
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel eco-friendly biocatalyst: soybean peroxidase immobilized onto activated carbon obtained from agricultural waste
J. A. Torres *a,
F. G. E. Nogueirab,
M. C. Silvac,
J. H. Lopesd,
T. S. Tavaresa,
T. C. Ramalhoa and
A. D. Corrêaa
*a,
F. G. E. Nogueirab,
M. C. Silvac,
J. H. Lopesd,
T. S. Tavaresa,
T. C. Ramalhoa and
A. D. Corrêaa
aDepartment of Chemistry, Universidade Federal de Lavras, Lavras, 37200-000, Brazil. E-mail: julianaarriel@hotmail.com
bDepartment of Chemistry Engineering, Universidade Federal de São Carlos, São Carlos, 13565-905, Brazil
cDepartment of Chemistry, Universidade Federal de Minas Gerais, Belo Horizonte, 31270-901, Brazil
dDepartment of Physical Chemistry, Universidade de Campinas, Campinas, 13083-970, Brazil
First published on 15th March 2017
Abstract
The immobilization of enzymes is an excellent alternative to overcome the drawbacks of using these biocatalysts in free form. This process plays a significant role in cost-effective recovery, increased catalyst productivity and in simplifying process operations. After the soybean peroxidase (SP) extraction, a residue at high carbon and low ash content is generated. This residue was used as carbonaceous precursor for production of carbon activated (AC) with high surface area (1603 m2 g−1). The AC produced was used as support for SP immobilization. The immobilization of SP was evaluated in different time conditions, enzyme load, pH and temperature. The samples, before and after immobilization, were characterized by thermogravimetric analysis, elemental analysis composition, specific surface area, X-ray powder diffraction, scanning electron microscopy and Fourier transform infrared spectroscopy. In addition, repeated applications of immobilized biocatalyst were made in order to evaluate its operational stability and capacity to recover the reaction medium, in which was observed that after a decline in activity from the first to the second cycle, it remained constant until the tenth application. In the context, the process of material obtainment constitutes a clean route for the development of more sustainable biocatalysts capable of applications in various areas.
Introduction
Enzymes are biocatalysts widely used in several areas, including the synthetic, pharmaceutical chemistry, wastewater bioremediation, fabrication of high performance biosensors, among others.1–5 They exhibit a high catalytic activity and selectivity, acting as an environmentally efficient catalyst without the need to work at high pressures, temperatures and harsh chemical environments. However, their use still is hampered due to some limitations related to their low stability front a pH, temperature, loss of activity due to the changes in environmental conditions and presence of the interferents, as well as the impossibility of reuse. The high cost associated with their production and purification, their fragile nature highly dependent on the reaction conditions and high loads required for commercial production are factors that hamper their free-form application.6–8 Due to these factors, the immobilization of enzymes has been an industrially and commercially viable alternative, solving the disadvantages of their use in solution related to stability and recovery of the reaction medium.The enzyme immobilization process may lead to an improved specificity and especially the catalyst stabilization, protecting the enzymes from denaturants agents and proteolysis.9,10 Several authors have describe processes of immobilization of different enzymes in various carriers which can be achieved by physical methods, that involve the entrapment of enzyme molecules within a porous matrix, and chemical methods, which, in turn, include enzyme attachment to the matrix by covalent bonds, cross-linking between enzyme and matrix, or enzyme cross-linking by multi-functional reagents.7,8,11–14 Among the various techniques used for the immobilization of enzymes, we can highlight adsorption, a simple, fast and economically viable process that can occur through physical interactions generated between the enzyme and the carrier. The physic-chemical parameters of the carrier that should be taken into an account are the surface area, pore structure and type of functional groups present on the surface. Adsorbed enzymes are shielded from aggregation, proteolysis and interaction with hydrophobic interfaces. In this process, the bond between the enzyme and the carrier is relatively weak compared to the chemical immobilization. That is very interesting, since the native structure of the enzyme is not altered enabling it to maintain its activity.7,11,15
In general, the carriers used for the enzyme immobilization by adsorption can be divided into both organic and inorganic origin. Among the various organic carriers used, the use of activated carbon can be emphasized, which presents a large surface area, a high pore volume and higher hydrothermal resistance compared to other porous materials. In addition, the immobilization process in this matrix is nontoxic, fast, inexpensive, easy to handle and stable against denaturation by oxidants.8,16
The search for different precursors for the synthesis of activated carbon has been increasing and the use of agro-industrial waste makes the process even more viable economically, besides meeting current environmental needs. Consequently, various types of precursors of vegetable origin have been researched, such as wood, apricot and/or cherry pits, wheat straw, bean and rice husks, nut husks, corn husks, among others.17 The soybean seed hulls are an abundant waste from soybean-processing industries and may offer an inexpensive and renewable additional source of activated carbons. Thus, conversion of this kind of waste in active carbon reduces the cost of waste disposal and provide an alternative to the existing commercial active carbon.18
Peroxidases (E.C.1.11.1.7) are enzymes that catalyze the reduction of peroxides, such as hydrogen peroxide and the oxidation of a variety of organic and inorganic compounds, such as, phenolic compounds, aromatic amines and dyes and, therefore, they have a great potential as bioremediation catalysts.19
Thus, the focus of this work was to synthesize an activated carbon from residue originating from enzymatic extraction of an agribusiness byproduct, optimize soybean peroxidase immobilization parameters in the activated carbon obtained and to evaluate the reuse of the immobilized biocatalyst in order to obtain a capable environmental friendly biocatalyst for applications in numerous technological areas.
Experimental
Obtention of soybean peroxidase (SP)
Initially the soybean seeds were immersed in distilled water for 24 h and then the husks were removed manually (Fig. 1). The process of obtaining the biocatalyst was conducted according to Silva et al.19 The peroxidase activity assay was based on the rate of guaiacol oxidation for the formation of tetraguaiacol (ε = 26.6 mM−1 cm−1). One unit of peroxidase activity at pH 6.0 represents the oxidation of 1 μmol of guaiacol × min−1.20 | ||
| Fig. 1 Removal of soybean hulls. | ||
Activated carbon preparation
The waste originating from the process of obtaining the soybean peroxidase was used as a carbonaceous precursor for synthesis of activated carbon. This waste was washed with distilled water successive times to remove excess phosphate buffer pH 6.5, dried at 60 °C for 24 h and then treated with a zinc chloride (ZnCl2, 97% P. A. Sigma-Aldrich) solution at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mass proportion (salt:raw material) and pyrolyzed in a tubular oven, under a N2 flow of 150 mL min−1, at 550 °C, for 3 h. The material was then washed with hot distilled water for 1 h to remove the excess activating agent and dried at 60 °C for 24 h.
:1 mass proportion (salt:raw material) and pyrolyzed in a tubular oven, under a N2 flow of 150 mL min−1, at 550 °C, for 3 h. The material was then washed with hot distilled water for 1 h to remove the excess activating agent and dried at 60 °C for 24 h.
Adsorption experiments (immobilization of soybean peroxidase)
For the immobilization process, 1 g of activated carbon was kept under constant stirring in a water bath with 20 mL of SP solution in buffer. The following parameters were evaluated: time (30 min to 300 min), enzyme load (0.05, 0.1, 0.25, 0.5 and 1 g), pH (3–6 citrate phosphate buffer, 7–9 tris–HCl buffer) and temperature (20 to 70 °C). To assist in understanding the enzyme immobilization process at different pH values, the pH corresponding to the point of zero charge (pHPCZ) for the AC was determined. The zeta potential of the dilute suspensions of the materials was measured in a Zeta Potential Meter (Malvern – Zeta Sizer model nano-ZS) in a pH range of 9.7 to 3.0, the pH value was adjusted by adding HCl (0.1 mol L−1) or NaOH (0.1 mol L−1). The immobilization procedure was stopped by separation of the immobilized material by centrifugation at 11000 × g, for 10 min at 25 °C. After the procedure, the material obtained was washed under vacuum with citrate-phosphate buffer pH 6.0 to remove excess unbound enzyme, and then dried at 4 °C overnight, and finally stored in a freezer. The immobilized material was called SP-AC.
Immobilization of SP was investigated by measurements of enzymatic activity according to Khan and Robinson.20 The immobilization yield was calculated from the difference between the initial and residual activity according to the following eqn (1):21,22
| φ(%) = (A0 − AF)/A0 × 100 | (1) |
Characterization
The specific surface area, total pore volume and average pore diameter were determined by physical N2 adsorption/desorption at 77 K using a Quantachrome Nova 1200 instrument. Prior to each analysis the materials were degassed at 250 °C under vacuum for 5 h. The total pore volume was calculated at a relative pressure at P/P0 ≈ 0.985.
000 × g for 2 min at 20 °C, washed with citrate phosphate buffer (0.1 mol L−1) pH 6.0 to remove excess unbound enzyme before the next cycle. The washing residue of SP-AC was collected and the residual enzymatic activity was determined. Then, the SP-AC was dispersed again in a fresh reaction medium.Results and discussion
Effect of time and enzyme load on the immobilization process
The effect of time on the enzyme immobilization process was evaluated in order to determine the equilibrium point, from which the yield of immobilization is maximum. According to Fig. 2a it can be observed that from 1 h the immobilization yield is not changed, keeping constant until 5 h (≈17.75%).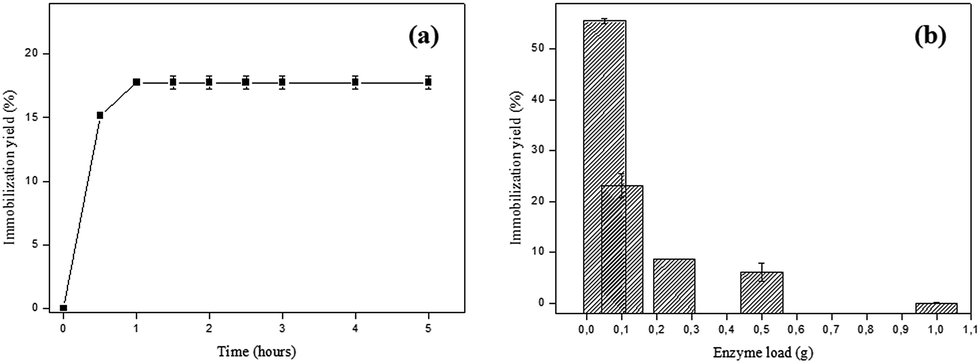 | ||
| Fig. 2 (a) Effect of time on the SP immobilization process in AC. Reaction conditions: initial enzyme load – SP 2.31 U; 1 g of activated carbon, pH 6.0, 30 °C; (b) effect of enzyme load on the SP immobilization process in AC. Reaction conditions: 1 g of activated carbon, pH 6.0, 30 °C for 1 h. | ||
At the beginning of the process there is a high number of available adsorption sites and the mass transfer is favored. The extent that SP molecules are adsorbed to the bare sites becomes scarce and therefore less available, causing the enzymes to become clustered decreasing the immobilization yield. This may explain why, after a certain time, the immobilization process becomes constant.8 Based on these data, the time of 1 h was maintained for the next immobilization experiments.
The second parameter evaluated was the variation of enzyme loading and the optimal enzyme load found was 0.05 g SP (in 20 mL phosphate citrate buffer pH 6.0), with immobilization yield of 55.52%, whereas using 1 g SP the yield was practically zero (Fig. 2b). The immobilization yield is greater at low enzyme loads, suggesting that lower yields may be associated with agglomeration of enzymes near the pore entrances and the active sites of the support used.24 Liu and coworkers16 found similar results which suggest that the resulting steric constraints could interfere in the dispersion of enzyme molecules on the activated carbon surface, which can be observed by the immobilization yield decrease.
Jain and coworkers25 also evaluated the effect of horseradish peroxidase (HRP) loadings on surface-modified mesoporous active carbon (SMAC) for different initial values of the enzyme:substrate ratio. For all ratios studied, the immobilization decreases with increasing enzyme:support ratio. Analyzing the commercial activated carbon as support for immobilization of horseradish peroxidase in a higher ratio (1 mg of HRP:0.1 g SMAC) the percentage of HRP loading was 49.9%.
Effect of pH and temperature on the immobilization process
In order to evaluate the optimum pH for the immobilization of SP on AC, the pH of the enzyme solution ranged from 3 to 9. The higher immobilization yield (86.17%) was obtained at pH 3.0, which may be related to the surface charge on the adsorbent and the enzyme (Fig. 3a).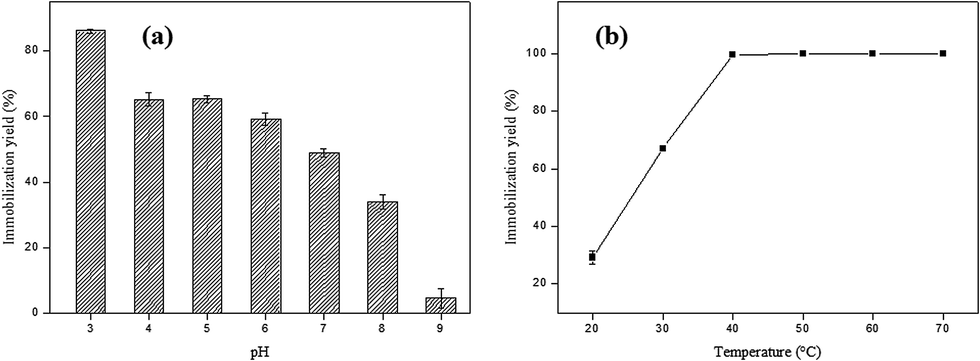 | ||
| Fig. 3 (a) Effect of pH on the SP immobilization process in AC. Reaction conditions: 1 g of activated carbon, 0.05 g of SP, 30 °C for 1 h; (b) effect of temperature on the SP immobilization process in AC. Reaction conditions: 1 g of activated carbon, 0.05 g of SP, pH 3.0, for 1 h. | ||
The functional group and delocalized electrons present in the activated carbon is that which will characterize the acidic or basic character of its surface. Thus, the modification of pH of the liquid phase strongly influences the surface properties of activated carbon. The surface of the adsorbent is neutral (surface charge equal to zero) when the solution pH is equal to point of zero charge (PCZ). The surface is negatively charged at pH values greater than pHPCZ (pH > pHPCZ), and positively charged at pH values lower than pHPCZ (pH < pHPCZ).41 The pH at the point of zero charge found for the AC obtained in this study was 7.36. Thus, at pH values in the 3 to 6 (pH < pHPCZ) range of the AC surface is positively charged, while at pH values 8 and 9, the AC surface is negatively charged (pH > pHPCZ).
Peroxidases are classified into three superfamilies: plant peroxidase, animal peroxidase and catalase peroxidase. The plant peroxidase superfamily is further subdivided into three classes: Class I, the intracellular peroxidases; Class II, which consists of extracellular fungal peroxidases and Class III, which comprises the plant class.26 However, the three peroxidases classes show low identity in their primary amino acid sequences and have distinct functions and reaction mechanisms. It is important to highlight that plant peroxidases contain isoforms different, and because of this factor, it is generally difficult to determining the isoelectric point of a specific isoform of peroxidase, mainly because protein purification is not straightforward.27 In this way, it is inferred that at pH values > pHPCZ where the immobilization yield was less pronounced, the enzyme was negatively charged, which can have led to an electrostatic repulsion between the adsorbent surface and the enzyme. On the other hand, at pH values < pHPCZ the observed effect was the opposite, with an increase in the immobilization percentage.
The next step in the evaluation of the optimal parameters consists in the evaluation of the effect of temperature on the immobilization of soybean peroxidase in activated carbon. This effect was evaluated at different temperatures and the pH maintained constant at 3.0.
The last parameter analyzed during the SP immobilization process in AC was temperature, and the results obtained are shown in Fig. 3b. An increase was observed of the immobilization yield with increasing the temperature from 20 to 50 °C, reaching 100% at 40 °C. After this temperature, the immobilization percentage remained constant up to 70 °C. At high temperatures the diffusion rate of the enzyme molecules increases through the external boundary layer and in the internal pores of the adsorbent as a function of the solution viscosity decrease, besides acquiring sufficient kinetic energy to interact with the active sites of the surface.28,29
From the results obtained in this manuscript, the best conditions for soybean peroxidase immobilization in activated carbon were obtained in a period of 1 h, with an enzyme: support ratio of 0.05:1 g, pH 3.0 (citrate-phosphate buffer) at temperature of 40 °C.
Characterization
The AC production from waste generated during the enzyme extraction process presents a high specific surface area 1603 m2 g−1 and total pore volume 0.847 cm3 g−1 with the average pore diameter obtained being 15.96 Å. In the adsorption process there is little control over the orientation of the enzyme in the pores of the support. However, some amino acid residues may interact more favorably with the pore walls by acquiring a certain orientation. In addition, the pore size can also force a certain orientation. The enzyme immobilization efficiency and its retention is directly dependent on enzyme size and pore diameter of the carrier.31
It is worth mentioning that the cost of supports for enzyme immobilization is one of the essential factors to know the economic feasibility of their use, which may be related to several factors, including the cost of the precursor used (natural agro-industrial byproducts, or synthesized products) and it availability.32 In addition, the obtaining of the catalyst described in this work complies with the principles of green chemistry,33 since the waste generated during the enzyme extraction process is repurposed, which makes it a process with zero residue generation.
 | ||
| Fig. 4 SEM images of precursor (a); activated carbon after activation with ZnCl2 (b); activated carbon after SP immobilization at different magnifications (c) and (d). | ||
The Fig. 4c and d show the micrographs of the activated carbon after SP immobilization. The presence of small structures on the SP-AC suggested that SP remains clustered after the immobilization procedure.
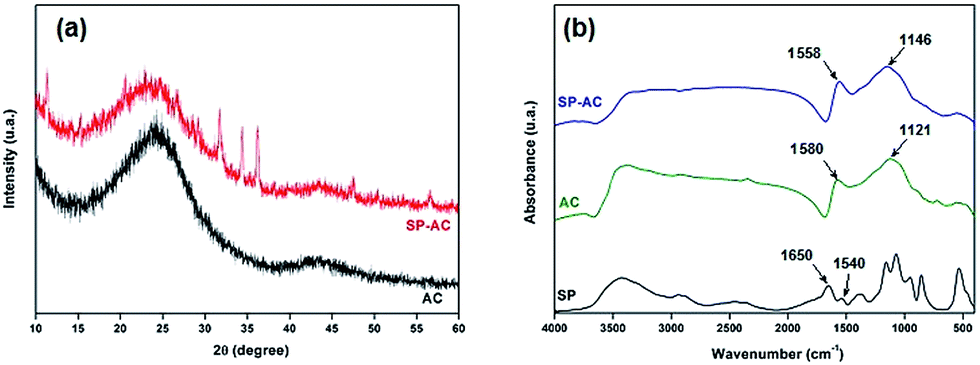 | ||
| Fig. 5 (a) DRX diffractograms and (b) FTIR spectra corresponding to pure AC, SP-AC and SP samples. The SP pure analyses were added to the comparison. | ||
The activated carbon and immobilized soybean peroxidase on activated carbon were characterized by FTIR spectroscopy. FTIR spectrum of pure SP enzyme displays absorbance bands between 3000 and 3500 cm−1 that are attributed to the N–H and O–H stretching, respectively, whereas the C–H asymmetric stretching mode is observed at 2930–2900 cm−1 (Fig. 5b). In addition, IR absorbance displays bands at 1640–1650 and 1540–1550 cm−1, which are attributed to the amide I and amide II absorbance bands, respectively.37,38 These regions consist mainly of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the backbone peptide bonds in proteins.39 The region between 1200 and 1400 cm−1 involves mainly C–H bending motions,40 whereas the 1260–1000 cm−1 and 700–400 cm−1 absorptions are attributed to phosphate groups (impurities in the activated carbon matrix) and C–C stretching modes, respectively. AC infrared spectrum exhibits a wide absorption at 3700–3200 cm−1 attributed to the presence of strong hydrogen bonds, i.e., O–H stretching or water adsorption. The position of this band at lower wave numbers indicate the presence of strong hydrogen bonds.38,42 Furthermore, the FTIR spectrum of AC displays absorptions at 1680–1480 cm−1 corresponding to the overlapping of stretching modes in aromatic skeletal and CC groups. The complex bands in the range from 1400 to 900 cm−1 are related to superposition of the following vibrational modes: C–H symmetric and asymmetric bending, C–O asymmetric stretching of aromatic ethers, esters and phenols, and C–O in carboxylic acids, alcohols, phenols and esters or the PO bond in phosphate esters. Similar absorptions bands were observed for the SP-AC sample, however, with some subtle spectral changes owing to the SP immobilization. The FTIR spectrum of SP-AC is characterized by the maximum displacement of the absorption band from 1580 cm−1 in AC to 1558 cm−1 in SP-AC. This red shift is associated with changes in the functional groups present on the activated carbon owing to SP immobilization. In fact, this spectral frequency has been attributed to overlapping of two absorptions ascribed to the (CO) stretching mode of the carbonyl group and associated with a combination of N–H and C–N stretching vibrations.37–39
O stretching vibration of the backbone peptide bonds in proteins.39 The region between 1200 and 1400 cm−1 involves mainly C–H bending motions,40 whereas the 1260–1000 cm−1 and 700–400 cm−1 absorptions are attributed to phosphate groups (impurities in the activated carbon matrix) and C–C stretching modes, respectively. AC infrared spectrum exhibits a wide absorption at 3700–3200 cm−1 attributed to the presence of strong hydrogen bonds, i.e., O–H stretching or water adsorption. The position of this band at lower wave numbers indicate the presence of strong hydrogen bonds.38,42 Furthermore, the FTIR spectrum of AC displays absorptions at 1680–1480 cm−1 corresponding to the overlapping of stretching modes in aromatic skeletal and CC groups. The complex bands in the range from 1400 to 900 cm−1 are related to superposition of the following vibrational modes: C–H symmetric and asymmetric bending, C–O asymmetric stretching of aromatic ethers, esters and phenols, and C–O in carboxylic acids, alcohols, phenols and esters or the PO bond in phosphate esters. Similar absorptions bands were observed for the SP-AC sample, however, with some subtle spectral changes owing to the SP immobilization. The FTIR spectrum of SP-AC is characterized by the maximum displacement of the absorption band from 1580 cm−1 in AC to 1558 cm−1 in SP-AC. This red shift is associated with changes in the functional groups present on the activated carbon owing to SP immobilization. In fact, this spectral frequency has been attributed to overlapping of two absorptions ascribed to the (CO) stretching mode of the carbonyl group and associated with a combination of N–H and C–N stretching vibrations.37–39
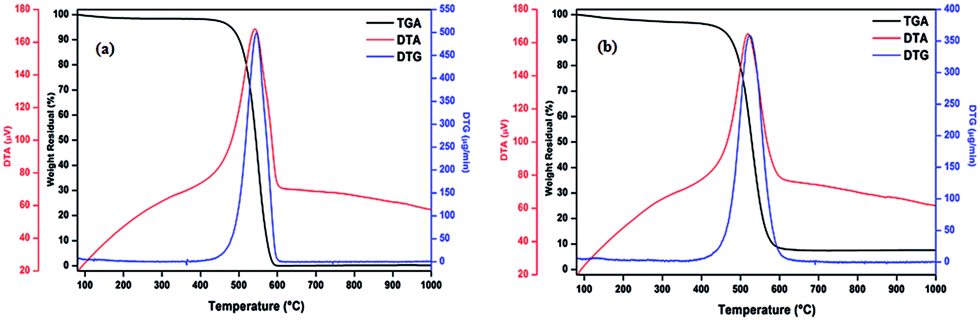 | ||
| Fig. 6 TGA/DTG/DTA curves for AC as-prepared (a) and SP-AC samples (b). | ||
The SP-AC was used in 10 cycles and the performance of the immobilized biocatalyst was analyzed as to determination the peroxidase activity at the end of each cycle. The results obtained are shown in Fig. 7. It was observed that the activity of the SP-AC undergoes a decline from the first to the second cycle, becoming constant until the last.
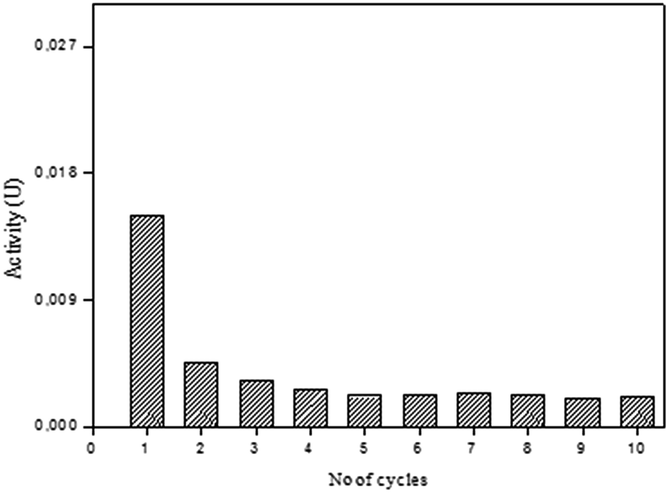 | ||
| Fig. 7 Operational stability of immobilized soybean peroxidase in active carbon in the activity assays. Reaction conditions: 20 mg of soybean peroxidase immobilized in activated carbon, citrate-phosphate buffer pH 6.0 (0.1 mol L−1), guaiacol (1% v/v) and hydrogen peroxide (0.3% v/v). | ||
To evaluate the amount of enzyme loss during the each cycle, the washing residue of SP-AC was collected and the residual enzymatic activity was determined. From the first cycle, no catalyst loss was detected, implying a strong bonding of SP on the support. Several authors suggest that the decrease in enzymatic activity after successive cycles is related to some enzyme leaching from the support.44,45 In this work, enzyme leaching after successive cycles was no detected, implying a strong bonding of SP on the activated carbon.
The decrease performance of the catalyst after each cycle may be attributed to the accumulation of the end-product polymer (formed during the oxidative polymerization of phenolic compounds by peroxidases) on the catalyst surface hindering the access of the substrate to the enzyme active site.4,46
Considering that the enzymes are not stable during storage, presenting an activity decrease over time, in addition to the impossibility of reuse in its free form, soybean peroxidase immobilized in activated carbon obtained from enzyme extraction waste was capable of maintaining the peroxidase activity over repeated uses, which is attractive from an economic point of view.
Conclusion
The immobilization of enzymes has become a topic of great interest in different technological areas. In this study, a support for soybean peroxidase immobilization was synthesized from the waste itself generated during the enzyme extraction process, characterizing a closed production cycle with a total utilization of biomass and minimal waste generation, which complies with green chemistry principles. The synthesized activated carbon exhibits a high surface area (1603 m2 g−1), an important feature during the immobilization process. An immobilization yield of 100% was achieved after optimizing the reaction parameters (enzyme:support ratio of 0.05:1 g, pH 3.0 (citrate-phosphate buffer) at temperature of 40 °C for a period of 1 h) and after 10 reuse cycles the immobilized soybean peroxidase still showed activity, a considerable aspect compared to biocatalysts in their free form, which cannot be reused. From the results obtained we concluded that soybean peroxidase immobilized on activated carbon is amenable to differentiated applications, such as in the biocatalysis, bioremediation, in clinical diagnostics, biosensor construction and other biotechnological industrial processes.
Acknowledgements
The authors acknowledge the use of the analytical instrumentation facility at Institute of Chemistry – University of Campinas, which is supported by the State of Paulo. This work was carried out with the support of the CAPES, CNPq and FAPEMIG.References
- J. Guo, T. Zhang, C. Hu and L. Fu, Nanoscale, 2015, 7, 1290 RSC.
- J. Lima-Ramos, W. Neto and J. M. Woodley, Top. Catal., 2014, 57, 301 CrossRef CAS.
- M. C. Silva, J. A. Torres, L. R. V. Sá, P. M. B. Chagas, V. S. Ferreira-Leitão and A. D. Corrêa, J. Mol. Catal. B: Enzym., 2013, 89, 122 CrossRef CAS.
- J. A. Torres, P. M. B. Chagas, M. C. Silva, C. D. Santos and A. D. Corrêa, Environ. Technol., 2015, 37, 1288 CrossRef PubMed.
- J. A. Torres, P. M. B. Chagas, M. C. Silva, C. D. Santos and A. D. Corrêa, Water Sci. Technol., 2016, 73, 39 CrossRef CAS PubMed.
- D. N. Tran and K. J. Balkus, ACS Catal., 2011, 1, 956 CrossRef CAS.
- T. Jesionowski, J. Zdarta and B. Krajewska, Adsorption, 2014, 20, 801 CrossRef CAS.
- P. Saranya, S. Ranjitha and G. Sekaran, RSC Adv., 2015, 5, 66239 RSC.
- G. Merle, M. Seon-lutz, J. H. Lopes and J. E. Barralet, Part. Part. Syst. Charact., 2014, 31, 1091 CrossRef CAS.
- J. H. Lopes, M. Guilhou, B. Marelli, F. G. Omenetto, D. L. Kaplan, J. E. Barralet and G. Merle, J. Mater. Chem. A, 2015, 3, 19282 CAS.
- F. B. O. Daoud, S. Kaddour and T. Sadoun, Colloids Surf., B, 2010, 75, 93 CrossRef CAS PubMed.
- U. Hanefeld, L. Cao and E. Magner, Chem. Soc. Rev., 2013, 42, 6211 RSC.
- R. C. Rodrigues, C. Ortiz, A. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290 RSC.
- S. A. Mohamed, A. A. Darwish and R. M. El-Shishtawy, Process Biochem., 2013, 48, 649 CrossRef CAS.
- S. Datta, L. R. Christena and Y. R. S. Rajaram, 3 Biotech., 2013, 3, 1 CrossRef.
- Y. Liu, Z. Zeng, G. Zeng, L. Tang, Y. Pang, Z. Li, C. Liu, X. Lei, M. Wu, P. Ren, Z. Liu, M. Chen and G. Xie, Bioresour. Technol., 2012, 115, 21 CrossRef CAS PubMed.
- L. C. A. Oliveira, E. Pereira, I. R. Guimaraes, A. Vallone, M. Pereira, J. P. Mesquita and K. Sapag, J. Hazard. Mater., 2009, 165, 87 CrossRef CAS PubMed.
- A. Demirbas, J. Hazard. Mater., 2009, 167, 1 CrossRef CAS PubMed.
- M. C. Silva, J. A. Torres, A. A. Castro, E. F. F. Cunha, L. C. A. Oliveira, A. D. Corrêa and T. C. Ramalho, J. Biomol. Struct. Dyn., 2016, 34, 1839 CAS.
- A. A. Khan and D. S. Robinson, Food Chem., 1994, 49, 407 CrossRef CAS.
- P. M. B. Chagas, J. A. Torres, M. C. Silva and A. D. Corrêa, Int. J. Biol. Macromol., 2015, 81, 568 CrossRef CAS PubMed.
- A. P. M. Tavares, C. G. Silva, G. Dračić, A. M. T. Silva, J. M. Loureiro and J. L. Faria, J. Colloid Interface Sci., 2015, 454, 52 CrossRef CAS PubMed.
- R. S. Bretzlaff and T. B. Bahder, Rev. Phys. Appl., 1986, 21, 833 CrossRef CAS.
- R. Kandasamy, L. J. Kennedy, C. Vidya, R. Boopathy and G. Sekaran, J. Mol. Catal. B: Enzym., 2010, 62, 59 CrossRef CAS.
- A. Jain, V. Ong, S. Jayaraman, R. Balasubramanian and M. P. Srinivasan, J. Supercrit. Fluids, 2016, 107, 513 CrossRef CAS.
- M. C. Silva, A. D. Corrêa and J. A. Torres, Quim. Nova, 2012, 35, 889 CrossRef CAS.
- C. Cosio and C. Dunand, J. Exp. Bot., 2009, 60, 391 CrossRef CAS PubMed.
- M. A. Ahmad and R. Alrozi, Chem. Eng. J., 2011, 171, 510 CrossRef CAS.
- D. Angin, Bioresour. Technol., 2014, 168, 259 CrossRef CAS PubMed.
- M. Gonçalves, M. C. Guerreiro, L. C. A. Oliveira, C. Solar, M. Nazarro and K. Sapag, Waste Biomass Valorization, 2013, 4, 395 CrossRef.
- D. N. Tran and K. J. Balkus, ACS Catal., 2011, 1, 956 CrossRef CAS.
- A. Bhatnagar, W. Hogland, M. Marques and M. Sillanpää, Chem. Eng. J., 2013, 219, 499 CrossRef CAS.
- I. Fechete, Y. Wang and J. C. Védrine, Catal. Today, 2012, 189, 2 CrossRef CAS.
- Q. Miao, Y. Tang, J. Xu, X. Liu, L. Xiao and Q. Chen, J. Taiwan Inst. Chem. Eng., 2013, 44, 458 CrossRef CAS.
- Y. Chen, X. Zhang, H. Zhang, X. Sun, D. Zhang and Y. Ma, RSC Adv., 2012, 2, 7747 RSC.
- P. Kalyani and A. Anitha, Port. Electrochim. Acta, 2013, 31, 165 CrossRef.
- H. Zheng and X. Jiang, Asian J. Chem., 2014, 26, 685 CrossRef CAS.
- M. Prokopijevic, O. Prodanovic, D. Spasojevic, Z. Stojanovic, K. Radotic and R. Prodanovic, Bioprocess Biosyst. Eng., 2014, 37, 799 CrossRef CAS PubMed.
- S. S. Karajanagi, A. A. Vertegel, R. S. Kane and J. S. Dordick, Langmuir, 2004, 20, 11594 CrossRef CAS PubMed.
- M. Khajehpour, J. L. Dashnau and J. M. Vanderkooi, Anal. Biochem., 2006, 348, 40 CrossRef CAS PubMed.
- M. Ghaedi, B. Sadeghian, A. A. Pebdani, R. Sahraei, A. Daneshfar and C. Duran, Chem. Eng. J., 2012, 187, 133 CrossRef CAS.
- K. Ramani, S. Karthikeyan, R. Boopathy, L. J. Kennedy, A. B. Mandal and G. Sekaran, Process Biochem., 2012, 47, 435 CrossRef CAS.
- I. V. Pavlidis, T. Vorhaben, T. Tsoufis, P. Rudolf, U. T. Bornscheuer, D. Gournis and H. Stamatis, Bioresour. Technol., 2012, 115, 164 CrossRef CAS PubMed.
- Y. Cao, L. Wen, F. Svec, T. Tan and Y. Lv, Chem. Eng. J., 2016, 286, 272 CrossRef CAS.
- R. Roto, Y. Yusran and A. Kuncaka, Appl. Surf. Sci., 2016, 377, 30 CrossRef.
- M. C. Silva, J. A. Torres, F. G. E. Nogueira, T. S. Tavares, A. D. Corrêa, L. C. A. Oliveira and T. C. Ramalho, RSC Adv., 2016, 6, 83856 RSC.
| This journal is © The Royal Society of Chemistry 2017 |