
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, biological evaluation and molecular docking of spirofurochromanone derivatives as anti-inflammatory and antioxidant agents†
D. Ashok *a,
E. V. L. Madhuria,
M. Sarasijab,
S. Sree Kanthc,
M. Vijjulathac,
Malini D. Alaparthid and
S. R. Sagurthi*d
*a,
E. V. L. Madhuria,
M. Sarasijab,
S. Sree Kanthc,
M. Vijjulathac,
Malini D. Alaparthid and
S. R. Sagurthi*d
aGreen and Medicinal Chemistry Lab, Department of Chemistry, Osmania University, Hyderabad, Telangana, India-500007. E-mail: ashokdou@gmail.com
bDepartment of Chemistry, Satavahana University, Karimnagar, Telangana, India-505001
cMolecular Modeling and Medicinal Chemistry Group, Department of Chemistry, University College of Science, Osmania University, Hyderabad, Telangana, India-500007
dDepartment of Genetics, Drug Design & Molecular Medicine Lab, Osmania University, Hyderabad, Telangana, India-500007. E-mail: drsomeswar@osmania.ac.in
First published on 12th May 2017
Abstract
A series of 2′-substituted-3′-methylspiro[cyclohexane-1,7′-furo[3,2-g]chroman]-5′(7′H)-one, 5a–i and 7a–u have been synthesized using an eco-friendly approach to attain good yields in a shorter reaction time. The structures of novel compounds were characterized by IR, 1H NMR, 13C NMR and mass spectrometry analysis. All the synthesized compounds were evaluated for their biological activity. Compounds 5a, 5b, 5c, 5d, 5e, 7g, 7h, 7j, 7l, 7n and 7q were found to have better anti-inflammatory activity in the albumin denaturation technique. All compounds exhibited good antioxidant activity by DPPH radical scavenging assay and most compounds showed activity in a hydrogen peroxide assay. Molecular docking scores as well biological assays results suggested that compound 7h has better anti-inflammatory activity among the synthesized compounds.
1 Introduction
Reactions occurring in several types of tissue injuries, infections, immunologic stimulation as a defense against foreign or altered endogenous substances is termed as inflammation. The release of chemical mediators from injured tissue and migrating cells triggers inflammation. NSAIDs (Non-Steroidal Anti-Inflammatory Drugs) will be generally prescribed to overcome such physiological disturbances. The activity of NSAIDs in inflammatory diseases does not seem to be only due to the inhibition of the production of endogenous prostaglandins, but also by preventing the denaturation of proteins (which act as antigens and auto immune disease).1 Grant et al.2 stated that non-steroidal anti-inflammatory molecules will stabilize bovine serum albumin (BSA) when exposed to a few degree rises in temperature. Selecting therapeutically interesting molecules without the use of animals or living organisms used to be a challenge to the scientific community, which was well addressed by Williams et al.3 by developing a technique of stabilizing thermal immunogenic bovine serum albumin by natural products. It is also revealed that a range of extracts and compounds with various biological properties can convey the stability to the protein.4 Thus, rather than just detecting non-steroidal anti-inflammatory agents, the bovine serum albumin denaturation assay could have a wider application.Biaryl units as molecular components has attracted enormous interest and their syntheses have been widely carried out and have yielded innumerable compounds with diverse biological activities such as: anti-inflammatory, antimicrobial, antifungal, antiproliferative, antidiabetic, immunosuppressant, analgesic, antioxidant.5 The Suzuki cross-coupling reaction, which represents an attractive and alternative methods that use organometallic species for the construction of unsymmetrical biaryl compounds, involves air and moisture stable organoboranes that possess relatively low toxicity and also has broad functional group tolerance. Catalysts used in the Suzuki reaction have been traditionally based on homogeneous palladium phosphine complexes.5 The study for improving the reactivity of Suzuki reactions, water or aqueous-organic mixtures has received attention due to the ability of the base to dissolve in water for activating arylboronic acids.6
Benzofurans are interesting oxygen containing heterocycles which are ubiquitous in nature and show a wide range of biological activities,7 as analgesics,8 antidepressants,9 antitumor agent.10 Similarly chromanone derivatives, in particular 2-spiro-chroman-4(1H)-ones are embodied in many bio-active molecules (Fig. 1) and possess various biological activities which include antiarrhythmic,11 anti-HIV,12 antidiabetic, ACC inhibitor,13 vanilloid receptor antagonist, growth hormone secretagogues,14 histamine receptor antagonist, antiviral15 and anti-inflammatory.16 Spirochromanone ring system represents an important class of naturally occurring substances characterized by highly pronounced biological properties.
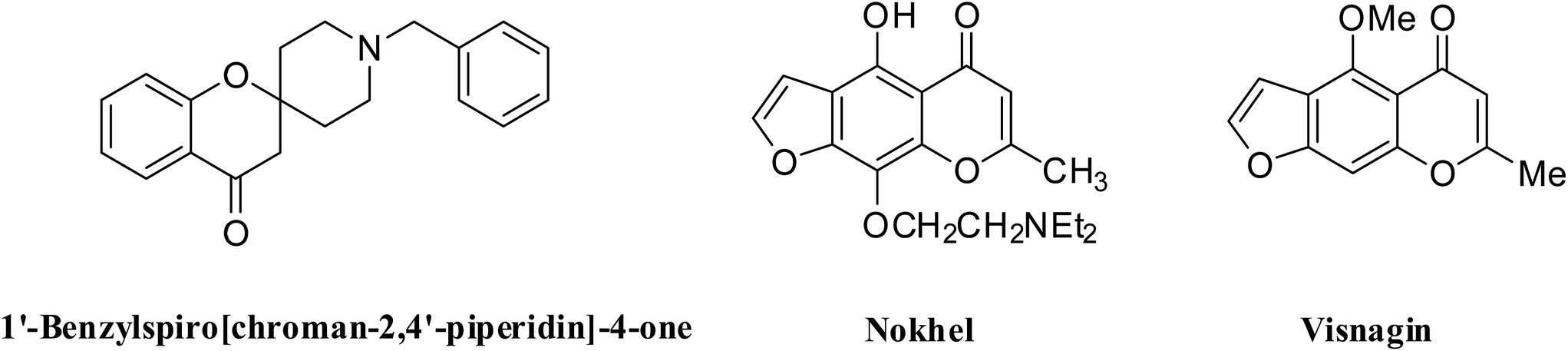 | ||
| Fig. 1 Bioactive molecules. | ||
The combination of two or more pharmacophores or chemical entities either linked with one another or fused together to create a new molecule is referred to as molecular hybridization,17 which aims to combat drug resistance and enrich existing arsenals of anti-infective agents.18 The selection of the pharmacophores is based upon their known bioprofiles, with the hope that the resulting hybrid molecules may exhibit synergistic or additive pharmacological activities.19
Potential availability of benzofuran and spirochromanone made us to synthesize some new spirofurochromanone-biaryl derivatives using Suzuki coupling by the combination of two green chemistry principles, namely microwave assisted synthesis and aqueous phase reactions. Hybrid compounds containing benzofuran and spirochromanone moieties, called spirofurochromanone, due to combined effect may exhibit better biological profile. Enabling technologies for organic syntheses such as microwave, aqueous phase reactions or green solvents have changed organic chemistry in terms of efficiency, work-up and speed. Microwave assisted organic synthesis leads to rate enhancement with excellent reproducibility, improved yields and less side reactions compared to conventional heating.20
In continuation to our efforts21–23 to synthesize new biaryl heterocyclic derivatives using eco-friendly approach, we have synthesized spirofurochromanone derivatives 5a–i and 7a–u from compounds 3a–c using microwave irradiation and conventional heating. All the compounds were well characterised by spectral data. Synthesized spirofurochromanone were screened for in vitro scavenging activity utilizing hydrogen peroxide and DPPH assay. These tested compounds shown high scavenging activity when compared with standard ascorbic acid.
2 Results and discussions
2.1 Chemistry
The desired spirofurochromanone scaffolds were synthesized in three steps as outlined (Scheme 1). Selective synthesis of monospiro chromanone derivatives 3a–c were achieved from diacetyl resorcinol 1 with cyclic alkanones 2a–c involving Kabbe condensation with pyrrolidine as base and toluene as solvent.24 Further reaction of 3a–c with 4′-substituted phenacyl bromide 4a–c in presence of potassium carbonate as base resulted in spirofurochromanone 5a–i with good yields in conventional heating and microwave irradiation methods as shown in Table 1. Finally the target compounds 7a–u were synthesized by reaction of compound 5d–f with arylboronic acids 6a–g in conventional heating and microwave irradiation methods (Scheme 2).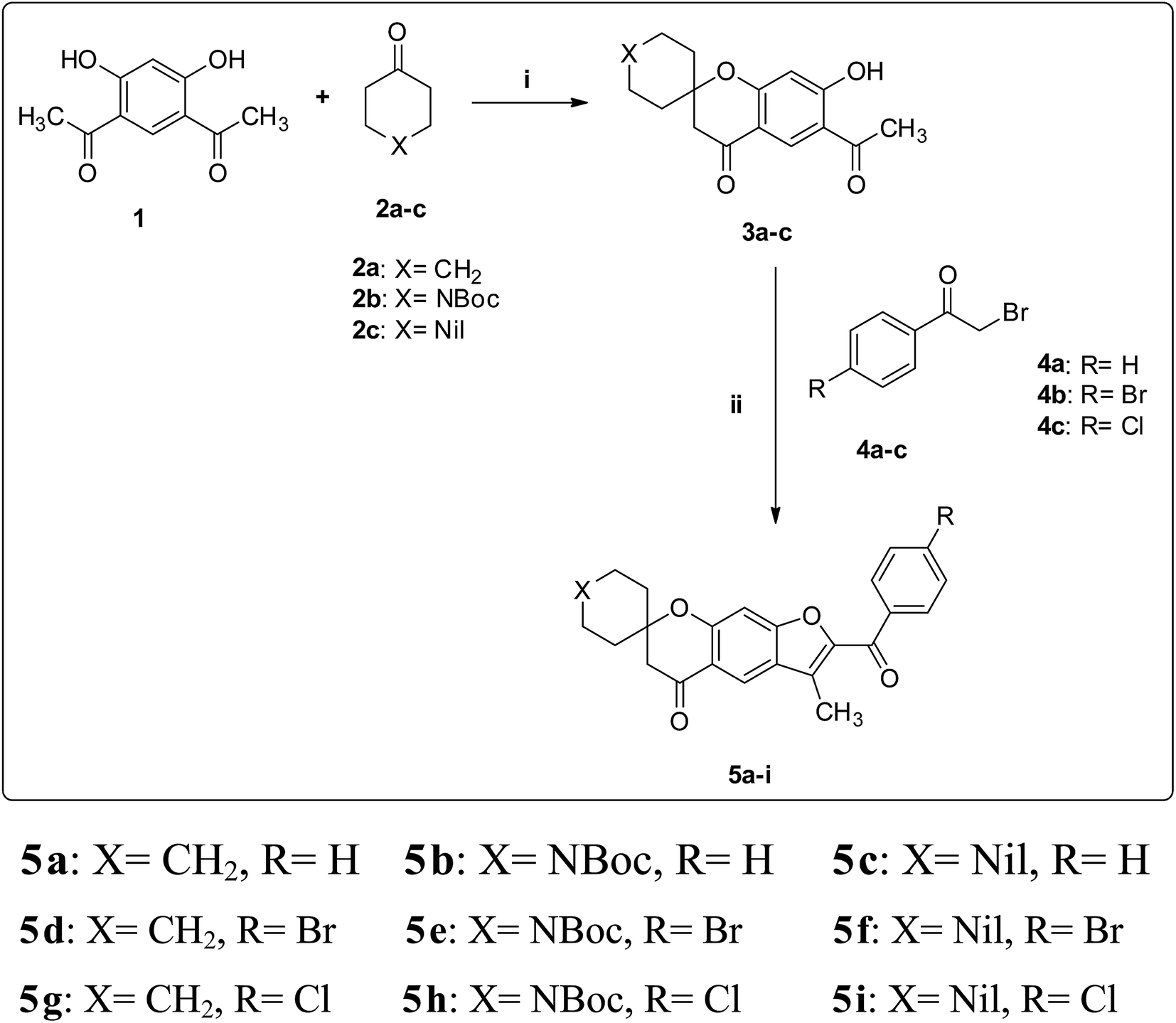 | ||
| Scheme 1 Synthesis of compounds 5a–i. Reagents and conditions: (i) toluene, pyrrolidine, reflux, 3 h. (ii) (method a) K2CO3, acetone, Δ (method b) K2CO3, MWI. | ||
| Entry | X | R | Mp, °C | Conventional heating | Microwave irradiation | ||
|---|---|---|---|---|---|---|---|
| Time, h | Yielda, % | Time, min | Yielda, % | ||||
| a Isolated yield. | |||||||
| 5a | CH2 | H | 128–130 | 3 | 57 | 5 | 82 |
| 5b | NBoc | H | 220–222 | 4 | 57 | 8 | 84 |
| 5c | Nil | H | 78–70 | 3 | 35 | 5 | 71 |
| 5d | CH2 | Br | 148–150 | 3 | 58 | 5 | 83 |
| 5e | NBoc | Br | 224–226 | 4 | 70 | 8 | 80 |
| 5f | Nil | Br | 130–132 | 3 | 50 | 5 | 77 |
| 5g | CH2 | Cl | 126–128 | 3 | 70 | 5 | 79 |
| 5h | NBoc | Cl | 218–220 | 4 | 72 | 8 | 77 |
| 5i | Nil | Cl | 125–127 | 3 | 55 | 5 | 80 |
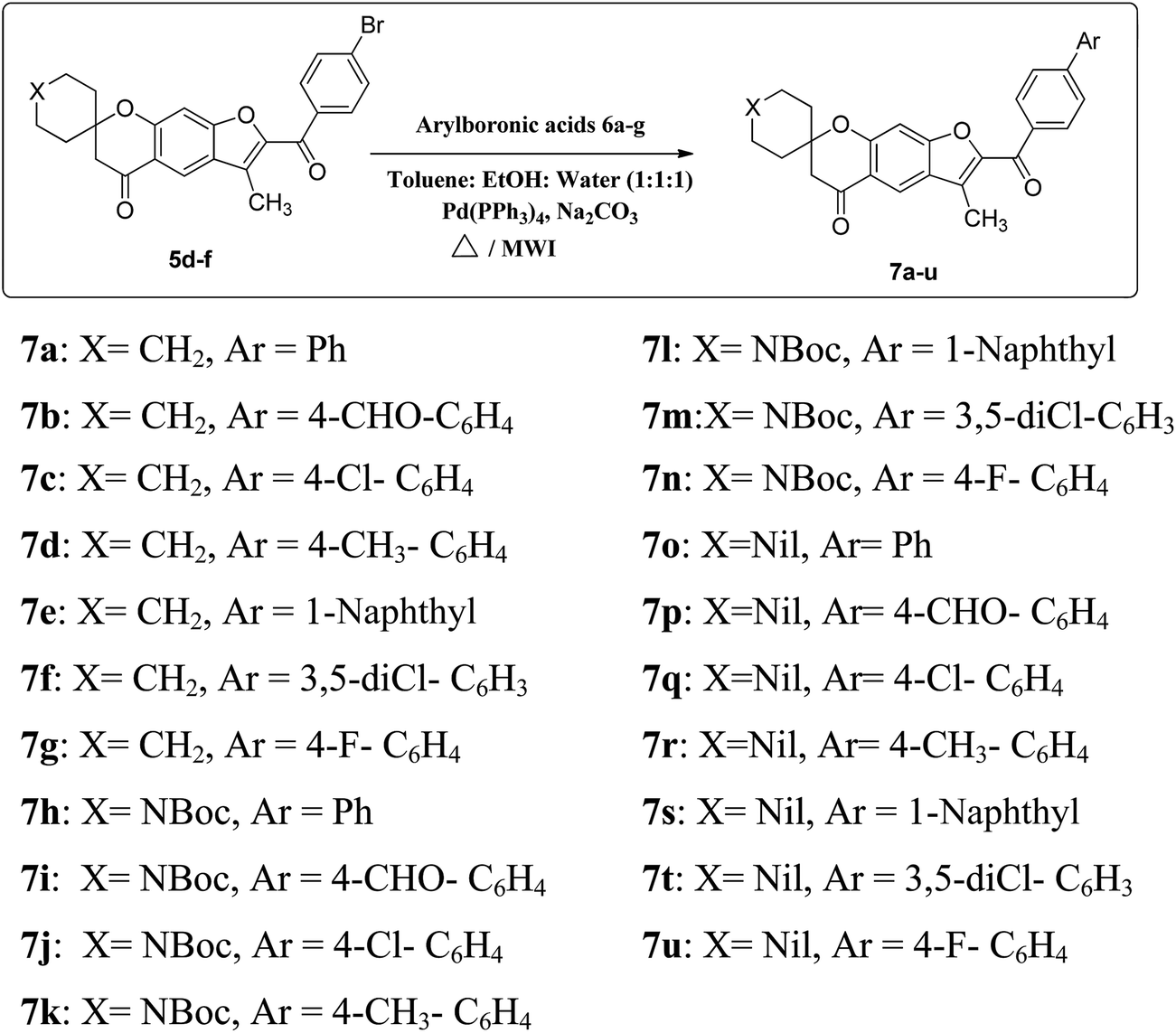 | ||
| Scheme 2 Synthesis of compounds 7a–u. | ||
In our initial attempts in conventional method for the synthesis of compound 7a from 5d, based on the previous experience of Suzuki coupling reactions in our research group,25 we started with 10 mol% Pd(PPh3)4 as catalyst, Na2CO3 as a base and 1 equiv. of phenylboronic acid 6a as a model reaction at 80 °C for 10 h under N2 atmosphere in DMF, ethanol as well as in dioxane separately, which resulted in very poor yields. Among them ethanol was found to be a better solvent with relatively better yield. Literature reports shows that addition of water as co-solvent to the polar aprotic solvents greatly improves the rate of reaction.26 Thus we decided to check with combination of solvents with water. Using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solvent water mixture, the reaction proceeded well furnishing improved yields in shorter time as shown in Table 2. The Suzuki coupled products were obtained in shorter time and high yield in 1:1:1 solvent mixture of ethanol, toluene and water. Using the same solvents, reaction was carried out in a sealed tube by conventional heating and the yields are listed in Table 2. Encouraged by the results obtained, we subsequently attempted to carry out the reaction under mild conditions and reduced reaction time by the application of microwave irradiation.
:1 solvent water mixture, the reaction proceeded well furnishing improved yields in shorter time as shown in Table 2. The Suzuki coupled products were obtained in shorter time and high yield in 1:1:1 solvent mixture of ethanol, toluene and water. Using the same solvents, reaction was carried out in a sealed tube by conventional heating and the yields are listed in Table 2. Encouraged by the results obtained, we subsequently attempted to carry out the reaction under mild conditions and reduced reaction time by the application of microwave irradiation.
| Entry | Solvent | Conventional heating | Conventional heating in sealed pressure tube | Microwave irradiation | |||
|---|---|---|---|---|---|---|---|
| Time, h | Yielda, % | Time, h | Yielda, % | Time, min | Yielda, % | ||
| a Isolated yield. | |||||||
| 7a | DMF | 10 | 10 | 8 | 20 | 40 | 40 |
| 7a | DMF/water (1:1) |
9 | 25 | 7 | 30 | 25 | 40 |
| 7a | Dioxane | 10 | 17 | 8 | 26 | 40 | 37 |
| 7a | Dioxane/water (1:1) |
9 | 40 | 7 | 45 | 25 | 55 |
| 7a | EtOH | 10 | 30 | 8 | 38 | 40 | 45 |
| 7a | EtOH/water (1:1) |
9 | 35 | 7 | 40 | 25 | 57 |
| 7a | EtOH/toluene/water (1:1:1) |
8 | 70 | 6 | 75 | 20 | 83 |
Microwave irradiation at 180 W, keeping similar conditions the reactions were completed within few minutes and increased yields of the product were obtained. Having established the optimal conditions for the reaction, compounds 5d, 5e and 5f were subjected to react with different substituted arylboronic acids 6a–g to give 7a–u. The comparative studies based on the optimization of both methods are reflected in Table 3. All the compounds synthesized were well characterized by spectral data.
| Entry | X | Ar | Mp, °C | Sealed tube, conventional heating | Microwave irradiation | ||
|---|---|---|---|---|---|---|---|
| Time, h | Yielda, % | Time, min | Yielda, % | ||||
| a Isolated yield. | |||||||
| 7a | CH2 | Ph | 172–174 | 6 | 75 | 20 | 83 |
| 7b | CH2 | 4-CHO-C6H4 | 168–170 | 6 | 74 | 22 | 82 |
| 7c | CH2 | 4-Cl-C6H4 | 186–188 | 6 | 74 | 20 | 84 |
| 7d | CH2 | 4-Me-C6H4 | 190–192 | 6 | 76 | 20 | 87 |
| 7e | CH2 | 1-Naphthyl | 152–154 | 6 | 75 | 20 | 86 |
| 7f | CH2 | 3,5-DiCl-C6H3 | 178–180 | 6 | 74 | 20 | 85 |
| 7g | CH2 | 4-F-C6H4 | 170–172 | 6 | 77 | 22 | 84 |
| 7h | NBoc | Ph | 205–207 | 7 | 76 | 23 | 82 |
| 7i | NBoc | 4-CHO-C6H4 | 210–212 | 7 | 71 | 25 | 80 |
| 7j | NBoc | 4-Cl-C6H4 | 218–220 | 7 | 74 | 23 | 83 |
| 7k | NBoc | 4-Me-C6H4 | 224–227 | 7 | 77 | 23 | 85 |
| 7l | NBoc | 1-Naphthyl | 147–148 | 7 | 73 | 23 | 84 |
| 7m | NBoc | 3,5-DiCl-C6H3 | 207–208 | 7 | 74 | 23 | 86 |
| 7n | NBoc | 4-F-C6H4 | 208–210 | 7 | 75 | 23 | 87 |
| 7o | Nil | Ph | 118–120 | 6 | 74 | 20 | 86 |
| 7p | Nil | 4-CHO-C6H4 | 182–184 | 6 | 76 | 21 | 88 |
| 7q | Nil | 4-Cl-C6H4 | 138–140 | 6 | 75 | 20 | 83 |
| 7r | Nil | 4-Me-C6H4 | 126–128 | 6 | 74 | 20 | 85 |
| 7s | Nil | 1-Naphthyl | 170–172 | 6 | 75 | 20 | 84 |
| 7t | Nil | 3,5-DiCl-C6H3 | 187–188 | 6 | 76 | 20 | 88 |
| 7u | Nil | 4-F-C6H4 | 138–140 | 6 | 73 | 20 | 85 |
2.2 Biological activity
All the newly synthesized compounds 5a–i and 7a–u were screened for their anti-inflammatory and antioxidant activities. The in vitro anti-inflammatory and antioxidant activities of the synthesized 5a–i and 7a–u are tabulated (Tables 4 and 5).| Compound | Anti-inflammatory activitya IC50 (μM mL−1) | Compound | Anti-inflammatory activitya IC50 (μM mL−1) |
|---|---|---|---|
| a Values are mean ± SD of three replicates. | |||
| 5a | 110.58 ± 0.78 | 7g | 97.39 ± 0.76 |
| 5b | 77.94 ± 1.00 | 7h | 77.11 ± 0.66 |
| 5c | 132.0 ± 1.02 | 7i | 142.31 ± 0.64 |
| 5d | 105.61 ± 0.59 | 7j | 108.97 ± 0.85 |
| 5e | 98.46 ± 0.90 | 7k | 120.48 ± 0.88 |
| 5f | 159.17 ± 1.00 | 7l | 108.49 ± 0.74 |
| 5g | 127.55 ± 0.86 | 7m | 152.77 ± 0.89 |
| 5h | 140.48 ± 1.29 | 7n | 116.10 ± 1.10 |
| 5i | 152.81 ± 0.92 | 7o | 130.92 ± 0.94 |
| 7a | 154.07 ± 0.71 | 7p | 137.42 ± 0.76 |
| 7b | 118.3 ± 0.95 | 7e | 102.82 ± 0.70 |
| 7c | 164.09 ± 1.15 | 7r | 172.04 ± 0.93 |
| 7d | 140.85 ± 1.02 | 7s | 168.42 ± 0.74 |
| 7e | 170.41 ± 0.74 | 7t | 145.33 ± 0.91 |
| 7f | 128.99 ± 0.87 | 7u | 127.59 ± 0.75 |
| Aspirin | 116.48 ± 0.98 | ||
| Compound | DPPHa IC50 (μg mL−1) | Compound | DPPHa IC50 (μg mL−1) |
|---|---|---|---|
| a Values are mean ± SD of three replicates. | |||
| 5a | 8.16 ± 1.41 | 7g | 3.93 ± 1.06 |
| 5b | 6.84 ± 1.51 | 7h | 63.92 ± 1.02 |
| 5c | 8.81 ± 1.24 | 7i | 56.17 ± 1.17 |
| 5d | 2.64 ± 1.14 | 7j | 10.16 ± 1.25 |
| 5e | 10.16 ± 1.39 | 7k | 6.84 ± 1.32 |
| 5f | 8.81 ± 1.06 | 7l | 4.86 ± 1.56 |
| 5g | 12.4 ± 0.98 | 7m | 6.84 ± 1.38 |
| 5h | 8.81 ± 1.15 | 7n | 37.04 ± 1.43 |
| 5i | 3.93 ± 1.07 | 7o | 66.2 ± 1.55 |
| 7a | 39.64 ± 1.16 | 7p | 39.64 ± 1.17 |
| 7b | 6.84 ± 1.31 | 7q | 76.64 ± 0.93 |
| 7c | 11.03 ± 1.89 | 7r | 47.5 ± 1.01 |
| 7d | 13.5 ± 1.04 | 7s | 47.49 ± 1.23 |
| 7e | 11.03 ± 1.12 | 7t | 80.59 ± 1.19 |
| 7f | 6.84 ± 1.29 | 7u | 66.16 ± 1.32 |
| Ascorbic acid | 145.4 ± 0.27 | ||
2.2.2.1 DPPH radical-scavenging assay. The DPPH (2,2-diphenyl-1-picrylhydrazyl) radical-scavenging assay is usually used to evaluate the abilities of new compounds to capture free radicals by producing the reduced form DPPH-H through a hydrogen-donating action. It was performed in order to determine the antioxidant potential.27
DPPH is a stable nitrogen-centered free radical. Its reaction rates correlate directly with antioxidant activity, the higher the rate, the more effective the antioxidant.28 A freshly prepared DPPH solution shows a deep purple color with an absorption maximum at 517 nm. When the purple color changes to yellow, it leads to decreased absorbance. This is because of the antioxidant molecule reducing the DPPH free radical through donation of hydrogen atom. Instantaneous or concomitant decrease in absorbance would be indicative of potent antioxidant activity by the compound.29
In this study, we investigated the antioxidant properties of spirofurochromanone derivatives using DPPH scavenging activity with respect to the standard ascorbic acid and using a spectrometric assay.
2.2.2.2 Hydrogen peroxide scavenging assay. Several reactive species are known to produce in excess during the inflammatory processes, the ROS peroxyl radical (ROO˙), HO˙, O2˙−, H2O2, and HOCl play important roles in their pathophysiological conditions, hydrogen peroxide (H2O2) is a biologically important, non-radical reactive oxygen species (ROS) that can influence several cellular processes, which makes them potential targets for the therapy of inflammation.30 In vitro antioxidant activities of synthesized compounds were evaluated against hydrogen peroxide and were compared with standard ascorbic acid. Our results reveal that these compounds exhibit better radical scavenging activities. The results of antioxidant activity of spirofurochromanone are shown in Table 6.
| S. no. | Compound | IC50a value (μg mL−1) | Compound | IC50a value (μg mL−1) |
|---|---|---|---|---|
| a Values are mean ± SD of three replicates. | ||||
| 1 | 5a | 112.05 ± 0.88 | 7g | 96.65 ± 0.79 |
| 2 | 5b | 73.67 ± 0.15 | 7h | 44.73 ± 0.49 |
| 3 | 5c | 25.12 ± 0.26 | 7i | 42.26 ± 0.41 |
| 4 | 5d | 83.14 ± 0.13 | 7j | 80.05 ± 0.16 |
| 5 | 5e | 98.55 ± 0.18 | 7k | 42.26 ± 0.41 |
| 6 | 5f | 41.11 ± 1.23 | 7l | 82.21 ± 0.61 |
| 7 | 5g | 68.6 ± 0.54 | 7m | 78.05 ± 0.65 |
| 8 | 5h | 77.05 ± 0.34 | 7n | 43.04 ± 0.50 |
| 9 | 5i | 51.42 ± 0.69 | 7o | 49.51 ± 0.17 |
| 10 | 7a | 103.95 ± 0.9 | 7p | 93.12 ± 0.23 |
| 11 | 7b | 70.93 ± 0.38 | 7q | 35.51 ± 0.07 |
| 12 | 7c | 32.11 ± 0.38 | 7r | 35.51 ± 0.07 |
| 13 | 7d | 62.05 ± 0.74 | 7s | 62.03 ± 0.77 |
| 14 | 7e | 51.21 ± 0.35 | 7t | 78.05 ± 0.22 |
| 15 | 7f | 37.11 ± 0.41 | 7u | 76.5 ± 0.24 |
| Ascorbic acid | 77.13 ± 0.87 | |||
2.2.3.1 Anti-inflammatory activity. In the series of compounds 5a–i cyclohexylspiro compounds exhibited mild anti-inflammatory activity, N-Bocpiperidinylspiro compound 5b has better activity with IC50 value 77.9 and the activity decreases with the change in the substitution at 4th carbon of 2′-benzoyl group with bromo (5e, IC50 value 98.4) or chloro (5h, IC50 value 140.4) groups. Among the Suzuki cross coupled products 7a–u, N-Bocpiperidinylspiro derivatives 7h–n show better activity when compared to cyclohexylspiro derivatives 7a–g and cyclopentylspiro derivatives 7o–u. Among N-Bocpiperidinylspiro compounds 7h (IC50 value 77.1) exhibited better activity when compared to other substitutions on the biaryl group.
2.2.3.2 Antioxidant activity.
2.2.3.2.1 DPPH radical scavenging activity. Compounds 5a–i, showed good antioxidant activity compared with the standard ascorbic acid. Compounds 5d and 5i have better activity while 5a, 5c, 5f, 5h have almost same activity. Cyclopentylspiro derivatives 7o–u among the biaryl series of compounds 7a–u show mild antioxidant activity whereas the series of N-Bocpiperidinylspiro and cyclohexylspiro compounds exhibit better activity. 7a–g compounds have better IC50 values ranging from 3 to 39 owing to the presence of the substituent on the phenyl group of biaryl compound.
2.2.3.2.2 Hydrogen peroxide scavenging assay. Compounds 5b–c and 5f–i, with chlorine and bromine as substituent show better activity as compared with the standard ascorbic acid. Among the Suzuki cross coupled products compared to cyclohexylspiro derivative, N-Bocpiperidinylspiro derivative and cyclopentylspiro derivative show better activity without any substituent on the biaryl group. In the series of cyclohexylspiro derivatives 7b–f exhibits better activity out of which presence of chloro group (7c, IC50 value 32.11 and 7f, IC50 value 37.11) on the biaryl moiety exhibits good activity. Among N-Bocpiperidinylspiro derivatives biaryl moieties with CHO, Me and F as substituents exhibit good activity (7i, IC50 value 42.26, 7k IC50 value 42.26, 7n IC50 value 43.04 respectively). Chloro and methyl substituents on the biaryl groups of the cyclopentylspiro derivatives exhibit good activity (7q IC50 value 35.51 and 7r IC50 value 35.51).
2.3 Molecular docking analysis
BSA consists of 583 amino acid residues present in three homologous α-helical domains (I, II, III). Each domain comprises sub-domain A and sub-domain B. Subdomains IIA, IIIA and IB among them are known as Sudlow's site I, II and III, respectively (ESI).31–33 To determine the binding interaction of the molecules with bovine serum albumin molecular docking analysis was performed on five binding cavities obtained from site map. Analysis of dock score reveals that the binding affinity of these molecules is more towards the site-5 and site-1 as compared to the other hydrophobic cavities, site-5 and site-1 correspond to drug binding site I and site II.31–39 Dock score of molecules at site-5 and site-1 along with the dock score of the molecules which were docked into COX-2 (one of the target for anti-inflammatory) to check the possible mode of interaction is provided in Table 8 (the dock score for molecules at all site is provided in Table 7). Binding energy of the molecules at site-5 ranged between −7.342 to −4.974 kcal mol−1, and for site-1 it ranged between −7.259 to −4.254 kcal mol−1 few molecules showed lower and positive binding energies at site-2, site-3 and site-4.| Molecule | Dock score in kcal mol−1 | ||||
|---|---|---|---|---|---|
| Site 1 | Site 2 | Site 3 | Site 4 | Site 5 | |
| 5a | −5.04465 | −3.76925 | −3.22027 | −1.97144 | −5.34381 |
| 5b | −4.91124 | −3.69292 | −2.36801 | −1.32099 | −6.55207 |
| 5c | −5.17773 | −3.79194 | −3.39834 | 0.802319 | −6.12478 |
| 5d | −5.14163 | −4.0252 | −2.04788 | 0.990484 | −6.02642 |
| 5e | −4.25431 | −3.73157 | −2.89809 | −1.93373 | −4.97476 |
| 5f | −5.33577 | −3.83444 | −3.39073 | 0.022163 | −5.15616 |
| 5g | −6.90317 | −3.67625 | −2.97721 | 1.069312 | −5.77175 |
| 5h | −4.28098 | −3.25556 | −3.15456 | 0.50295 | −6.799 |
| 5i | −6.28092 | −3.9708 | −3.3511 | −0.73673 | −6.35011 |
| 7a | −5.66842 | −4.50547 | −3.2195 | −2.63023 | −6.64511 |
| 7b | −5.24438 | −3.09017 | −3.44921 | −0.71678 | −7.00437 |
| 7c | −5.91122 | −3.45821 | −2.0643 | −2.46974 | −7.34291 |
| 7d | −5.85966 | −3.22219 | −1.79908 | −1.40511 | −6.77104 |
| 7e | −7.25977 | −4.21203 | 0.528595 | −2.32848 | −6.20783 |
| 7f | −6.07993 | −3.94704 | −1.57723 | −3.23294 | −6.81214 |
| 7g | −6.13347 | −4.7289 | −3.18466 | 1.503116 | −7.29907 |
| 7h | −6.74828 | −3.78363 | −3.95033 | −3.43081 | −6.0202 |
| 7i | −5.42021 | −3.15689 | −2.20818 | −3.40362 | −6.66791 |
| 7j | −6.89574 | −3.61415 | −2.67466 | −0.57856 | −6.67635 |
| 7k | −6.17567 | −4.04447 | −1.78585 | −1.43157 | −6.52291 |
| 7l | −6.59588 | −4.43139 | −0.44855 | −2.04492 | −5.46365 |
| 7m | −4.5574 | −3.97825 | −1.5371 | −2.85352 | −5.66012 |
| 7n | −7.01527 | −5.64609 | −1.0642 | −2.98835 | −6.32379 |
| 7o | −5.5679 | −4.59082 | −3.14312 | −0.16728 | −6.54276 |
| 7p | −5.22365 | −2.67248 | −3.32493 | −1.96602 | −6.74707 |
| 7q | −6.75491 | −4.53187 | −2.50503 | −2.28726 | −6.61332 |
| 7r | −6.02244 | −3.42691 | −3.08999 | −2.80693 | −6.5254 |
| 7s | −6.41682 | −3.98382 | −2.45205 | −0.77139 | −6.29255 |
| 7t | −5.87745 | −3.0637 | −3.17249 | −2.34786 | −6.92566 |
| 7u | −6.50778 | −4.66838 | −3.28726 | −2.36884 | −6.51469 |
| Molecule | Dock score in kcal mol−1 | Molecule | Dock score in kcal mol−1 | ||||
|---|---|---|---|---|---|---|---|
| BSA | COX-2 | BSA | COX-2 | ||||
| Site 1 | Site 5 | Site 1 | Site 5 | ||||
| 5a | −5.044 | −5.343 | −8.415 | 7g | −6.133 | −7.299 | −9.378 |
| 5b | −4.911 | −6.552 | −9.807 | 7h | −6.748 | −6.020 | −9.549 |
| 5c | −5.177 | −6.124 | −7.794 | 7i | −5.420 | −6.667 | −5.600 |
| 5d | −5.141 | −6.026 | −8.562 | 7j | −6.89574 | −6.676 | −8.509 |
| 5e | −4.254 | −4.974 | −8.998 | 7k | −6.17567 | −6.522 | −5.329 |
| 5f | −5.335 | −5.156 | −7.424 | 7l | −6.59588 | −5.463 | −5.522 |
| 5g | −6.903 | −5.771 | −7.689 | 7m | −4.5574 | −5.660 | −9.519 |
| 5h | −4.280 | −6.799 | −7.055 | 7n | −7.01527 | −6.323 | −5.839 |
| 5i | −6.280 | −6.350 | −6.919 | 7o | −5.5679 | −6.542 | −8.001 |
| 7a | −5.668 | −6.645 | −7.839 | 7p | −5.22365 | −6.747 | −8.249 |
| 7b | −5.244 | −7.004 | −7.655 | 7q | −6.75491 | −6.613 | −9.686 |
| 7c | −5.911 | −7.342 | −8.111 | 7r | −6.02244 | −6.525 | −9.150 |
| 7d | −5.859 | −6.771 | −9.257 | 7s | −6.41682 | −6.292 | −9.478 |
| 7e | −7.259 | −6.207 | −5.922 | 7t | −5.87745 | −6.925 | −8.230 |
| 7f | −6.079 | −6.812 | −4.634 | 7u | −6.50778 | −6.514 | −9.276 |
Fig. 2 shows the interaction of molecule 7h with protein at site-5. The molecule showed three hydrogen bond interactions with amino acid residues Leu 480, Val 481 and Arg 208. Two electrostatic interactions were also seen between the π electron cloud of fused aromatic ring and residues Lys 350 and Glu 353. A hydrophobic interaction is also seen with Leu 326. Fig. 3 represents the hydrogen bond interaction with Leu 480, Val 481, Arg 208 and electrostatic interaction with Lys 350, Glu 353 of molecule 7h at site-5 of BSA. Fig. 4 represents the docked conformation of molecule 7h showing π–π interaction with Tyr 341 and hydrophobic interaction Gly 512 and Ala 513 at active site of COX-2. Fig. 5 shows the ligand π–π interaction with Tyr 341 and hydrophobic interaction with Gly 512, Ala 513 of molecule 7h docked at active site of COX-2.
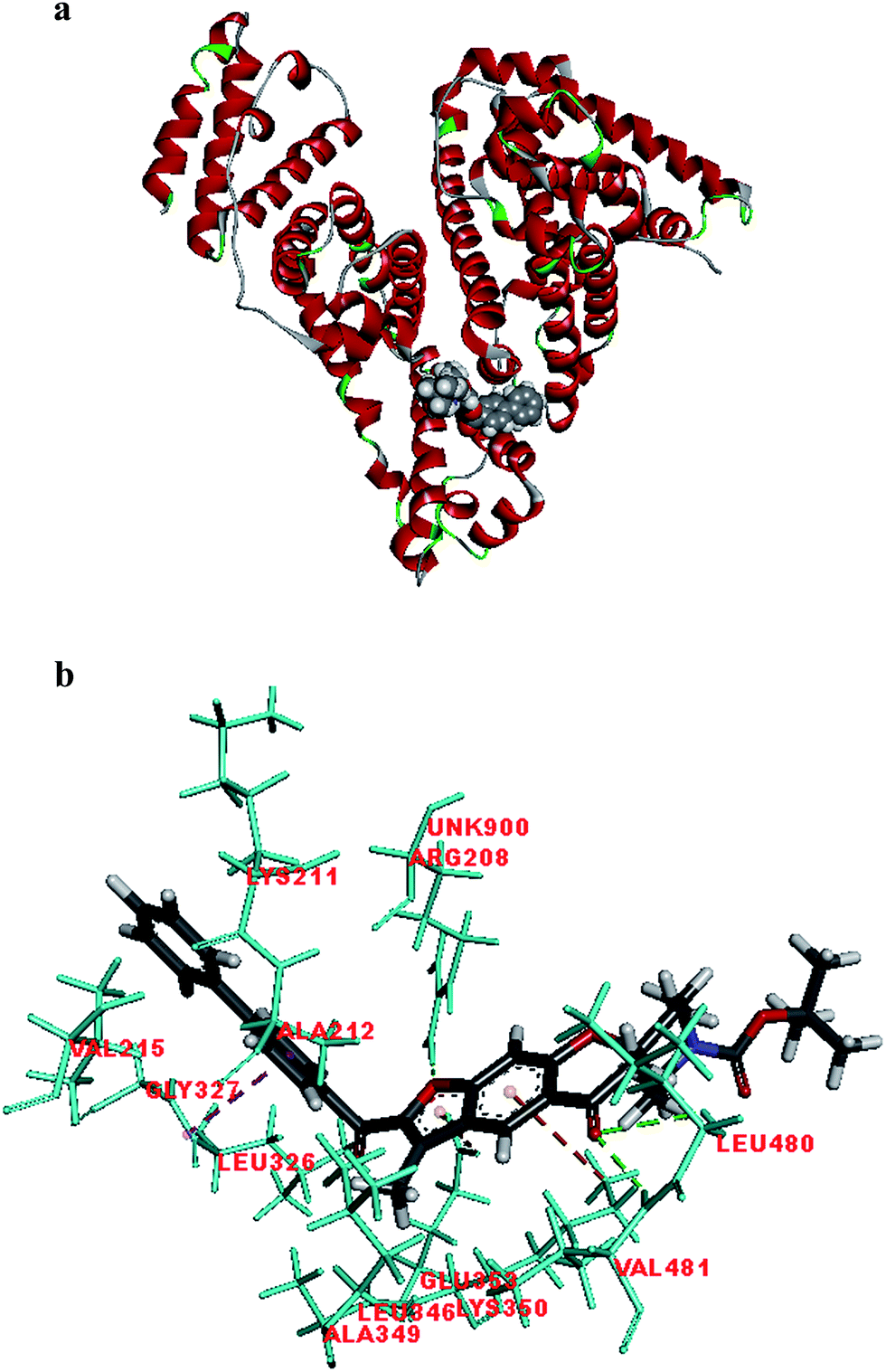 | ||
| Fig. 2 (a) Molecule 7h docked at site-5 of BSA. (b) Molecular interaction of molecule 7h showing hydrogen bond interaction with Leu 480, Val 481 and Arg 208, electrostatic interaction with Lys 350 and Glu 353. | ||
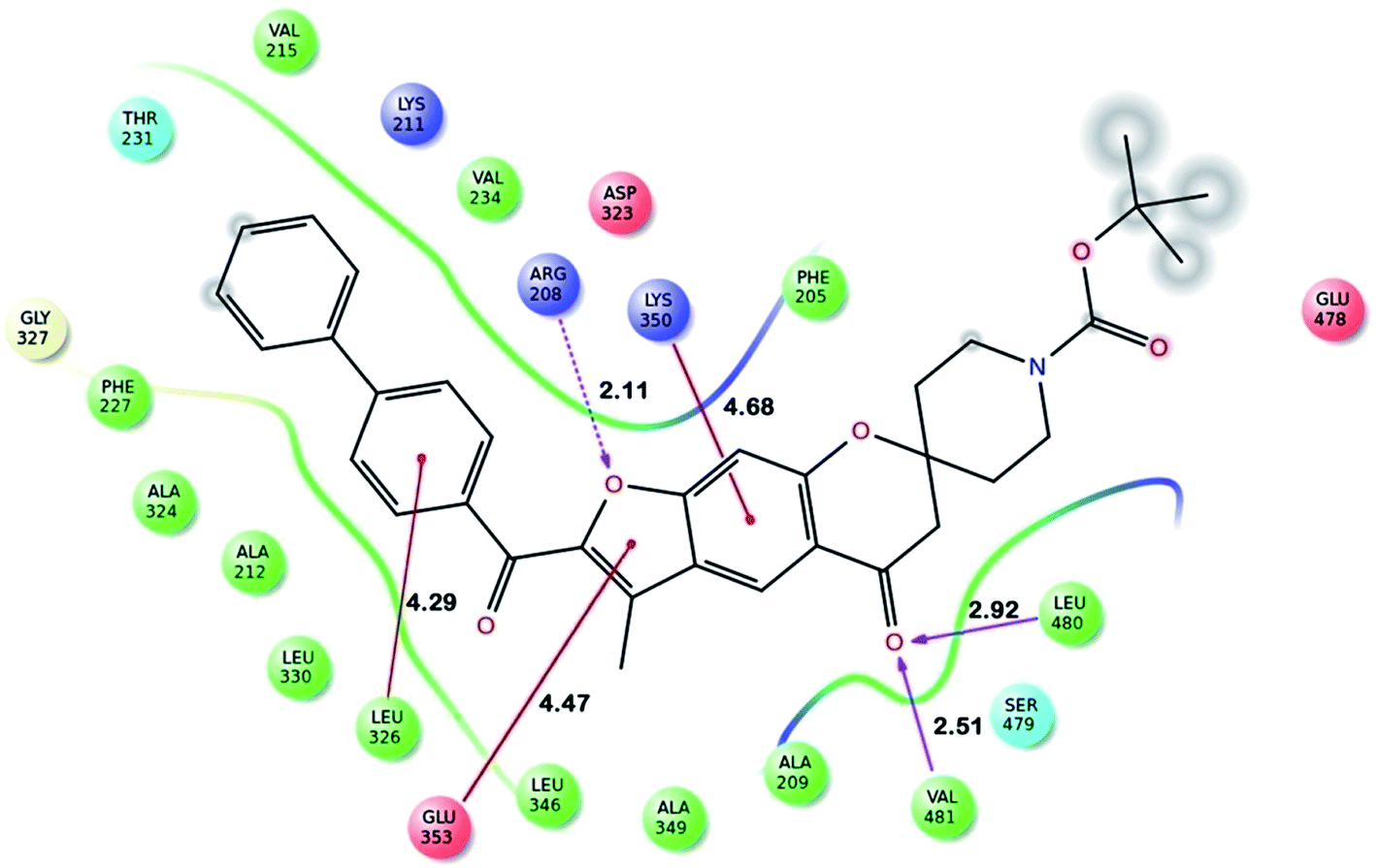 | ||
| Fig. 3 Ligand interaction diagram of molecule 7h docked at site-5 of BSA showing hydrogen bond interaction with Leu 480, Val 481 and Arg 208, electrostatic interaction with Lys 350 and Glu 353 [distance are mentioned in Å]. | ||
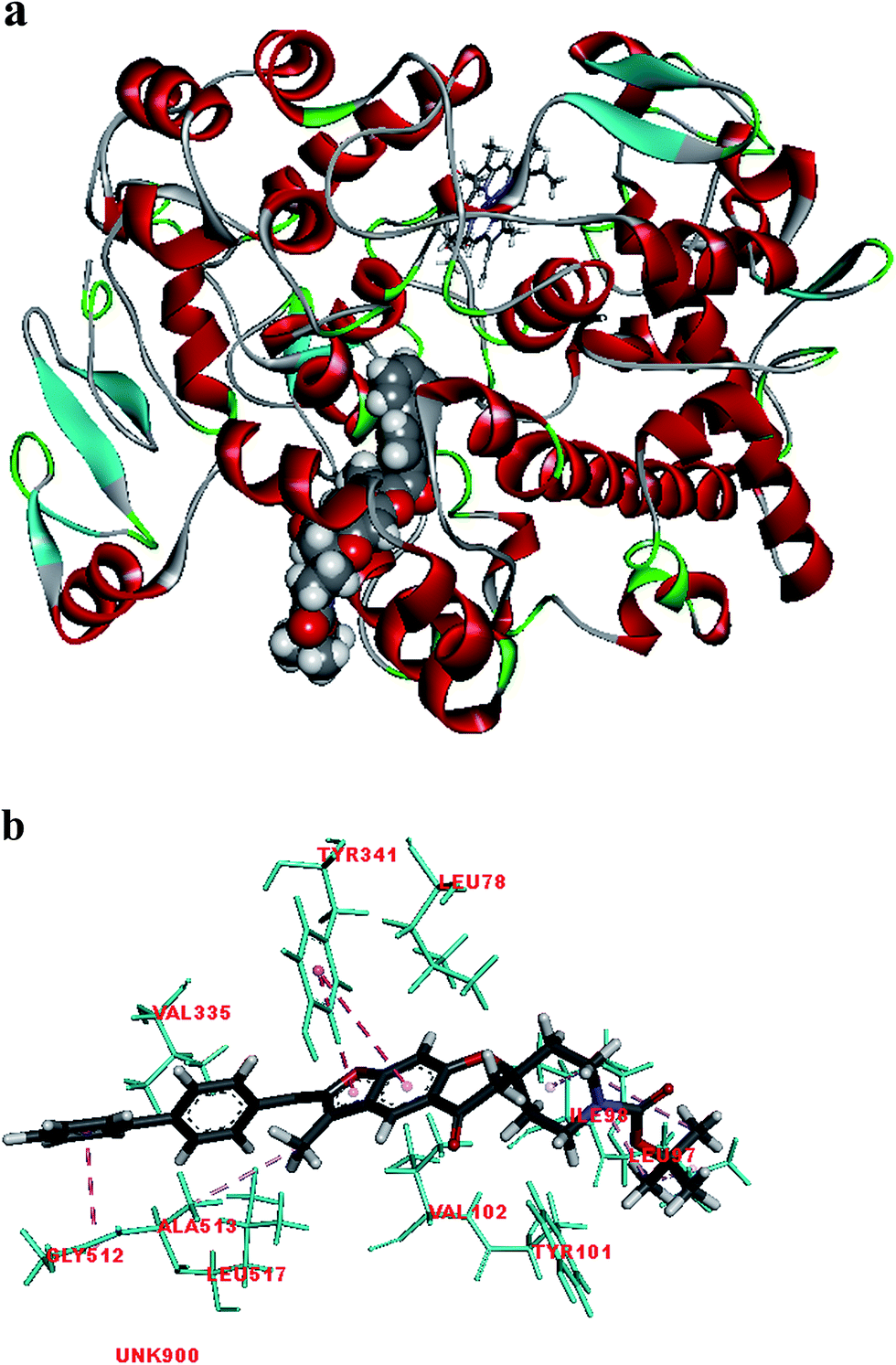 | ||
| Fig. 4 (a) Molecule 7h docked at active site of COX-2. (b) Molecular interaction of molecule 7h showing π–π interaction with Tyr 341 and hydrophobic interaction Gly 512 and Ala 513. | ||
Po/w) between −1 and 6.5 and solubility (logS) greater than −7. Pmdck greater than 5 and logBB greater than −3, these parameter tells us about the ability of the drug to pass through blood brain barrier. Majority of the molecules have the properties in range, partition coefficient of molecules ranged within the acceptable limits expect for two molecules 7l and 7m that had values greater than 7. Cell permeability and blood brain barrier permeability for all molecules is in permissible range. Water solubility and oral absorption for half of the molecules is 100%. The values invariably imply that these molecules are potential drug molecules and can be further optimized for better activity (Fig. 5).
| Molecule | QPlogPo/wa |
QPlogSb |
QPlogBBc |
QPPMDCKd | Percent human oral absorptione |
|---|---|---|---|---|---|
| a Predicted octanol/water partition coefficient logP (acceptable range—2.0–6.5).b Predicted aqueous solubility in mol L−1 (acceptable range—6.5–0.5).c Predicted blood brain barrier permeability (acceptable range—3–1.2).d Predicted apparent MDCK cell permeability in nm s−1 (acceptable range in nm s−1) (acceptable range: <25 is poor and >500 is great).e Percentage of human oral absorption (acceptable range: <25 is poor and >80% is high). |
|||||
| 5a | 4.022 | −5.314 | −0.453 | 643.955 | 100 |
| 5b | 4.601 | −6.753 | −0.995 | 280.644 | 100 |
| 5c | 3.756 | −4.981 | −0.46 | 616.566 | 100 |
| 5d | 4.604 | −6.194 | −0.289 | 1709.413 | 100 |
| 5e | 5.19 | −7.652 | −0.841 | 744.903 | 81.033 |
| 5f | 4.309 | −5.807 | −0.285 | 1679.254 | 100 |
| 5g | 4.528 | −6.082 | −0.299 | 1589.697 | 100 |
| 5h | 5.111 | −7.531 | −0.849 | 692.753 | 80.568 |
| 5i | 4.23 | −5.687 | −0.294 | 1561.757 | 100 |
| 7a | 5.711 | −7.481 | −0.612 | 643.259 | 100 |
| 7b | 4.579 | −6.945 | −1.543 | 103.589 | 96.205 |
| 7c | 6.212 | −8.234 | −0.456 | 1588.787 | 100 |
| 7d | 6.033 | −8.079 | −0.638 | 643.102 | 100 |
| 7e | 6.65 | −8.631 | −0.646 | 642.485 | 95.54 |
| 7f | 6.721 | −9.017 | −0.302 | 3911.802 | 95.966 |
| 7g | 5.951 | −7.857 | −0.505 | 1164.676 | 100 |
| 7h | 6.285 | −8.904 | −1.183 | 280.243 | 87.434 |
| 7i | 5.149 | −8.358 | −2.202 | 45.132 | 67.651 |
| 7j | 6.795 | −9.687 | −1.036 | 692.527 | 90.425 |
| 7k | 6.612 | −9.519 | −1.219 | 280.159 | 89.347 |
| 7l | 7.236 | −10.104 | −1.234 | 280.245 | 93.003 |
| 7m | 7.295 | −10.439 | −0.886 | 1704.24 | 93.351 |
| 7n | 6.525 | −9.28 | −1.078 | 507.476 | 88.843 |
| 7o | 5.448 | −7.157 | −0.623 | 615.852 | 100 |
| 7p | 4.316 | −6.621 | −1.547 | 99.175 | 94.352 |
| 7q | 5.954 | −7.926 | −0.468 | 1521.395 | 100 |
| 7r | 5.775 | −7.772 | −0.651 | 615.837 | 100 |
| 7s | 6.387 | −8.307 | −0.657 | 615.107 | 100 |
| 7t | 6.418 | −8.605 | −0.297 | 3842.145 | 94.06 |
| 7u | 5.688 | −7.533 | −0.516 | 1115.143 | 100 |
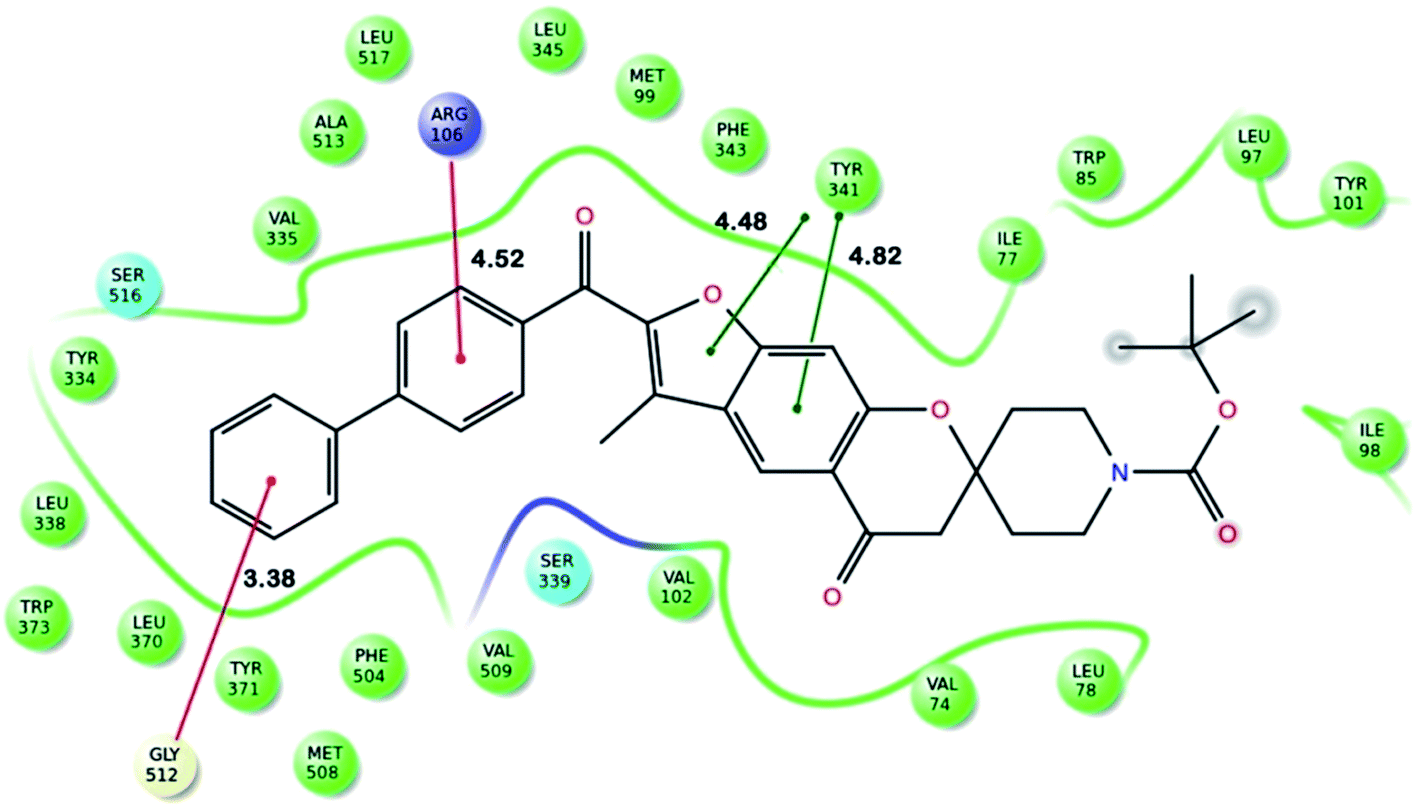 | ||
| Fig. 5 Ligand interaction diagram of molecule 7h docked at active site of COX-2 showing π–π interaction with Tyr 341 and hydrophobic interaction Arg 106, Gly 512 and Ala 513 [distance are mentioned in Å]. | ||
3 Conclusion
In conclusion, we herein report the selective synthesis of monospiro chromanone derivatives 3a–c from which a series of spirofurochromanones were synthesized following microwave irradiation method and their anti-inflammatory and antioxidant activities were determined. The reaction protocol requires cheap starting materials and is carried out under mild conditions. Compounds 5a, 5b, 5c, 5d, 5e, 7g, 7h, 7j, 7l, 7n and 7q were found to be promising with IC50 values ranging from 77.11 to 116.10 μM mL−1 indicates that they are potential anti-inflammatory agents. The binding mode of the synthesized compounds with protein active site was predicted using in silico docking. All the compounds 5a–i and 7a–u were docked well into the binding pocket of the target protein at all the five sites. The compounds showed good docking at site I and site V.4 Experimental section
4.1 General methods
All the Suzuki reactions were performed under nitrogen atmosphere using oven dried apparatus. Reactions were monitored by thin layer chromatography (TLC) on silica gel plates (60 F254), visualizing with ultraviolet light. Column chromatography was performed on silica gel (60–120 mesh) using distilled hexane, ethyl acetate. 1H NMR and 13C NMR spectra were determined in CDCl3 solution by using 400 and 100 MHz spectrometers, respectively (Instrument Bruker Avance II 400 MHz). Proton chemical shifts (δ) are relative to tetramethylsilane (TMS, δ = 0.00) as internal standard and expressed in ppm. Spin multiplicities are given as s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet) as well as bs (broad singlet). Coupling constants (J) are given in hertz. Mass spectra were recorded on GCMS-QP 1000 EX mass spectrometer. Infrared spectra were recorded on a Shimadzu FT-IR-8400s spectrometer. Melting points were determined using Stuart SMP3 melting-point apparatus and are uncorrected. CEM discover microwave reaction vessel equipped with a magnetic stirrer was used for microwave irradiated reactions. The antioxidant property was carried out by using Shimadzu UV-2450 spectrophotometer and the Perkin Elmer Lambda 750 UV-Visible Spectrophotometer was used to calculate the percentage inhibition for anti inflammatory activity. Bovine serum albumin used for the anti-inflammatory activity was purchased from Sigma Aldrich.4.2 General experimental procedure 3a–c
A solution of compound 1 (1 mmol) and pyrrolidine (1 mmol) in toluene was refluxed for 10 min. To the above solution various cycloalkanones 2a–c (1.5 mmol) were added and the reaction mixture was stirred at 90 °C for 4 h, the completion of reaction was monitored by TLC. Compound from the resulting solution was precipitated using 15% NaOH solution, the precipitate was filtered, solid was neutralized with dil. HCl and washed with water, dried in air and recrystallised from ethanol to afford pure monospiro compound 3a–c.4.3 General experimental procedure 5a–i
4.3.2.1 2′-Benzoyl-3′-methylspiro[cyclohexane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (5a). White solid; M.F: C24H22O4; mp: 128–130 °C; yield: 82%; IR (KBr, cm−1): 3082, 2931, 1649, 1618, 1475; 1H NMR (400 MHz, CDCl3) δ 8.27 (s, 1H), 8.04–8.02 (d, J = 7.0 Hz, 2H), 7.63–7.59 (t, J = 7.0 Hz, 1H), 7.52–7.50 (t, J = 7.0 Hz, 2H), 7.07 (s, 1H), 2.77 (s, 2H), 2.59 (s, 3H), 2.03–2.01 (m, 2H), 1.75–1.68 (m, 2H), 1.56–1.51 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 192.1, 185.3, 160.0, 158.9, 149.0, 137.7, 132.6, 129.5, 128.3, 127.7, 124.0, 120.9, 118.9, 100.5, 80.6, 48.3, 34.9, 25.1, 21.4, 10.0; MS (ESI mass): m/z, 375 [M + H]+.
4.3.2.2 tert-Butyl-7-benzoyl-6-methyl-4-oxo-4,8-dihydro-3H-spiro[cyclopenta[g]chromene-2,4′-piperidine]-1′-carboxylate (5b). White solid; M.F: C28H29NO6; mp: 220–222 °C; yield: 84%; IR (KBr, cm−1): 2972, 1683, 1620, 1246, 1153; 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 8.04–8.02 (d, J = 7.5 Hz, 2H), 7.63–7.61 (t, J = 7.5 Hz, 1H), 7.54–7.50 (t, J = 7.5 Hz, 2H), 7.10 (s, 1H), 3.88 (bs, 2H), 3.24 (bs, 2H), 2.79 (s, 2H), 2.60 (s, 3H), 2.02–2.06 (m, 2H), 1.69–1.62 (m, 2H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 191.0, 185.2, 159.3, 158.9, 154.6, 149.2, 137.6, 132.7, 129.5, 128.3, 127.5, 124.5, 121.1, 118.7, 100.6, 79.8, 78.5, 48.1, 34.1, 28.4, 9.9; MS (EI mass): m/z, 475 [M]+.
4.3.2.3 2′-Benzoyl-3′-methylspiro[cyclopentane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (5c). White solid; M.F: C23H20O4; mp: 68–70 °C; yield: 71%; IR (KBr, cm−1): 2982, 1687, 1618, 1230, 1145; 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 8.04–8.02 (d, J = 8.5 Hz, 2H), 7.63–7.59 (m, J = 8.5 Hz, 1H), 7.54–7.50 (t, J = 8.5 Hz, 2H), 7.02 (s, 1H), 2.90 (s, 2H), 2.59 (s, 3H), 2.13–2.08 (m, 2H), 1.93–1.87 (m, 2H), 1.77–1.66 (m, 4H); MS (ESI mass): m/z, 361 [M + H]+.
4.3.2.4 2′-(4-Bromobenzoyl)-3′-methylspiro[cyclohexane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (5d). White solid; M.F: C24H21BrO4; mp: 148–150 °C; yield: 83%; IR (KBr, cm−1): 3265, 2927, 1689, 1620, 1336, 1244; 1H NMR (400 MHz, CDCl3) δ 8.28 (s, 1H), 7.95–7.93 (d, J = 8.5 Hz, 2H), 7.67–7.65 (d, J = 8.5 Hz, 2H), 7.07 (s, 1H), 2.77 (s, 2H), 2.62 (s, 3H), 1.75–1.64 (m, 3H), 1.57–1.51 (m, 7H); 13C NMR (100 MHz, CDCl3) δ 192.1, 183.9, 160.2, 158.8, 148.7, 136.3, 131.1, 128.4, 127.8, 123.9, 121.0, 119.0, 100.5, 80.7, 48.2, 34.9, 25.0, 21.4, 10.0; MS (ESI mass): m/z, 453 [M + H]+.
4.3.2.5 tert-Butyl-2-(4-bromobenzoyl)-3-methyl-5-oxo-5,6-dihydrospiro[furo[3,2-g]chromene-7,4′-piperidine]-1′-carboxylate (5e). White solid; M.F: C28H28BrNO6; mp: 224–226 °C; yield: 80%; IR (KBr, cm−1): 2974, 1683, 1620, 1246, 1151; 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 7.95–7.93 (d, J = 8.7 Hz, 2H), 7.68–7.66 (d, J = 8.7 Hz, 2H), 7.10 (s, 1H), 3.88 (bs, 2H), 3.23 (bs, 2H), 2.80 (s, 2H), 2.62 (s, 3H), 2.06–2.02 (m, 2H), 1.70–1.66 (m, 2H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 191.0, 183.9, 159.4, 158.8, 154.6, 148.9, 136.1, 131.6, 131.1, 128.2, 127.9, 124.3, 121.2, 118.7, 100.6, 79.9, 78.6, 48.0, 34.1, 28.3, 10.0; MS (ESI mass): m/z, 552 [M – H]+.
4.3.2.6 2′-(4-Bromobenzoyl)-3′-methylspiro[cyclopentane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (5f). White solid; M.F: C23H19BrO4; mp: 130–132 °C; yield: 77%; IR (KBr, cm−1): 2958, 1689, 1616, 1222, 1143; 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 7.95–7.92 (d, J = 8.7 Hz, 2H), 7.67–7.65 (d, J = 8.7 Hz, 2H), 7.01 (s, 1H), 2.90 (s, 2H), 2.62 (s, 3H), 2.13–2.08 (m, 2H), 1.93–1.87 (m, 2H), 1.75–1.67 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 192.1, 183.9, 160.9, 158.7, 148.7, 136.3, 131.6, 131.1, 128.4, 127.8, 123.9, 121.3, 119.0, 100.6, 90.7, 47.1, 37.5, 23.8, 10.0; MS (ESI mass): m/z, 437 [M – H]+; HRMS: 438.0446.
4.3.2.7 2′-(4-Chlorobenzoyl)-3′-methylspiro[cyclohexane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (5g). White solid; M.F: C24H21ClO4; mp: 126–128 °C; yield: 79%; IR (KBr, cm−1): 2929, 1691, 1618, 1328, 1244, 1143; 1H NMR (400 MHz, CDCl3) δ 8.28 (s, 1H), 8.03–8.01 (d, J = 8.5 Hz, 2H), 7.50–7.48 (d, J = 8.5 Hz, 2H), 7.07 (s, 1H), 2.77 (s, 2H), 2.62 (s, 3H), 1.78–1.65 (m, 3H), 1.56–1.51 (m, 7H); 13C NMR (100 MHz, CDCl3) δ 192.1, 183.7, 160.2, 158.9, 148.7, 139.1, 135.9, 131.0, 128.6, 123.9, 121.0, 119.0, 100.5, 80.7, 48.2, 34.9, 29.6, 25.0, 21.4, 10.0; MS (ESI mass): m/z, 409 [M + H]+.
4.3.2.8 tert-Butyl-2-(4-chlorobenzoyl)-3-methyl-5-oxo-5,6-dihydrospiro[furo[3,2-g]chromene-7,4′-piperidine]-1′-carboxylate (5h). White solid; M.F: C28H28ClNO6; mp: 218–220 °C; yield: 77%; IR (KBr, cm−1): 2922, 1683, 1620, 1244, 1151; 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 8.03–8.01 (d, J = 8.5 Hz, 2H), 7.51–7.49 (d, J = 8.5 Hz, 2H), 7.10 (s, 1H), 3.89 (bs, 2H), 3.23 (bs, 2H), 2.91 (s, 2H), 2.62 (s, 3H), 2.12–2.03 (m, 2H), 1.76–1.67 (m, 2H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 191.0, 183.7, 159.4, 158.8, 154.6, 148.9, 139.2, 135.7, 131.0, 130.1, 128.7, 124.3, 121.2, 118.7, 100.6, 99.6, 79.9, 78.5, 48.0, 47.5, 28.3, 10.0; MS (ESI mass): m/z, 508 [M – H]+.
4.3.2.9 2′-(4-Chlorobenzoyl)-3′-methylspiro[cyclopentane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (5i). White solid; M.F: C23H19ClO4; mp: 125–127 °C; yield: 80%; IR (KBr, cm−1): 2954, 1691, 1616, 1246, 1145; 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 8.03–8.00 (d, J = 8.5 Hz, 2H), 7.51–7.48 (d, J = 8.5 Hz, 2H), 7.02 (s, 1H), 2.90 (s, 2H), 2.62 (s, 3H), 2.13–2.08 (m, 2H), 1.92–1.87 (m, 2H), 1.75–1.66 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 192.1, 183.7, 160.8, 158.7, 148.8, 139.1, 135.8, 131.0, 128.6, 128.3, 123.9, 121.2, 119.0, 100.6, 90.6, 47.1, 37.5, 23.8, 10.0; MS (ESI mass): m/z, 393 [M – H]+.
4.4 General experimental procedure 7a–u
:1:1 of toluene, ethanol, water (9 mL) was introduced into a round bottomed flask and heated it at 80 °C for 8 h with constant stirring under nitrogen atmosphere. The resulting solution was diluted with ice water and was extracted with ethyl acetate; the combined organic layers were washed with brine, dried over Na2SO4. The solvent was removed under reduced pressure, and the crude thus obtained was purified using column chromatography to afford titled compound 7(a–u).
4.4.1.1 Sealed tube under conventional heating. A mixture of 5d–f (1.0 mmol), arylboronic acid 6a–g (1.3 mmol), Na2CO3 (3.00 mmol), and mixture of 1
:1:1 of toluene, ethanol, water (9 mL) are degassed in a sealed pressure tube and Pd(PPh3)4 (115 mg, 0.10 mmol) was added followed by degassing and heated it at 80 °C for 6–7 h with constant stirring. The resulting solution was diluted with ice water and was extracted with ethyl acetate; the combined organic layers were washed with brine, dried over Na2SO4. The solvent was removed under reduced pressure, and the crude thus obtained was purified using column chromatography (using 5–10% petroleum ether/ethyl acetate) to afford titled compound 7a–u.
:1:1 of toluene, ethanol, water (9 mL) are degassed in a sealed pressure tube and Pd(PPh3)4 (115 mg, 0.10 mmol) was added followed by degassing. This was introduced into CEM discover microwave reaction vessel equipped with a magnetic stirrer. The vessel was sealed and then placed into the microwave cavity. Initial microwave irradiation of 180 W was used, the temperature being ramped from room temperature to the desired 80 °C temperature. Once this was reached the reaction mixture was heated at this temperature for appropriate time. The resulting solution was diluted with ice water and was extracted with ethyl acetate; the combined organic layers were washed with brine, dried over Na2SO4. The solvent was removed under reduced pressure, and the crude thus obtained was purified column chromatography to afford titled compound 7a–u.
4.4.2.1 2′-([1,1′-Biphenyl]-4-carbonyl)-3′-methylspiro[cyclohexane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (7a). White solid; M.F: C30H26O4; mp: 172–174 °C; yield: 83%; IR (KBr, cm−1): 2929, 1687, 1622, 1290, 1246, 1151; 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 8.16–8.14 (d, J = 8.5 Hz, 2H), 7.76–7.74 (d, J = 8.5 Hz, 2H), 7.68–7.66 (d, J = 7.2 Hz, 2H), 7.51–7.47 (t, J = 7.2 Hz, 2H), 7.44–7.40 (t, J = 7.2 Hz, 1H), 7.01 (s, 1H), 2.78 (s, 2H), 2.63 (s, 3H), 2.04–2.00 (m, 2H), 1.75–1.69 (m, 4H), 1.56–1.51 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 192.2, 184.7, 160.0, 158.9, 149.1, 145.4, 140.0, 136.3, 130.23, 128.9, 128.2, 127.8, 127.3, 127.0, 124.0, 118.9, 100.5, 80.7, 48.3, 34.9, 25.1, 21.4, 10.1; MS (ESI mass): m/z, 451 [M + H]+.
4.4.2.2 4′-(3′-Methyl-5′-oxo-5′,6′-dihydrospiro[cyclohexane-1,7′-furo[3,2-g]chromen]-2′-ylcarbonyl)-[1,1′-biphenyl]-4-carbaldehyde (7b). White solid; M.F: C31H26O5; mp: 168–170 °C; yield: 82%; IR (KBr, cm−1): 2929, 1687, 1625, 1292, 1220, 1145; 1H NMR (400 MHz, CDCl3) δ 10.09 (s, 1H), 8.30 (s, 1H), 8.19–8.17 (d, J = 8.5 Hz, 2H), 8.02–8.00 (d, J = 8.5 Hz, 2H), 7.85–7.83 (d, J = 8.2 Hz, 2H), 7.80–7.78 (d, J = 8.2 Hz, 2H), 7.10 (s, 1H), 2.78 (s, 2H), 2.65 (s, 3H), 2.04–2.00 (m, 2H), 1.78–1.69 (m, 4H), 1.57–1.51 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 192.2, 191.7, 184.4, 160.1, 158.9, 148.9, 145.8, 143.7, 137.3, 135.7, 130.3, 128.2, 127.9, 127.3, 123.9, 121.0, 118.9, 100.5, 80.7, 48.2, 34.8, 29.6, 25.0, 21.4, 10.0; MS (ESI mass): m/z, 479 [M + H]+.
4.4.2.3 2′-(4′-Chloro-[1,1′-biphenyl]-4-carbonyl)-3′-methylspiro[cyclohexane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (7c). White solid; M.F: C30H25ClO4; mp: 186–188 °C; yield: 84%; IR (KBr, cm−1): 2924, 1689, 1616, 1296, 1143; 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 8.15–8.13 (d, J = 8.5 Hz, 2H), 7.72–7.70 (d, J = 8.7 Hz, 2H), 7.61–7.59 (d, J = 8.5 Hz, 2H), 7.47–7.45 (d, J = 8.7 Hz, 2H), 7.09 (s, 1H), 2.78 (s, 2H), 2.63 (s, 3H), 2.04–2.00 (m, 2H), 1.78–1.68 (m, 4H), 1.56–1.51 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 192.2, 184.5, 160.1, 158.9, 149.0, 144.0, 138.4, 136.6, 134.4, 130.3, 129.1, 128.5, 127.9, 126.8, 124.0, 120.9, 118.9, 100.5, 80.7, 48.2, 34.8, 25.0, 21.4, 10.0; MS (ESI mass): m/z, 485 [M + H]+.
4.4.2.4 3′-Methyl-2′-(4′-methyl-[1,1′-biphenyl]-4-carbonyl)spiro[cyclohexane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (7d). White solid; M.F: C30H25ClO4; mp: 190–192 °C; yield: 87%; IR (KBr, cm−1): 2931, 1689, 1616, 1294, 1143; 1H NMR (400 MHz, CDCl3) δ 8.28 (s, 1H), 8.14–8.12 (d, J = 8.5 Hz, 2H), 7.74–7.72 (d, J = 8.5 Hz, 2H), 7.59–7.57 (d, J = 8.0 Hz, 2H), 7.31–7.29 (d, J = 8.0 Hz, 2H), 7.10 (s, 1H), 2.78 (s, 2H), 2.62 (s, 3H), 2.42 (s, 3H), 2.04–2.00 (m, 2H), 1.78–1.65 (m, 4H), 1.56–1.51 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 192.2, 184.7, 160.0, 158.9, 149.1, 145.3, 138.1, 137.0, 136.0, 130.2, 129.6, 127.6, 127.1, 126.7, 124.0, 120.8, 118.8, 100.5, 80.6, 48.2, 34.8, 25.0, 21.4, 21.1, 10.0; MS (ESI mass): m/z, 465 [M + H]+; HRMS: 464.19812.
4.4.2.5 3′-Methyl-2′-(4-(naphthalen-1-yl)benzoyl)spiro[cyclohexane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (7e). White solid; M.F: C34H28O4; mp: 152–154 °C; yield: 86%; IR (KBr, cm−1): 2929, 1689, 1622, 1292, 1145; 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 8.21–8.19 (d, J = 8.5 Hz, 2H), 7.95–7.90 (m, 3H), 7.68–7.65 (d, J = 8.5 Hz, 2H), 7.59–7.46 (m, 4H), 7.13 (s, 1H), 2.78 (s, 2H), 2.67 (s, 3H), 2.04–2.01 (m, 2H), 1.75–1.65 (m, 4H), 1.54–1.47 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 192.2, 184.8, 160.0, 158.9, 149.1, 145.4, 139.1, 136.4, 133.7, 131.2, 130.0, 129.7, 128.3, 127.9, 125.9, 125.6, 125.3, 124.0, 120.9, 118.9, 100.5, 80.6, 48.3, 34.8, 25.0, 21.4, 10.0; MS (ESI mass): m/z, 501 [M + H]+.
4.4.2.6 2′-(3′,5′-Dichloro-[1,1′-biphenyl]-4-carbonyl)-3′-methylspiro[cyclohexane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (7f). White solid; M.F: C30H24Cl2O4; mp: 178–180 °C; yield: 85%; IR (KBr, cm−1): 2929, 1687, 1627, 1292, 1147; 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 8.16–8.14 (d, J = 8.5 Hz, 2H), 7.70–7.68 (d, J = 8.5 Hz, 2H), 7.54–7.53 (d, J = 1.7 Hz, 2H), 7.40 (t, J = 1.7 Hz, 1H), 7.09 (s, 1H), 2.78 (s, 2H), 2.64 (s, 3H), 2.04–2.00 (m, 2H), 1.75–1.66 (m, 4H), 1.54–1.51 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 192.1, 184.3, 160.1, 158.9, 148.9, 142.4, 137.3, 135.5, 130.3, 128.2, 125.7, 123.9, 121.0, 118.9, 100.5, 80.7, 48.2, 34.8, 29.6, 25.0, 21.4, 10.0; MS (ESI mass): m/z, 519 [M + H]+.
4.4.2.7 2′-(4′-Fluoro-[1,1′-biphenyl]-4-carbonyl)-3′-methylspiro[cyclohexane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (7g). White solid; M.F: C30H25FO4; mp: 160–162 °C; yield: 84%; IR (KBr, cm−1): 2931, 1685, 1614, 1336, 1246, 1149; 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 8.15–8.13 (d, J = 8.5 Hz, 2H), 7.71–7.68 (d, J = 8.5 Hz, 2H), 7.65–7.62 (m, 2H), 7.20–7.16 (t, J = 8.7 Hz, 2H), 7.09 (s, 1H), 2.78 (s, 2H), 2.63 (s, 3H), 2.04–2.00 (m, 2H), 1.75–1.68 (m, 4H), 1.55–1.51 (m, 2H), 1.27–1.24 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 192.2, 184.6, 164.2, 161.7, 160.0, 158.9, 149.0, 144.3, 136.3, 130.3, 128.9, 127.8, 126.8, 124.0, 120.9, 118.8, 115.7, 100.5, 80.7, 48.2, 34.8, 25.0, 21.4, 10.0; MS (ESI mass): m/z, 469 [M + H]+.
4.4.2.8 tert-Butyl-2-([1,1′-biphenyl]-4-carbonyl)-3-methyl-5-oxo-5,6-dihydrospiro[furo[3,2-g]chromene-7,4′-piperidine]-1′-carboxylate (7h). White solid; M.F: C34H33NO6; mp: 204–207 °C; yield: 82%; IR (KBr, cm−1): 2992, 1683, 1618, 1246, 1151; 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 8.15–8.13 (d, J = 8.5 Hz, 2H), 7.76–7.74 (d, J = 8.5 Hz, 2H), 7.69–7.67 (d, J = 7.2 Hz, 2H), 7.52–7.48 (t, J = 7.2 Hz, 2H), 7.44–7.40 (t, J = 7.2 Hz, 1H), 7.12 (s, 1H), 3.90 (bs, 2H), 3.23 (bs, 2H), 2.80 (s, 2H), 2.64 (s, 3H), 2.07–2.03 (m, 2H), 1.70–1.63 (m, 2H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 191.2, 184.7, 159.3, 154.7, 149.3, 145.4, 139.9, 138.2, 130.3, 129.7, 128.9, 128.2, 127.3, 127.0, 126.7, 121.2, 120.9, 118.7, 100.6, 79.9, 78.5, 48.3, 34.9, 28.4, 21.4, 10.0; MS (ESI mass): m/z, 552 [M + H]+; HRMS: 551.23011.
4.4.2.9 tert-Butyl-2-(4′-formyl-[1,1′-biphenyl]-4-carbonyl)-3-methyl-5-oxo-5,6-dihydrospiro[furo[3,2-g]chromene-7,4′-piperidine]-1′-carboxylate (7i). White solid; M.F: C35H33NO7; mp: 210–212 °C; yield: 80%; IR (KBr, cm−1): 2924, 1689, 1627, 1419, 1153; 1H NMR (400 MHz, CDCl3) δ 10.09 (s, 1H), 8.32 (s, 1H), 8.19–8.17 (d, J = 8.2 Hz, 2H), 8.02–8.00 (d, J = 8.2 Hz, 2H), 7.85–7.79 (m, 4H), 7.12 (s, 1H), 3.91 (bs, 2H), 3.24 (bs, 2H), 2.80 (s, 2H), 2.65 (s, 3H), 2.07–2.03 (m, 2H), 1.69–1.64 (m, 2H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 191.8, 191.1, 184.5, 159.4, 154.7, 149.2, 145.8, 143.8, 137.2, 135.8, 130.4, 127.9, 127.3, 124.4, 121.3, 118.8, 100.6, 79.9, 78.6, 48.1, 29.7, 28.4, 22.7, 21.4, 14.1, 10.1; MS (ESI mass): m/z, 580 [M + H]+.
4.4.2.10 tert-Butyl-2-(4′-chloro-[1,1′-biphenyl]-4-carbonyl)-3-methyl-5-oxo-5,6-dihydrospiro[furo[3,2-g]chromene-7,4′-piperidine]-1′-carboxylate (7j). White solid; M.F: C34H32ClNO6; mp: 218–220 °C; yield: 83%; IR (KBr, cm−1): 2924, 1683, 1618, 1246, 1151; 1H NMR (400 MHz, CDCl3) δ 8.32 (s, 1H), 8.15–8.13 (d, J = 8.5 Hz, 2H), 7.72–7.70 (d, J = 8.5 Hz, 2H), 7.1–7.59 (d, J = 8.7 Hz, 2H), 7.47–7.45 (d, J = 8.7 Hz, 2H), 7.12 (s, 1H), 3.89 (bs, 2H), 3.25 (bs, 2H), 2.80 (s, 2H), 2.64 (s, 3H), 2.07–2.01 (m, 2H), 1.66–1.70 (m, 2H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 191.1, 184.6, 159.4, 158.9, 154.7, 149.3, 144.2, 138.4, 136.5, 134.4, 130.3, 129.2, 128.5, 126.9, 124.5, 121.2, 118.7, 100.6, 79.9, 78.6, 48.1, 34.1, 29.7, 28.4, 14.1, 10.0; MS (ESI mass): m/z, 586 [M + H]+.
4.4.2.11 tert-Butyl-3-methyl-2-(4′-methyl-[1,1′-biphenyl]-4-carbonyl)-5-oxo-5,6-dihydrospiro[furo[3,2-g]chromene-7,4′-piperidine]-1′-carboxylate (7k). White solid; M.F: C35H35NO6; mp: 224–227 °C; yield: 85%; IR (KBr, cm−1): 2974, 1683, 1620, 1246, 1151; 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 8.14–8.12 (d, J = 8.2 Hz, 2H), 7.74–7.72 (d, J = 8.2 Hz, 2H), 7.59–7.57 (d, J = 8.0 Hz, 2H), 7.31–7.29 (d, J = 8.0 Hz, 2H), 7.12 (s, 1H), 3.90 (bs, 2H), 3.24 (bs, 2H), 2.80 (s, 2H), 2.63 (s, 3H), 2.42 (s, 3H), 2.07–2.03 (m, 2H), 1.70–1.65 (m, 2H), 1.48 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 191.1, 184.7, 159.3, 158.9, 154.7, 149.4, 145.5, 138.2, 137.0, 135.9, 130.3, 129.7, 127.4, 127.1, 126.8, 124.5, 121.1, 118.6, 100.6, 79.9, 78.5, 48.1, 34.1, 29.7, 28.4, 21.2, 10.0; MS (ESI mass): m/z, 566 [M + H]+.
4.4.2.12 tert-Butyl-3-methyl-2-(4-(naphthalen-1-yl)benzoyl)-5-oxo-5,6-dihydrospiro[furo[3,2-g]chromene-7,4′-piperidine]-1′-carboxylate (7l). White solid; M.F: C38H35NO6; mp: 147–148 °C; yield: 84%; IR (KBr, cm−1): 2972, 1673, 1624, 1243, 1192; 1H NMR (400 MHz, CDCl3) δ 8.33 (s, 1H), 8.21–8.18 (d, J = 8.2 Hz, 2H), 7.95–7.91 (t, J = 8.0 Hz, 3H), 7.68–7.66 (d, J = 8.2 Hz, 2H), 7.59–7.46 (m, 4H), 7.16 (s, 1H), 3.90 (bs, 2H), 3.24 (bs, 2H), 2.80 (s, 2H), 2.67 (s, 3H), 2.07–2.04 (m, 2H), 1.70–1.64 (m, 2H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 191.1, 184.9, 159.3, 158.9, 154.7, 149.3, 145.5, 139.1, 136.4, 133.8, 131.2, 130.1, 129.7, 128.3, 127.7, 126.9, 126.4, 126.0, 125.6, 125.3, 124.5, 121.2, 118.7, 100.7, 79.9, 78.6, 48.1, 34.1, 29.7, 28.4, 22.6, 10.0; MS (ESI mass): m/z, 602 [M + H]+.
4.4.2.13 tert-Butyl-2-(3′,5′-dichloro-[1,1′-biphenyl]-4-carbonyl)-3-methyl-5-oxo-5,6-dihydro spiro[furo[3,2-g]chromene-7,4′-piperidine]-1′-carboxylate (7m). White solid; M.F: C34H31Cl2NO6; mp: 207–208 °C; yield: 86%; IR (KBr, cm−1): 2924, 1691, 1616, 1244, 1151; 1H NMR (400 MHz, CDCl3) δ 8.32 (s, 1H), 8.16–8.14 (d, J = 8.5 Hz, 2H), 7.70–7.68 (d, J = 8.5 Hz, 2H), 7.54–7.53 (d, J = 1.7 Hz, 2H), 7.40–7.15 (d, J = 1.7 Hz, 1H), 7.12 (s, 1H), 3.89 (bs, 2H), 3.25 (bs, 2H), 2.80 (s, 2H), 2.64 (s, 3H), 2.07–2.03 (m, 2H), 1.70–1.64 (m, 2H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 191.1, 184.4, 159.4, 158.9, 154.7, 149.1, 142.9, 142.6, 137.3, 135.5, 130.4, 128.0, 127.0, 125.8, 124.4, 121.3, 118.8, 100.6, 79.9, 78.6, 48.1, 34.2, 29.7, 28.4, 10.0; MS (ESI mass): m/z, 620 [M + H]+.
4.4.2.14 tert-Butyl-2-(4′-fluoro-[1,1′-biphenyl]-4-carbonyl)-3-methyl-5-oxo-5,6-dihydrospiro[furo[3,2-g]chromene-7,4′-piperidine]-1′-carboxylate (7n). White solid; M.F: C34H32FNO6; mp: 208–210 °C; yield: 87%; IR (KBr, cm−1): 2922, 1681, 1622, 1244, 1153; 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 8.15–8.13 (d, J = 8.5 Hz, 2H), 7.71–7.69 (d, J = 8.5 Hz, 2H), 7.65–7.62 (m, 2H), 7.20–7.16 (t, J = 8.7 Hz, 2H), 7.12 (s, 1H), 3.90 (bs, 2H), 3.24 (bs, 2H), 2.80 (s, 2H), 2.64 (s, 3H), 2.07–2.03 (m, 2H), 1.70–1.66 (m, 2H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 191.1, 184.6, 159.3, 158.8, 154.7, 149.3, 144.5, 136.2, 132.1, 132.0, 130.3, 129.0, 128.9, 128.4, 126.8, 124.5, 121.2, 118.6, 116.0, 115.8, 100.6, 79.9, 78.6, 48.1, 28.4, 10.0; MS (ESI mass): m/z, 570 [M + H]+.
4.4.2.15 2′-([1,1′-Biphenyl]-4-carbonyl)-3′-methylspiro[cyclopentane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (7o). White solid; M.F: C29H24O4; mp: 118–120 °C; yield: 86%; IR (KBr, cm−1): 2962, 1689, 1616, 1234, 1151; 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 8.15–8.13 (d, J = 8.5 Hz, 2H), 7.76–7.74 (d, J = 8.5 Hz, 2H), 7.69–7.67 (d, J = 7.7 Hz, 2H), 7.51–7.47 (t, J = 7.7 Hz, 2H), 7.44–7.39 (m, 1H), 7.05 (s, 1H), 2.91 (s, 2H), 2.63 (s, 3H), 2.14–2.09 (m, 2H), 1.92–1.88 (m, 2H), 1.75–1.68 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 192.2, 184.8, 160.8, 158.8, 149.2, 145.5, 139.9, 136.3, 130.2, 128.9, 128.2, 127.7, 127.3, 127.0, 124.1, 121.2, 118.9, 100.7, 90.6, 47.1, 37.5, 23.8, 10.0; MS (ESI mass): m/z, 437 [M + H]+.
4.4.2.16 4′-(3′-Methyl-5′-oxo-5′,6′-dihydrospiro[cyclopentane-1,7′-furo[3,2-g]chromen]-2′-ylcarbonyl)-[1,1′-biphenyl]-4-carbaldehyde (7p). White solid; M.F: C30H24O5; mp: 182–184 °C; yield: 88%; IR (KBr, cm−1): 2958, 1693, 1622, 1290, 1213, 1143; 1H NMR (400 MHz, CDCl3) δ 10.09 (s, 1H), 8.32 (s, 1H), 8.19–8.16 (d, J = 8.5 Hz, 2H), 8.02–8.00 (d, J = 8.5 Hz, 2H), 7.85–7.83 (d, J = 8.2 Hz, 2H), 7.80–7.78 (d, J = 8.2 Hz, 2H), 7.04 (s, 1H), 2.91 (s, 2H), 2.65 (s, 3H), 2.14–2.09 (m, 2H), 1.93–1.88 (m, 2H), 1.77–1.68 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 192.1, 191.7, 184.5, 160.8, 158.8, 149.0, 145.8, 143.7, 137.3, 135.7, 130.3, 128.2, 127.9, 127.3, 124.0, 121.2, 119.0, 100.6, 90.6, 47.1, 37.5, 23.8, 10.0; MS (ESI mass): m/z, 465 [M + H]+.
4.4.2.17 2′-(4′-Chloro-[1,1′-biphenyl]-4-carbonyl)-3′-methylspiro[cyclopentane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (7q). White solid; M.F: C29H23ClO4; mp: 138–140 °C; yield: 83%; IR (KBr, cm−1): 2924, 1687, 1612, 1236, 1149; 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 8.15–8.13 (d, J = 8.5 Hz, 2H), 7.72–7.70 (d, J = 8.5 Hz, 2H), 7.61–7.59 (d, J = 8.7 Hz, 2H), 7.47–7.45 (d, J = 8.7 Hz, 2H), 7.04 (s, 1H), 2.91 (s, 2H), 2.63 (s, 3H), 2.13–2.08 (m, 2H), 1.92–1.88 (m, 2H), 1.76–1.68 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 192.2, 184.6, 160.8, 158.8, 149.1, 144.0, 138.4, 136.6, 134.4, 130.3, 129.1, 128.5, 127.9, 126.8, 124.0, 121.2, 119.0, 90.6, 47.1, 37.5, 23.8, 10.0; MS (ESI mass): m/z, 471 [M + H]+; HRMS: 470.12822.
4.4.2.18 3′-Methyl-2′-(4′-methyl-[1,1′-biphenyl]-4-carbonyl)spiro[cyclopentane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (7r). White solid; M.F: C30H26O4; mp: 126–128 °C; yield: 85%; IR (KBr, cm−1): 2958, 1691, 1620, 1290, 1147; 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 8.13–8.11 (d, J = 8.5 Hz, 2H), 7.74–7.72 (d, J = 8.5 Hz, 2H), 7.59–7.57 (d, J = 8.0 Hz, 2H), 7.31–7.29 (d, J = 8.0 Hz, 2H), 7.04 (s, 1H), 2.91 (s, 2H), 2.62 (s, 3H), 2.42 (s, 3H), 2.14–2.08 (m, 2H), 1.92–1.88 (m, 2H), 1.77–1.67 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 192.2, 184.7, 160.7, 158.7, 149.2, 145.4, 138.1, 137.0, 136.0, 130.2, 129.6, 128.3, 127.1, 126.7, 124.1, 121.1, 118.9, 100.6, 90.6, 47.1, 37.5, 23.8, 21.1, 10.0; MS (EI mass): m/z, 450 [M]+.
4.4.2.19 3′-Methyl-2′-(4-(naphthalen-1-yl)benzoyl)spiro[cyclopentane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (7s). White solid; M.F: C33H26O4; mp: 170–172 °C; yield: 84%; IR (KBr, cm−1): 2928, 1680, 1623, 1248, 1152; 1H NMR (400 MHz, CDCl3) δ 8.32 (s, 1H), 8.20–8.18 (d, J = 8.5 Hz, 2H), 7.95–7.90 (m, 3H), 7.67–7.65 (d, J = 8.5 Hz, 2H), 7.58–7.45 (m, 4H), 7.07 (s, 1H), 2.91 (s, 2H), 2.67 (s, 3H), 2.14–2.09 (m, 2H), 1.92–1.88 (m, 2H), 1.75–1.68 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 192.2, 184.8, 160.7, 158.8, 149.1, 145.4, 139.0, 136.4, 133.7, 131.1, 130.0, 129.6, 128.3, 127.8, 126.9, 126.3, 125.9, 125.6, 125.3, 124.1, 121.2, 118.9, 100.7, 90.6, 47.1, 37.4, 23.8, 10.0; MS (ESI mass): m/z, 487 [M + H]+.
4.4.2.20 2′-(3′,5′-Dichloro-[1,1′-biphenyl]-4-carbonyl)-3′-methylspiro[cyclopentane-1,7′-furo[3,2-g]chromen]-5′(6′H)-one (7t). White solid; M.F: C29H22Cl2O4; mp: 187–188 °C; yield: 88%; IR (KBr, cm−1): 2924, 1694, 1617, 1234, 1152; 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 8.16–8.13 (d, J = 8.5 Hz, 2H), 7.70–7.68 (d, J = 8.5 Hz, 2H), 7.53 (d, J = 1.7 Hz, 2H), 7.40 (d, J = 1.7 Hz, 1H), 7.04 (s, 1H), 2.91 (s, 2H), 2.64 (s, 3H), 2.14–2.08 (m, 2H), 1.92–189 (m, 2H), 1.76–1.69 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 192.1, 184.4, 160.8, 158.7, 148.9, 142.9, 142.4, 137.3, 135.5, 130.3, 128.2, 127.9, 126.9, 125.7, 124.0, 121.2, 119.0, 100.6, 90.6, 47.1, 37.5, 23.8, 10.0; MS (ESI mass): m/z, 505 [M + H]+.
4.4.2.21 2′-(4′-Fluoro-[1.1′-biphenyl]-4-carbonyl)-3′-methylspiro[cyclopentane-1.7′[3,2-g]chromen]-5′(6′H)-one (7u). White solid; M.F: C29H23FO4; mp: 138–140 °C; yield: 85%; IR (KBr, cm−1): 2934, 1675, 1614, 1326, 1236, 1149; 1H NMR (400 MHz, CDCl3) δ 8.23 (s, 1H), 8.07–8.05 (d, J = 8.5 Hz, 2H), 7.63–7.61 (d, J = 8.5 Hz, 2H), 7.58–7.55 (t, J = 8.7 Hz, 2H), 7.13–7.08 (t, J = 8.7 Hz, 2H), 6.97 (s, 1H), 2.84 (s, 2H), 2.56 (s, 3H), 2.06–2.01 (m, 2H), 1.86–1.81 (m, 2H), 1.51 (s, 2H), 1.28 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 192.2, 184.6, 160.7, 158.7, 149.1, 144.3, 136.3, 130.3, 128.8, 127.8, 126.8, 124.0, 121.2, 118.9, 116.0, 115.7, 100.6, 90.6, 47.1, 37.5, 29.6, 23.8, 10.0; MS (ESI mass): m/z, 455 [M + H]+.
5 Biological methodologies
5.1 Anti-inflammatory
The anti-inflammatory activity of synthesized compounds was studied by using inhibition of albumin denaturation technique according to Mizushima et al.41 and Sakat et al.42 followed with minor modifications. The reaction mixture consists of test compounds and 1% aqueous solution of bovine albumin fraction, at pH 7.4. The reaction mixture was adjusted using small amounts of 1 N HCl. The sample extracts were incubated at 37 °C for 20 min and then heated to 51° C for 20 min, after cooling the samples the turbidity was measured at 660 nm. The percentage inhibition of protein denaturation was calculated as follows:| Percentage inhibition = (abs control − abs sample) × 100/abs control with reference to coefficient to correlation coefficient value (r) of 0.946 |
5.2 Antioxidant
| (DPPH) + (H-A) → DPPH-H + (A) |
Antioxidants react with DPPH and reduce it to DPPH-H and as a consequence the absorbance decreases. The degree of discoloration indicates the scavenging potential of the antioxidant compounds or extracts in terms of hydrogen donating ability. The DPPH free radical scavenging assay was performed by following below method. 200 μL of 0.1 mM DPPH prepared in methanol was added to different concentrations of compounds (0.1 to 500 μM). The resulting mixture was incubated at room temperature in the dark for 15 minutes. Absorbance was observed at 517 nm. Ascorbic acid was taken as a positive control. The experiment was carried out in triplicates and percentage inhibition of the DPPH radical scavenging activity was calculated.
| % Inhibition = ((A0 − A1)/A0) × 100 |
6 Molecular modeling studies
To gain more insight into the interactions of spirofurochromanone derivatives 5a–i and 7a–u, molecular docking studies were performed. Crystal structure of bovine serum albumin in complex with 3,5-diiodosalicylic acid (PDB id: 4JK4)44 and COX-2 (PDB id: 3NTG)34 were downloaded from protein data bank (www.rcsb.org). Interactions of the molecules with the proteins were analyzed to identify their hypothetical binding mode. All the molecular modeling calculations were performed using Schrödinger Suite 2010 (ref. 35) on Linux platform. The protein was prepared using protein preparation module applying the default parameters, hydrogen atoms were added and unwanted water molecules were removed from the protein structure followed by hydrogen bond optimization and energy minimization. Sitemap36 analysis was performed on Bovine Serum Albumin (BSA) as it had many co-crystallized ligands in it. Five top binding cavities were selected and grid was generated around these (binding site are represented in the Table 7). In case of COX-2, grid was generated around the active site defined by the co-crystallized ligand. Receptor Van der Waals scaling for the nonpolar atoms was set to 0.9.37 Molecules were built using Maestro build panel and prepared by LigPrep OPLS_2005 force. GLIDE 5.6 was used for molecular docking. Low energy conformation of the ligands was selected and docked into the grid using extra precision (XP) docking mode. Further the absorption, distribution, metabolism and excretion (ADME) properties were calculated using Qikprop module in Schrodinger Suite.Acknowledgements
This work was supported by UGC (University Grants Commission) as UGC-MRP, Project sanction letter No: F. 43-211/2014 (SR). New Delhi, and is gratefully acknowledged. Our regards to CSIR, for their financial support to M. D. Alaparthi. We thank Head, Department of Chemistry, Osmania University, Hyderabad for providing laboratory facilities. We also thank CFRD and IICT analytical team for providing spectral analytical facilities.References
- P. Ningappa Tatti, S. Anitha, S. Shashidhara, M. Deepak and B. Sanjeevkumar, Int. J. Pharm. Sci., 2012, 3, 2314 Search PubMed.
- N. H. Grant, H. E. Alburnand and C. Kryzanauska, Biochem. Pharmacol., 1970, 715 CrossRef CAS.
- L. A. D. Williams, A. O'Connar, L. Latore, O. Dennis, S. Ringer and J. A. Whittaker, et al., West Indian Med. J., 2008, 57, 327 CAS.
- L. A. D. Williams, West Indian Med. J., 2009, 58, 191 Search PubMed.
- N. T. S. Phan, D. H. Brown and P. Styring, Tetrahedron Lett., 2004, 4, 7915 CrossRef.
- Q. N. Chun Liu, F. Bao and J. Qiu, Green Chem., 2011, 13, 1270 Search PubMed.
- S. I. El-Desoky, F. A. Badria, M. A. Abozeid, E. A. Kandeel and A. H. Abdel-Rahman, Med. Chem. Res., 2013, 22, 2105 CrossRef CAS.
- P. Hezky, I. Krejci, S. Radl, J. Urbankova and P. Vachal, Collect. Czech. Chem. Commun., 2000, 75, 280 Search PubMed.
- D. Dauzonne, J. M. Gillardin, F. Lepage and R. Pointet, Eur. J. Med. Chem., 1995, 30, 53 CrossRef CAS.
- J. Brady, Y. Hayakawa, M. J. Smyth and S. L. Nutt, J. Immunol., 2004, 172, 2048 CrossRef CAS.
- J. M. Elliott, H. G. Selnick, D. A. Claremon, J. J. Baldwin, S. A. Buhrow, J. W. Butcher, C. N. Habecker, S. W. King, J. J. Lynch Jr, B. T. Phillips, G. S. Ponticello, E. M. Radzilowski, D. C. Remy, R. B. Stein, J. I. White and M. B. Young, J. Med. Chem., 1992, 35, 3973 CrossRef CAS PubMed.
- J. D. Rizzo, M. T. Flavin, A. Khilevich, A. Kucherenko, A. K. Sheinkman, V. Vilaychack, L. Lin, W. Chen, E. M. Greenwood, T. Pengsuparp, J. M. Pezzuto, T. M. Hughes Shf, M. Cibulski, W. A. Boulanger, R. L. Shone and Z. Q. Xu, J. Med. Chem., 1996, 39, 1303 CrossRef PubMed.
- T. Yamakawa, H. Jona, K. Niiyama, K. Yamada, T. Iino, M. Ohkubo, H. Imamura, J. Shibata, J. Kusunoki and L. Yang, WO 2007011809 A1, 2007.
- G. Morriello, L. Yang, K. Prendergast, K. Cheng, T. Jacks, W. W. S. Chan, K. D. Schleim, R. G. Smith and A. A. Patchett, Bioorg. Med. Chem. Lett., 1998, 8, 107 CrossRef.
- S. B. S. Nikolic, M. Brands, U. Niewohner, A. Paessens, E. Graef, K. H. Schlemmer, K. Henninger, R. Endermann, O. Weber and D. Koletzki, US Pat. 20020082274, 2002.
- H. Abolhasani, S. Dastmalchi, M. Hamzeh-Mivehroud, B. Daraei and A. Zarghi, Med. Chem. Res., 2016, 25, 858 CrossRef CAS.
- J. Walsh and A. Bell, Curr. Pharm. Des., 2009, 15, 2970 CrossRef CAS PubMed.
- D. A. Viegas-Junior, V. da Silva Bolzani, E. J. Barreiro and C. A. Fraga, Curr. Med. Chem., 2007, 14, 1829 CrossRef PubMed.
- M. Eric, K. N. Guantai, T. J. Egan, J. Gut, P. J. Rosenthal, P. J. Smith and K. Chibale, Bioorg. Med. Chem., 2010, 18, 8243 CrossRef PubMed.
- J. M. Uwe Schön, S. Eichner and A. Kirschning, Tetrahedron Lett., 2008, 49, 3204 CrossRef.
- D. Ashok, K. Sudershan and M. Khalilullah, Green Chem. Lett. Rev., 2012, 5, 121 CrossRef CAS.
- D. Ashok and D. Shravani, Tetrahedron Lett., 2008, 49, 7227 CrossRef CAS.
- D. Ashok, D. Mohan Gandhi, A. Vikaskumar, G. Srinivas, M. Srinivas Reddy, S. SreeKanth and M. Vijjulatha, Med. Chem. Res., 2016, 25, 2882 CrossRef CAS.
- M. Mujahid, P. Yogeeswari, D. Sriram, U. M. V. Basavanag, E. Díaz-Cervantes, L. Córdoba-Bahena, J. Robles, R. G. Gonnade, M. Karthikeyan, R. Vyas and M. Muthukrishnan, RSC Adv., 2015, 5, 106448 RSC.
- D. Ashok, B. V. Lakshmi and M. Sarasija, Lett. Org. Chem., 2017, 13, 77 Search PubMed.
- R. B. Sanjay and B. W. Suresh, Beilstein J. Org. Chem., 2011, 7, 310 CrossRef PubMed.
- B. Bartolome, V. Nunez, M. Monagas and C. Gomez-Cordoves, Eur. Food Res. Technol., 2004, 218, 173 CrossRef CAS.
- J. S. Wright, Chem. Br., 2003, 39, 25 Search PubMed.
- K. R. Raghavendra, N. Renuka, V. H. Kameshwar, B. Srinivasan, K. Ajay Kumar and S. Sheena, Bioorg. Med. Chem. Lett., 2016, 26, 3621 CrossRef CAS PubMed.
- D. Costa, A. Gomes, S. Reis, J. L. F. C. Lima and E. Fernandes, Life Sci., 2005, 76, 2841 CrossRef CAS PubMed.
- D. C. Carter and J. X. Ho, Adv. Protein Chem., 1994, 45, 153 CrossRef CAS PubMed.
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1975, 11, 824 CAS.
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1976, 12, 1052 CAS.
- J. L. Wang, D. Limburg, M. J. Graneto, J. Springer, J. R. Hamper, S. Liao, J. L. Pawlitz, R. G. Kurumbail, T. Maziasz, J. J. Talley, J. R. Kiefer and J. Carter, Bioorg. Med. Chem. Lett., 2010, 20, 7159 CrossRef CAS PubMed.
- Schrödinger, LLC, 2010, New York, Ligprep, Glide 5.6, Sitemap, Qikprop.
- T. A. Halgren, J. Chem. Inf. Model., 2009, 49, 377 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks and R. B. Murphy, J. Med. Chem., 2004, 47, 1739 CrossRef CAS PubMed.
- G.-F. Shen, T.-T. Liu, Q. Wang, M. Jiang and J.-H. Shi, J. Photochem. Photobiol., B, 2015, 153, 380 CrossRef CAS PubMed.
- N. Rajendiran and J. Thulasidhasan, J. Lumin., 2016, 31, 1438 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3 CrossRef CAS.
- Y. Mizushima and M. Kobayashi, J. Pharm. Pharmacol., 1968, 20, 169 CrossRef CAS PubMed.
- S. Sakat, A. R. Juvekar and M. N. Gambhire, Int. J. Pharm. Pharm. Sci., 2010, 2, 146 Search PubMed.
- S. T. Chang, J. H. Wu, S. Y. Wang, P. L. Kang, N. S. Yang and L. F. Shyur, J. Agric. Food Chem., 2001, 49, 3420 CrossRef CAS PubMed.
- K. Zielinski, A. Bujacz, B. Sekula and G. Bujacz, Int. J. Biol. Macromol., 2013, 60C, 316 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01550j |
| This journal is © The Royal Society of Chemistry 2017 |