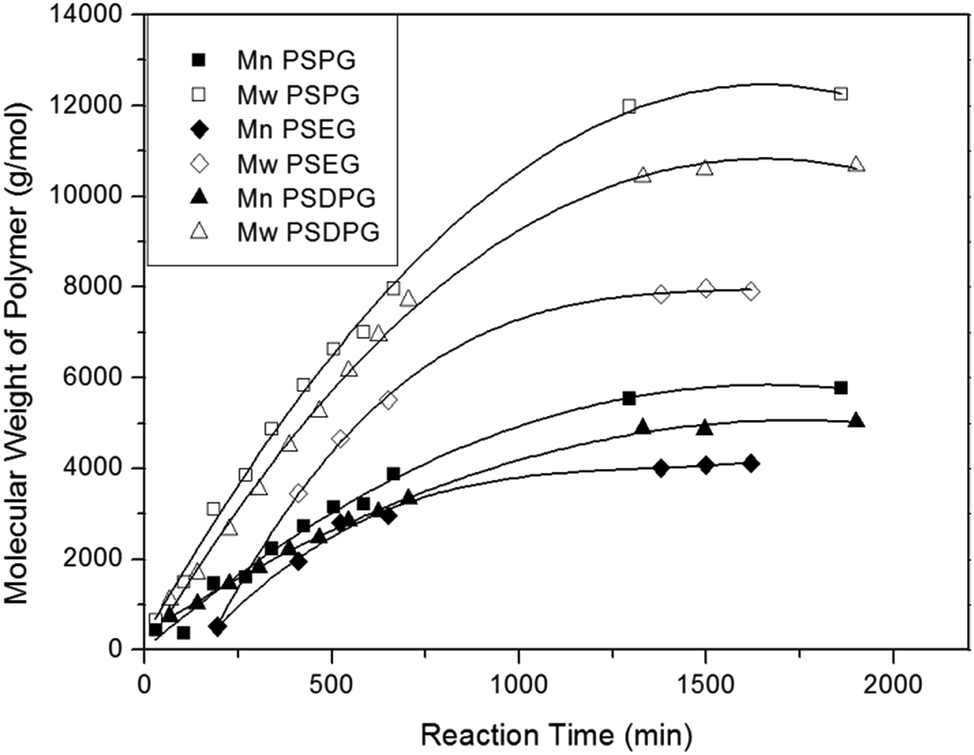 | ||
| Fig. 1 The kinetic plots of molecular weight versus reaction time of PSPG, PSDPG at 70 °C and PSEG at 60 °C. | ||
3.2. Phase behavior of poly(silyl ether)s in CO2
Analyzing the cloud point curves of poly(silyl ether)s and PPG (see in Fig. 2), we can find that incorporating a silicon unit into either PPG or into PEG decreased their cloud point pressure. For the derivative of PPG, the Pcloud of PSPG-2500 changed in the scope of 9.61 to 16.54 MPa with the concentration range of 0.2 to 1.0 weight percent (wt%), while the Pcloud of PPG-2000 increased from 18.66 to 24.59 MPa at the concentration of 0.2 to 0.6 wt%. The Pcloud of PSPG-2500 decreased over 9 MPa than that of PPG-2000. The Pcloud of PSPG-5000 was also lower than that of PPG-2000, though the number average molecular weight of PSPG-5000 was over twice that of PPG-2000. PSDPG-2000 exhibited approximately the same cloud point pressures with PSPG-2500, despite the fact that PSDPG-2000 has lower molecular weight. This result indicates that PSPG with more silicon content exhibited higher solubility in CO2 than PSDPG. Clearly, incorporating a silicon unit into PPG significantly enhanced its solubility in CO2. One can also find that poly(silyl ether)s based on PEG exhibited a similar result. PEG with a molecular weight of 2000 g mol−1 could not completely dissolve in CO2 at a concentration of 0.4 wt% and pressure of 30 MPa, while the cloud point pressures of PDSDEG-1900 appeared in the 14.8–17.9 MPa range for concentrations of 0.4–1.0 wt%, and the value of PDSDEG-2900 was also lower than that of PPG. PSEG-4300 exhibited similar phase behaviors with PSPG-5000. Higher pressure was required for PDSDEG-2900 to dissolve in CO2 than that required for PSPG-5000 and PSEG-4300; this may be explained from the possibility of two adjacent ethyoxyl units in the PDSDEG backbone causing stronger polymer–polymer interaction, which is unfavorable to the dissolution of PDSDEG. On comparing PSPG-2500 and PSPG-5000 with PDSDEG-1900, PSEG-4300 and PDSDEG-2900, we found that the derivatives of PPG showed higher solubility than that of PEG with the same silicon content. Considering the low intermolecular forces between the methyl groups of Si–CH3,39 we speculate that silicon unit contained in poly(silyl ether)s may decrease the polymer–polymer interactions of polyether resulting in higher solubility of poly(silyl ether)s in CO2. This is consistent with the fact that low surface tension and high chain flexibility of PDMS is responsible for its high solubility in CO2.42 In summary, the incorporation of silicon unit both significantly enhanced PPG and PEG solubility in CO2, and poly(silyl ether) based on PPG exhibit higher solubility in CO2 than that based on PEG with the same silicon content.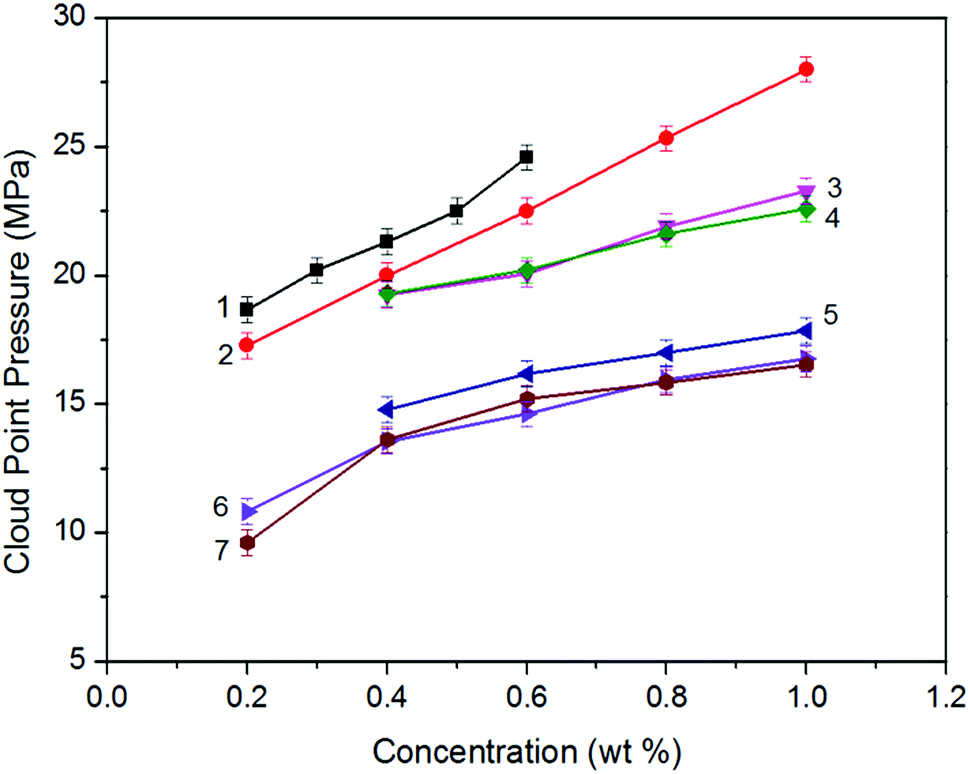 | ||
| Fig. 2 Phase behavior of mixtures of CO2 with (1) PPG-2000, (2) PDSDEG-2900, (3) PSPG-5000, (4) PSEG-4300, (5) PDSDEG-1900, (6) PSDPG-2000 and (7) PSPG-2500. The cloud point pressure values of all the samples were measured at 30 °C. | ||
Considering the excellent solubility of poly(silyl ether)s based on PPG, we further investigated the impact of molecular weight on the solubility of derivatives of PPG (Fig. 3A). It could be seen that PPG exhibited a steeper cloud point curve indicating that the solubility of PPG strongly depends on molecular weight. The Pcloud of PPG increased significantly from 11.9 to 21.3 MPa when the molecular weight increased from 1400 to 2040 g mol−1. In contrast, the Pcloud of PSPG increased only from 13.6 MPa to 24.9 MPa with a large increase of molecular weight from 2470 to 8480 g mol−1, and the Pcloud of PSDPG increased slightly from 13.6 MPa to 23.5 MPa with the increase of molecular weight from 1960 to 5060 g mol−1. Fig. 3A shows that silicon unit effectively decreased the molecular weight dependence of PPG solubility. We speculate that the strong molecular weight dependence of PPG solubility was attributed to its strong polymer–polymer interactions. Moreover, the high solubility of high molecular weight PDMS with inherently low surface tension in CO2 supports our viewpoint.13 This result is especially helpful to the design and utilization of high molecular weight CO2-philic materials. As far as we know, the study aimed to improve polyether solubility in CO2 has mostly focused on low molecular weight PPG (Mw ≤ 2000 g mol−1) via endgroup modification or side chain functionalization. For high molecular weight polyether, only poly(hexafluoropropylene oxide)12 and ether-carbonate copolymers31 exhibited excellent solubility in CO2.
 | ||
| Fig. 3 (A) Molecular weight dependence of the cloud point pressure of PPG, PSPG and PSDPG. The cloud point pressure values of the samples were measured at 30 °C with the concentration of 0.4 wt%. (B) Temperature dependence of the solubility of PPG-2000 and PSPG-3300 in CO2 at 0.4 wt% and 30 °C. | ||
The temperature dependence of the solubility of PPG and PSPG was studied, and the results are shown in Fig. 3B. It was observed that the Pcloud of both PPG and PSPG increased with the rise of temperature resulting from decreased solvability of CO2 toward PPG and PSPG due to the decreased density of CO2 with the rise of temperature.35
To get an insight into the phase behavior results of the poly(silyl ether)s, the polymer–polymer interactions were first assessed by surface tension (γ)5,40 which was measured with the pendant drop experiment (listed in Table 1). Surface tension values decreased from 30.1 mN m−1 at 26 °C (the reported value of PPG with 2025 g mol−1 is 31.5 mN m−1 at 20 °C (ref. 15)) for pure PPG-2000 to about 22 mN m−1 for PSPG and PSDPG, and the values decreased from 37.5 mN m−1 for PEG (M = 600 g mol−1)15 to about 24 mN m−1 for PDSDEG and to about 22.4 mN m−1 for PSEG-4300. It should be noted that PSPG with different molecular weights have nearly the same surface tension values; this is consistent with the relationship between surface tension and molecular weight, where surface tension changes little when the molecular weight exceeds about 2000–3000 g mol−1.15 As expected, silicon unit significantly weakened the polymer–polymer interactions of the polyether resulting from the reduced dispersion force and induction force after incorporating the silicon unit into the polyether backbone. Clearly, the lower surface tension is in favor of the result discussed in this study, which indicates that poly(silyl ether)s exhibit higher solubility in CO2 than PPG. We also noted that the change trend of the surface tension of PSDPG-2000, PSPG-2500, PDSDEG-1900 and PPG-2000 (samples with nearby molecular weight avoid the effect of molecular weight on the solubility) was consistent with the trend of solubility, which indicated that higher surface tension resulted in a higher cloud point pressure (Fig. 4). These results proved that polymer–polymer interactions play an important role on the dissolution of polyether in CO2. Therefore, the lower solubility of PDSDEG compared to PSPG can be accounted for by the higher surface tension of PDSDEG, and the similar phase behaviors of PSPG-5000 and PSEG-4300 may be explained by the nearby surface tension values.
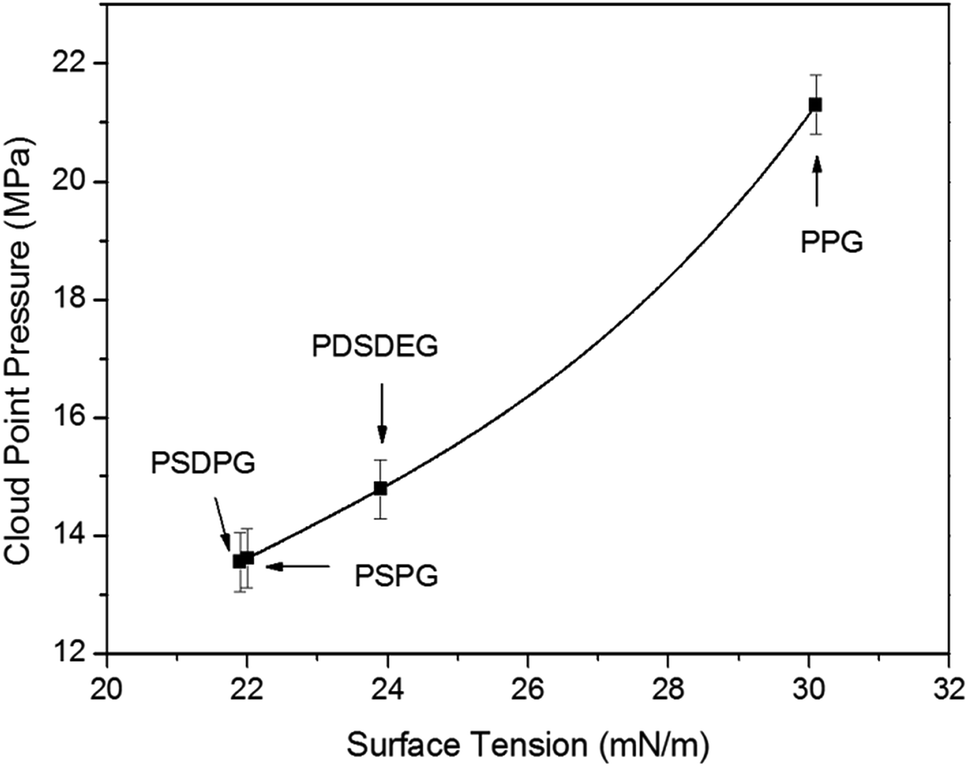 | ||
| Fig. 4 Relationship between cloud point pressure and surface tension values from PSPG-2500, PSDPG-2000, PDSDEG-1900 and PPG-2000. The cloud point pressure values of the samples were measured at 30 °C with 0.4 wt% concentration. | ||
Furthermore, the relative chain flexibility and free volume were evaluated with Tg,36 the values of Tg, as shown in Table 1. The Tg of PSEG and PDSDEG decreased to about −88 and −87 °C, respectively, compared to PEG (Tg = −64 °C),15 which stems from the increased free volume and remarkably decreased molecule interactions after incorporating the silicon unit. The lower Tg would improve the entropy of mixing and should be one of the driving forces for PSEG and PDSDEG dissolving in CO2. However, PSEG and PDSDEG appeared to be less soluble in CO2 than PSPG, as discussed previously, though the Tg values of PSEG and PDSDEG were lower than PSPG. Apparently, the improved entropy of mixing is not the main driving force for the enhanced solubility of poly(silyl ether)s in CO2. Polymer–CO2 interactions were then analyzed based on the work of Pauline Vitoux.43 The authors had reported the stabilization energy of methoxytrimethylsilane (MTMS) and propyl methyl ether (PME) with CO2 using MP2 method with aug-cc-VDZ basis set where the special functional groups are Si–O–C group and C–O–C group (of PEG) respectively. The stabilization energy of MTMS–CO2 complex (−3.59 kcal mol−1) was slightly higher (0.16 kcal mol−1) than PME–CO2 complex (−3.43 kcal mol−1). Given that the solution of PDMS in CO2 exhibited upper critical solution temperature (UCST) type behavior that stems from weak solute–solvent interactions,44 we believe that polyether–CO2 interactions were not significantly changed by the silicon unit and cannot dominate the special phase behavior of poly(silyl ether)s in CO2. These results further confirm that the high solubility of poly(silyl ether)s in CO2 is mainly attributed to lower polymer–polymer interactions.
4. Conclusions
In conclusion, a series of poly(silyl ether)s were prepared by condensation polymerization and hydrosilation polymerization. Upon introducing a silicon unit, the cloud point pressure of PEG and PPG was significantly decreased and the molecular weight dependence of PSPG and PSDPG solubility in CO2 compared with PPG was weakened. It should be also mentioned that PSPG with more silicon contents exhibited higher solubility in CO2 than PSDPG. Incorporating a silicon unit into the PEG and PPG backbone is an advisable and convenient approach to enhance their solubility in CO2, and the CO2-philicity of poly(silyl ether) can be altered based on the comonomers. The higher solubility of poly(silyl ether)s in CO2 compared to PPG is mainly attributed to lower polymer–polymer interactions demonstrated by the surface tension measurements and glass transition temperature tests. This study would be conducive to the development of novel functional materials based on polyether, such as surfactants, stabilizers, CO2 adsorbent and CO2-philic polymer membrane.Acknowledgements
This research was supported by the National Science Foundation of China (No. 21074042, 50773028), and major science and technology research project of Jilin province (No. 20140203012G X).References
- J. B. McClain, D. E. Betts, D. A. Canelas, E. T. Samulski, J. M. DeSimone, J. D. Londono, H. D. Cochran, G. D. Wignall, D. Chillura-Martino and R. Triolo, Science, 1996, 274, 2049–2052 CrossRef CAS PubMed.
- M. F. Kemmere and T. Meyer, Supercritical Carbon Dioxide in Polymer Reaction Engineering, John Wiley &VCH, Weinheim, 2005, pp. 1–12 Search PubMed.
- F. K. Christopher and M. A. McHugh, Chem. Rev., 1999, 99, 565–602 CrossRef.
- E. Girard, T. Tassaing, C. Ladaviere, J. D. Marty and M. Destarac, Macromolecules, 2012, 45, 9674–9681 CrossRef CAS.
- M. L. O'Neill, Q. Cao, M. Fang, K. P. Johnston, S. P. Wilkinson, C. D. Smith, J. L. Kerschner and S. H. Jureller, Ind. Eng. Chem. Res., 1998, 37, 3067–3079 CrossRef.
- F. Rindfleisch, T. P. DiNoia and M. A. McHugh, J. Phys. Chem., 1996, 100, 15581–15587 CrossRef CAS.
- E. J. Beckman, Chem. Commun., 2004, 1885–1888 RSC.
- H. Lee, J. W. Pack, W. X. Wang, K. J. Thurecht and S. M. Howdle, Macromolecules, 2010, 43, 2276–2282 CrossRef CAS.
- B. Tan, C. L. Bray and A. I. Cooper, Macromolecules, 2009, 42, 7945–7952 CrossRef CAS.
- J. M. DeSimone, Z. Guan and C. S. Elsbernd, Science, 1992, 257, 945–947 CAS.
- J. B. McClain, D. Londono, J. R. Combes, T. J. Romack, D. A. Canelas, D. E. Betts, G. D. Wignall, E. T. Samulski and J. M. DeSimone, J. Am. Chem. Soc., 1996, 118, 917–918 CrossRef CAS.
- T. Hoefling, D. Stofesky, M. Reid, E. J. Beckman and R. M. Enick, J. Supercrit. Fluids, 1992, 5, 237–241 CrossRef CAS.
- Y. Xiong and E. Kiran, Polymer, 1995, 36, 4817–4826 CrossRef CAS.
- Z. Bayraktar and E. Kiran, J. Appl. Polym. Sci., 2000, 75, 1397–1403 CrossRef CAS.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, John Wiley & Sons, New York, 4th edn, 1998, vol. 6, pp. 226–528 Search PubMed.
- S. Kilic, S. Michalik, Y. Wang, J. K. Johnson, R. M. Enick and E. J. Beckman, Macromolecules, 2007, 40, 1332–1341 CrossRef CAS.
- G. B. Jacobson, C. T. Lee, K. P. Johnston and W. Tumas, J. Am. Chem. Soc., 1999, 121, 11902–11903 CrossRef CAS.
- G. B. Jacobson, C. T. Lee, S. R. P. da Rocha and K. P. Johnston, J. Org. Chem., 1999, 64, 1207–1210 CrossRef CAS.
- X. Fan, V. K. Potluri, M. C. McLeod, Y. Wang, J. C. Liu, R. M. Enick, A. D. Hamilton, C. B. Roberts, J. K. Johnson and E. J. Beckman, J. Am. Chem. Soc., 2005, 127, 11754–11762 CrossRef CAS PubMed.
- S. R. P. Da Rocha, K. L. Harrison and K. P. Johnston, Langmuir, 1999, 15, 419–428 CrossRef.
- S. Mawson, M. Z. Yates, M. L. O'Neill and K. P. Johnston, Langmuir, 1997, 13, 1519–1528 CrossRef CAS.
- D. Bratton, M. Brown and S. M. Howdle, Chem. Commun., 2004, 808–809 RSC.
- D. H. Builes, A. Tercjak and I. Mondragon, Polymer, 2012, 53, 3669–3676 CrossRef CAS.
- E. Torino, E. Reverchon and K. P. Johnston, J. Colloid Interface Sci., 2010, 348, 469–478 CrossRef CAS PubMed.
- S. F. Wang, X. Q. Li, H. Wu, Z. Z. Tian, Q. P. Xin, G. W. He, D. D. Peng, S. L. Chen, Y. Yin, Z. Y. Jiang and M. D. Guiver, Energy Environ. Sci., 2016, 9, 1863–1890 CAS.
- M. B. Miller, D. R. Luebke and R. M. Enick, Energy Fuels, 2010, 24, 6214–6219 CrossRef CAS.
- T. Sakaguchi, F. Katsura, A. Iwase and T. Hashimoto, Polymer, 2014, 55, 1459–1466 CrossRef CAS.
- S. C. Li, Z. Wang, C. X. Zhang, M. M. Wang, F. Yuan, J. X. Wang and S. C. Wang, J. Membr. Sci., 2013, 436, 121–131 CrossRef CAS.
- H. Q. Lin and B. D. Freeman, J. Mol. Struct., 2005, 739, 57–74 CrossRef CAS.
- A. Gonzalez, M. Iriarte, P. J. Iriondo and J. J. Iruin, Polymer, 2003, 44, 7701–7708 CrossRef CAS.
- T. Sarbu, T. Styranec and E. J. Beckman, Nature, 2000, 405, 165–168 CrossRef CAS PubMed.
- Z. Shen, M. A. McHugh, J. Xu, J. Belardi, S. Kilic, A. Mesiano, S. Bane, C. Karnikas, E. J. Beckman and R. M. Enick, Polymer, 2003, 44, 1491–1498 CrossRef CAS.
- B. Tan, H. M. Woods, P. Licence, S. M. Howdle and A. I. Cooper, Macromolecules, 2005, 38, 1691–1698 CrossRef CAS.
- C. D. Wick, J. I. Siepmann and D. N. Theodorou, J. Am. Chem. Soc., 2005, 127, 12338–12342 CrossRef CAS PubMed.
- G. G. Zhou, J. G. Chen, M. X. Wang, M. Zhang, J. L. Guo, S. K. Shen, Z. T. Liu, Z. W. Liu, J. Q. Jiang and J. Lu, Green Chem., 2015, 17, 4489–4498 RSC.
- C. Drohmann and E. J. Beckman, J. Supercrit. Fluids, 2002, 22, 103–110 CrossRef CAS.
- L. Hong, D. Tapriyal and R. M. Enick, J. Chem. Eng. Data, 2008, 53, 1342–1345 CrossRef CAS.
- S. Li, Y. Q. Li and J. X. Wang, Fluid Phase Equilib., 2007, 253, 54–60 CrossRef CAS.
- C. Burger and F.-H. Kreuzer, in Silicon in Polymer Synthesis, ed. H. R. Kricheldorf, Spring-Verlag, Hamburg, 1996, Polysiloxanes and Polymers Containing Siloxane Groups, p. 122 Search PubMed.
- E. Girard, T. Tassaing, S. Camy, J. S. Condoret, J. D. Marty and M. Destarac, J. Am. Chem. Soc., 2012, 134, 11920–11923 CrossRef CAS PubMed.
- M. Padmanaban, M. A. Kakimoto and Y. Imai, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 2997–3005 CrossRef CAS.
- E. Girard, T. Tassaing, J. D. Marty and M. Destarac, Chem. Rev., 2016, 116, 4125–4169 CrossRef CAS PubMed.
- P. Vitoux, T. Tassaing, F. Cansell, S. Marre and C. Aymonier, J. Phys. Chem. B, 2009, 113, 897–905 CrossRef CAS PubMed.
- R. Fink, D. Hancu, R. Valentine and E. J. Beckman, J. Phys. Chem. B, 1999, 103, 6441–6444 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01587a |
| This journal is © The Royal Society of Chemistry 2017 |