
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of fentanyl acrylic derivatives†
Shaohua Liab,
Devora Cohen-Karniab,
Marina Kovaliovab,
Nestor Tomyczb,
Boyle Chengb,
Donald Whitingb and
Saadyah Averick *ab
*ab
aNeuroscience Disruptive Research Lab, Allegheny Health Network Research Institute, Allegheny General Hospital, Pittsburgh, Pennsylvania 15212, USA. E-mail: Saadyah.Averick@ahn.org; Tel: +1-412-359-4943
bNeuroscience Institute, Allegheny Health Network, Allegheny General Hospital, Pittsburgh, Pennsylvania 15212, USA
First published on 5th April 2017
Abstract
The synthesis of novel fentanyl acrylate derivatives via bromo-fentanyl using Heck coupling is described. The synthesis is concise and represents an efficient and useful method for functionalizing fentanyl for medicinal chemistry investigations. The agonistic and analgesic activities are evaluated by Mu opioid receptor activation and hot plate assays in mice.
Pain has become the most common reason for patients to seek advice from healthcare professionals. Opioids and their synthetic analogs belong to the most potent analgesics currently used for moderate to severe chronic pain treatment.1–3 The primary opioid receptor involved in pain perception is the Mu opioid receptor (MOR), making Mu opioid agonists, such as morphine, methadone, codeine and fentanyl commonly used opioids.4–8 A tremendous amount of synthetic work has been reported with the aim of altering the potency, selectivity, and bioavailability of these opioids.6,8–13
Fentanyl is a potent synthetic MOR agonist that belongs to the class of compounds known as 4-anilidopiperidines.14–19 Its use is generally restricted to post-surgical acute pain management and transdermal patches due to the high risk for overdoes and death.20–28 Due to fentanyl's hydrophobicity it has a short therapeutic half-life of 15 minutes which creates challenges in its use for managing post-surgical pain.29–32 Although fentanyl analogues have been synthesized and evaluated, new fentanyl derivatives with improved half-life and lower risk for overdose are still needed.
An apparent gap in the literature regarding PEG-conjugates of fentanyl encouraged us to investigate a parent derivative with a synthetic handle to facilitate the preparation of a small fentanyl derivative library with diverse functional groups and properties.15,33,34 Considering recent achievements in PEG-drug conjugates including opioids, PEG-fentanyl derivatives have the potential for improved properties by increasing hydrophilicity and molecular weight.35–38 Therefore, herein we report the synthesis of a fentanyl-aryl halide derivative: Fen-Br that allows for the synthesis of novel PEO(PEG)-fentanyl conjugates and also provides a unique linking group chemistry while activating MOR both in vitro and in vivo.
We prepared Fen-Br derivative in one step from commercially available starting materials. Initial derivatives in our campaign were prepared by direct palladium catalysed cross-coupling of acrylates with Fen-Br using Pd(OAc)2 and dppe. Upon refinement of the reaction conditions, Fen-Br was coupled with tert-butyl acrylate (Fen-Acry-tBu) followed by trifluoroacetic acid mediated hydrolysis to yield Fen-Acry-OH. The use Fen-Acry-OH dramatically simplifies both synthesis and purification of the desired derivatives. A small library of fentanyl derivatives was then prepared (Scheme 1). The commonality in the library is a shared linking group between the fentanyl and the new moiety.
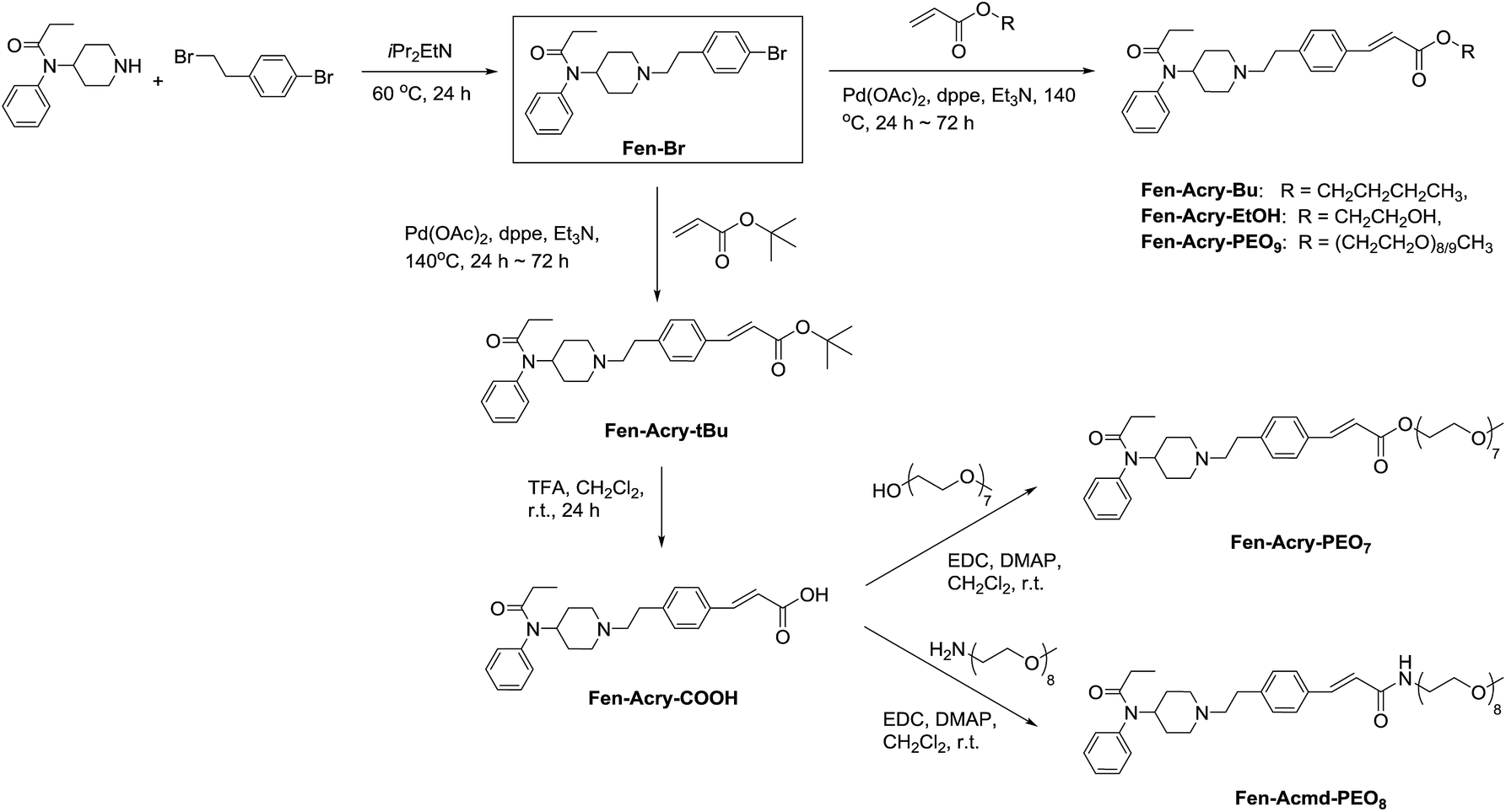 | ||
| Scheme 1 Synthesis of Fen-Br and its derivatives through Heck coupling reactions. | ||
We prepared a small library of fentanyl analogs by utilizing a novel “rigid” acrylate linking group between fentanyl and the hydrophilic moiety. The unique acrylate linking chemistry allowed for the retention of Mu opioid receptor agonist properties both in vitro and in vivo while dramatically increasing molecular weight, polar surface area, and hydrophilicity.
Fen-Br was prepared from commercially available reagents by a substitution reaction of norfentanyl and bromoethyl phenyl bromide. Pure Fen-Br was isolated as HCl salt by precipitation from ethyl ether, with a 95% yield in gram scale. Fen-Br was characterized by 1H NMR and RP-HPLC, as shown in Fig. 1a. Identical peaks at ∼7.38, 7.08, and 7.02 ppm are attributed to the phenyl protons (full 1H NMR spectrum is presented in Fig. S1 in ESI†).
 | ||
| Fig. 1 1H NMR and RP-HPLC spectra of (a) Fen-Br and (b) Fen-Acry-OH. | ||
The initial synthetic approach was performed by directly reacting Fen-Br and acrylates using Pd(OAc)2/dppe. Fen-Acry-EtOH, Fen-Acry-Bu, Fen-Acry-tBu, and Fen-Acry-PEO9 were obtained with good yields and high purity.
The initial synthetic was performed by directly reacting Fen-Br and acrylates using Pd(OAc)2 and dppe. Fen-Acry-EtOH, Fen-Acry-Bu, Fen-Acry-tBu, and Fen-Acry-PEO9 were obtained with good yields and high purity. However, this synthetic route requires preparation of variable acrylates prior to the Heck coupling reaction, which would complicate the purification of the products. An alternate route was designed and performed by synthesizing Fen-Acry-tBu by Heck coupling reaction followed by deprotection of the tert-butyl group. The deprotection by TFA afforded Fen-Acry-OH in quantitative yield as determined by proton NMR and RP-HPLC analysis (Fig. 1b). New proton shifts at ∼7.6 and ∼6.4 ppm were observed for the acrylate functional group. The Fen-Acry-OH also exhibited higher polarity by eluting earlier in RP-HPLC analysis (Fig. 1b). The resulting compound Fen-Acry-OH was used in a variety of additional coupling reactions to afford esters and amides. As a demonstration of concept we prepared Fen-Acry-PEO7 and Fen-Acmd-PEO8.
To quantify the influence of chemical modification of fentanyl on its physiochemical and biological properties we computationally characterized the library for impacts on molecular weight, polar surface area, and log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P. The impact on Mu opioid agonist activity was determined by measuring changes in cAMP levels in a live cell assay with CHO cells expressing the MOR (CHO-MORs) (Fig. 2). Our results indicate that the incorporation of the “rigid” linking group allows for a dramatic increase (1.2–3 times) in molecular weight and hydrophilicity while still ensuring Mu opioid agonist activity (Table 1).
P. The impact on Mu opioid agonist activity was determined by measuring changes in cAMP levels in a live cell assay with CHO cells expressing the MOR (CHO-MORs) (Fig. 2). Our results indicate that the incorporation of the “rigid” linking group allows for a dramatic increase (1.2–3 times) in molecular weight and hydrophilicity while still ensuring Mu opioid agonist activity (Table 1).
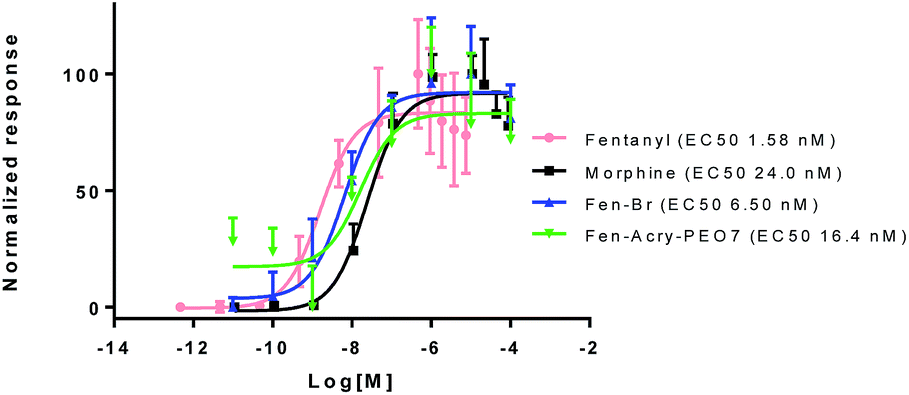 | ||
| Fig. 2 Activation of MOR by synthesized fentanyl analogs using cAMP-Glo™ Assay. | ||
| Name | Structure | Predicted PSA | PSA/PSA-fentanyl | logP |
MW (g mol−1) | MW/fentanyl | EC50 (nM) |
|---|---|---|---|---|---|---|---|
| Fentanyl | 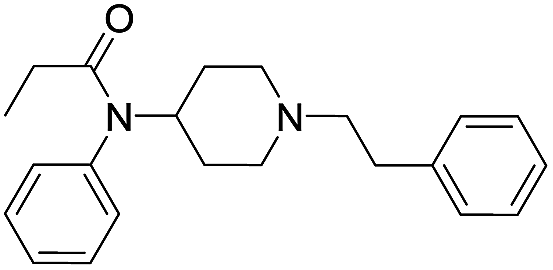 |
23.55 | 1.00 | 3.79 | 336.48 | 1.0 | 1.58 ± 0.04 |
| Fen-Br | 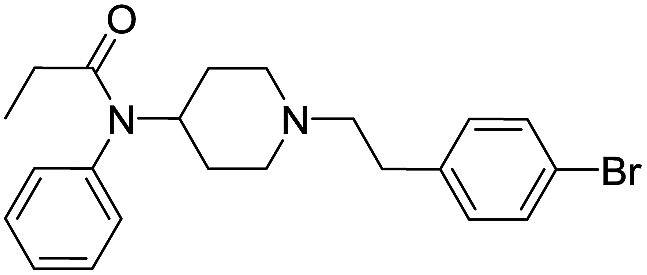 |
23.55 | 1.00 | 4.60 | 415.38 | 1.2 | 6.54 ± 0.15 |
| Fen-Acry-OH | 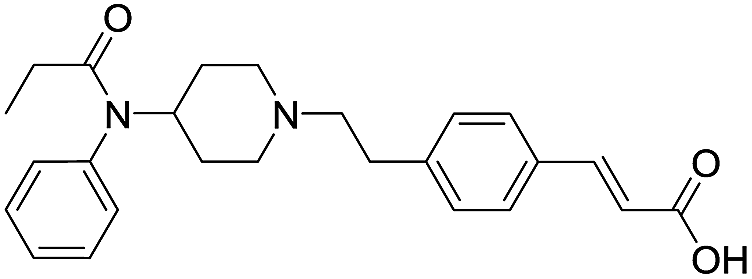 |
60.85 | 2.58 | 3.69 | 406.53 | 1.2 | 129.50 ± 3.28 |
| Fen-Acry-EtOH |  |
70.08 | 2.98 | 3.44 | 450.58 | 1.3 | 22.86 ± 0.85 |
| Fen-Acry-Bu | 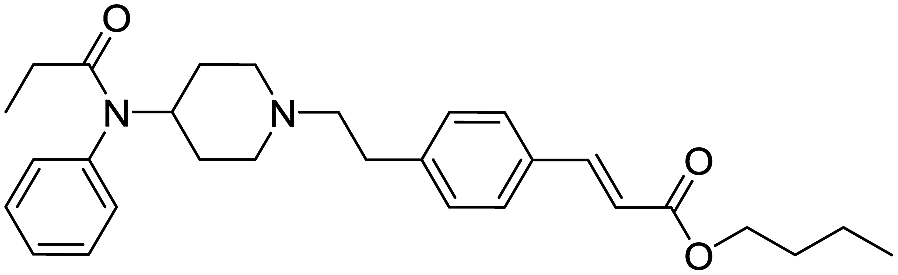 |
49.85 | 2.12 | 5.82 | 462.63 | 1.4 | 15.01 ± 0.43 |
| Fen-Acry-tBu | 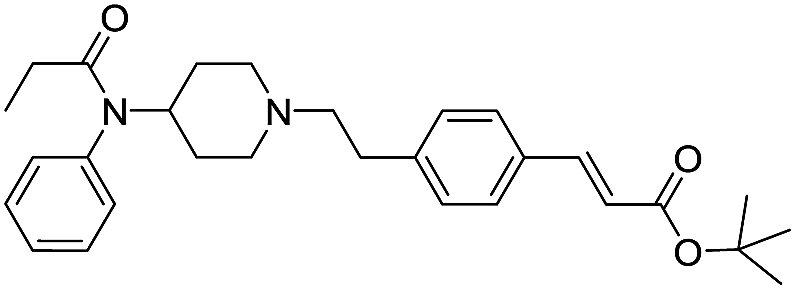 |
49.85 | 2.12 | 5.57 | 462.63 | 1.4 | 53.53 ± 1.61 |
| Fen-Acry-PEO7 | 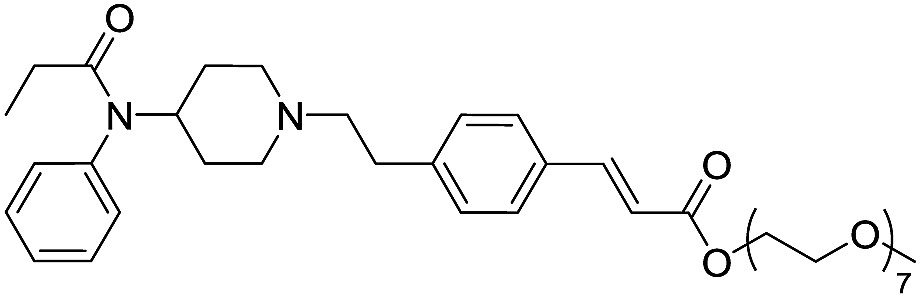 |
114.49 | 4.86 | 2.95 | 728.92 | 2.2 | 16.43 ± 0.72 |
| Fen-Acry-PEO9 | 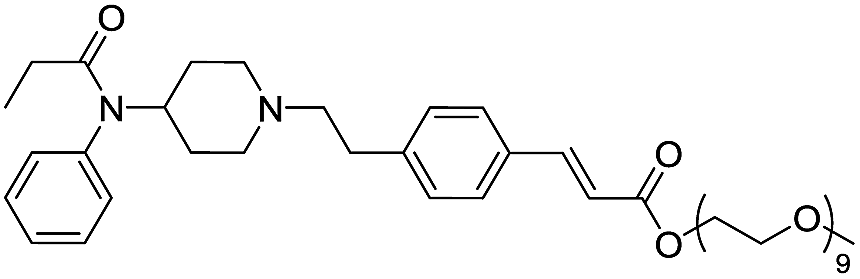 |
132.96 | 5.65 | 2.55 | 817.03 | 2.4 | 7.40 ± 0.93 |
| Fen-Acmd-PEO8 | 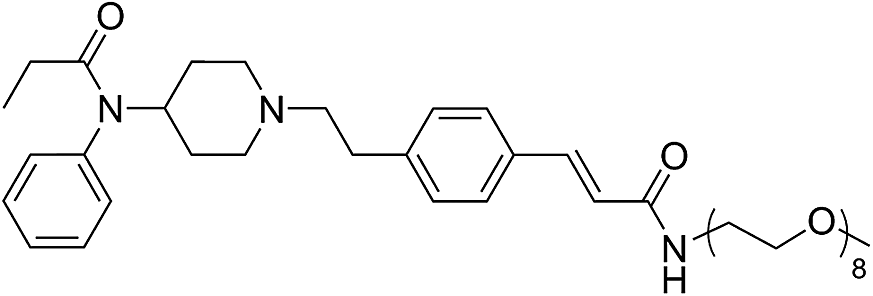 |
126.49 | 5.37 | 2.41 | 772.40 | 2.3 | 10.02 ± 1.21 |
MOR activation assays were used to determine the EC50 values of the Fen-Br derivatives. cAMP inhibition studies were performed by incubation of the Fen-Br derivatives at a range of concentrations (10−12 to 10−4 M), with forskolin treated CHO-MOR cells. cAMP levels were determined using a cAMP-Glo kit from Promega using manufactures instructions. DAMGO (EC50 2.71 ± 0.07 nM), morphine (EC50 24.03 ± 0.401 nM) and fentanyl (EC50 1.58 ± 0.04 nM) were used as references.
All compounds were tested and most of their EC50 values were determined within the range of 6–50 nM, excluding Fen-Acry-OH. Fen-Acry-PEO7, Fen-Acry-PEO9 and Fen-Acmd-PEO8 have EC50 of 16.4, 7.4 and 10.0 nM respectively, which are an order of magnitude less effective than fentanyl and still more active than morphine (EC50 24.03 ± 0.401 nM) (Fig. 2). In addition, their relatively low ClogP, and increased weight are predicted to have longer circulation half-lives with the potential for minimal blood brain barrier (BBB) permeability. Our assay shows that the Fen-Acry-OH is two orders of magnitude less active than fentanyl. This derivative would be deprotonated at physiological pH thus likely to carry a negative charge which may be a contributor for poor activation of MOR. Fen-Acry-tBu has relatively weaker interaction with the receptor (EC50 53.53 ± 1.61) possibly due to steric hindrance of the tert-butyl bulky group. With the preparation of Fen-Acry-PEO9 we have identified a strong lead compound for further evaluation. Although Fen-Acmd-PEO8 shows good activity (EC50 10.02 ± 1.21 nM) we decided to concentrate on acrylate analogs in this communication due to their preferable synthesis and purification conditions. We selected seven acrylate derivatives for in vivo studies. Despite the high EC50 of Fen-Acry-OH we decided to test it as a potential metabolite of PEG-fentanyl conjugates.
The in vivo antinociceptive properties of selected acrylate derivatives were evaluated using an in vivo hot plate withdrawal assay (Fig. 3). Hot plate withdrawal assay is commonly used to corroborate analgesia due to its sensitivity and is largely employed to evaluate opioids. All animal care was in compliance with the Guide for the Care and Use of Laboratory Animals prepared by the Institute of Laboratory Animal Resources and published by the National Institutes of Health (NIH publication no. 86-23) and approved by Institutional Animal Care and Use Committee (IACUC) at Allegheny Health Network. Male CD-1 mice (n = 10) weighing 30 g were dosed subcutaneously 30 minutes prior to placement on a 55 °C hot plate and withdrawal latencies were measured (jumping or hind-paw licking) within a 30 second time frame. The maximum possible effects (MPE) at a 95% confidence interval were calculated according to the following equation: MPE = 100 × (timelatency − timesaline)/(30 s − timesaline).
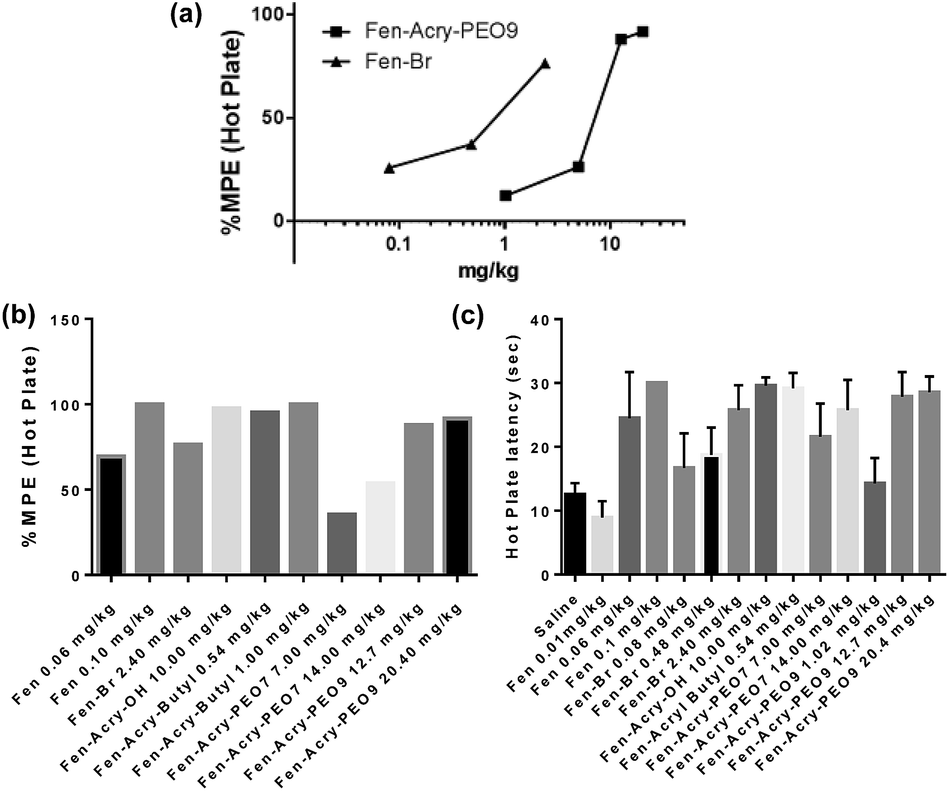 | ||
| Fig. 3 Hot plate test: (a) each point shows the % of MPE induced by Fen-Br and Fen-Acry-PEO9; (b) each column shows the % of MPE induced by Fen-acrylate derivatives; (c) each column indicates latency of withdrawal (s) mean ± SEM (n = 10) at 30 min post dose of Fen-acrylate derivatives in different concentrations. | ||
The MPE for Fen-Br and Fen-Acry-Bu were 73% at 2.4 mg kg−1 and 94% at 0.54 mg kg−1 respectively compared to an MPE of 65% at 0.06 mg kg−1 of fentanyl. The PEG-fentanyl hydrophobic conjugates Fen-Acry-PEO9 and Fen-Acry-PEO7 had antinociceptive properties with MPE of 87% at 12.7 mg kg−1 and 54% at 14.0 mg kg−1. These results indicate that acrylate linking group can be used to prepare compounds with acceptable in vivo activity. The high dose required for the Fen-Acry-PEO9 can be explained by the relatively low permeability of these compounds as well as being restricted to peripheral MORs which prevents neural MOR binding. All the tested compounds exhibit two orders of magnitude lower activity than fentanyl, and are still well within the range of known opioids,4,39 introducing a new family of active compounds. Notably, the rigid linking group chemistry has activity that translates well from in vitro to in vivo and is not dependent on the polarity of the linked group i.e. butyl vs. PEO.
While opioids elicit their therapeutic effect by binding both central and peripheral receptors within the human nervous system and some soft tissues, the binding of opioids across the BBB to the brainstem neuronal receptors is the primary mechanism of opioid addiction.40–42 Our novel fentanyl derivatives have been designed to have an intrinsically lower BBB permeability than the parent compound due to an increased molecular weight, polar surface area, and hydrophilicity. In this manuscript we report the synthesis and characterization of these novel hydrophilic opioid receptor agonists and in future work we will study the BBB permeability in a rodent model.
These results will be further expounded upon in future publications focusing on the preparation of a library of hydrophilic opioid derivatives with full SAR tables for different Fen-Br isomers and different length PEO oligomers. Furthermore, we found that assuming a constant blood volume of 75 ml kg−1 (calculated in mol L−1) the fentanyl and Fen-Br derivate exhibit an MPE50 about 3000 times their EC50 data as generated in cells. This data can be used to assist in predicting in vivo activity of novel fentanyl derivatives.
Conclusions
In conclusions, a series of fentanyl derivatives containing “rigid” linkers were synthesized from a new parent compound, Fen-Br, through Heck Pd catalyzed cross coupling. These compounds were evaluated in vitro by Mu opioid receptor activation in a live cell cAMP inhibition assay and most resulted in EC50 values within the range of 6–50 nM. Optimized hydrophilic compounds present lower EC50 values (higher activity) than morphine. Select compounds were evaluated in vivo in a hot plate withdrawal assay, demonstrating positive results. Future studies will explore different rigid linking groups accessible through Pd catalyzed cross coupling reactions (i.e. acrylamide, alkene, alkyne) as well as the effect on different length PEO on MOR activation.Acknowledgements
We gratefully thank the Allegheny Health Network Research Institute and the Neuroscience Institute for start-up funds to conduct this research.Notes and references
- I. Kissin, Anesth. Analg., 2010, 110, 780–789 CAS.
- J. Woodcock, J. Witter and R. A. Dionne, Nat. Rev. Drug Discovery, 2007, 6, 703–710 CrossRef CAS PubMed.
- D. M. Zimmerman and J. D. Leander, J. Med. Chem., 1990, 33, 895–902 CrossRef CAS PubMed.
- C. Andrews and C. Prys-Roberts, J. Clin. Anesthesiol., 1983, 1, 97–112 CAS.
- P. W. H. Peng and A. N. Sandler, Anesthesiology, 1999, 90, 576–599 CrossRef CAS PubMed.
- R. R. Petrov, R. S. Vardanyan, Y. S. Lee, S. W. Ma, P. Davis, L. J. Begay, J. Y. Lai, F. Porreca and V. J. Hruby, Bioorg. Med. Chem. Lett., 2006, 16, 4946–4950 CrossRef CAS PubMed.
- A. Poklis, J. Toxicol., Clin. Toxicol., 1995, 33, 439–447 CrossRef CAS PubMed.
- R. S. Vardanyan and V. J. Hruby, Future Med. Chem., 2014, 6, 385–412 CrossRef CAS PubMed.
- P. A. Janssen, C. J. Niemegeers and J. G. Dony, Arzneimittelforschung, 1963, 13, 502–507 CAS.
- W. B. Wright, H. J. Brabander and R. A. Hardy, J. Org. Chem., 1961, 26, 485–490 CrossRef CAS.
- L. V. Kudzma, S. A. Severnak, M. J. Benvenga, E. F. Ezell, M. H. Ossipov, V. V. Knight, F. G. Rudo, H. K. Spencer and T. C. Spaulding, J. Med. Chem., 1989, 32, 2534–2542 CrossRef CAS PubMed.
- R. Vardanyan, V. K. Kumirov, G. S. Nichol, P. Davis, E. Liktor-Busa, D. Rankin, E. Varga, T. Vanderah, F. Porreca, J. Lai and V. J. Hruby, Bioorg. Med. Chem., 2011, 19, 6135–6142 CrossRef CAS PubMed.
- P. T. Bremer, A. Kimishima, J. E. Schlosburg, B. Zhou, K. C. Collins and K. D. Janda, Angew. Chem., 2016, 55, 3772–3775 CrossRef CAS PubMed.
- M. P. Davis, Expert Rev. Neurother., 2011, 11, 1197–1216 CrossRef CAS PubMed.
- R. Weibel, D. Reiss, L. Karchewski, O. Gardon, A. Matifas, D. Filliol, J. A. Becker, J. N. Wood, B. L. Kieffer and C. Gaveriaux-Ruff, PLoS One, 2013, 8, e74706 CAS.
- T. N. Riley, D. B. Hale and M. C. Wilson, J. Pharm. Sci., 1973, 62, 983–986 CrossRef CAS PubMed.
- F. Janssens, J. Torremans and P. A. Janssen, J. Med. Chem., 1986, 29, 2290–2297 CrossRef CAS PubMed.
- B. S. Lin, L. V. Kudzma and H. K. Spencer, US Pat., 4 791 120 A, 1988.
- J. R. Bagley and H. K. Spencer, US Pat., 4 900 738 A, 1988.
- G. Hadley, S. Derry, R. A. Moore and P. J. Wiffen, Cochrane Database Syst Rev., 2013, 10, Cd010270 Search PubMed.
- W. Jeal and P. Benfield, Drugs, 1997, 53, 109–138 CrossRef CAS PubMed.
- R. B. Muijsers and A. J. Wagstaff, Drugs, 2001, 61, 2289–2307 CrossRef CAS PubMed.
- R. Benyamin, A. Trescot, S. Datta, R. Buenaventura, R. Adlaka, N. Sehgal, S. Glaser and R. Vallejo, Pain Physician, 2008, 11, S105–S120 Search PubMed.
- R. C. Dart, H. L. Surratt, T. J. Cicero, M. W. Parrino, S. G. Severtson, B. Bucher-Bartelson and J. L. Green, N. Engl. J. Med., 2015, 372, 241–248 CrossRef PubMed.
- R. L. DuPont, J. Psychoact. Drugs, 2010, 42, 127–132 CrossRef PubMed.
- M. J. Edlund, B. C. Martin, J. E. Russo, A. DeVries, J. B. Braden and M. D. Sullivan, Clin. J. Pain, 2014, 30, 557–564 Search PubMed.
- J. A. Gwira Baumblatt, C. Wiedeman, J. R. Dunn, W. Schaffner, L. J. Paulozzi and T. F. Jones, JAMA Intern. Med., 2014, 174, 796–801 CrossRef PubMed.
- L. J. Paulozzi and G. W. Ryan, Am. J. Prev. Med., 2006, 31, 506–511 CrossRef PubMed.
- L. E. Edinboro, A. Poklis, D. Trautman, S. Lowry, R. Backer and C. M. Harvey, J. Forensic Sci., 1997, 42, 741–743 CAS.
- M. I. Jumbelic, Am. J. Forensic Med. Pathol., 2010, 31, 18–21 CrossRef PubMed.
- C. Naumann, S. Erdine, A. Koulousakis, J.-P. Van Buyten and M. Schuchard, Neuromodulation, 1999, 2, 92–107 CrossRef CAS PubMed.
- T. L. Yaksh, S. Hassenbusch, K. Burchiel, K. R. Hildebrand, L. M. Page and R. J. Coffey, Pain Med., 2002, 3, 300–312 CrossRef PubMed.
- J. Riggs-Sauthier, B.-L. Deng and T. A. Riley, US Pat., 8 946 285 B2, 2016.
- J. Riggs-Sauthier, B. L. Deng and T. A. Riley, EP Pat., 2 628 489 A1, 2013.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS PubMed.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS PubMed.
- R. Greenwald, J. Controlled Release, 2001, 74, 159–171 CrossRef CAS PubMed.
- X. Pang, H.-L. Du, H.-Q. Zhang, Y.-J. Zhai and G.-X. Zhai, Drug Discovery Today, 2013, 18, 1316–1322 CrossRef CAS PubMed.
- R. O. Girón, R. Abalo, C. Goicoechea, M. I. Martín, L. F. Callado, C. Cano, P. Goya and N. Jagerovic, Life Sci., 2002, 71, 1023–1034 CrossRef.
- D. F. Wu, Y. S. Kang, U. Bickel and W. M. Pardridge, Drug Metab. Dispos., 1997, 25, 768–771 CAS.
- H. Pajouhesh and G. R. Lenz, NeuroRx, 2005, 2, 541–553 CrossRef PubMed.
- A. Reichel, Chem. Biodiversity, 2009, 6, 2030–2049 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR spectra, RP-HPLC traces, reaction conditions, and methods. See DOI: 10.1039/c7ra01346a |
| This journal is © The Royal Society of Chemistry 2017 |