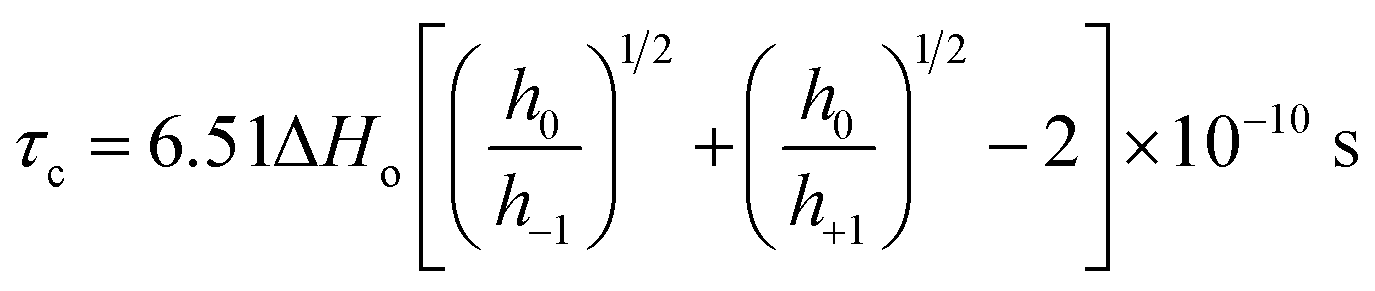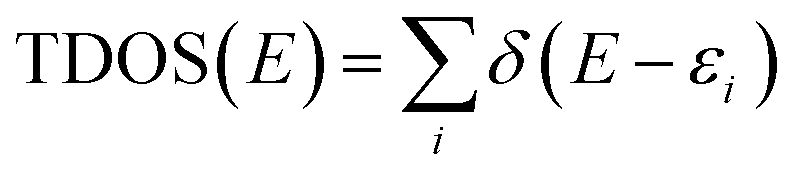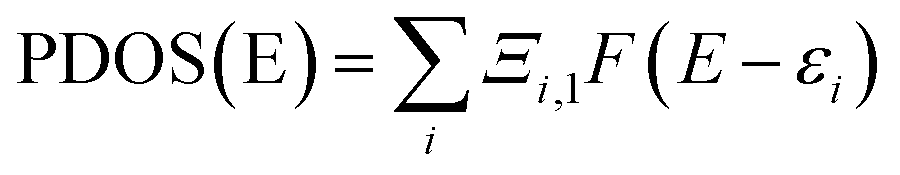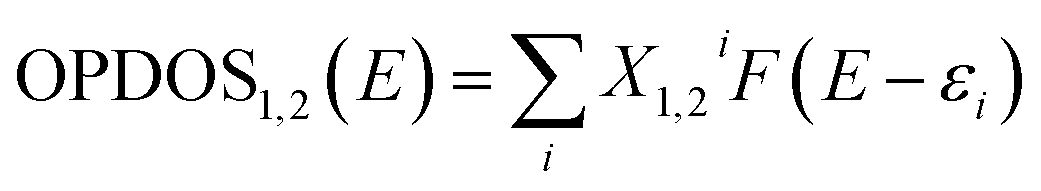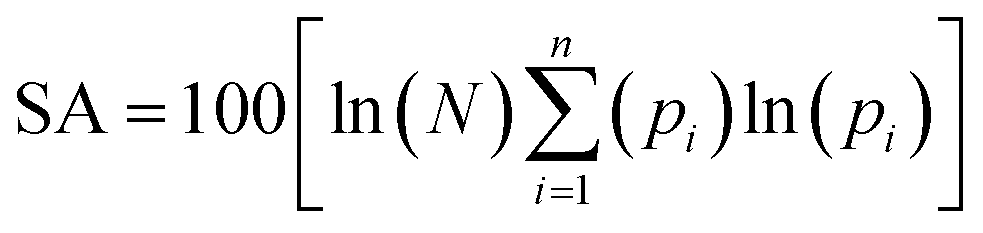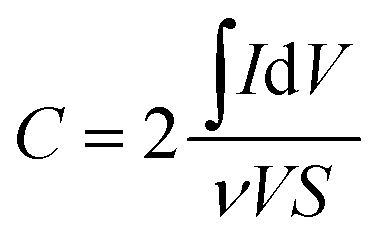DOI:
10.1039/C6RA27411K
(Paper)
RSC Adv., 2017,
7, 16623-16636
Synthesis, characterization and theoretical studies on novel organic–inorganic hybrid ion–gel polymer thin films from a γ-Fe2O3 doped polyvinylpyrrolidone–N-butylpyridinium tetrafluoroborate composite via intramolecular thermal polymerization†
Received
27th November 2016
, Accepted 26th January 2017
First published on 15th March 2017
1. Introduction
Research on new kinds of hybrid organic–inorganic hybrid materials based on a polymer and ILs is gaining unprecedented importance for developing batteries and supercapacitors that can offer a highly efficient and sustainable way of energy storage. Highly ionic-conductive polymers when combined with inorganic compounds can improve the charge storage capability, electrode lifetime, rate capabilities, voltage, mechanical as well as thermal stability of electrochemical devices.1 Therefore, development of new kinds of polymer ion–gel based inorganic–organic hybrid materials for charge storage applications inherent with properties like good electrical conductivity, flexibility, superior charge storage capacity, high coulombic efficiency, high ionic conductivity at ambient temperature and good thermal stability via low cost routes is very crucial for helping us to face the current scenario of energy crisis. Polymer–ionic liquid ion–gel electrolytes and thin films have been widely explored during the past few years for applications like heterogeneous catalysis, as photonic crystal gas sensors for detecting analytes like glucose and ammonia, for transport and separation of binary gas mixtures, in proton exchange membrane fuel cells (PEMFCs), lithium ion batteries, solid state flexible supercapacitors and dye-sensitized solar cells (DSSCs).2–9 The most commonly studied polymer–ionic liquid matrix includes combinations of polymers like poly(4-vinylphenol), poly(ethylene oxide) (PEO) and poly(vinylidene fluoride) (PVDF) and ionic liquids (ILs) of alkyl pyrrolidinium, alkyl imidazolium and alkyl sulfonium based cations with different types of anions.10–17 However, not much have been studied on the scope of pyridinium based ILs in combination with polyvinylpyrrolidone (PVP) for charge storage applications. Among the conjugated polymers, polyvinylpyrrolidone (PVP) deserves a special attention due to its easy processability, thermal stability and flexibility, moderate electrical conductivity and ability to permit faster ionic mobility compared to other semicrystalline polymers, due to its inherent amorphous nature.18,19
This research work is driven by the need to explore the following aspects of PVP-based ion–gel polymers;
(a) The amorphism of PVP that permits faster ion mobility and good flexibility can be combined with the benefit of high ionic conductivity and wide electrochemical window of ILs for developing flexible high ionic conducting polymer systems for solid state fuel cells and batteries.
(b) Studying the electronic and atomic or molecular level interactions between PVP and ILs can help in understanding the overall nature of these polymer–IL blends in order to tune them for specific applications. The PVP pyrrolidone rings possess highly polar proton accepting carbonyl group that creates a favourable environment for bpy[BF4] to perfectly blend among the polymer chains resulting in a highly conducting medium for electrons.
(c) Generally, ion–gel polymers consisting of IL molecules trapped inside a flexible polymer matrix exhibits large capacitance values.20–23 Investigating the capacitance properties of this PVP–IL system will be a new contribution to the polymer ion–gel research and indeed very interesting.
(d) Researchers have already reported that doping iron oxide like materials having high dielectric constant can improve the efficiency of electronic/opto-electronic devices and sensors by improving the charge storage capacity, minimizing the leakage currents and the problem of quantum tunneling through dielectric barriers.24,25 Hence, more insight can be obtained on improving the charge storage property of this PVP–IL material by doping γ-Fe2O3 like inorganics.
Many ILs have been successfully used as a plasticizer or medium to carry out free radical polymerization reactions or to dissolve or disperse polymers and these abilities are being governed by the forces of dispersion, hydrogen-bonding, n–p, p–p, dipolar, hydrophobic and ionic interactions between ILs and polymers.26–28 Pyridinium based ILs are highly versatile in comparison with their imidazolium based counterparts and have experimentally been proved to facilitate more rapid intermolecular electron transfers (ET).29–32 Studies of solvent-mediated and photoinduced ET in pyridinium based ILs suggested that the favorable reduction potential of the pyridinium cations may even permit electron migration to occur via a secondary reduction of one or more solvent cations.30,33,34 This property arises from the tendency of negative charges like excess electrons to preferentially migrate over the aromatic ring which has strong π-type Lewis acidity character which is also favoured by the accompanying anion.35 Earlier, the studies on ET process in bpy[BF4] by pulse radiolysis pointed out that the IL not only acts as solvent but also as active solute in solvent-mediated reactions, while the BF4− in bpy[BF4] were not active and did not react with solvated electrons.36 Other observations from laser flash photolysis (LFP) studies suggest that solvent mediation increases both rates and efficiencies for ET processes in pyridinium based ILs.30 As a polymer dopant, bpy[BF4] which has low viscosity can significantly enhance the room temperature conductivity of IL–polymer composites.6–8
Therefore, in this paper we have studied a new type of inorganic–organic hybrid ion–gel polymer from γ-Fe2O3 doped PVP–bpy[BF4] composite having good flexibility, low charge transfer resistance, high specific capacitance, high energy density and better charge–discharge characteristics than pure PVP or PVP–bpy[BF4] composite. The immense potentiality of iron oxide based MNPs have already attracted immense experimental and theoretical research attention in various fields.37 PVP coated γ-Fe2O3 MNPs were doped within the newly synthesized PVP–IL thin film for improving the charging–discharging properties and energy density of the PVP–IL thin film. Here, we have investigated the following aspects: (a) intermolecular interaction between PVP and IL through electron transfer (ET) from PVP to IL, (b) the ESR activity of the PVP–IL composite leading to complete cross-polymerisation of the PVP chains under aerobic heating conditions to form a amorphous thin film (Fig. 1(c) and (e)), (c) DFT analysis of PVP–IL binding energy, changes in enthalpy and entropy values before and after binding, plot of density of states (DOS) and molecular orbitals of PVP–bpy[BF4] system, (d) changes in band gap and electron–hole recombination rate of PVP–IL before heating, after heating and after doping γ-Fe2O3, (e) influence of γ-Fe2O3 MNPs on the specific capacitance, energy density and charging–discharging property of the newly synthesised PVP–IL thin film and (f) thermal stability of PVP–IL ion–gel.
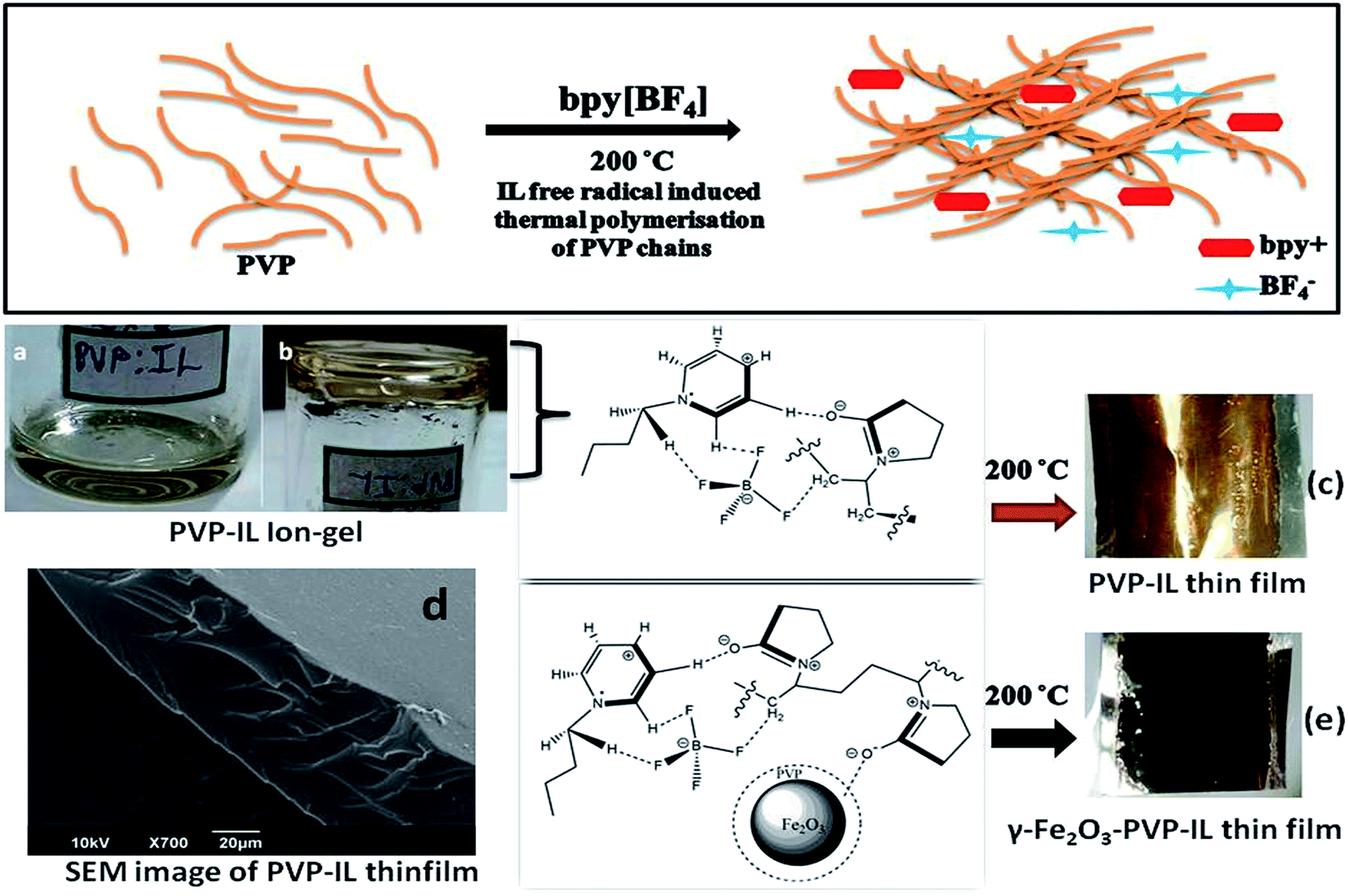
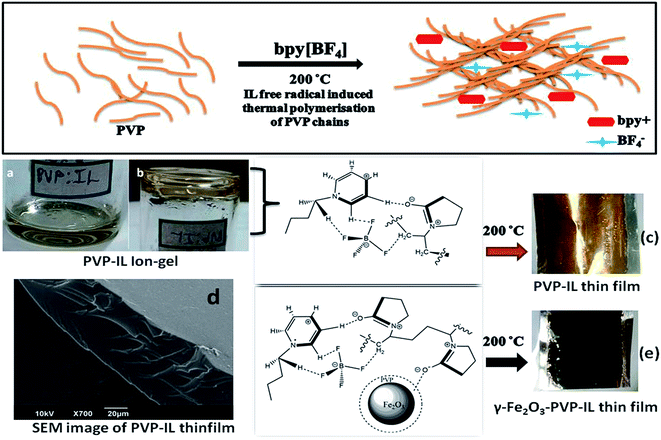 |
| | Fig. 1 Schematic representation of bpy+˙ cation radical initiated thermal polymerization in PVP chains at 200 °C is shown above. (a) Picture showing the nature of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 PVP–IL mixture (b) inverted container with PVP–IL mixture without flowing down (c) yellow coloured PVP–IL thin film coated on an Al foil after heating at 200 °C (d) SEM image of PVP–IL thin film formed at 200 °C (e) γ-Fe2O3 doped magnetic PVP–IL thin film coated on an Al foil. :1 PVP–IL mixture (b) inverted container with PVP–IL mixture without flowing down (c) yellow coloured PVP–IL thin film coated on an Al foil after heating at 200 °C (d) SEM image of PVP–IL thin film formed at 200 °C (e) γ-Fe2O3 doped magnetic PVP–IL thin film coated on an Al foil. | |
2. Experimental
Materials
N-Butylpyridinium tetrafluoroborate (bpy[BF4]) of stated purity >95%, was purchased from TCI Chemicals Pvt. Ltd. (India). Polyvinylpyrrolidone (PVP K-30) powder having average Mw 40000 and tetramethylammonium hydroxide (25 wt% in H2O) was procured from Sigma Aldrich. Acetone for analysis from EMPARTA® ACS was used as solvent. Iron(III) chloride anhydrous (98%) and iron(II) sulphate heptahydrate (99.5%) was purchased from Acros Organics. About 30% GR ammonia solution from Merck was used.
Synthesis
Fabrication of PVP–IL thin film. PVP in the form of a white powder was completely soluble in bpy[BF4]. PVP–IL composite thin film was prepared as follows. 0.29 g of PVP dissolved in 5 mL acetone was mixed with 0.29 g of bpy[BF4] (IL) which was liquid at room temperature and heated at 30 °C with constant stirring for 1 h till a purely colorless, uniform and highly viscous ion–gel was formed after the complete removal of acetone (shown in Fig. 1(a) and (b)). This material was coated on glass plate or Al foil cleaned over using acetone in an ultrasonic bath which was then heated in an air oven at 200 °C to obtain yellow amorphous polymer ion–gel thin film (Fig. 1(c)) without using any binder molecules. The 1:1 mixture of PVP and IL heated at different temperatures: 25, 50, 100, 150, and 200 °C are denoted as PVP–IL 25, PVP–IL 50, PVP–IL 100, PVP–IL 150 and PVP–IL 200 respectively.
Synthesis of PVP coated γ-Fe2O3 MNPs. PVP capped iron oxide nanoparticles was prepared by the following method. To a solution of 1 g PVP dissolved in 40 mL deionized water, 2 mmol FeSO4 and 4 mmol FeCl3 was added. To this, 5–10 mL of 30% aqueous ammonia was added till the pH was about 10. The mixture was heated for 45 min at 50 °C. The PVP coated γ-Fe2O3 MNPs were separated washed with ethanol and deionised water and dried at 80 °C for 1/2 an hour. The PVP capping over γ-Fe2O3 MNPs has two important effects. One is to confine the size of MNPs to nano dimensions and the other is the effective blending of iron oxide nanoparticles with the PVP–IL mixture. The HR-TEM images of the PVP coated γ-Fe2O3 nanoparticles thus synthesized is shown in Fig. S1 in ESI.†
Fabrication of γ-Fe2O3 MNPs doped PVP–IL thin film. For the preparation of magnetic γ-Fe2O3 MNPs doped PVP–IL thin film (γ-Fe2O3–PVP–IL), 0.1 g of the PVP coated γ-Fe2O3 MNPs synthesised using the above method was dispersed in acetone by ultrasonication for 1 h. This was mixed with 0.29 g PVP and 0.29 g bpy[BF4] dissolved in 5 mL acetone. The mixture was slowly heated at 30 °C with constant stirring for 1 h till the acetone was completely removed. The black thick viscous fluid obtained was coated on a clean glass plate or Al foil and heated at 200 °C for fabricating magnetic thin film (Fig. 1(e)). Since the MNPs were initially coated with a thin layer of PVP during the co-precipitation reaction, the effective blending of MNPs within the cross-polymerized PVP–IL matrix was possible.
Characterization techniques
Scanning electron micrograph of PVP–IL 200 thin film was attained with the help of JOEL JSM-6390 SEM instrument. The high resolution transmission electron micrograph (HR-TEM) and SAED pattern of the PVP coated γ-Fe2O3 MNPs were obtained using JEOL JEM-2100 microscope at an accelerating voltage of 200 kV. The ESR spectra of PVP–IL composite at different temperatures was recorded using JEOL JES-FA200 ESR Spectrometer (Japan) with X band and Q band, of E standard frequency range 8.75–9.65 GHz and sensitivity 7 × 109 spins/0.1 mT. Due to the liquid nature, the PVP–IL composite was filled in a capillary tube sample holder and heated at different temperatures for taking ESR measurements. The UV/vis spectra were collected from a Thermo Scientific EVOLUTION 201 spectrometer that uses a 546.1 nm mercury line and can measure in the wavelength range from 190 to 1100 nm. The fluorescence spectra were measured with an HORIBA (TCSPC) Fluorolog FL-1057 spectrometer. The FT-IR spectra were obtained using a Perkin Elmer Spectrum 400 FT-IR/FT-FIR spectrometer. Raman spectra, Raman imaging and AFM imaging was done using integrated WITec Raman-AFM Alpha300 RA-Confocal Raman spectrometer. The alpha300 RA consists of the Raman microscopy system (alpha300 R) and Atomic Force Microscopy (alpha300 A) for chemical imaging and nanoscale surface characterization that can facilitate a comprehensive understanding of the thin film samples. The solution state C-13 NMR analysis was done using NMR 400 MHz – Bruker Spectrometer in D2O and solid state C-13 NMR was done using ECX-II JEOL-400 MHz with glycine as an internal standard. The XRD analysis of γ-Fe2O3–MNPs and γ-Fe2O3–MNPs doped PVP–IL thin film was done with the help of XPert PRO PANalytical X-ray diffractometer using Cu Kα radiation, in the 2θ range of 20–80° and magnetic properties was analysed using Lakeshore VSM 7410 Vibration Sample Magnetometer. For the analysis of thermal properties, TG-DTG of the samples was taken using NETZSCH STA 449 F3 Jupiter thermal analyser. Electrochemical measurements were executed by Biologic SP-200 electrochemical workstation. Electrochemical impedance spectroscopic (EIS) measurements and cyclic voltammetric measurements were done by Biologic SP-200 workstation (Fig. S2 in ESI†). The frequency range in EIS measurements studied varied from 100 kHz to 100 mHz. The voltage between two electrodes was kept at 0.05 V during the measurements. Leakage current was measured using chronoamperometry under the same experimental setup under a constant potential of 0.35 and 0.75 V.
For all the electrochemical measurements, customized three electrode system was used for the measurements in which the pencil graphite rod with 2 mm diameter was the working electrode, Pt wire as counter electrode and Ag–AgCl as reference electrode. For the working electrode preparation, electrochemically cleaned pencil graphite rods with same length and diameter were adopted for all the samples. The working electrode have been polished using both alumina and diamond powder and were subjected to electrochemical pretreatment by applying voltage from 1.2 to −1.2 V in 1 M H2SO4 until superficial cycles were obtained. After this electrochemical pre-treatment, the upper portion of the pencil graphite rod was tightly covered with a Teflon band as shown in Fig. S2 in ESI† and the lower circular surface was coated with the samples by drop casting and drying at high temperatures. For this, the 1:1 mixture of PVP–IL ion–gel was coated over the lower circular circumference of the graphite rod and heated at 150 and 200 °C for measuring the electrical properties of PVP–IL 150 and PVP–IL 200 thin films. All the measurements were done using 1 M tetramethyl ammonium hydroxide in methanol as electrolyte.
Computational details
The molecular structures optimization, binding energy and thermochemistry calculations were carried out using DFT with 6311G/B3LYP and 6311++G (d,p)/B3LYP level of theories using Gaussian 09.38 The calculations were done with the help of Polish Grid Infrastructure. The results from G 09 in the form of formatted checkpoint (fch.) files were analysed using Multiwfn software (Version 3.3.8). Multifunctional Wavefunction Analyzer or Multiwfn is a free software developed by Tian Lu and his team at the Beijing Kein Research Center for Natural Sciences.39
3. Results and discussion
Identification of free radical involvement from ESR spectra
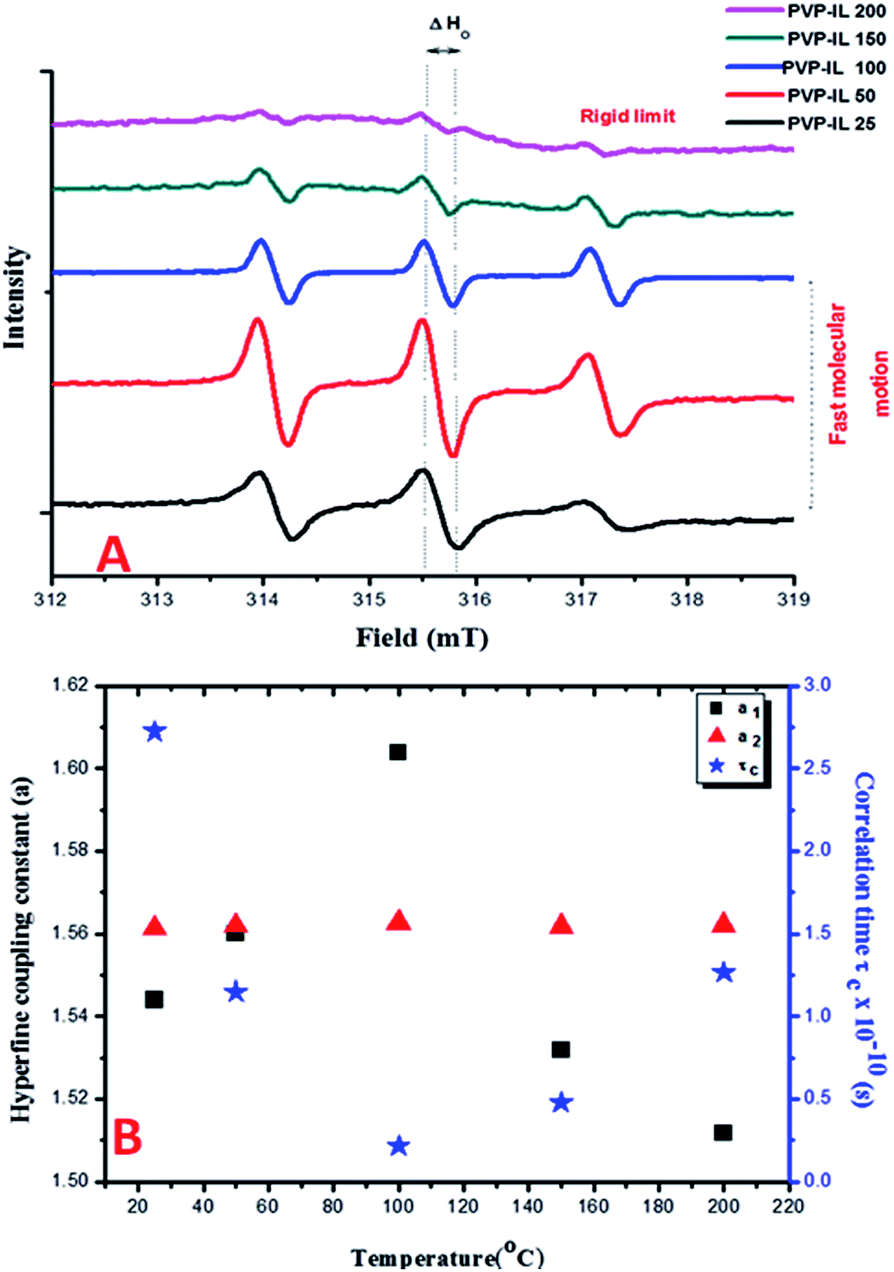
The ESR spectra of spin probed non-functionalized PVP and blends of PVP with other polymers such as PEO exhibiting a single line ESR spectrum have been previously reported in the literature.40–42 From our observation of the continuous wave (CW) X-band ESR spectra of the 1:1 mixture of PVP–IL ion–gel measured at different temperatures (25, 50, 100, 150 and 200 °C) in Fig. 2(A), the ESR signal of PVP was split into three lines in the presence of bpy[BF4] reflecting the strong intermolecular interaction between them. This ESR activity was suspected due to the spontaneous formation of 1-butylpyridinium radical ions (bpy+˙) from bpy[BF4] without the help of any induced process like pulse radiolysis or LFP, that hold the key to the free radical induced cross polymerization of PVP chains at high temperature. The nature of the cation radical present in the PVP–IL composite and thin film was determined from ESR spectra. Fig. 2(A) shows the ESR lines exhibited by PVP–IL matrix with relative intensity 1:1:1 showing the strong presence of an unpaired electron in the composite. The ratio of the relative intensity of the ESR lines indicates that the splitting is caused by a single nucleus of spin value I = 1. Hence, it was inferred that the electron-nuclear hyperfine interaction from 14N atom (I = 1) gave rise to the ESR splitting and the free electron was confined to the electron deficient N atom in bpy+ cation of bpy[BF4]. In the 1:1 PVP–IL mixture, the IL free radical ion could exhibit a fast tumbling motion between temperature range of 25 to 100 °C, averaging all the anisotropies. The ESR spectra of radicals in liquids is highly dependent on the rate of this molecular tumbling.43 However, the question is whether in a 1:1 PVP–bpy[BF4] mixture of high viscosity, will the rate of molecular tumbling (τ−1) be large or small compared to the anisotropies in the hyperfine interaction. The anisotropies are more effectively averaged out at 100 °C than at 25 °C when the viscosity is lowered and the molecules could undergo faster kinetic motion. The changes in the H-bonding interaction between polar PVP carbonyl group and the ring H at the 3rd position of the bpy+ cation (showed in Fig. 3) at different temperatures accounts for the variation in line width of the hyperfine multiplets denoted by ΔHo in Fig. 2(A). In the PVP–IL composite, the ion transport occurs as a result of the combined effect of IL ion movement, local motion of the PVP polymer segments and the changes in the co-ordination sites between the PVP and bpy[BF4]. After cross-polymerisation of the PVP–IL matrix at 200 °C, the local relaxation process of the PVP chains that experienced a liquid like degrees of freedom became restricted. This resulted in the trapping of IL radical ions in between the PVP polymer network and changes in the line intensity of the ESR spectra. Therefore, anisotropies showed up in the ESR spectra recorded at 200 °C. The new type of PVP–IL 200 thin film was also ESR active. At temperatures up to 50 °C, the intensity of the ESR peak increased considerably. As the integrated intensity of the spectrum is proportional to the concentration of radicals in the sample, it is quite evident from Fig. 2(A) that above 50 °C, a drastic reduction in the peak intensity was observed due to the increased number of radicals consumed for free radical initiated cross polymerization of the PVP chains.
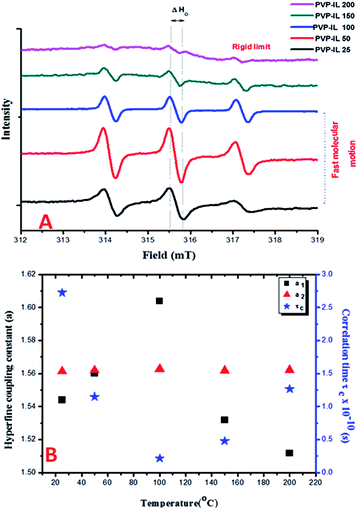 |
| | Fig. 2 (A) The X-band CW ESR spectra of 1:1 mixture of PVP and bpy[BF4] measured at 25, 50, 100, 150 and 200 °C. (B) The variation in the hyperfine coupling constants (a1 and a2) and correlation time of the free radical ion embedded in PVP matrix with respect to temperature. | |
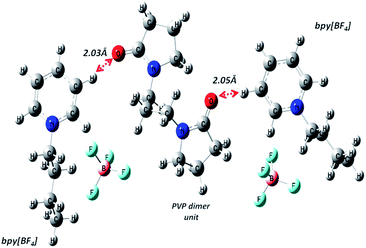 |
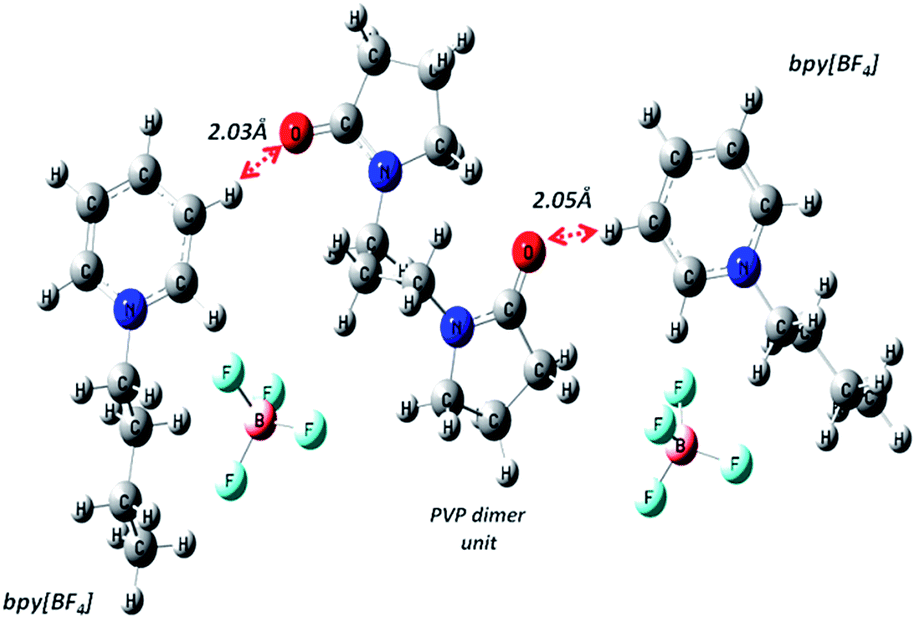
| | Fig. 3 Geometry optimized structure of PVP dimer and two bpy[BF4] molecules showing close interaction between carbonyl O atom of PVP and H atom of pyridinium cation ring. | |
Fig. 2(B) shows the plot of temperature vs. the hyperfine coupling constants (a) and correlation times (τc) determined from the spacing of the lines and the height of the intensity peaks indicating the way in which the unpaired electron spin density is distributed in the molecule. Correlation times (τc) for the free radical ions in PVP matrix at different temperatures were obtained using the expression (1) given below:44
| |
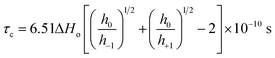 | (1) |
where
h+1,0,−1 refer to the heights of the three spin-label lines as shown in Scheme 1 in ESI.
† The value of
τc decreases from 25 to 100 °C till before the onset of cross-polymerization and then increases. Therefore the rate of molecular tumbling increases from 25 to 100 °C and then decreases subsequently after cross polymerization. Previously, there have been reports of metal atoms of Na, Li or Cs when dissolved in pyridine gave yellow solution with ESR activity in frozen state due to the formation of pyridine radical anion which rapidly reacted at room temperature to form 4,4′-bipyridyl radical anion.
45 The PVP–IL system, in our observation was similar to this provided that the electron supplying agent was a polymer, not a metal. The relatively comparable energy levels of PVP and bpy[BF
4] would have favoured an effortless electron migration from PVP to pyridinium moiety. The possibility of electron hopping from electron rich PVP oxygen atom to the electron deficient bpy
+ cation ring
via H-bond mediated route favoured by the geometrical orientation of pyridinium and pyrrolidone rings obtained theoretical supported from the DFT optimized PVP–IL molecular structures obtained from Gaussian 09 using 6311++G (d,p)/B3LYP level of theory (as shown in
Fig. 3). The type of interaction stabilizes the negative charge density on the O atom in PVP forming a partial O⋯H bond through a stable resonance structure involving the shifting of lone pair electrons of the adjacent N atom in PVP, thus increasing the spin density on the N atom of bpy
+ cation.
DFT analysis of PVP–bpy[BF4] intermolecular interaction
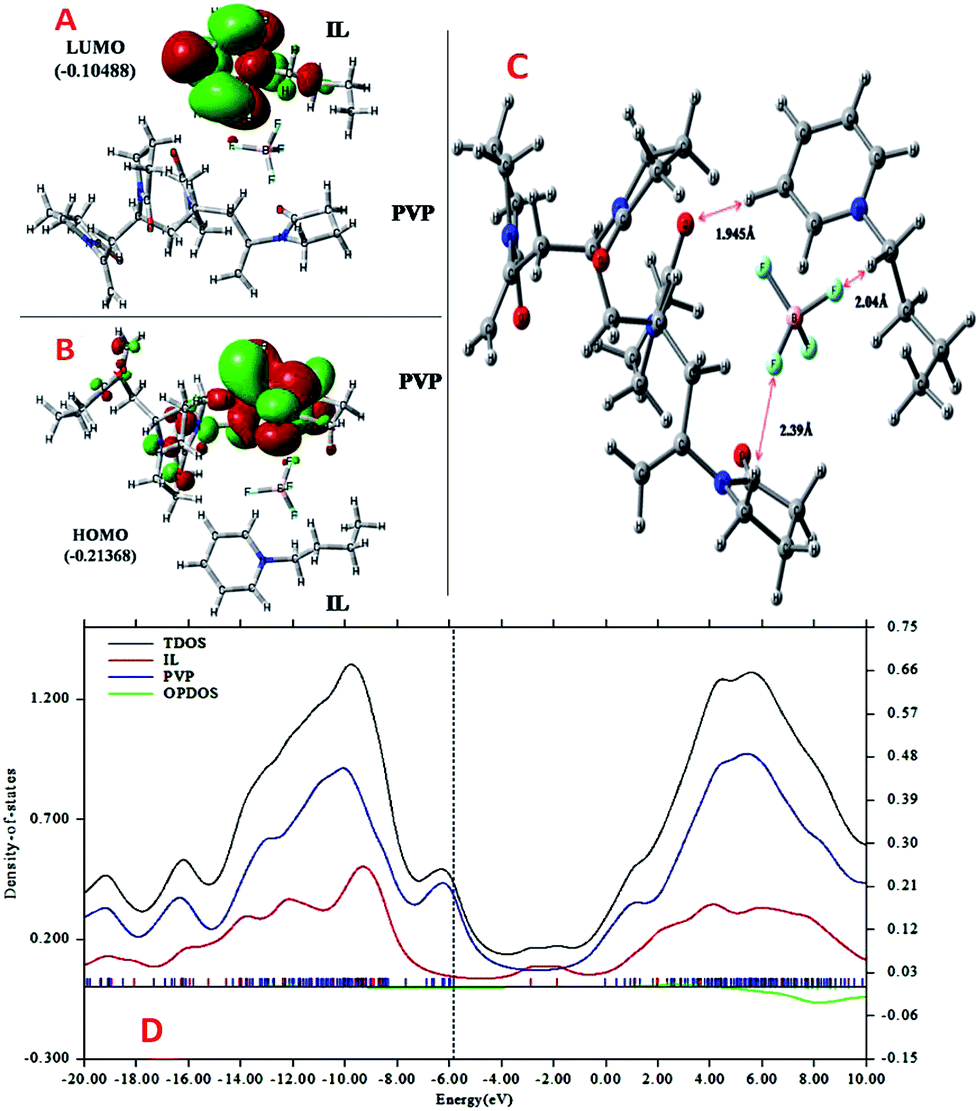
More detailed understanding of the nature of the interaction in this PVP–IL blend can be understood from the structure of its HOMO in Fig. 4(A) and LUMO in Fig. 4(B). A major portion of the HOMO of the IL–PVP tetramer system is contributed by the pyrrolidone ring of the PVP structure where as the LUMO is contributed by the bpy+ ring of the IL. This also explains the possibility of charge transfer from PVP HOMO to IL LUMO in the PVP–IL system. The electron deficient pyridinium ring can accept electrons from the electron rich pyrrolidone rings making it a highly conducting medium for transport of free electrons. Thus, in addition to the ESR data, theoretical results also point towards a proton coupled-electron transfer (PCET) process occurring in PVP–IL mixture which involves the concerted transfer of an electron and proton to or from a substrate.
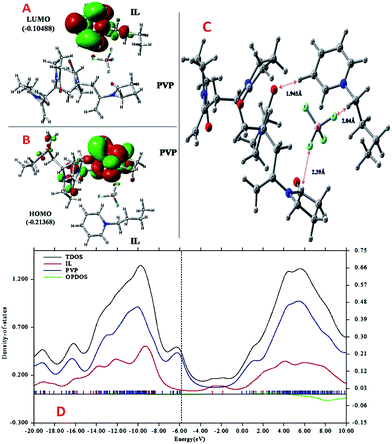 |
| | Fig. 4 ((A) and (B)) The structure and energy in atomic units (a.u.) of HOMO and LUMO (C) analysis of intermolecular distance and H-bonding interactions in the DFT optimized (D) the plot of density of states (DOS) versus energy (in eV) the bpy[BF4]–PVP tetramer system. | |
From structure optimization of the PVP tetramer unit, it was observed that the pyrrolidone rings attached to the carbon chain skeleton was in staggered position in order to minimize the mutual steric hinderance between the rings. The DFT optimized structure of a staggered conformer of PVP tetramer binding to a bpy[BF4] molecule obtained from Gaussian 09 using Density Functional Theory (DFT) at 6311G/B3LYP level of theory is shown in Fig. 4(C). The binding energy of this PVP–IL system was calculated to be 2180137 kJ mol−1 (830.37 hartrees) from the expression:46 ΔE = EPVP–IL − (EPVP + 2EIL). The bound state of tetramer PVP–bpy[BF4] complex was extremely stable. The enthalpy and free energy change when a PVP tetramer unit binds to a single IL molecule was calculated to be 199.102 kJ mol−1 and −10.929 kJ mol−1 from the thermochemistry data. It is evident that both the cation and anion of the bpy[BF4] effectively bound to the PVP chain either through O⋯H or H⋯F routes. The three different types of halogen–hydrogen bonding: (a) between O-atom of pyrrolidone ring in PVP and H-atom of pyridinium ring in bpy[BF4] (b) intra molecular H-bond between F atom of tetrafluoroborate ion and H atom of n-butyl chain attached to pyridinium ring in the bpy[BF4] (c) H-bond between F atom of bpy[BF4] and H atom of pyrrolidone ring in PVP was observed. The highly ionic nature of the IL ions providing attractive electrostatic interactions between the polymer chain and ions also enhance the stability of the PVP–IL bound systems.
Though the exact nature of density of states (DOS) for organic semiconductors is not known precisely and is a matter of intense investigation,47 the DOS plot for such systems can be used as a valuable tool for studying the nature of their electron structure and transport properties. The original total density of states (TDOS) of an isolated system such as an ionic liquid molecule can be written as:39
| |
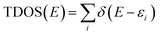 | (2) |
where ‘
ε’ is Eigen value set of single-particle Hamilton, ‘
δ’ is Dirac delta function. By replacing ‘
δ’ by broadening function
F(
x), such as Gaussian, Lorentzian and pseudo-Voigt function, broadened TDOS can be obtained. Here, we have used Lorentzian function to plot the DOS curves. The plot of broadened partial DOS (PDOS) and overlap DOS (OPDOS) for the ionic liquid (bpy[BF
4]) and PVP tetramer unit was obtained from Multiwfn after defining them as fragments 1 and 2 respectively. The PDOS function of the fragments is defined as:
| |
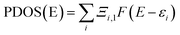 | (3) |
where
Ξi,1 is the composition of the fragment in orbital
i. The OPDOS between fragment 1 and 2 is defined as:
| |
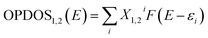 | (4) |
where
X1,2i is the composition of total cross term between fragment 1 and 2 in orbital
i.
39 Fig. 4(D) shows the TDOS and OPDOS plots for the bpy[BF
4]–PVP tetramer system. The plots help in visualizing orbital composition analysis in the PVP–IL composite. From the curves corresponding to TDOS, PDOS (red line for fragment 1 corresponding to IL, blue line for fragment 2 corresponding to PVP) and OPDOS (green line), the characters of each orbital can be identified. The degenerate energy levels cannot be distinguished due to the overlapping of the energy levels. The height of black curve (TDOS) in
Fig. 4(D) clearly shows how dense the energy levels are distributed. The region in the plot where blue curve of PDOS is high and nearly approaching black line above it indicates that the orbitals of PVP has significant contribution to corresponding MOs. The region where the green curve is positive on the
Y-axis indicate that the corresponding MOs are favourable for bond formation between IL and PVP and for regions of green curve is negative indicates that the corresponding MOs are unfavorable for bonding. The vertical dashed line indicates the position of HOMO level. The orbitals to the right end of this vertical line contribute the high-energy state orbitals which are of antibonding character. The occupation of these high-energy state orbitals renders the molecule to be highly unstable or broken. From the analysis of OPDOS curves in these plots, it is understood that the MOs participating in electron exchange between the IL and PVP are those having energy less than 4.5 eV approximately. This is the region were the OPDOS curve is positive after which it begins to proceed towards negative region. During the critical point (CP) analysis of the PVP–IL system (in
Fig. 5) using multiwfn, the Poincaré–Hopf relationship given in the expression
(5) was verified which indicated that all CPs may have been found.
| | |
n(3,−3) − n(3,−1) + n(3,+1) − n(3,+3) = 1
| (5) |
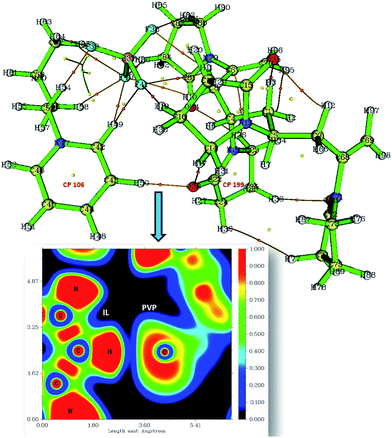 |
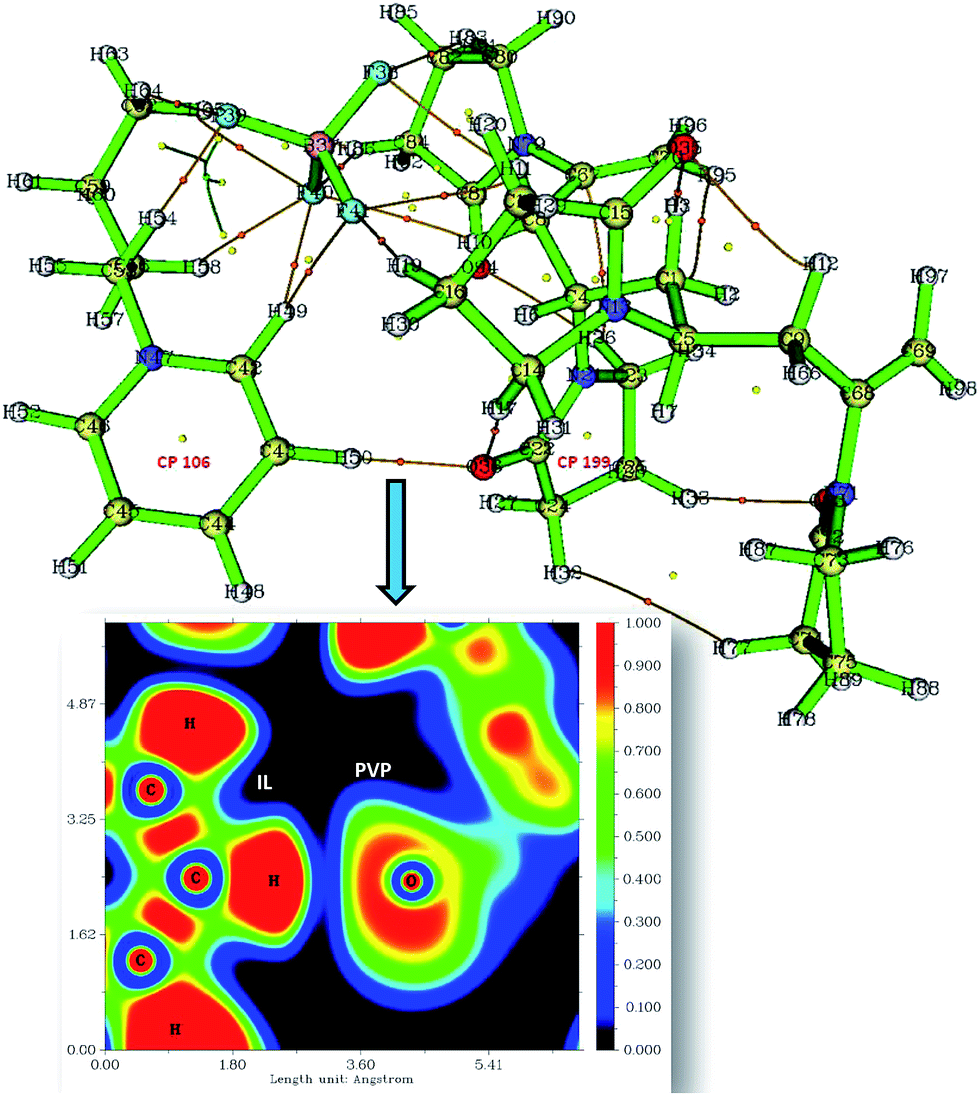
| | Fig. 5 The image shows the CPs (yellow and orange spots) and CP paths generated after critical point analysis of PVP–IL system and the plot of electron density distribution between O36 of PVP and H50 of bpy[BF4] along the C–H⋯O plane (orange spots-bond critical points (BCP), yellow spot-ring critical points (RCP)). The inset figure shows the plot of electron localization function (ELF plot). | |
Critical points (CPs) at which gradient norm of function value is zero (except at infinity) are four types: nuclear critical point (NCP) or (3,−3), bond critical point (BCP) or (3,−1), ring critical point (RCP) or (3,+1) and cage critical point (CCP) or (3,+3), depending on the number of Eigen values of Hessian matrix of real space function which are negative. The number of critical points of each of these types in PVP–IL system was (3,−3): 98, (3,−1): 122, (3,+1): 26 and (3,+3): 1. The (3,+1) CP is in the case of a centre of a ring in pyridinium and pyrrolidone rings having one negative and two positive curvatures, called the ring critical point (RCP). In the ring plane, there will be two positive curvatures; (1) from the ring centre where the electron density is low (example: CPs at the centre of the pyridinium ring-CP106 and at the adjacent pyrrolidone ring-CP199), (2) towards the rim where the electron density is high. The electron density decreases in the direction perpendicular to the plane of the ring. The Shannon aromaticity index was calculated for CP106 and CP199 attached to each other by H-bond between the pyrrolidone O atom (O36) and pyridinium ring H atom at 3rd position (H50) (see Fig. 5). The electron density at CP106 and CP199 was 0.02073 and 0.03695 respectively.
The Shannon Aromaticity (SA) index, a measure of spatial localization of electron density is defined using the expression (6) given below:48
| |
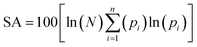 | (6) |
where
N is the number of considered bond critical points (BCPs) in the ring and
pi is the normalized probability electron density at the
ith BCP. The range of SA values between 0.003 < SA < 0.005 is proposed as the boundary between aromatic and anti-aromatic compounds. The calculated SA value from Multiwfn using the B3LYP/6311++G(d,p) level of theory indicated the antiaromaticity (SA = 0.04) of the bpy
+˙ ring in the PVP–IL bound system. The total Shannon entropy of this ring which is the sum of the local information entropies of the constituted bonds in the ring was calculated to be 0.6531. The electron density distribution between IL H
50 and PVP O
36 is shown in the plot of ELF (electron localization function) in
Fig. 5 indicating the overlap of electron density (blue region) of these atoms. These observations provide sufficient theoretical evidence for the phenomenon of electron hoping from PVP to bpy
+ cation.
Apart from H-bonding, other factors affecting the successful interaction between the PVP and IL are the IL anion–cation interaction energy, cation–cation stacking interaction and anion size. Cation–cation orientation in the IL bulk is mainly influenced by the aromatic stacking interactions such as edge-face, offset stacking and face–face stacking. Strength of off-set stacking is smaller in ILs containing BF4− like larger anion compared to that containing single-atomic anion such as Cl.49 This is because, the larger anions causing steric effect among the ions mostly occupy positions above and below the cationic ring having a delocalized positive charge. The weak cation–cation and anion–cation interactions in bpy[BF4] can result in a well distribution of the IL ions among the PVP chains.
Spectroscopic characterization
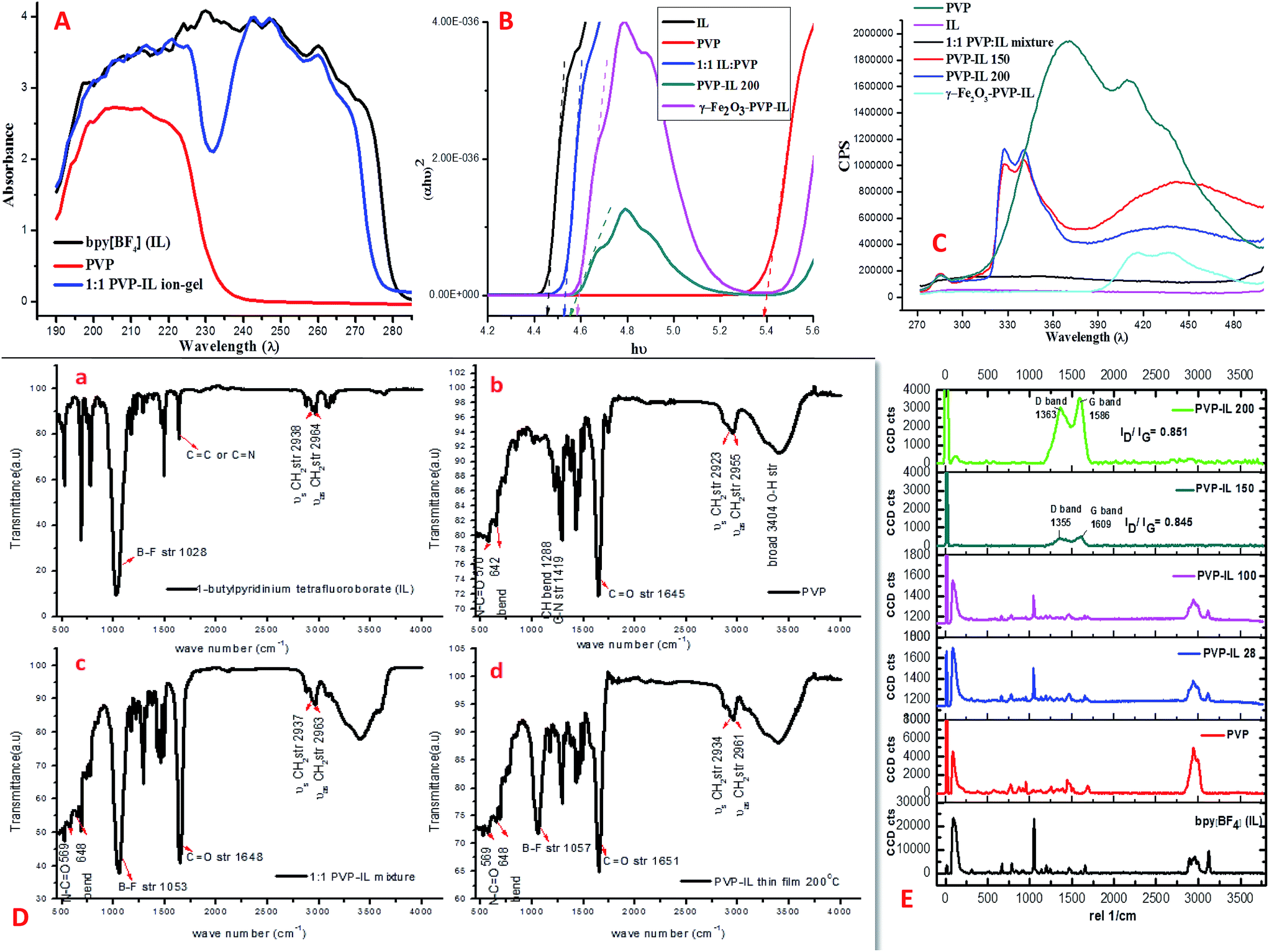
UV-visible spectra and band gap analysis. The charge transfer transitions occurring between PVP and IL in 1:1 PVP–IL mixture is evident from their UV-vis absorption spectra shown in Fig. 6(A). The UV-vis absorption spectra of bpy[BF4] exhibits a single broad absorption peak ranging from 190 to 280 nm (black line) while the PVP shows only a single broad absorption peak between 190 and 230 nm (red line) as a result of π–π* transitions from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. The 1:1 mixture of PVP–IL composite shows two different broad absorption peaks at the range of 190–230 nm and 240–275 nm (blue line). The first peak due to π–π* transitions from CO bonds of PVP showed a small shift towards higher wavelength region in 1:1 PVP–IL mixture which indicates the proper blending of IL with the PVP chain. The absence of any absorption band at 232 nm in the 1:1 PVP–IL blend indicates the strong binding between PVP and IL causing the prevention of certain absorptive transitions in IL due to the binding of PVP.
O bonds. The 1:1 mixture of PVP–IL composite shows two different broad absorption peaks at the range of 190–230 nm and 240–275 nm (blue line). The first peak due to π–π* transitions from CO bonds of PVP showed a small shift towards higher wavelength region in 1:1 PVP–IL mixture which indicates the proper blending of IL with the PVP chain. The absence of any absorption band at 232 nm in the 1:1 PVP–IL blend indicates the strong binding between PVP and IL causing the prevention of certain absorptive transitions in IL due to the binding of PVP.
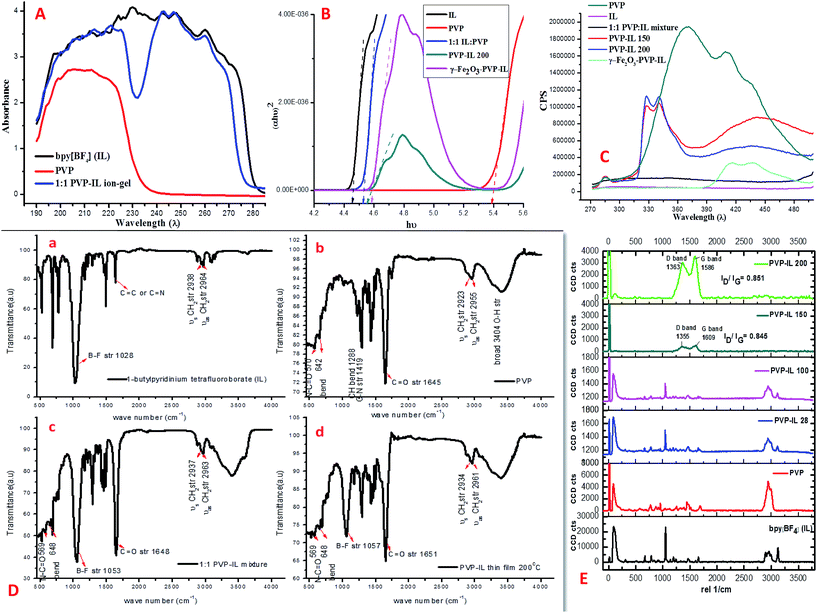 |
| | Fig. 6 (A) UV absorbance spectra of bpy[BF4] (IL), polyvinylpyrrolidone (PVP) and 1:1 mixture of IL and PVP. (B) Kubelka–Munk (K–M) plot of bpy[BF4] (IL), PVP, 1:1 mixture of PVP–IL ion–gel, PVP–IL 200 and γ-Fe2O3–PVP–IL showing variation in band gap. (C) Fluorescence spectra of PVP, bPy[BF4] (IL), 1:1 mixture of PVP–IL ion–gel, PVP–IL 150, PVP–IL 200 and γ-Fe2O3–PVP–IL at 260 nm excitation wavelength. (D) FTIR spectra of (a) bpy[BF4], (b) PVP, (c) 1:1 PVP–IL ion–gel and (d) PVP–IL 200. (E) Raman spectra of bPy[BF4] (IL), PVP, 1:1 mixture of PVP–IL composite, PVP–IL 100, PVP–IL 150, PVP–IL 200, γ-Fe2O3–PVP–IL at 260 nm excitation wavelength. | |
The energy band gap (Eg) was determined for PVP, IL, 1:1 PVP–IL mixture, PVP–IL 200 thin film and γ-Fe2O3–PVP–IL thin film using the conventional Tauc equation given below:50,51
where
α is the absorption coefficient;
h is the Planck constant;
ν is the photon frequency,
hν is the photon energy;
A is the constant; where
n is a constant (
n = 1/2, 3/2, 2, or 3 for allowed direct, forbidden direct, allowed indirect, and forbidden indirect transitions respectively).
Fig. 6(B) shows the Kubelka–Munk (K–M) plot of the materials showing variation in band gap obtained by plotting (
αhν)
2 versus hν. The band gap values obtained by extrapolating the linear portion of the plot to (
αhν)
2 = 0 on the
X-axis is shown by the dotted arrow lines. For PVP and bpy[BF
4] the band gap was calculated to be 5.35 eV and 4.44 eV respectively. In the 1
:
1 PVP–IL mixture, the bandgap value was 4.51 eV which is intermediate to the values of its constituent materials. This dramatic change in 1
:
1 PVP–IL mixture is due to the presence of bpy
+˙ radicals and [BF
4]
− anions trapped in between the PVP chains to promote the ET process among them. The decreased in PVP optical band gap in presence of IL indicates an improvement in the optical response of the PVP–IL mixture which may become useful in optical sensors. On heating the 1
:
1 PVP–IL composite at 200 °C, the band gap value slightly increases to 4.9 eV from 4.51 eV which indicates certain chemical transformations occurring in the PVP matrix that might have caused due to the change in the bpy
+˙ radical ion concentration and also due to the breakage of weak H-bondings between the PVP and IL molecules. However, incorporating γ-Fe
2O
3 MNPs into this thin film again decreased the band gap value to 4.55 eV thus improving the conductivity of the material.
Fluorescence spectra. The presence of IL in PVP–IL matrix can lead to localized states which traps or slows down the carriers. It was observed that the fluorescence intensity of 1:1 PVP–IL, PVP–IL 150, PVP–IL 200 and γ-Fe2O3–PVP–IL significantly decreased in comparison with the pure PVP indicating an increase in the electron–hole separation process. This can be attributed to the transfer of electron from PVP to the IL during photo excitation resulting in the acceleration of electron–hole separation rate. In Fig. 6(C), bpy[BF4] shows no fluorescence characteristics in the range of 280–480 nm region whereas, PVP shows strong broad peaks with multiple maxima at 370, 410 and 436 nm due to the electronic transitions involving the CO functional group. The PVP blue emission band at about 436 nm occurs due to the radiative relaxation of electrons from its LUMO to HOMO.52 In the 1:1 PVP–IL mixture, the fluorescence of PVP is totally quenched by the trapped states created by the IL preventing the direct band-to-band recombination in PVP. The fluorescence quenching was a result of the electron transfer from PVP to IL cation. This reveals that when the carboxylate oxygen co-ordinates with the bpy+ cation, the nitrogen lone pair in pyrrolidone ring becomes conjugated with the adjacent CO group. As a result, no fluorescence is shown by the 1:1 PVP–IL mixture. These results elucidate the following inferences on the whole electron–hole recombination process of these materials. The absorption of a photon of energy greater than the band gap energy of PVP would result in the formation of a conduction band electron and a valance band hole. The conduction band electrons are trapped by the intermediate energy states of the bpy+ cations in the IL which resulted in the formation of bpy+˙ radical ions. The greater the formation rate of bpy+˙ radical ions, the higher is the separation efficiency of electron–hole pairs in the PVP–IL matrix. However, in PVP–IL 150 and PVP–IL 200, the fluorescence peaks once quenched reappeared with a strong red shift to 327 and 340 cm−1. This showed a breakage in the intermolecular electron hopping pathway between PVP and IL resulting in a small increase in the electron–hole recombination rate. Doping γ-Fe2O3 MNPs in PVP–IL 200 again improved the separation efficiency of the electron–hole pairs. The fluorescence spectra of PVP–IL 200 and γ-Fe2O3–PVP–IL measured at 523 nm excitation wavelength can be seen in Fig. S3 in ESI.†
FTIR spectra. FTIR spectra of bpy[BF4], PVP, 1:1 PVP–IL mixture and PVP–IL 200 is given in Fig. 6(D). The strong band shown at 1028 cm−1 in the spectrum of bpy[BF4] belongs to B–F bond stretching.53 After mixing bpy[BF4] with PVP, the IL–PVP composite exhibited the characteristic absorption bands of both the constituents. Shift of the PVP CO peak at 1645 cm−1 to 1651 cm−1 in the 1:1 PVP–bpy[BF4] composite indicated strong coulombic or intra molecular H-bonding interaction between the CO group of PVP and 1-butylpyridinium cation or [BF4−] anion. From the FTIR spectra of IL, PVP, 1:1 PVP–IL mixture and PVP–IL 200, it was evident that almost all the functional group remained unchanged after heating except a small change occurred in the relative intensity of B–F stretching band compared to the CO peak intensity. In addition to this, position of B–F band and CO band at 1053 and 1648 cm−1 in the 1:1 PVP–IL mixture slightly shifted to 1057 and 1651 cm−1 respectively in the heat treated sample. From the summarized spectra features listed above, it can be seen that stronger molecular interaction exists between polymers and dopant. Both the FTIR and C-13 NMR spectra (in Fig. S4 and S5 in ESI†) supports the weak binding interaction between PVP and IL and the trapping of IL molecules in between the highly cross-polymerized PVP matrix. The possible mechanism of free-radical induced polymerization of PVP in presence of IL is shown in Scheme 2 in ESI.†
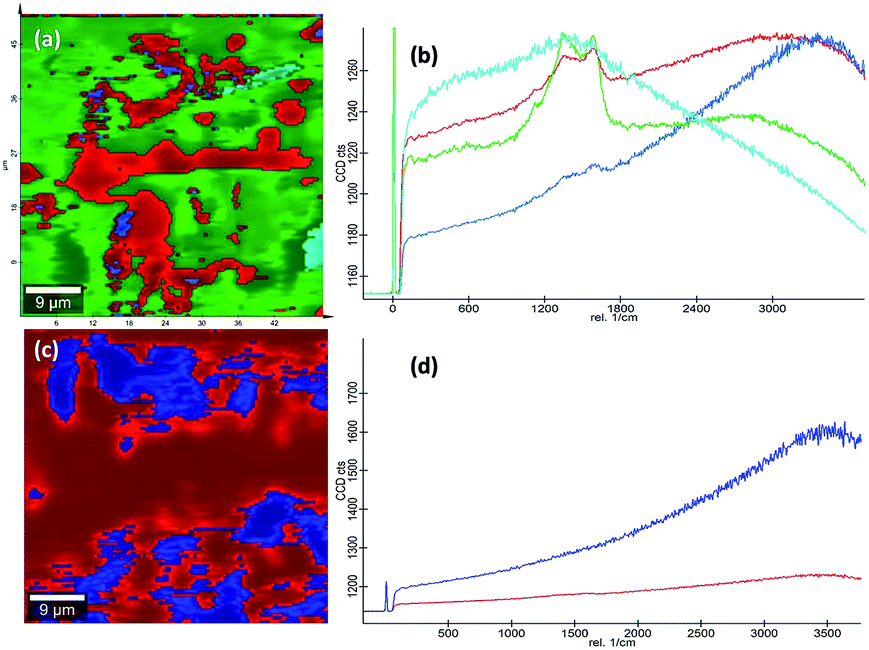
Raman spectra. Raman spectroscopy which uses monochromatic laser beam to interact with molecular vibrational modes and phonons is a very powerful noninvasive method for studying carbon-related materials. The Raman spectra of bpy[BF4] (IL), pure PVP (K30), 1:1 mixture of PVP and IL prepared at room temperature (28 °C), PVP–IL 100, PVP–IL 150 and PVP–IL 200 is shown in Fig. 6(E). Till 100 °C, the interaction between PVP and IL molecules were of a physical nature similar to that occurred in the 1:1 PVP–IL mixture prepared at room temperature. The chemical transformation in the PVP–IL composite occurs between 100 and 150 °C. The Raman spectra of the PVP–IL 150 and PVP–IL 200 exhibited strong broad D and G bands similar to that of graphitic oxide bands. In PVP–IL 150, D and G band occurred at 1355 and 1609 cm−1 respectively, which became more intense and shifted to 1363 and 1586 cm−1 respectively in PVP–IL 200. The red shift experienced by the G band can be the result of increase in thickness or number of layers after cross-polymerization.54 Previously, Maghsoumi et al. reported the formation of D and G peaks in the Raman spectra of polycyclic aromatic hydrocarbons.55 The contribution to the G peak may arise from the CC sp2 stretch vibrations of olefinic or conjugated carbon chains and thus does not necessarily prove that the film is an a-C film consisting only of graphite or fused benzene rings.56 Here, the G peak cannot solely claim its origin from the olefinic groups of the bpy+ moiety imbedded in the film. Instead, the thin film showed broad D and G bands suggesting the formation of amorphous carbon material. The ID/IG ratio of the composite heated at 150 °C and 200 °C is 0.845 and 0.851 respectively. From the ID/IG ratio obtained, the level of disorder can be characterized since it is directly related to the average distance between defects (LD).57 It is reported that at a “high” defect density, ID/IG will begin to decrease as an increasing defect density results in a more amorphous carbon structure, attenuating all Raman peaks.58 The resultant D and G bands are mainly caused by “sp2 hybridised amorphous carbon” phases. In a high-defect-density regime, a full breakdown of the carbon lattice occurs.59,60In addition to this, the presence of nitrogen centres in between the carbon chains along with the trapped IL ions after cross-polymerisation reaction elevated the degree of defective sites. This resulted in the enhancement of D and G bands corresponding to amorphous carbon material suppressing all the other Raman peaks. This observation is also supported in another study reported on polymer electrolyte films of PVP and 1-ethyl-3-methylimidazolium tetrafluoroborate ([EMIM][BF4]) in which the XRD data revealed that the inclusion of IL moieties increased the amorphous nature of PVP membranes.19 More detailed observations on the chemical composition of the PVP–IL 200 and γ-Fe2O3–PVP–IL thin films fabricated at 200 °C can be made from the 2D Raman spectral images shown in Fig. 7(a) and (c). The images show the presence of localized amorphous regions in the thin films which can enhance the low temperature conductivity of the material. The corresponding spectra in Fig. 7(b) and (d) mainly indicate the presence of sp2 amorphous carbon phase in the material. The Raman spectral intensity of γ-Fe2O3 doped PVP–IL thin film was observed to be higher than that of the undoped PVP–IL thin film.
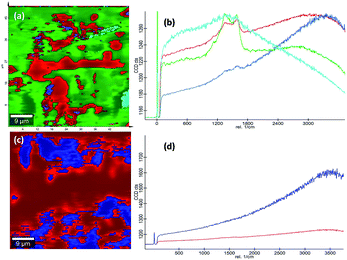 |
| | Fig. 7 (a and c) 2D Raman image and (b and d) the corresponding spectra of PVP–IL 200 and γ-Fe2O3–PVP–IL thin film. | |
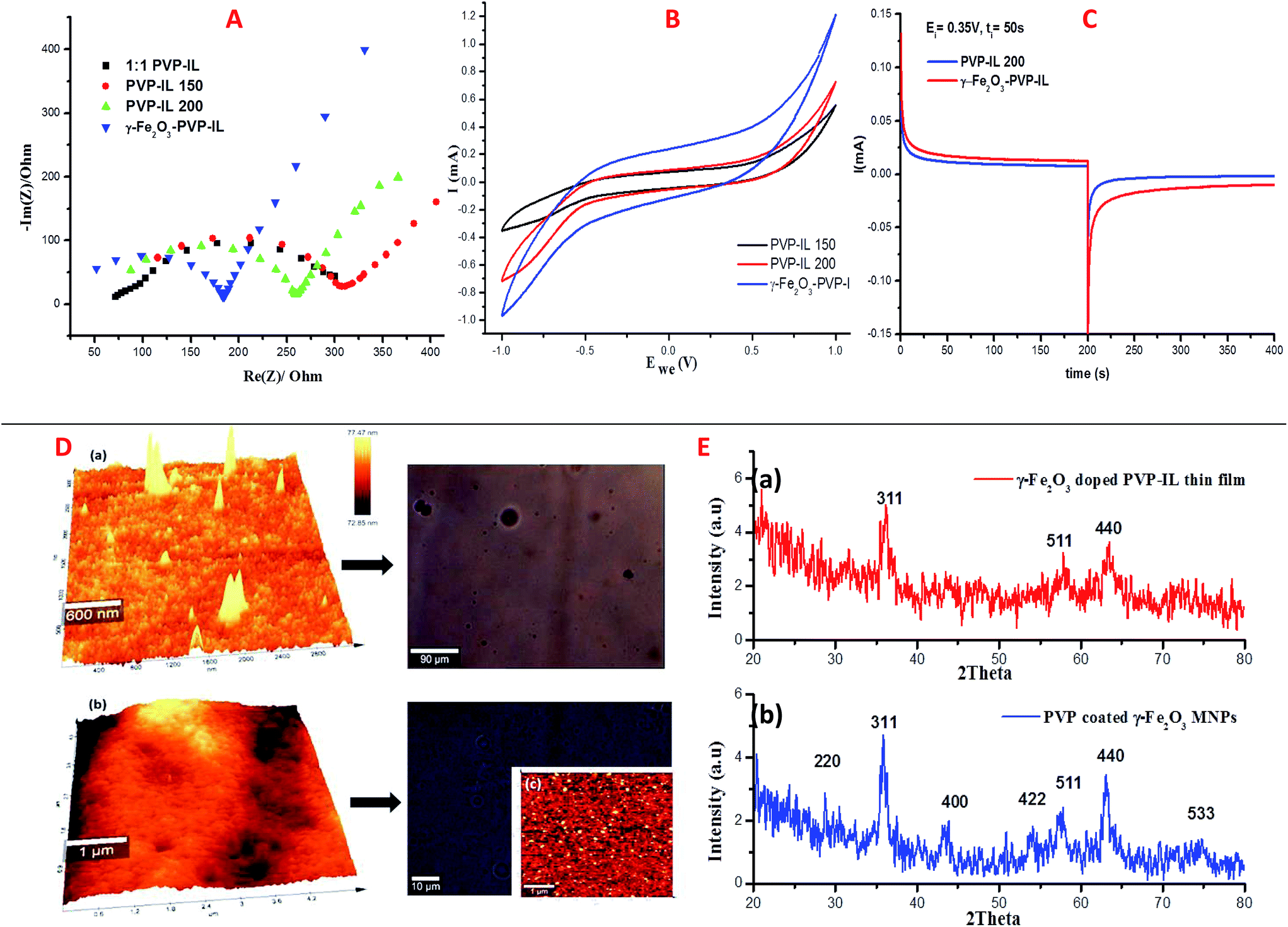
Electrochemical characterization
Using three-electrode system, electrochemical impedance spectroscopy (EIS) measurements of 1:1 PVP–IL composite, PVP–IL 150, PVP–IL 200 and γ-Fe2O3–PVP–IL were performed under potentiostatic control. Nyquist impedance plots in Fig. 8(A) indicates that on heating the 1:1 PVP–IL composite, the material exhibits capacitive properties. The radius of the semicircle in the plot for the γ-Fe2O3 doped PVP–IL became smaller than that for IL–PVP 200 and PVP–IL 150 which is characteristic of a decrease in the solid state interface layer resistance and the charge transfer resistance on the surface.61 From the Nyquist plot, the value of the ionic conductivity was calculated from the expression σ = l/RbA where, l is the thickness of the sample coating, A is the area of surface of graphite rod (A = πr2, where r = 1 mm), and Rb is the bulk resistance of the material denoted by the intercept on the X-axis of the EIS spectra.9 The ionic conductivity for PVP–IL 200 and γ-Fe2O3–PVP–IL was calculated to be approximately 331.7 and 450.66 μS cm−1. The new ion–gel polymer thus obtained was highly conductive than that of the PVP base material due to the trapped bpy+ and BF4− ions in between the chains. From the EIS spectra, γ-Fe2O3–PVP–IL shows more efficient separation of electron–hole pairs and higher charge transfer efficiency than PVP–IL 200 and PVP–IL 150. This is also evident from the CV curve of the materials shown in Fig. 8(B). The area of the CV curve under constant scan rates varies in the order: PVP–IL 150 < PVP–IL 200 < γ-Fe2O3–PVP–IL which indicates an increase in the capacitance property of the material. Specific capacitance values of PVP–IL 200 and γ-Fe2O3–PVP–IL measured using three electrode system at a scan rate of 10 mV s−1 was 2.575 and 92.215 F g−1 respectively. The areal capacitance of PVP–IL 200 and γ-Fe2O3–PVP–IL was calculated to be 0.1907 and 8.229 F cm−2 respectively from the three-electrode CV study using the expression given below:| |
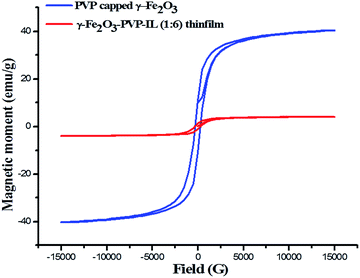
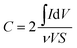 | (8) |
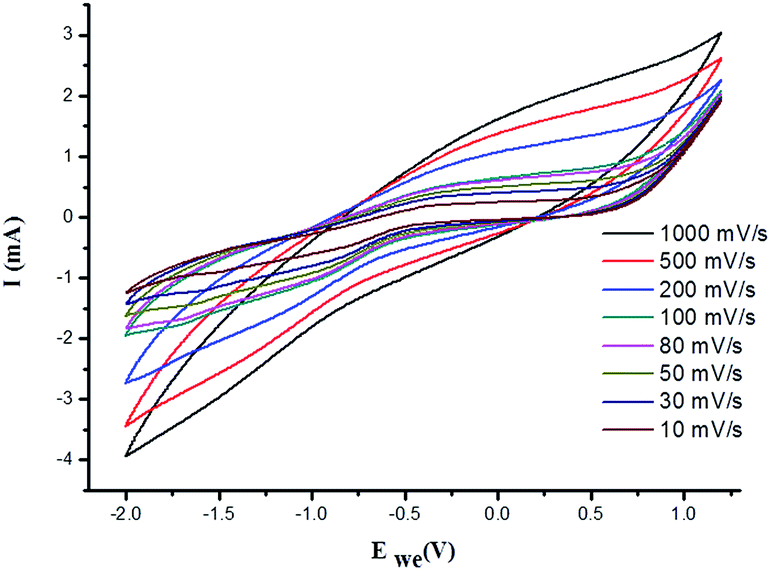
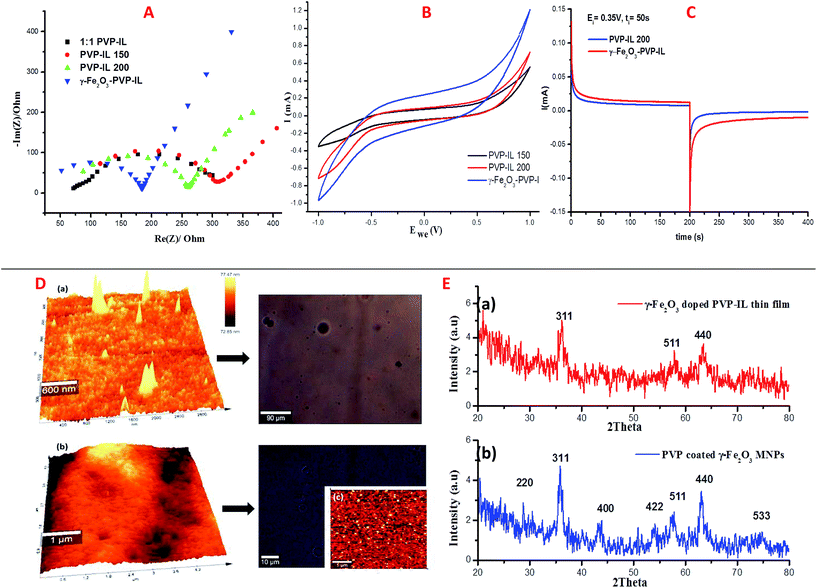
where, I is the current in the CV curve, ν is the potential scanning rate, V is the potential window, and S is the active materials surface area of the single electrodes (cm2).62 At about 7 Hz, the capacitance value of PVP–IL 200 was above 20 μF and that of γ-Fe2O3–PVP–IL was above 30 μF (shown in Fig. S6(A) in ESI†). Doping γ-Fe2O3 in PVP–IL 200 in the weight ratio 1:3:3 (γ-Fe2O3:PVP:IL) increased the energy density of the PVP–IL 200 from 1.386 × 10−6 to 2.049 × 10−4 W h kg−1. The CV curves measured at different scan rates for γ-Fe2O3–PVP–IL and PVP–IL 200 is shown in Fig. 9 and S6(B) in ESI† respectively. The area of the scan curves increased with the scan rate. Fig. 8(C) shows the charge–discharge behaviour of PVP–IL 200 and γ-Fe2O3–PVP–IL coated over the graphite electrode measured using chronoamperometric technique under a constant potential of 0.35 V applied for 50 s. The charging and discharging behavior of the materials measured using chronoamperometric technique under a constant potential of 0.75 V applied for 10 s is also shown in Fig. S7 in ESI.† Current I(t) flowing through the material after time t is the sum of charge current, absorption current and leakage current. Doping γ-Fe2O3 could reduce the leakage current and improve the charge dispensing property of the material. This also shows that doping high dielectric γ-Fe2O3 could significantly improve the charge storage capacity as well as charge–discharge property of the PVP–IL 200 thin film.
 |
| | Fig. 8 (A) Nyquist impedance plot of 1:1 mixture of PVP–IL, PVP–IL 150, PVP–IL 200 and γ-Fe2O3–PVP–IL; (B) comparison of room temperature CV curves of PVP–IL 150, PVP–IL 200 and γ-Fe2O3–PVP–IL measured at 20 mV s−1; (C) charge–discharge profiles for PVP–IL 200 and γ-Fe2O3–PVP–IL measured using chronoamperometric technique at a constant potential of 0.35 V (Ei) for 50 s (ti); (D) (a) 3D AFM image of PVP–IL 200 and its optical microscope image (b) 3D AFM image of γ-Fe2O3–PVP–IL thin film prepared at 200 °C and its optical microscope image (c) phase image of γ-Fe2O3–PVP–IL thin film shown in the inset of the optical microscope image showing uniform dispersion of γ-Fe2O3 MNPs in the thermally polymerized PVP–IL matrix. (E) (a) XRD of γ-Fe2O3–PVP–IL thin film and (b) PVP coated γ-Fe2O3 MNPs. | |
 |
| | Fig. 9 Cyclic voltammetric curves of γ-Fe2O3–PVP–IL measured at different scanning rates. | |
Morphological and magnetic studies
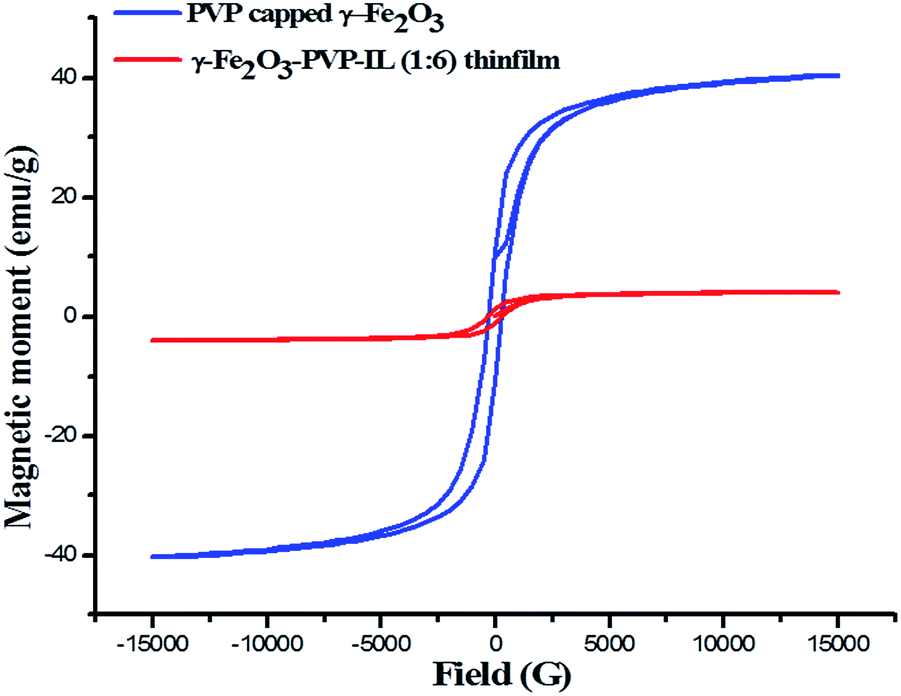
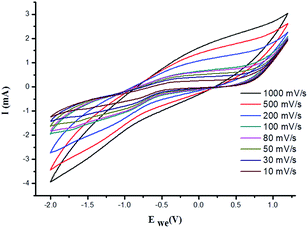
The X-ray diffraction patterns of PVP coated γ-Fe2O3 MNPs and γ-Fe2O3–PVP–IL thin film is shown in Fig. 8(E)(a) and (b) respectively in which the peak intensity was slightly masked due to the amorphous nature of PVP. All the peak of PVP coated γ-Fe2O3 MNPs in Fig. 8(E)(b) matched with the JCPDS: 39-1346 file for γ-Fe2O3 maghemite phase of iron oxide. The 3D AFM image and optical microscopy image of thin films obtained from 1:1 mixture of PVP and bpy[BF4] before and after doping PVP coated γ-Fe2O3 is shown in Fig. 8(D)(a) and (b) respectively. The AFM phase image shown in Fig. 8(D)(c) indicates the uniform incorporation of MNPs within the PVP–IL matrix while preparing γ-Fe2O3–PVP–IL thin film. Reducing the amount of IL would enhance the physical characteristics of PVP powder whereas increasing the amount of IL would give a liquid like material with very low viscosity which would make it rather difficult for making thin film. The yellow color of the film was caused by the auxochromic groups in the pyridinium cationic ring. The M(H) curves of PVP coated γ-Fe2O3 MNPs and a comparative M(H) curves of γ-Fe2O3–PVP–IL magnetic thin film at 301 and 363 K shown in Fig. 10 and S8 in ESI† respectively exhibit slight hysteresis property. The saturation magnetization of PVP coated γ-Fe2O3 MNPs was 40.438 emu g−1 and that of 1:6 wt% γ-Fe2O3 doped PVP–IL magnetic thin film was 4.064 emu g−1 at 301 K.
 |
| | Fig. 10 Room temperature M–H curve of γ-Fe2O3 MNPs and γ-Fe2O3 doped PVP–IL magnetic thin film. | |
Thermal stability analysis
The previously reported higher thermal stability of PVP and bpy[BF4] above 200 °C was an added advantage for preparing thin films for high temperature applications.63,64 The TG-DTG analysis of 1:1 PVP–IL composite done in nitrogen atmosphere at a heating rate of 10 °C min−1 is given in Fig. 11. A small inflection at 92.2 °C was observed due to the escape of adsorbed water molecules in the 1:1 PVP–IL mixture due to the high binding affinity of ILs for water molecules.65–67 Another major weight loss between 300–450 °C was observed as a result of the thermal degradation of the organic material. Pure PVP have been reported to have higher thermal stability of about 405 °C (678 K) and exhibited only one major mass lose curve between 405–505 °C (678–778 K).63 But, this degradation stability range of pure PVP slightly diminished in the presence of the ionic liquid.
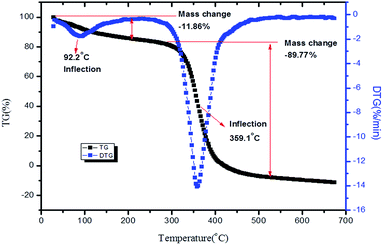 |
| | Fig. 11 TG-DTG of 1:1 ion–gel mixture of PVP and bpy[BF4]. | |
While choosing IL for preparing ion–gel composites, their thermal behaviour, intrinsic reactivity, acidic nature and instability of anions have to be considered. Also, in ILs, the glass-transition temperature (Tg) indicates the cohesive energy within the ions which increases with attractive coulombic and van der Waals interactions and decreases with repulsive Pauli forces.68 Therefore, ILs with a lower Tg value has lower cohesive energy within the ions and hence higher ionic mobility. Bandreś et al. reported that bpy[BF4] exhibited a glass transition at 192 ± 0.5 K with ΔCp = 67 ± 5 J mol−1 K−1, the exothermic peaks corresponding to the cold crystallization at around 235 K with ΔHcc ≈ 5 kJ mol−1 and also the complex peaks corresponding to melting at 252.3, 255, 268.1, and 272.5 K with ΔHm ≈ 10.5 kJ mol−1.69 The Tonset i.e., the temperature at which thermal decomposition begins for bpy[BF4] was reported to be at 610 K (373 °C) from TG analysis.64 The TG-DTA graphs of PVP–IL 200 thin film and γ-Fe2O3–PVP–IL thin film prepared at 200 °C is also shown in Fig. S9 and S10 in ESI.†
4. Conclusion
Research on solid polymer–ionic liquid electrolytes made from solid dielectric materials such as polyvinylpyrrolidone (PVP) is of high interest due to its many practical applications in photonics and many electronic devices. Here, we have proposed a facile low cost synthesis of a novel organic–inorganic hybrid ion–gel polymer thin film from γ-Fe2O3 magnetic nanoparticles, polyvinylpyrrolidone (PVP) and the ionic liquid, N-butylpyridinium tetrafluoroborate via intramolecular thermal polymerization. We have also studied the nature of intermolecular interaction between bpy[BF4] (IL) and PVP using many experimental and theoretical methods. The electron hoping phenomenon occurring from PVP to ionic liquid which kick started the ESR activity of the PVP–IL ion–gel composite and the PVP–IL thin films was supported by our experimental and theoretical investigations. The bpy[BF4] modified the spectral and electrochemical properties of PVP and also induced free radical initiated cross-polymerization among the PVP chains at high temperatures. The PVP–IL ion–gel through cross polymerization can be fabricated into a thin film of desired thickness and shape for tuning its desired properties or can be doped with inorganic γ-Fe2O3 nanoparticles to further modify its electrochemical properties. The modified inorganic–organic hybrid γ-Fe2O3 doped PVP–IL thin film was found to be superior to the PVP–IL thin film.
Acknowledgements
We greatly acknowledge the financial assistance given by DST (DST/IND/POL/P-06/2014), Govt. of India and the Polish Ministry of Science and Higher Education through the Indo-Poland joint research project 2015–17. The Gaussian 09 calculations were done with the help of Polish Grid Infrastructure (https://docs.cyfronet.pl/display/PLGDoc/Chemia%3A+InSilicoLab+for+Chemistry). We also acknowledge Jawaharlal Nehru Scholarship for Doctoral Studies and CSIR for providing financial assistance for PhD programme. We acknowledge the analytical facilities provided by Theoretical Unit of Dept. Of Chemistry, CMS College (Kottayam), NMR Research Centre at IISc Bangalore for solid state C-13 NMR, SAIF at IIT Madras for VSM and TG-DTG, SAIF at IIT Bombay for ESR, SAIF at Mahatma Gandhi University, Kottayam for AFM-Confocal Raman and Fluorescence spectra measurements and IIRBS (Mahatma Gandhi University, Kottayam) for solution state C-13 NMR.
References
- P. Sengodu and A. D. Deshmukh, RSC Adv., 2015, 5, 42109–42130 RSC.
- Y.-S. Ye, J. Rick and B.-J. Hwang, J. Mater. Chem. A, 2013, 1, 2719–2743 CAS.
- N. L. Smith, Z. Hong and S. A. Asher, Analyst, 2014, 139, 6379–6386 RSC.
- S. U. Hong, D. Park, Y. Ko and I. Baek, Chem. Commun., 2009, 7227–7229 RSC.
- B. Zhang, L. Zhang, Q. Wu, Q. Wang, B. Song, W. Wu, B. Lu and T. Ren, RSC Adv., 2014, 4, 20506–20515 RSC.
- J. Zhao, X. Shen, F. Yan, L. Qiu, S. Lee and B. Sun, J. Mater. Chem., 2011, 21, 7326–7330 RSC.
- X. Chen, J. Zhao, J. Zhang, L. Qiu, D. Xu, H. Zhang, X. Han, B. Sun, G. Fu and Y. Zhang, J. Mater. Chem., 2012, 22, 18018–18024 RSC.
- T. W. J. Bandara, P. Ekanayake, M. L. Dissanayake, I. Albinsson and B.-E. Mellander, J. Solid State Electrochem., 2010, 14, 1221–1226 CrossRef CAS.
- Y.-k. Ahn, B. Kim, J. Ko, D.-J. You, Z. Yin, H. Kim, D. Shin, S. Cho, J. Yoo and Y. S. Kim, J. Mater. Chem. A, 2016, 4, 4386–4391 CAS.
- K.-F. Lai, W.-Y. Su, W.-T. Chang and S.-H. Cheng, Int. J. Electrochem. Sci., 2013, 8, 7959–7975 CAS.
- X. Wei, L. Yu, X. Jin, D. Wang and G. Z. Chen, Adv. Mater., 2009, 21, 776–780 CrossRef CAS.
- A. S. Fisher, M. B. Khalid, M. Widstrom and P. Kofinas, J. Power Sources, 2011, 196, 9767–9773 CrossRef CAS.
- A. R. Polu and H.-W. Rhee, Int. J. Hydrogen Energy, 2016 DOI:10.1016/j.ijhydene.2016.04.160.
- M. Li, L. Yang, S. Fang and S. Dong, J. Membr. Sci., 2011, 366, 245–250 CrossRef CAS.
- A. N. Mondal, B. P. Tripathi and V. K. Shahi, J. Mater. Chem., 2011, 21, 4117–4124 RSC.
- J. Ko, S. J. Lee, K. Kim, E. Lee, K.-H. Lim, J.-M. Myoung, J. Yoo and Y. S. Kim, J. Mater. Chem. C, 2015, 3, 4239–4243 RSC.
- M. J. Park, I. Choi, J. Hong and O. Kim, J. Appl. Polym. Sci., 2013, 129, 2363–2376 CrossRef CAS.
- N. Vijaya, S. Selvasekarapandian, S. Karthikeyan, M. Prabu, N. Rajeswari and C. Sanjeeviraja, J. Appl. Polym. Sci., 2013, 127, 1538–1543 CrossRef CAS.
- A. Saroj, R. Singh and S. Chandra, Mater. Sci. Eng., B, 2013, 178, 231–238 CrossRef CAS.
- J. H. Cho, J. Lee, Y. Xia, B. Kim, Y. He, M. J. Renn, T. P. Lodge and C. D. Frisbie, Nat. Mater., 2008, 7, 900–906 CrossRef CAS PubMed.
- K. H. Lee, M. S. Kang, S. Zhang, Y. Gu, T. P. Lodge and C. D. Frisbie, Adv. Mater., 2012, 24, 4457–4462 CrossRef CAS PubMed.
- M. Shin, J. H. Song, G. H. Lim, B. Lim, J. J. Park and U. Jeong, Adv. Mater., 2014, 26, 3706–3711 CrossRef CAS PubMed.
- S. Thiemann, S. Sachnov, S. Porscha, P. Wasserscheid and J. Zaumseil, J. Phys. Chem. C, 2012, 116, 13536–13544 CAS.
- A. Bahari, M. Babaeipour and B. Soltani, J. Mater. Sci.: Mater. Electron., 2016, 27, 2131–2137 CrossRef CAS.
- A. Bahari, M. Roodbari Shahmiri, M. Derahkshi and M. Jamali, J. Nanostruct., 2012, 2, 313–316 Search PubMed.
- N. Winterton, J. Mater. Chem., 2006, 16, 4281–4293 RSC.
- A. Miyata and H. Miyafuji, J. Wood Sci., 2014, 60, 438–445 CrossRef CAS.
- V. Strehmel, A. Laschewsky, H. Wetzel and E. Görnitz, Macromolecules, 2006, 39, 923–930 CrossRef CAS.
- K. Ghandi, Green Sustainable Chem., 2014, 2014, 43349 Search PubMed.
- R. C. Vieira and D. E. Falvey, J. Am. Chem. Soc., 2008, 130, 1552–1553 CrossRef CAS PubMed.
- R. C. Vieira and D. E. Falvey, J. Phys. Chem. B, 2007, 111, 5023–5029 CrossRef CAS PubMed.
- D. Behar, P. Neta and C. Schultheisz, J. Phys. Chem. A, 2002, 106, 3139–3147 CrossRef CAS.
- X. Li, M. Liang, A. Chakraborty, M. Kondo and M. Maroncelli, J. Phys. Chem. B, 2011, 115, 6592–6607 CrossRef CAS PubMed.
- G. Zhu, L. Zhang, Y. Wang, X. Xu and X. Peng, J. Mol. Liq., 2016, 213, 289–293 CrossRef CAS.
- Z. Wang, L. Zhang, R. I. Cukier and Y. Bu, Phys. Chem. Chem. Phys., 2010, 12, 1854–1861 RSC.
- H. Fu, Z. Xing, X. Cao and G. Wu, Chin. Sci. Bull., 2013, 58, 1882–1886 CrossRef CAS.
- A. Joseph and S. Mathew, ChemPlusChem, 2014, 79, 1382–1420 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, available: http://www.gaussian.com/g_tech/g_ur/m_citation.htm, accessed, 2014, 9.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- K. N. Kumar, K. Sivaiah and S. Buddhudu, SOLID STATE PHYSICS: Proceedings of the 58th DAE Solid State Physics Symposium 2013, AIP Publishing, 2014, vol. 1591, pp. 893–894 Search PubMed.
- V. Torchilin, T. Levchenko, K. Whiteman, A. Yaroslavov, A. Tsatsakis, A. Rizos, E. Michailova and M. Shtilman, Biomaterials, 2001, 22, 3035–3044 CrossRef CAS PubMed.
- J. Fraissard and H. A. Resing, Magnetic Resonance in Colloid and Interface Science: Proceedings of a NATO Advanced Study Institute and the Second International Symposium Held at Menton, Springer Science & Business Media, France, 2012 Search PubMed.
- H. M. McConnell, J. Chem. Phys., 1956, 25, 709–711 CrossRef CAS.
- T. Stone, T. Buckman, P. Nordio and H. McConnell, Proc. Natl. Acad. Sci. U. S. A., 1965, 54, 1010–1017 CrossRef CAS.
- C. D. Schmulbach, C. C. Hinckley and D. Wasmund, J. Am. Chem. Soc., 1968, 90, 6600–6602 CrossRef CAS.
- G. García, M. Atilhan and S. Aparicio, Phys. Chem. Chem. Phys., 2015, 17, 13559–13574 RSC.
- D. Basu, Charge transport in polymer semiconductors, ProQuest, 2007 Search PubMed.
- E. Shakerzadeh, E. Tahmasebi and M. Oftadeh, Comput. Theor. Chem., 2013, 1017, 31–36 CrossRef CAS.
- B. Qiao, C. Krekeler, R. Berger, L. Delle Site and C. Holm, J. Phys. Chem. B, 2008, 112, 1743–1751 CrossRef CAS PubMed.
- F. M. Pontes, A. J. Chiquito, W. B. Bastos, M. A. Pereira-da-Silva, A. Marcelo and E. Longo, J. Mater. Chem. C, 2016, 9331–9342 RSC.
- Z. Jiang, C. Zhu, W. Wan, K. Qian and J. Xie, J. Mater. Chem. A, 2016, 4, 1806–1818 CAS.
- K. Manzoor, S. Vadera, N. Kumar and T. Kutty, Solid State Commun., 2004, 129, 469–473 CrossRef CAS.
- R. L. Amster and R. C. Taylor, Spectrochim. Acta, 1964, 20, 1487–1502 CrossRef CAS.
- A. Gupta, G. Chen, P. Joshi, S. Tadigadapa and P. Eklund, Nano Lett., 2006, 6, 2667–2673 CrossRef CAS PubMed.
- A. Maghsoumi, L. Brambilla, C. Castiglioni, K. Müllen and M. Tommasini, J. Raman Spectrosc., 2015, 46, 757–764 CrossRef CAS.
- J. Schwan, S. Ulrich, V. Batori, H. Ehrhardt and S. Silva, J. Appl. Phys., 1996, 80, 440–447 CrossRef CAS.
- I. Childres, L. A. Jauregui, W. Park, H. Cao and Y. P. Chen, New Dev. Photon Mater. Res., 2013, 1, 1–20 CrossRef.
- M. M. Lucchese, F. Stavale, E. M. Ferreira, C. Vilani, M. Moutinho, R. B. Capaz, C. Achete and A. Jorio, Carbon, 2010, 48, 1592–1597 CrossRef CAS.
- L. Cancado, K. Takai, T. Enoki, M. Endo, Y. Kim, H. Mizusaki, A. Jorio, L. Coelho, R. Magalhaes-Paniago and M. Pimenta, Appl. Phys. Lett., 2006, 88, 163106 CrossRef.
- L. G. Cançado, A. Jorio, E. M. Ferreira, F. Stavale, C. Achete, R. Capaz, M. Moutinho, A. Lombardo, T. Kulmala and A. Ferrari, Nano Lett., 2011, 11, 3190–3196 CrossRef PubMed.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2009, 4, 380–386 CrossRef PubMed.
- K. Wang, H. Wu, Y. Meng, Y. Zhang and Z. Wei, Energy Environ. Sci., 2012, 5, 8384–8389 CAS.
- H. M. Kamari, M. G. Naseri and E. B. Saion, Metals, 2014, 4, 118–129 CrossRef.
- Y. Cao and T. Mu, Ind. Eng. Chem. Res., 2014, 53, 8651–8664 CrossRef CAS.
- N.-N. Wang, Q.-G. Zhang, F.-G. Wu, Q.-Z. Li and Z.-W. Yu, J. Phys. Chem. B, 2010, 114, 8689–8700 CrossRef CAS PubMed.
- M. G. Freire, C. M. Neves, K. Shimizu, C. E. Bernardes, I. M. Marrucho, J. A. Coutinho, J. N. C. Lopes and L. s. P. N. Rebelo, J. Phys. Chem. B, 2010, 114, 15925–15934 CrossRef CAS PubMed.
- M. G. Freire, P. J. Carvalho, R. L. Gardas, L. s. M. Santos, I. M. Marrucho and J. A. Coutinho, J. Chem. Eng. Data, 2008, 53, 2378–2382 CrossRef CAS.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206–237 CrossRef CAS PubMed.
- I. Bandrés, F. M. Royo, I. Gascón, M. Castro and C. Lafuente, J. Phys. Chem. B, 2010, 114, 3601–3607 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of additional characterizations. See DOI: 10.1039/c6ra27411k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Marilyn Mary Xavierb,
Gaweł Żyłac,
P. Radhakrishnan Nairb,
A. S. Padmanabhanad and
Suresh Mathew
a,
Marilyn Mary Xavierb,
Gaweł Żyłac,
P. Radhakrishnan Nairb,
A. S. Padmanabhanad and
Suresh Mathew