
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chitosan microspheres as a template for TiO2 and ZnO microparticles: studies on mechanism, functionalization and applications in photocatalysis and H2S removal†
Enrico Colombo,
Wu Li,
Sukhvir Kaur Bhangu and
Muthupandian Ashokkumar *
*
School of Chemistry, University of Melbourne, VIC 3010, Australia. E-mail: masho@unimelb.edu.au
First published on 31st March 2017
Abstract
In the past few decades, several studies have used nanoparticles for photocatalytic reactions. For practical applications, the use of nanomaterials may not be advantageous due to the difficulties in removing them from processed liquids, and they also pose possible health risks. We have developed a procedure that uses chitosan to convert nanosized photocatalysts into micron sized materials without losing their efficiency or surface area. Additionally, the synthetic method offers the possibility of adding functionality like macroporosity and doping carbon or gold nanoparticles. The synthesis involves ultrasonic emulsification of tetradecane in an aqueous chitosan solution containing photocatalytic nanoparticles. Tetradecane-core chitosan/photocatalyst composite-shelled microspheres were produced during the ultrasonic emulsification process. Calcination of these microspheres resulted in the formation of carbon doped micron-sized photocatalytic particles with macroporous structure. Detailed characterisations were carried out using dynamic light scattering, zeta potential, SEM, TEM, BET, XRD and XPS measurements. A possible mechanism for the formation of micron-sized photocatalytic particles is proposed. The photocatalytic efficiencies of synthesised microparticles and their reusability through filtration were evaluated and compared with that of starting nanomaterials using two organic dyes as model pollutants. One of the catalysts (gold doped photocatalyst) was also tested for H2S gas removal by adsorption. The regeneration of the adsorbent was also achieved after room temperature photocatalysis. The results indicate that the synthetic procedure can be used to produce macroscale photocatalysts with a size range of 10–20 μm as efficient as the starting nanopowders (size range of 20–50 nm).
1 Introduction
Chitosan (CS), a natural, inexpensive co-polymer (2-glucosamine and N-acetyl-2-glucosamine), has been shown useful in various applications in industry,1 medicine,2 catalysis3 and the controlled release or delivery of food4 and drugs.5 Its structural features allow for its easy dissolution, controlled precipitation as a function of solution pH, and simple structural and chemical modifications. In particular, the use of chitosan as an adsorbent or combined with semiconductors for photocatalysis has been of interest6–8 in wastewater treatment9–11 and air pollution control.12–14 Moreover, composite materials containing gold, chitosan, and TiO2 have been studied for their potential use as sensors for different molecules such as glucose,15 dopamine,16 H2S17 or organophosphate.18Chitosan can be used as an adsorbent or combined with other materials to be used in catalysis applications. Zhang et al.7 used TiO2-chitosan as an adsorbent for Ag+ and the photodegradation of rhodamine B. The synthesis adopted led to the formation of particles in the range 50–500 μm, which can be easily removed by filtration at the end of the treatment. Unfortunately, the efficiency recorded was modest, around 80% of dye was degraded in 6 h of irradiation, particularly, considering the amount of catalyst (0.2 g), rhodamine B (40 ml, 10 ppm), and lamp power (20 W UV lamp 365 nm) used. El Kadib et al. have reported on a synthetic procedure to produce large particles using chitosan and organic precursors of metal oxides.19 The particles were in the millimetres range, which would be of great advantage for their removal at the end of photodegradation. However, no study on photocatalytic performance was carried out.
Working with a nanodispersion system may result in a better efficiency owing due to a higher surface area. However, this approach cannot be used in an industrial environment, where filtration costs would be significantly high. To overcome the filtration problem, catalysts can be deposited on a substrate. Fisher20 et al. have studied chitosan as supporting material for TiO2 for the coating of the internal lumen of glass bottles. The system was used for wastewater treatment, but the degradation efficiency was hardly comparable to systems where TiO2 powder was dispersed in a medium. In terms of efficiency, working with suspensions is still a better choice.
An interesting approach to overcome the filtration problem is the use of magnetic nanoparticles, which can be easily removed from the medium applying a magnetic field. Xin et al. have used magnetic Fe3O4/TiO2 particles (400 nm) for the degradation of pollutants in water.21 However, the use of this technology on a large scale is difficult to be implemented. When large volumes are used two main problems arise. The first is that a strong magnetic field is needed for the removal process, which can result in an increase of the costs of this technology.22 Second, a separation unit will be required, increasing the cost further.23 Instead, several studies have shown that the use of larger particles in continuous flow systems is of great advantage for scaling-up of the process.24,25 Bianchi et al.26 have compared the efficiency of different sized TiO2 using gas and liquid phase systems. The photocatalytic degradation of pollutants like acetone and toluene in the gas phase was studied with four TiO2 samples possessing different sizes, viz., two samples of 20 and 25 nm and two samples of 130 and 192 nm. The results showed that the efficiency of the degradation follows the catalyst size (higher degradation for smaller particles). Some studies have shown that it is possible to use catalysts with a larger particle size and obtain higher degradation efficiency. For instance, Nezamzadeh-Ejhieh et al. have compared the photocatalytic decolorization ability of CuO/zeolite composite particles possessing different sizes in the range 88–360 nm.27 In this case, they reached an increase in decolorization of about 50% using the larger size particles.
Although it may be a great result tripling the size of the catalysts without losing their efficiency, particles with sizes below 1000 nm are still difficult to be implemented in an industrial wastewater treatment process. For this reason, Li et al.28 have evaluated the photocatalytic efficiency of TiO2 microspheres with a size larger than 30 μm. The results showed that the degradation efficiency of this catalyst was about 30% higher than that obtained with an equivalent nanosized used as reference (20 nm). However, due to the synthetic procedure, doping of the catalyst was not possible, limiting its absorption only in the UV range. The use of dopants to reduce this limitation has been widely studied. For example, gold NPs can be used to influence the energetics by improving the photo-induced charge separation, which ultimately increases the performance of the catalysts.29
Apart from reducing the band gap, gold nanoparticles can be used in many ways. Particularly, in the past few years, gold, chitosan, and TiO2 composite materials have been of considerable interest, due to the ability of gold nanoparticles to interact with different functional groups. One of the most common examples is the capacity in making interaction with thiol groups. In the gas phase, the adsorption is only physical but can be converted into chemical by simply increasing the temperature.30 This feature has been used in gas detectors, particularly for H2S, where absorption undergoes a redshift as a function of the amount of H2S adsorbed.31 Sui et al.32 have studied gold nanoparticles for RSH removal. They observed that gold can be used for thiol removal from gas streams if the gold nanoparticles are supported on TiO2 and not on ZnO (due to the formation of ZnS). Moreover, the temperature to regenerate the adsorbent was reduced from 350 °C (for conventional adsorbent like zeolite) to around 200 °C.33
In our study, we have developed a new procedure to produce visible light active micron-sized photocatalysts with efficiency comparable to that of the corresponding nanomaterial. The effects of doping and gold nanoparticles decoration were explored and applied to two different systems: photodegradation of pollutant in aqueous medium; and H2S adsorption in the gas phase. A detailed mechanism for the formation of micron-size photocatalysts is proposed. The photocatalytic degradation efficiencies of the catalysts were evaluated using organic dyes (negatively charged metanil yellow and positively charged methylene blue) as model pollutants. The recyclability of the catalysts was explored to make the overall process viable for industrial applications.
2 Experimental
2.1 Materials
P25 was purchased from Degussa. Salicylic acid (>99%), chitosan with low molecular weight (CAS number 9012-76-4; MW not reported) and 75–85% degree of deacetylation, glutaraldehyde grade II solution (25%), coumarin (>99%), PBS (phosphate buffered saline tablets), tetradecane (>99%), 6% alumina doped zinc oxide with a size < 50 nm (>97%), metanil yellow (∼70%), sodium hydroxide (>99%), gold(III) chloride trihydrate (>99.9%), sodium sulfide nonahydrate (>98%) hydrochloric acid (>36%) and methylene blue trihydrate (>82%) were purchased from Sigma-Aldrich Co. LLC (Australia). All the reagents were used without further purification.2.2 Chitosan solution
Chitosan was used in order to create photocatalyst microspheres during the emulsification process. The microspheres' shell, mixed with photocatalyst nanoparticles, was then used as a template for the creation of micrometric structures before calcination. Chitosan (CS) is soluble in acidic aqueous solutions,34 and an aqueous salicylic acid solution was chosen to dissolve CS. Salicylic acid has a low solubility in water (0.7 g in 100 ml of water at 25 °C). However, the acid reacts with chitosan forming salicylate, which is highly soluble (125 g in 100 ml of water). Indeed, it was demonstrated in our previous study that using amphiphilic acids to produce microspheres allows to generate more stable structures in the size range of 1–4 μm.34A stock solution was prepared at a concentration of 10 mg ml−1. 1.0 g of chitosan (CS) powder and 0.858 g of salicylic acid were dispersed in 100 ml of MilliQ water. Then, the solution was left stirring overnight. Appropriate dilutions were made in order to obtain a solution with a final concentration of 1 mg ml−1.
2.3 Catalysts preparation
Five catalysts were used: P25, 6% Al2O3–ZnO, TiO2/C-Micro, TiO2/C-Micro-Au, and ZnO-Micro. Alumina-doped ZnO was chosen to look at the possibility of applying the synthetic technique on already doped photocatalysts and evaluating the use of a different nano size as a starting material. The samples TiO2/C-Micro and ZnO-Micro were prepared using a similar procedure: 30 ml of 1 mg ml−1 chitosan solution was added to 70 ml of MilliQ water, and then 0.150 g of P25 or 6% Al2O3–ZnO was dispersed using an ultrasonic bath for 5 minutes. Then, 2 ml of tetradecane was added as oil phase and the entire mixture was sonicated for 1 minute at 41.8 W power (calorimetrically measured) to create an oil-in-water emulsion. As shown in our previous work,34 when an oil-in-water emulsion is formed in this condition, chitosan moves to the oil/water interface, forming a microcapsule. After sonication, NaOH solution was added until pH 8, and then the cross-linker was added. In case of glutaraldehyde, 20 ml of aqueous solution (0.25%) was added drop-by-drop under continuous stirring. The molar ratio of glutaraldehyde![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NH2–chitosan in the final solution was about 3. When gold was used, 20 ml of 0.9 mg ml−1 gold solution (0.0457 mmol) was added in order to have a molar ratio of gold:chitosan about 25. Chitosan had a composition of 80% 2-glucosamine and 20% N-acetyl-2-glucosamine with an average monomeric molecular weight of 169.4 g mol−1. Using this information, the moles of CS–NH2 were calculated, which were around 0.177 mmol.
:NH2–chitosan in the final solution was about 3. When gold was used, 20 ml of 0.9 mg ml−1 gold solution (0.0457 mmol) was added in order to have a molar ratio of gold:chitosan about 25. Chitosan had a composition of 80% 2-glucosamine and 20% N-acetyl-2-glucosamine with an average monomeric molecular weight of 169.4 g mol−1. Using this information, the moles of CS–NH2 were calculated, which were around 0.177 mmol.
Ten minutes after the cross-linker addition, the solution was left resting for 2 h in order to permit the separation of the aggregates formed. Then, the aggregates were collected and washed three times and placed in an oven to dry overnight at 70 °C. Once dried, the powder was gently milled with a mortar before the calcination. For both samples, the calcination was carried out for 2.5 hours to reach 450 °C and 10 hours at 450 °C in air atmosphere.
The powders obtained after calcination were collected, dispersed in water and sonicated in an ultrasonic bath for an hour. Then, the samples were centrifuged at 2000 rpm for 10 minutes in order to separate the un-encapsulated nano from the microparticles. The samples were washed at least ten times until no particles dispersion was observed. Between each washing, the sample was left in an ultrasonic bath for at least 30 minutes. Finally, the micro powder was collected and dried overnight. The average yield (expressed as “gmicro size/gnano powder used”) was around 70%.
2.4 Characterisation
The particle size distribution was determined using a Malvern Mastersizer 2000 Laser diffraction system (Malvern Instruments Ltd., Malvern, UK). A refractive index of 2.6 for TiO2, 2.01 for ZnO and 1.33 for water as a dispersant was used. Particle size distribution presented is an average of three independent measurements and volume mean particle diameter (D[4,3]) was obtained.The morphologies of the samples were observed using a high-resolution field emission scanning electron microscope (Quanta 200 FEI) without sputter coating pre-treatment. High-resolution Transmission Electron Microscopy (HRTEM; FEI Tecnai F20) with selected area electron diffraction (SAED) was used to identify the structure of TiO2 microparticles and, for the band-gap calculation, the samples were characterised by UV/Vis spectrophotometer (CARY 50 Bio UV-Visible) in the wavelength range 200 to 800 nm.
To investigate the surface area, the catalysts were degassed at 50 °C on a vacuum line for 16 h. A standard multipoint Brunauer–Emmett–Teller (BET) method was used to calculate the specific surface area. The pore size distributions of the materials were derived from the adsorption branches of the isotherms by the Barrett–Joyner–Halenda (BJH) model.
Diffraction data were collected using a Bruker D8 Advance X-ray Diffractometer (XRD) with Ni-filtered Cu Kα-radiation (1.54 Å). Data were collected between 5 and 85° 2θ, with a step size of 0.02° and a scan rate of 0.5 s per step.
X-ray Photoelectron Spectroscopy (Kratos Axis ULTRA; Thermo Scientific) was used to investigate the C 1s on the catalysts surfaces. Data was acquired using a Kratos Axis ULTRA X-ray Photoelectron Spectrometer incorporating a 165 mm hemispherical electron energy analyzer. The incident radiation was monochromatic Al Kα X-rays (1486.6 eV) at 150 W (15 kV, 15 mA). Survey scans were taken at an analyzer pass energy of 160 eV over 1200–0 eV binding energy range with 1.0 eV steps and a dwell time of 100 ms. Base pressure in the analysis chamber was 1.0 × 10−9 torr and during sample analysis 1.0 × 10−8 torr.
The zeta potential of the particles was measured using a Zetasizer Nano ZS (Malvern Instruments, UK). Phosphate buffer (0.1 M) at a pH of 6.8 was used as the dilutant. The solution was diluted approximately 1:1000 and placed inside a disposable polycarbonate folded zeta potential cell cuvette (ATA Scientific, DTS1070).
Experiments for the determination of hydroxyl radicals during photocatalysis were carried out using a 10−3 M solution of coumarin with a catalyst concentration of 0.05 g L−1, as showed by Xiang et al.35 The fluorescence spectra were acquired using a Shimadzu RF-5301PC fluorescence spectrophotometer (Shimadzu) equipped with a xenon lamp and 1.0 cm optical length quartz cell. All the measurements were carried out after dilution in PBS (Phosphate Buffered Saline) solution to a final concentration of 6.25 10−5 M. Samples were excited at 332 nm, and the fluorescence spectra were recorded from 350 to 600 nm.
2.5 Photocatalytic activity measurements
The photocatalytic degradation efficiencies of the catalysts were evaluated using organic dyes (negatively charged: metanil yellow and positively charged: methylene blue) as model pollutants. Photocatalytic reactions in air-saturated (no gas purging) aqueous medium were carried out in a reactor equipped with a cooling jacket, quartz window, and magnetic stirring (Fig. S1†). The amount of photocatalyst powder used in all cases was 0.05 g L−1 dispersed in 180 ml of the dye solution. The concentration of dye was 0.04 mM for metanil yellow (MY) and 0.06 mM for methylene blue (MB). An ORIEL 450 W arc lamp was used as light source and placed in front of the quartz window on the side of the reactor (22.9 W cm−2) (emission spectra shown in Fig. S2†). To avoid the heating of the solution during illumination, cold water was circulated through the jacket around the wall of the reactor. The reaction temperature was maintained at 22 °C. The dye concentration was monitored by UV-Vis spectrophotometer at regular intervals. For the catalyst regeneration in the H2S experiments, 0.5 g of TiO2/C-Micro-Au was dispersed in 180 ml of MilliQ water, and the reaction was monitored using a pH meter at different time intervals for a total reaction time of 2 hours. Routine studies, such as the influence of pH or kinetic studies with respect to photocatalyst quantity or light intensity, were not carried out since the aim of this study was the synthesis of micro photocatalytic particles with comparable efficiencies to those of nanoparticles. In all experiments, the catalysts were collected with a vacuum filtration using a fritted filter with a pore diameter between 1 and 1.6 μm.2.6 Hydrogen sulfide adsorption
The experiments were carried out at room temperature in a sealed chamber (Fig. S3†) (volume 3.5 L) connected to an Industrial Scientific MX6-Ibrid detector equipped with H2S sensor and internal pump, which recirculated the gas in the chamber at a rate of 500 ml min−1. H2S was generated in situ by the reaction between 20 ml 2.0 mM sodium sulfide solution and 1 ml of 0.1 M hydrochloric acid solution. The hydrogen sulfide concentration was measured at different time intervals for a total time of 24 hours.The experiments were divided into three sets. The first one was carried out without the catalyst in order to evaluate the efficiency of the sealed chamber and to evaluate the consistency of the amount of H2S generated. In the second one, TiO2/C-Micro or P25 (0.1 g), was used to assess possible H2S adsorption onto TiO2 surface. The third experiment was carried out in the presence of 0.1 g of TiO2/C-Micro-Au in order to quantify the maximum amount of H2S adsorbed. Experiments were repeated at least three times to check for reproducibility.
3 Results and discussions
To investigate chitosan microspheres as a template for synthesising microcatalysts, two nanomaterials were chosen: 6% Al2O3–ZnO (50 nm) and TiO2 (25 nm).36 As shown in our previous work, chitosan microspheres produced via an ultrasonic emulsification process are very stable. It is possible to enhance their mechanical strength and stability by the addition of a cross-linker. Their size can be controlled by the choice of the acid for the dissolution of chitosan.12 For this reason, salicylic acid was chosen. Its amphiphilicity allows to obtain highly stable microspheres and with a size range of 1–4 μm. After the microspheres formation, the addition of glutaraldehyde or gold chloride was investigated to evaluate the possibility to retain the microspheres spherical shape or the introduction of different functionality, such as deposition of gold NPs on the microspheres, or both.Separate analyses were carried out not only to characterise the final materials, but also to evaluate the mechanism of the formation of microstructures. The samples were named TiO2/C-Micro, ZnO-Micro and TiO2/C-Micro-Au. The first two are the microparticles obtained from P25 and 6% Al2O3–ZnO using glutaraldehyde. The third one comes from the synthesis that used P25 and gold chloride.
3.1 Characterisation
Fig. 1 shows the size distribution obtained with Mastersizer 2000 for all samples synthesized.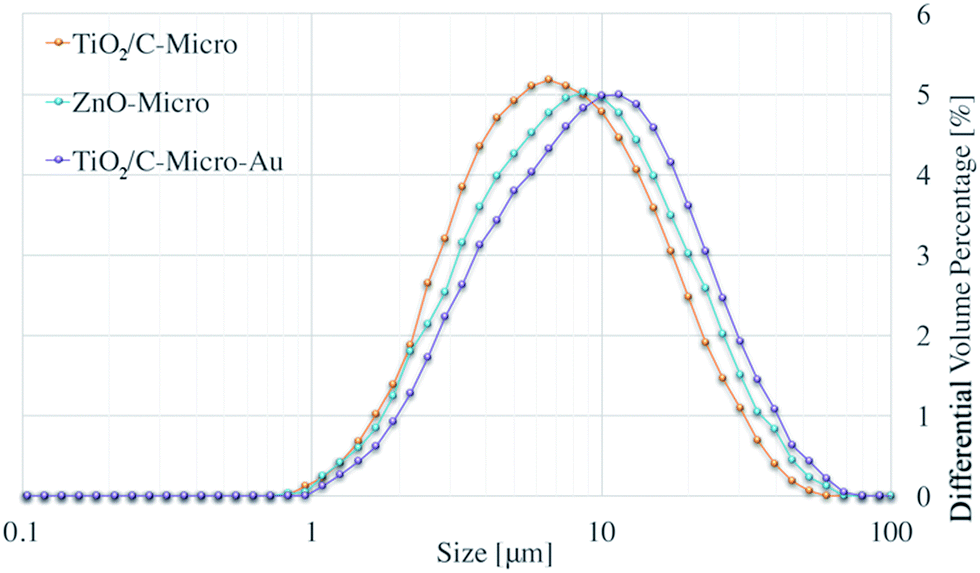 | ||
| Fig. 1 Size distribution of TiO2/C-Micro, ZnO-Micro and TiO2/C-Micro-Au. | ||
Average sizes of ∼8 μm (D[4,3]) and ∼10 μm (D[4,3]) can be observed for TiO2/C-Micro and ZnO-Micro, respectively. ZnO-Micro sample has relatively larger particles. This increment can be associated with the relatively larger size of the precursor particles, which were almost twice larger than the TiO2 nanoparticles. Instead, the use of gold leads to a greater difference. The size D[4,3] of TiO2/C-Micro-Au is about 23 μm.
The large size obtained in all the samples can be explained by the synthetic procedure adopted. It must be noted that the chitosan shell formed after NaOH addition is a gel, which allows structural modifications. When glutaraldehyde or gold chloride was added, the solution was kept under stirring for about 10 minutes. After this time, the solution was left standing for the aggregates to float to the surface, leading to the formation of larger aggregates. In case of gold, as observed by Corma et al.37 during the resting period, the aggregates change colour from white to pinkish due to the intrinsic reduction property of chitosan. In addition, chitosan has also been used for its property in stabilization of colloidal gold through its amino groups, which remain embedded in chitosan matrix.37
SEM and TEM analysis were also carried out to confirm the shape of the particles obtained and the presence of gold nanoparticles (in Fig. 2).
 | ||
| Fig. 2 SEM images of ZnO-Micro (A1 and A2), TiO2/C-Micro (B1 and B2) and TIO2/C-Micro-Au (C1–C3). TEM images of TiO2/C-Micro-Au (C5 and C6). | ||
The images collected show that the particles are in the size range observed in the Mastersizer. The particles possess a spherical shape in case of TiO2/C-Micro (Fig. 2B1) and TiO2/C-Micro-Au (Fig. 2C1). However, instead of a hollow structure, a rich macroporosity, within a diameter of 1–4 μm (Fig. 2B2 and C2), can be observed. ZnO-Micro (Fig. 2A1) does not possess a spherical shape, but macro-porosity can also be observed (Fig. 2A2). Also, the particles show a surface typical of materials characterized by a high surface area (Fig. 2B2 and C3). The TEM images in Fig. 2C5 and C6 show the gold particles on TiO2 surface. From TEM measurements, an average size of 10 ± 5 nm was recorded.
To confirm the direct fusion of gold NPs on the microparticles, 1 g of TiO2/C-Micro-Au was dispersed in 20 ml of water, sonicated for an hour in a sonic bath and filtered through a 0.22 μm filter. Then, the filtered solution was analysed by UV/Vis. No absorbance from gold was detected, which confirm the fusion of the particles on the microparticles and not just physical interactions.
XRD analyses (Fig. 3) of P25, TiO2/C-Micro, 6% Al2O3/ZnO and ZnO-Micro, were carried out. Both P25 and TiO2/C-Micro show that anatase phase is predominant (2θ = 25°) over rutile phase (2θ = 27°).14 For ZnO samples, the sharp peaks at 2θ of 32°, 34° and 37° show a high degree of crystallinity, which could be correlated to hexagonal ZnO.38
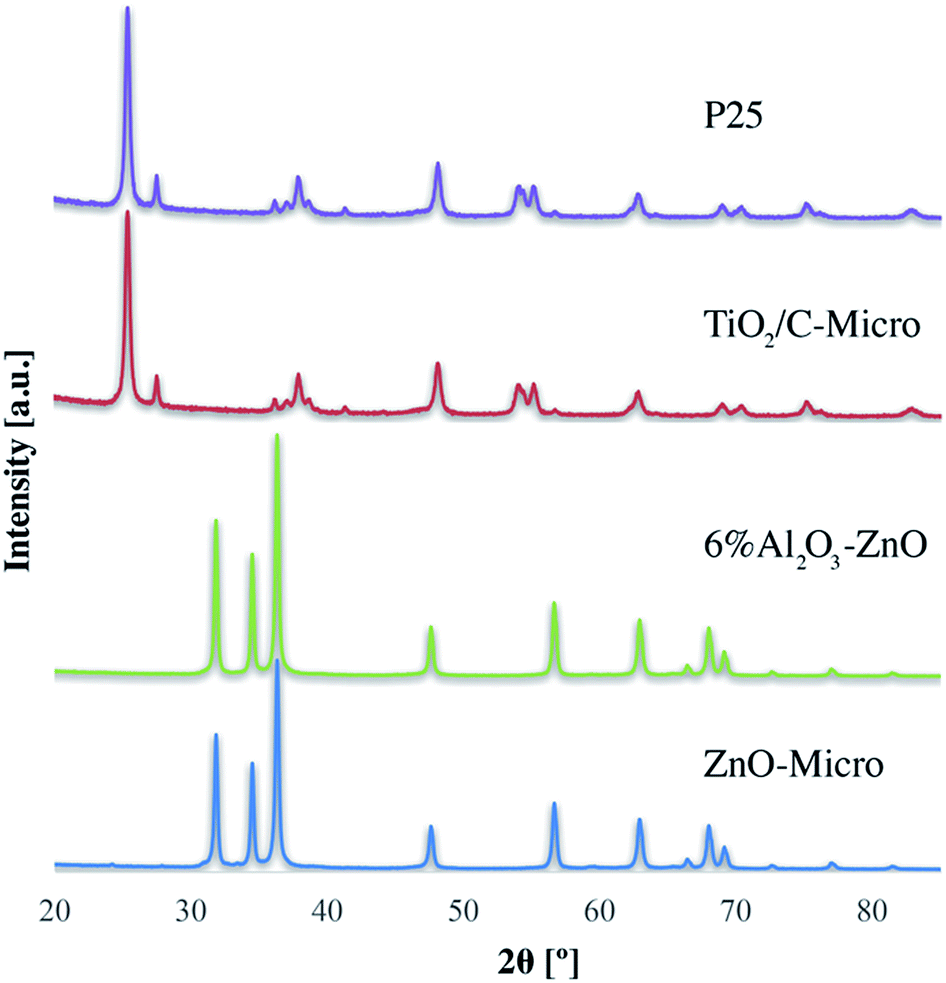 | ||
| Fig. 3 XRD data for the four catalysts, TiO2/C-Micro, P25, ZnO-Micro and 6% Al2O3–ZnO. | ||
XRD data and calculations (using Scherrer equation, Table S1†) show that the crystal size and phase remained almost unchanged, with a slight increase in crystal size after the nanoparticles were converted in microparticles (Table S1†). In other words, in the micro samples, fusion between TiO2–TiO2 nanoparticles during the calcination process cannot be the only reason of microparticles formation, suggesting that the particles are held together by means of other forces, such as TiO2–C–TiO2 fusion.
All three samples were analyzed to evaluate their composition and confirm the presence of gold in TiO2/C-Micro-Au via XPS analyses (Fig. 4).
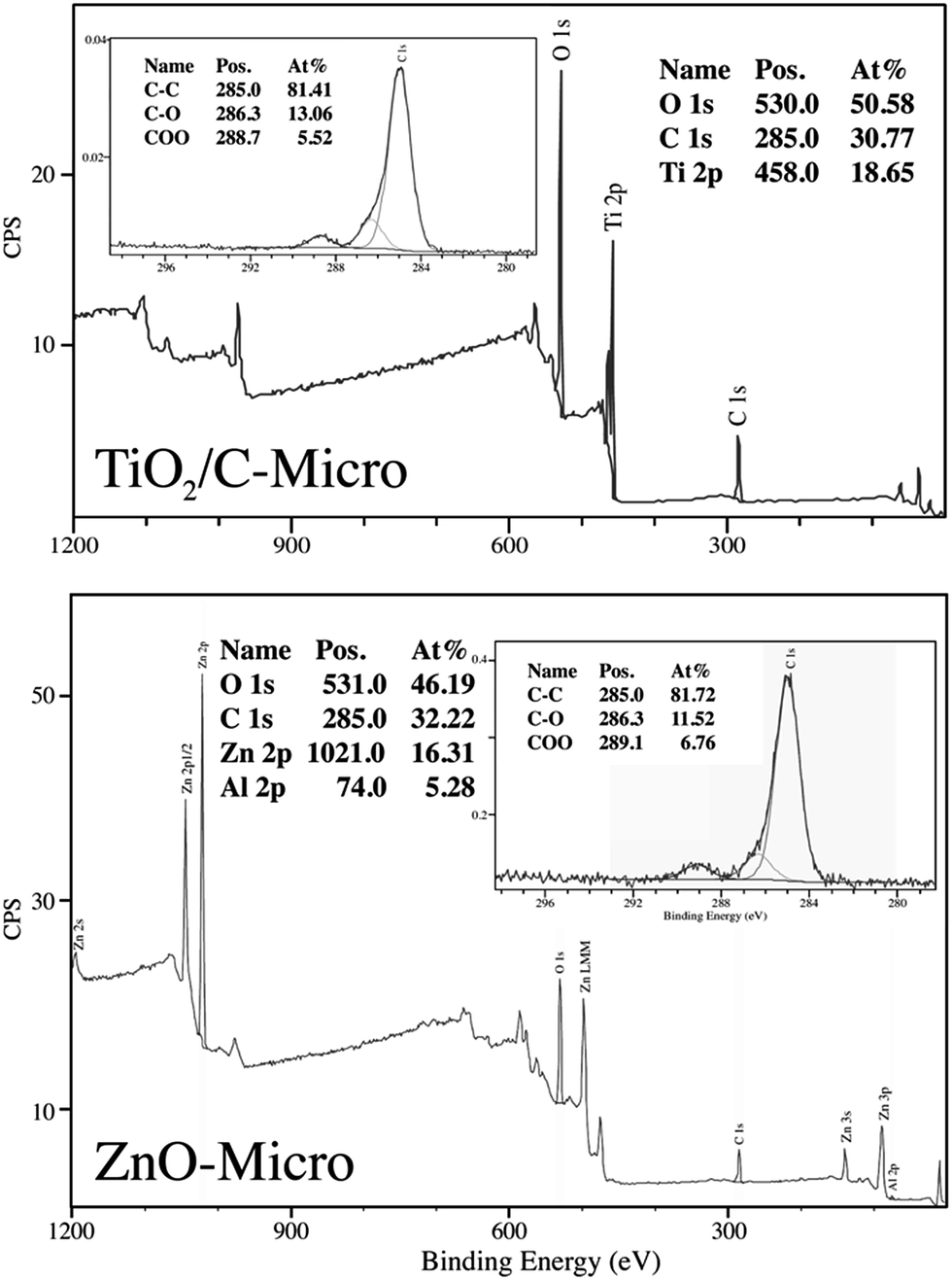 | ||
| Fig. 4 XPS analyses of TiO2/C-Micro and ZnO-Micro. | ||
As expected, the presence of TiO2 (Fig. 4 top) in TiO2/C-Micro and TiO2/C-Micro-Au, and Al2O3/ZnO (Fig. 4 bottom) in ZnO-Micro is observed. The gold content in the TiO2/C-Micro-Au sample was found be around about 1%, as shown by the signal at 83 eV (Au 4f). However, in all samples, a significant amount of carbon is observed as well. In particular, the second main peak at 285 eV is ascribed as residual carbon or adventitious hydrocarbon from the XPS instrument itself, or both.39 Generally, the adventitious hydrocarbon is around 10–15%.40 When the percentage is higher, it is an indication of carbon presence due to incomplete degradation of the polymer during calcination.41,42 Similar data were also observed by Irie et al. during the characterization of carbon doped TiO2.43 Unborn carbon can act as cementing between nanoparticles, TiO2–C–TiO2, holding together the final macro structure. In addition, Asahi et al. have studied the effect of carbon doped onto O sites.44 The results showed that C-doping into TiO2 lattice contributed to the formation of impurity states in the band gap, leading to a red-shift. A shift also observed for all samples in our study due to carbon doping (Fig. 5).
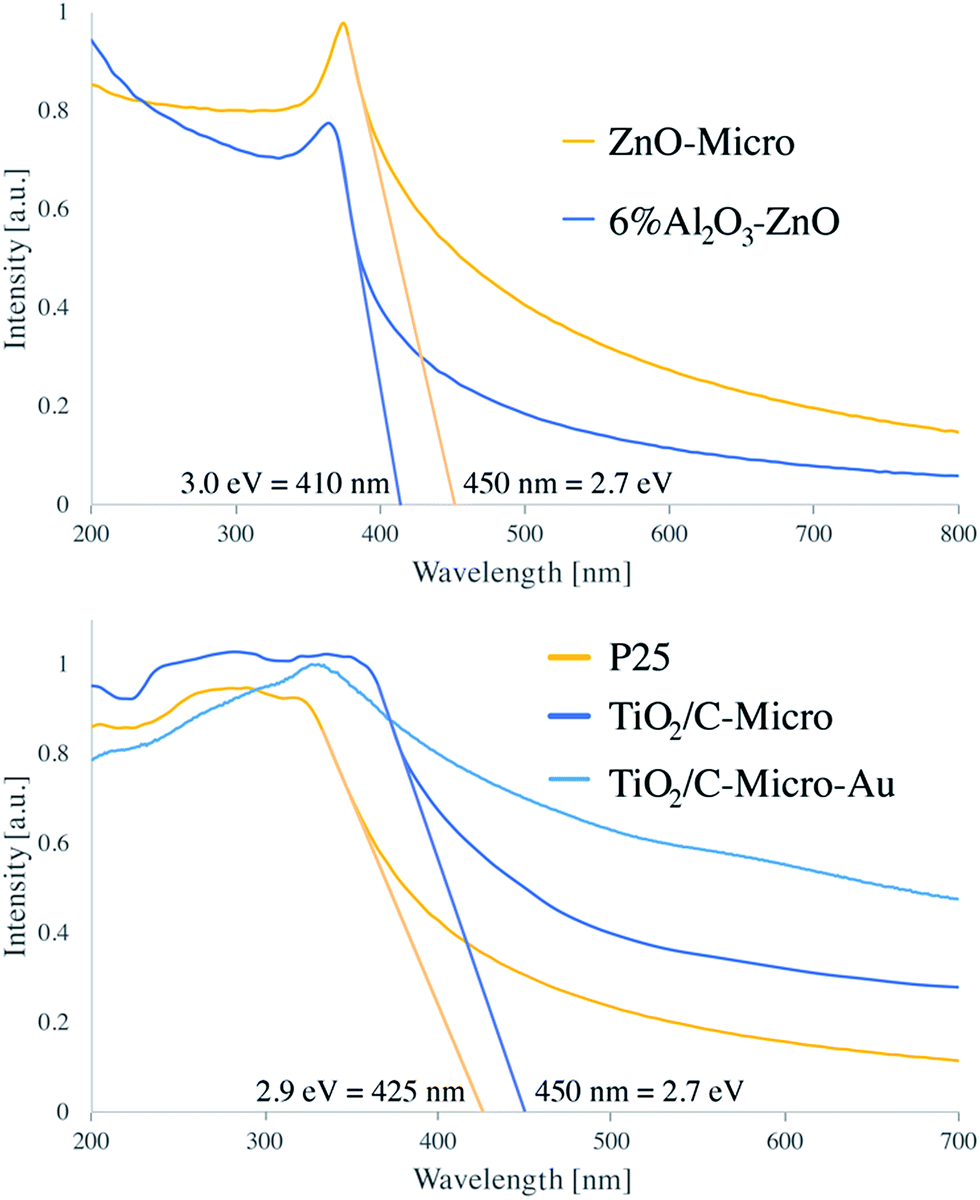 | ||
| Fig. 5 Adsorption spectra of 6% Al2O3–ZnO, ZnO-Micro, P25, TiO2/C-Micro and TiO2/C-Micro-Au. | ||
P25 has been reported to have a band gap around 3.1 eV.12 Instead, ZnO band gap is reported to be 3.2 eV (ref. 45) and 3.2/3.3 eV for 5% Al2O3/ZnO.46 From our analysis, both samples showed a consistent difference with literature values, with 2.9 eV for P25 and 3.0 eV for 6% Al2O3/ZnO. However, leaving aside the absolute values, a comparison between the different samples can still be made. In both micro samples, similar reductions in band gap values were recorded: 2.7 eV, in agreement with carbon doped photocatalyst.43 Moreover, the zeta potential analysis shows a shift in the PZC (point zero charge) for all catalysts (Fig. S4†). P25 has been reported to have the PZC at around pH 6.47 Instead, both TiO2 sample synthesised have a PZC at acidic pH with TiO2/C-Micro-Au slightly more acidic, between pH 4 and 5, and ZnO-Micro has the PZC at around 8. For TiO2, the shift from 6 to 4.5 is in agreement with carbon doping, as shown by Kuang et al.47 As expected, TiO2/C-Micro-Au shows a more notable increase in absorption with a small shoulder at 550–600 nm typical of gold NPs decoration.29 The increase in absorption is due to two main factors. The first one is the presence of residual carbon during the calcination; the second one is due to the noble metal NPs deposited on the surface, which work as light trapping, scattering, and concentration centres. In this way, light can be concentrated and ‘folded’ into a semiconductor layer, thereby enhancing the absorption, as explained by Wang et al.48 The particles were also analysed via fluorescence microscopy, which shows a weak fluorescence in the green (Fig. S5†), as also observed in previous studies when gold nanoparticles are in the range of 5–10 nm.49
To have a complete characterisation, BET analyses were also carried out. Fig. S6† shows the comparison in term of pore volume. Instead, the BET data, the size range of microsamples (via DLS analyses) and the size range of nanosamples (via TEM analyses), are presented in Table 1 (sizes obtained with other techniques are reported in Table S1†).
| Sample | Size | SBET [m2 g−1] | Vp [cm3 g−1] | dp [cm3 g−1] |
|---|---|---|---|---|
| P25 | 25 nm | 60 | 0.2 | 15 |
| TiO2/C-Micro | 8 μm | 55 | 0.5 | 31 |
| TiO2/C-Micro-Au | 23 μm | 52 | 0.4 | 29 |
| 6% Al2O3–ZnO | 50 nm | 26 | 0.1 | 16 |
| ZnO-Micro | 10 μm | 22 | 0.15 | 26 |
From the data, it can be observed that the synthetic procedure generates large particles without significant loss in surface area. Moreover, the pore distributions (Fig. S6†) show that there is a decrease in nano-porosity and an increase in mesoporosity for all the catalysts analyzed. Mainly, it does not seem that the choice between glutaraldehyde and gold results in a significant change regarding of pore distribution and surface area. TiO2/C-Micro-Au shows a slightly smaller mesoporosity compared to TiO2/C-Micro. The difference is also reflected in the slight reduction in surface area. P25 and 6% Al2O3/ZnO also show a mesoporosity even if their size is below 50 nm. This result can be correlated to small groups of particles physically aggregated during sample analyses. The synthesized micro powders did not have such particle aggregation and the pore volume data better reflect the actual porosity of the samples.
3.2 Mechanism
The analytical data discussed suggests the formation of similar structures independent of the choice to use glutaraldehyde or gold chloride. A mechanism could be proposed for the formation of microparticles using chitosan microspheres as a template (Fig. 6).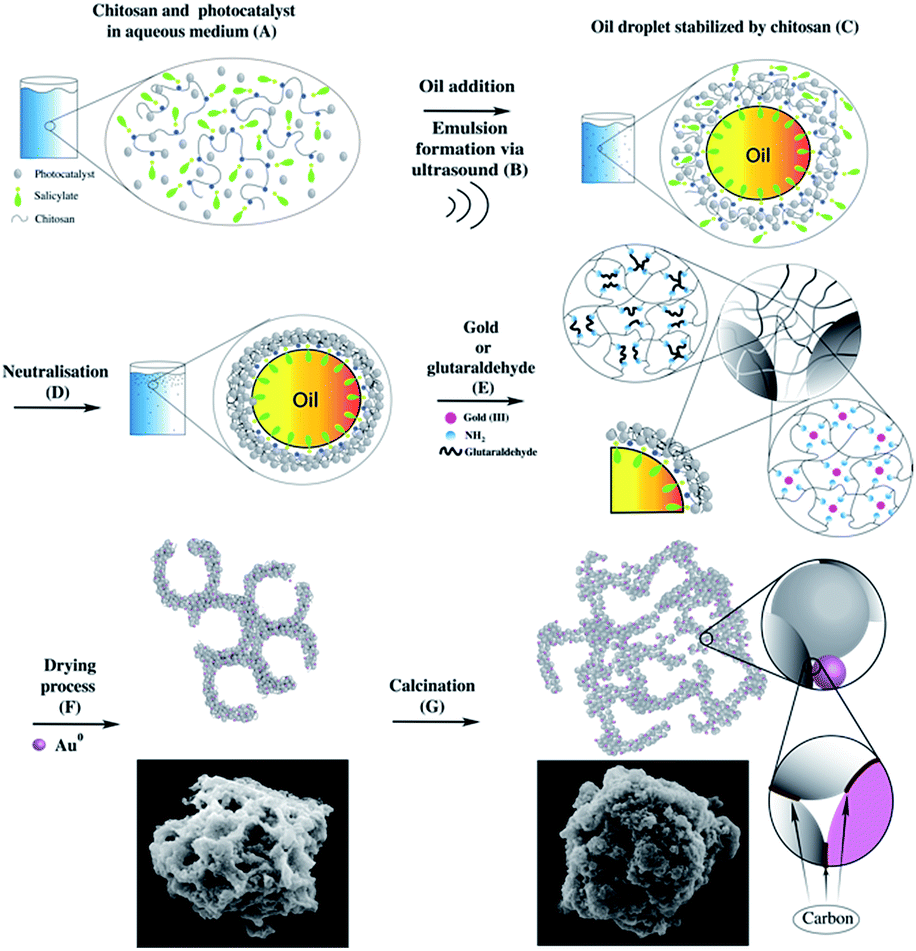 | ||
| Fig. 6 Microparticles formation mechanism: TiO2 dispersion in chitosan solution (A), oil droplets stabilized by chitosan (C), neutralisation (D), gold or glutaraldehyde addition and effects (E), drying (F) and calcination (G). | ||
In (A) is shown the graphical representation of the behaviour of TiO2 nanoparticle and chitosan dissolved in aqueous salicylic acid solution. When chitosan powder is dispersed in an acidic environment (in this case salicylic acid), the amine protonation permits the unfolding of the polymer coil, resulting in its solubilisation.34 After the addition of oil (tetradecane), acoustic cavitation, generated by ultrasound, (B) breaks up the nonpolar phase and spreads it into the medium as droplets. At this point, the oil droplets are stabilized by the polymer chain (C), with the amphiphilic counter anion at the oil/aqueous solution interface (C). The amphiphilicity of the counter anion is a key factor. With high amphiphilicity, the resulting microspheres are smaller, more stable and with a narrower size distribution.34 With the addition of NaOH (D), chitosan loses its charges and form microspheres characterised by a stronger shell with TiO2 NPs embedded within it.
After these first steps, the resulting microsphere shell is still not hard enough. For this reason, organic molecules, like glutaraldehyde, or metal compounds, like gold, can be used to provide additional strength to the microsphere shells. For the former, it is expected a Schiff base reaction between chitosan amino groups and the –CHO group present in the glutaraldehyde, for the latter, chitosan itself can promote a first metal reduction,37 followed by the amine stabilisation of the metal nanoparticles. Independent of the nature of the compound used to strength the shell, the result is the formation of a rigid network between the polymer chains inter and intra microspheres (E). Then, during the drying process, the shell is broken to permits the escape of the oil from the inside of the microspheres. At the same time, the rise in temperature completes the reduction of the gold ions. The result is a microstructure characterized by many macropores and kept together by the polymer (F), which can also be decorated with gold NPs (depending on the compound chosen in the step “E”). In the final stage (G), calcination maintains the structure burning the excess of polymer. However, some of the carbon cannot burn probably due to an inner location which prevents the combustion. As a result, this carbon undergoes to carbonization, and it may work as a cementing material between particles.
The photocatalytic efficiency of these particles was tested in two different phases: aqueous and gas. In aqueous phase, the catalysts were dispersed in a water medium and the efficiency, in terms of pollutant degradation under light irradiation, was measured. In the gas phase, the catalysts were tested to evaluate the possibility to use them for H2S adsorption and subsequent regeneration of the adsorbent activity via photocatalysis.
3.3 Photocatalytic efficiency
The catalysts were tested for their photocatalytic efficiency using two organics dyes: metanil yellow and methylene blue (Fig. 7 and 8). It can be seen in Fig. 7 that metanil yellow has a negligible degradation without photocatalysts and under photo-irradiation. After 5 hours, its concentration decreases only about 3%. Instead, methylene blue (Fig. 8) is dramatically sensitive to photo-irradiation. After 3.5 hours, its concentration is almost halved. However, due to catalysts surface charges, a comparison between anionic (metanil yellow) and cationic (methylene blue) pollutants need to be carried out for a better understanding of the catalysts' performance.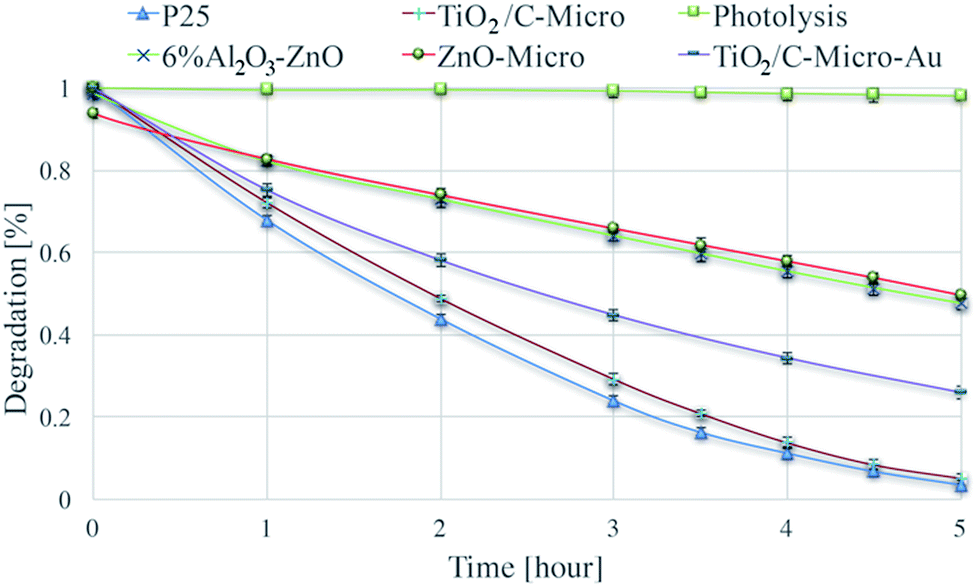 | ||
| Fig. 7 Degradation of metanil yellow via photolysis and via photodegradation with P25, TiO2/C-Micro, TiO2/C-Micro-Au, 6% Al2O3–ZnO and ZnO-Micro. | ||
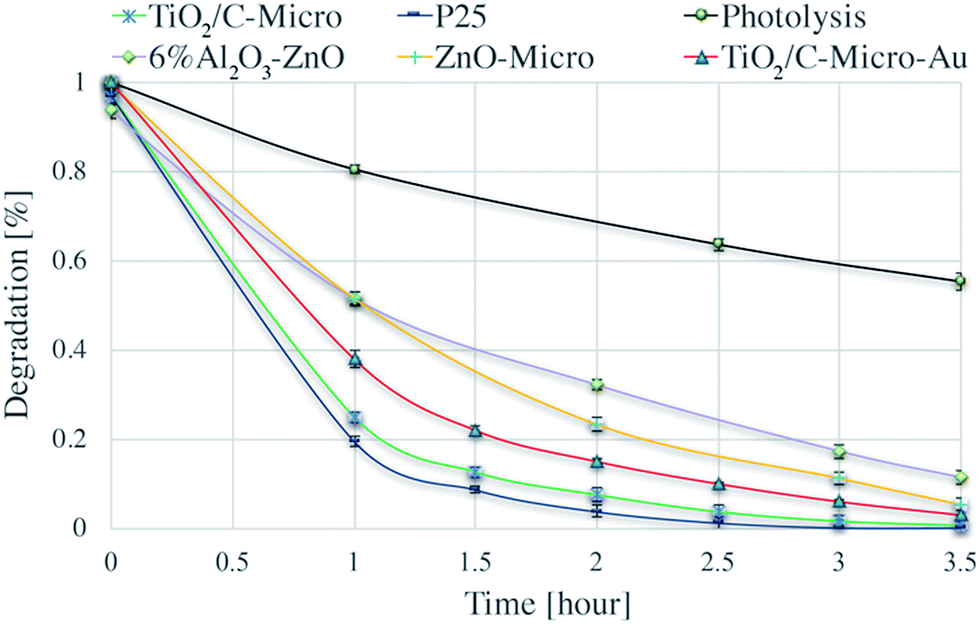 | ||
| Fig. 8 Degradation of methylene blue via photolysis and via photodegradation with P25, TiO2/C-Micro, TiO2/C-Micro-Au, 6% Al2O3–ZnO and ZnO-Micro. | ||
Dark adsorption experiments were also carried out. The solution pH and absorbance values were monitored. After overnight dark adsorption, the pH values of the solutions were found unaffected at 6.4. From ξ potential analyses, at the working pH, TiO2 surface is negatively charged, and anionic molecules do not adsorb due to electrostatic repulsion (Fig. S4†). Instead, when a cationic dye is used, methylene blue (Fig. 8), a slight decrease in concentrations is observed, around 3%.
For the 6% Al2O3–ZnO sample the situation is more complicated. The surface charge is positive at the pH used (Fig. S4†). Hence, no adsorption for methylene blue and adsorption for metanil yellow should be expected. However, the behaviour exhibited by the nanopowder is considerably different than anticipated. In effect, the nanopowder showed a strong adsorption for methylene blue (8%) and a little adsorption for metanil yellow (2%). This behaviour can be explained by impurities or specific adsorption of ions in the starting sample, which can lead to a modification of surface charge surface. Degen et al. have analysed a similar case.50 They found that these impurities are generally zinc, lead, sulphate and chloride ions. Instead, the micro size exhibits the behaviour expected suggesting a removal of those contaminants. That can be correlated to the procedures to produce it, where the catalyst has been heated and washed several times removing any possible impurities.
Two key observations can be made from Fig. 7 and 8:
(i) The three TiO2 samples are more efficient than ZnO.
(ii) The nano and micro size samples show a similar efficiency.
Point (i) can be easily explained by the combination of surface areas and band gap values. Indeed, ZnO samples have a smaller surface area and a larger band gap, which lead to a less amount of radicals produced (i.e., less dye degraded). Point (ii) needs a more elaborated explanation. The similar photocatalytic efficiency between the two sizes of catalysts cannot be correlated to the differences in surface area, which is very similar between the two P25/TiO2/C-Micro and 6% Al2O3–ZnO/ZnO-Micro samples. For the nanocatalysts, it is possible to assume that the surface area is similar to the irradiated surface. However, for the microparticles, this assumption cannot be made. For micro powders, reasoning on surface area alone cannot lead to a complete understanding of the observed results. A reduction in the surface area should lead to a lower production of radicals. However, the amounts of hydroxyl radicals generated are similar for TiO2/C-Micro:P25 and 6% Al2O3–ZnO:ZnO-Micro (Fig. S7†), suggesting that the lower band gap in the micro catalysts allows the absorption of additional photons to compensate for the lower surface area effect. In summary, a balance between larger size, lower surface area and smaller band gap results in the maintenance of the overall catalyst efficiency. Moreover, as in the case of the micro powders, surface roughness may play a role in improving the efficacy. Roughness leads to reflection of photons, which could result in increased photon absorption.51 In particular, this effect is enhanced when the pores are large. Reflected photons can easily penetrate into micropores and be used for radicals formation. Moreover, microsize pores permit a better adsorption of reactants. Pollutant molecules can easily penetrate into the pores and react with radicals. As observed by Kimura et al. in their studies on mesoporous films of TiO2 during photodegradation of methylene blue,52 an increase in photodegradation is possible with an increase in porosity.
An interesting observation can be made on TiO2/C-Micro-Au. It can be observed from Fig. 7 and 8 that the degradation recorded for TiO2/C-Micro-Au was 10–15% lower compared to that of TiO2/C-Micro. In effect, several studies report an increasing in photocatalytic activity after the decoration with gold nanoparticles on photoactive materials.53 The comparison is generally between the same catalyst with and without decoration, leaving all the other variables constant (i.e., shape, size, surface area, etc.). In this case, such reasoning cannot be made. TiO2/C-Micro-Au was synthesized with a procedure similar to that of TiO2/C-Micro, and particles with similar shape were obtained. However, the size of TiO2/C-Micro-Au is greater than that of TiO2/C-Micro (Table S1†), leading to a lower surface area. The result is that a balance between surface area and band-gap is not enough to observe an efficiency equal to that of P25. It can be noted that an increase in size from 10 to 20 μm (reducing the surface area) results in 10–15% decrease in photocatalytic activity.
In order to evaluate the stability and recycling capacity of the catalyst, micro catalyst powders were filtered after the photocatalytic experiments, washed several times, dried at 70 °C overnight and reused in subsequent experiments. The process was repeated three times. After each cycle, the size distribution and photocatalytic efficiency were measured. The size of the catalysts, as well as their photocatalytic degradation efficiencies, were not significantly affected, confirming the stability and reusability of the catalysts prepared.
3.4 H2S removal
It was shown before that the gold nanoparticles did not increase the photocatalytic efficiency. However, Au deposition can be of great advantage for other applications. Sui et al.32 have studied the mechanism of H2S adsorption using gold nanoparticles supported by metal oxides, and their regeneration at high temperature. The aim of our study was to investigate if H2S adsorption was possible also with TiO2/C-Micro-Au. Moreover, regeneration tests at room temperature using photocatalysis were also carried out, which explain the importance of micro sized particles. Fig. 9 shows the H2S adsorption experimental results.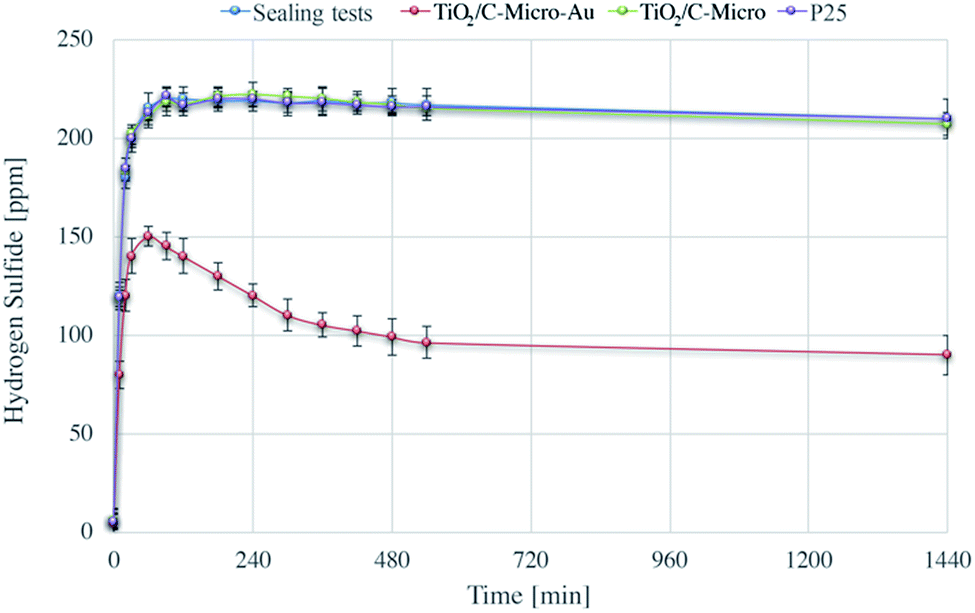 | ||
| Fig. 9 H2S sealing test and adsorption experiments using TiO2/C-Micro, TiO2/C-Micro-Au, and P25. | ||
The sharp increase in H2S level in the first few minutes is due to the generation of H2S in the sealed chamber. The amount of gas produced is small (around 220 mg m−3, which would result, assuming a perfect behaviour, in an increasing of pressure of about 1%) and does not influence the total pressure significantly inside the chamber. Moreover, the data shows that the chamber has a negligible loss of gas during the 24 hours (less than 2% in 6 h and around 5% after 24 h) due to leakage.
Beck et al. have studied the H2S adsorption on TiO2, they observed that TiO2 alone is not able to adsorb this molecule on its surface.54 This result was also confirmed by our experiments in which the adsorption on TiO2/C-Micro and P25 was not observed in a contact time of 24 hours.
TiO2/C-Micro-Au shows a remarkable degree of adsorption. Most of the H2S is adsorbed in the first minutes of contact (around 80 ppm). Also, an additional quantity is adsorbed more slowly in the next 6 hours (30 ppm) reaching a plateau around 8/10 h (100 ppm). This behaviour is probably due to inner sites inside the micro catalyst structure that prevent a fast diffusion of the gas phase. In total, an adsorption of around 120 ppm was observed, which correspond to an adsorption of 4.2 mg g−1.
In Fig. 10 are reported the XPS analyses of the fresh TiO2/C-Micro-Au (A), immediately after the adsorption experiment (B) and after four weeks (C).
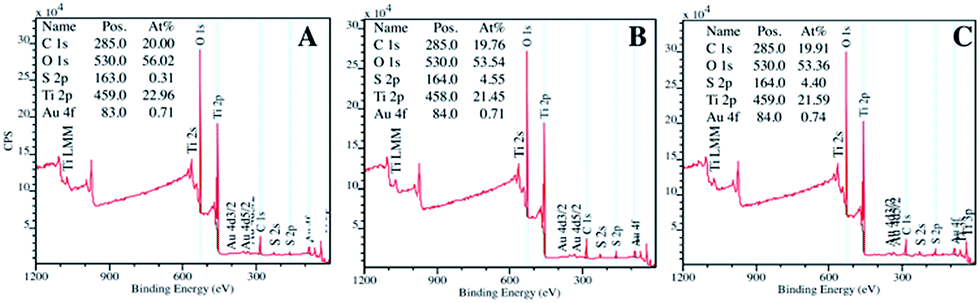 | ||
| Fig. 10 XPS analyses of TiO2/C-Micro-Au before adsorption (A), after adsorption (B) and after four weeks from adsorption (C). | ||
These analyses confirm the adsorption of sulfur, which increased from 0.3% (Fig. 10A) to around 4.5% after adsorption (Fig. 10B). The quantity of sulfur did not change significantly during the four weeks' stock period (Fig. 10C), demonstrating that the material is suitable to be stored once used, without the risk of releasing H2S.
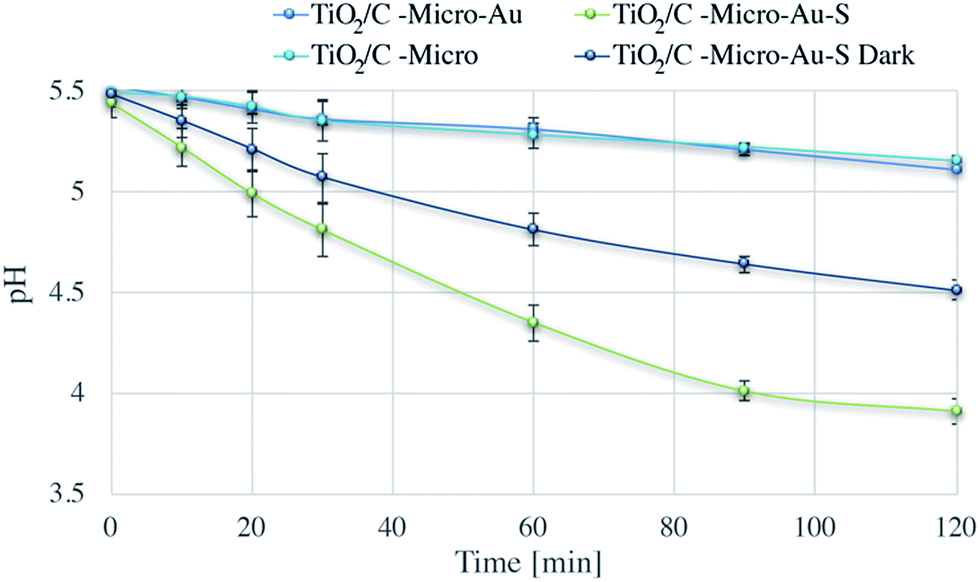 | ||
| Fig. 11 Degradation via photocatalysis of H2S followed pH-metrically. | ||
Dark and under irradiation tests were performed for the fresh catalyst (TiO2/C-Micro-Au) and the one exposed to hydrogen sulphide (TiO2/C-Micro-Au-S). Also in this case, TiO2/C-Micro was used for comparison.
During photocatalysis, a modest reduction in pH was observed for both TiO2/C-Micro and the TiO2/C-Micro-Au that was not exposed to H2S. Instead, the dark dispersion in water of TiO2/C-Micro-Au-S led to a pH decrease of 1 unit after two hours, which is expected due to spontaneous reaction with oxygen dissolved in water.56 Furthermore, the photocatalysis of TiO2/C-Micro-Au-S produced a lower pH value, about 3.9, which can be a confirmation of H2S decomposition carried out by radicals photocatalytically generated. In order to verify this hypothesis, XPS analyses of the catalyst were performed in the middle and at the end of the photodegradation (Fig. 12).
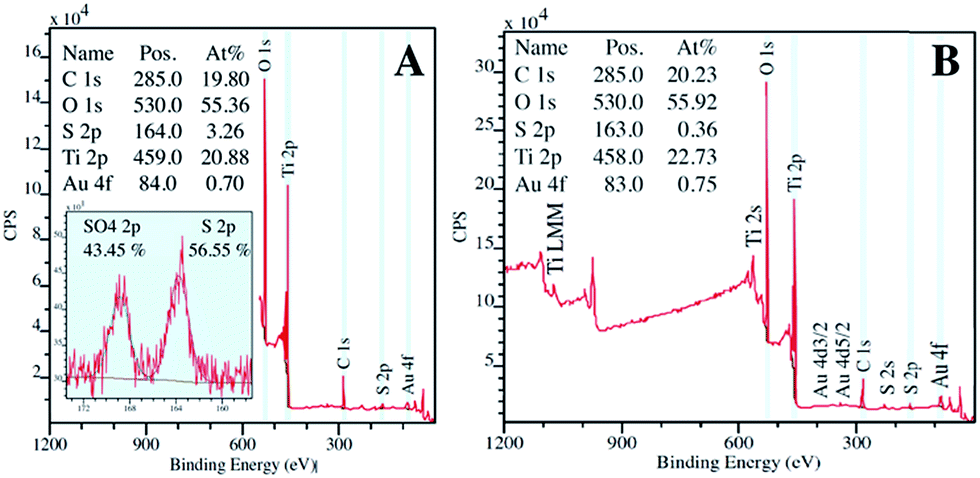 | ||
| Fig. 12 XPS analysis of TiO2/C-Micro-Au-S after 1 (A) and 2 (B) hours of irradiation. | ||
Fig. 12A shows XPS spectra of the sample after one hour of reaction. The analysis reveals the presence of sulfur in both reduced and oxidized states, confirming the decomposition of the molecule. Moreover, after 2 hours of reaction (Fig. 12B), a return to a sulfur quantity close to zero was observed.
Thanks to its micro scale, the collection of the adsorbent after photo-regeneration was easy. In effect, a simple fritted filter (pore size 1/1.6 μm) was used to collect and wash the catalyst before reuse. The adsorption experiments and the regenerations were repeated for ten times to evaluate the recycling property of the catalyst. The results are shown in Fig. S8.† TiO2/C-Micro-Au lost 10% activity after the first regeneration and an additional 5% after the second one. This deactivation can be due to the inability of regenerate most inner sites, where radicals have more difficulties to diffuse. However, after the fourth regeneration, the adsorbent seems to reach a plateau, maintaining an efficiency of around 80% of the initial one, demonstrating a room temperature renewability.
4 Conclusions
We have developed a simple methodology to synthesise micron-sized photocatalyst particles without compromising the large surface area required for high photocatalytic activity. In addition, we have shown the versatility of the method in which the use of different molecules permits to add functionality such as doping or metal NPs decoration. At the end of the characterization, a mechanism was proposed for the microparticles formation, and a new approach for H2S removal, and its decomposition, was also evaluated.ZnO-Micro, TiO2/C-Micro, and TiO2/C-Micro-Au were characterised by light-scattering, ξ potential, SEM, TEM, absorption spectroscopy, BET, XRD and XPS analyses. The synthetic procedure resulted in an increase in the size of the catalytic particles from 20/50 nm to 10–20 μm. Moreover, the catalyst showed an increase in mesoporosity, band gap reduction and the formation of macro size pores in the range 1–4 μm. The photocatalytic efficiency of these catalysts was evaluated using methylene blue and metanil yellow as model pollutants. In both cases, micro samples showed similar photocatalytic activities to those of respective nanoparticles, and an enhancement four times higher than commercial micro titania available in the market.12 The synthetic procedure developed can be used to produce highly efficient micron size photocatalysts for wastewater treatment applications overcoming the problems associated with recyclability and with the removal of the catalysts by filtration following treatment. Moreover, TiO2/C-Micro-Au shows potential for H2S adsorption in the gas phase and sequential photodegradation in an aqueous medium. The quantity that can be adsorbed is modest, but the room temperature regeneration and the small loss in activity after ten cycles make this catalyst valuable for further studies.
Acknowledgements
EC acknowledges the University of Melbourne for the award of MIRS/MIFRS and financial support from the Australian Research Council (ARC-DP) is also gratefully acknowledged. A special thanks to Dr Meifang Zhou for the TEM analyses.Notes and references
- B. N. Estevinho, F. Rocha, L. Santos and A. Alves, Trends Food Sci. Technol., 2013, 31, 138–155 CrossRef CAS.
- M. Colone, S. Kaliappan, A. Calcabrini, M. Tortora, F. Cavalieri and A. Stringaro, J. Mol. Genet. Med., 2016, 10(200), 1747–0862 Search PubMed.
- G. Paradossi, E. Chiessi, F. Cavalieri, D. Moscone and V. Crescenzi, Polym. Gels Networks, 1998, 5, 525–540 CrossRef.
- M. Tortora, F. Cavalieri, P. Mosesso, F. Ciaffardini, F. Melone and C. Crestini, Biomacromolecules, 2014, 15, 1634–1643 CrossRef CAS PubMed.
- Q. Pan, Y. Lv, G. R. Williams, L. Tao, H. Yang, H. Li and L. Zhu, Carbohydr. Polym., 2016, 72(1), 52–59 Search PubMed.
- Z. Zainal, L. K. Hui, M. Z. Hussein, A. H. Abdullah and I. M. Hamadneh, J. Hazard. Mater., 2009, 164, 138–145 CrossRef CAS PubMed.
- X. Zhang, X. Zhao and H. Su, Korean J. Chem. Eng., 2011, 28, 1241–1246 CrossRef CAS.
- C. E. Zubieta, P. V. Messina, C. Luengo, M. Dennehy, O. Pieroni and P. C. Schulz, J. Hazard. Mater., 2008, 152, 765–777 CrossRef CAS PubMed.
- M. N. Chong, B. Jin, C. W. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- S.-Y. Lee and S.-J. Park, J. Ind. Eng. Chem., 2013, 19, 1761–1769 CrossRef CAS.
- T. Mano, S. Nishimoto, Y. Kameshima and M. Miyake, Chem. Eng. J., 2015, 264, 221–229 CrossRef CAS.
- C. L. Bianchi, E. Colombo, S. Gatto, M. Stucchi, G. Cerrato, S. Morandi and V. Capucci, J. Photochem. Photobiol., A, 2014, 280, 27–31 CrossRef CAS.
- M. Gallus, V. Akylas, F. Barmpas, A. Beeldens, E. Boonen, A. Boréave, M. Cazaunau, H. Chen, V. Daële, J. F. Doussin, Y. Dupart, C. Gaimoz, C. George, B. Grosselin, H. Herrmann, S. Ifang, R. Kurtenbach, M. Maille, A. Mellouki, K. Miet, F. Mothes, N. Moussiopoulos, L. Poulain, R. Rabe, P. Zapf and J. Kleffmann, Build. Environ., 2015, 84, 125–133 CrossRef.
- C. L. Bianchi, C. Pirola, F. Galli, G. Cerrato, S. Morandi and V. Capucci, Chem. Eng. J., 2015, 261, 76–82 CrossRef CAS.
- Y. Zhang, G. Chang, S. Liu, W. Lu, J. Tian and X. Sun, Biosens. Bioelectron., 2011, 28, 344–348 CrossRef CAS PubMed.
- Q. Huang, H. Zhang, S. Hu, F. Li, W. Weng, J. Chen, Q. Wang, Y. He, W. Zhang and X. Bao, Biosens. Bioelectron., 2014, 52, 277–280 CrossRef CAS PubMed.
- A. Y. Mironenko, A. A. Sergeev, A. E. Nazirov, E. B. Modin, S. S. Voznesenskiy and S. Y. Bratskaya, Sens. Actuators, B, 2016, 225, 348–353 CrossRef CAS.
- Y. Qu, H. Min, Y. Wei, F. Xiao, G. Shi, X. Li and L. Jin, Talanta, 2008, 76, 758–762 CrossRef CAS PubMed.
- A. El Kadib, K. Molvinger, T. Cacciaguerra, M. Bousmina and D. Brunel, Microporous Mesoporous Mater., 2011, 142, 301–307 CrossRef CAS.
- M. B. Fisher, D. A. Keane, P. Fernández-Ibáñez, J. Colreavy, S. J. Hinder, K. G. McGuigan and S. C. Pillai, Appl. Catal., B, 2013, 130–131, 8–13 CrossRef CAS.
- T. Xin, M. Ma, H. Zhang, J. Gu, S. Wang, M. Liu and Q. Zhang, Appl. Surf. Sci., 2014, 288, 51–59 CrossRef CAS.
- X. Zheng, D. Li, X. Li, L. Yu, P. Wang, X. Zhang, J. Fang, Y. Shao and Y. Zheng, Phys. Chem. Chem. Phys., 2014, 16, 15299–15306 RSC.
- X. Qu, P. J. J. Alvarez and Q. Li, Water Res., 2013, 47, 3931–3946 CrossRef CAS PubMed.
- G. E. Romanos, C. Athanasekou, F. Katsaros, N. Kanellopoulos, D. Dionysiou, V. Likodimos and P. Falaras, J. Hazard. Mater., 2012, 211, 304–316 CrossRef PubMed.
- J. Ryu, W. Choi and K.-H. Choo, Water Sci. Technol., 2005, 51, 491–497 CAS.
- C. L. Bianchi, S. Gatto, C. Pirola, A. Naldoni, A. Di Michele, G. Cerrato, V. Crocellà and V. Capucci, Appl. Catal., B, 2014, 146, 123–130 CrossRef.
- A. Nezamzadeh-Ejhieh and M. Karimi-Shamsabadi, Appl. Catal., A, 2014, 477, 83–92 CrossRef CAS.
- X. Li, H. Liu, L. Cheng and H. Tong, Environ. Sci. Technol., 2003, 37, 3989–3994 CrossRef CAS PubMed.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943–4950 CrossRef CAS PubMed.
- V. De Renzi, Surf. Sci., 2009, 603, 1518–1525 CrossRef CAS.
- Z. Zhang, Z. Chen, S. Wang, C. Qu and L. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 6300–6307 CAS.
- R. Sui, K. L. Lesage, S. K. Carefoot, T. Fürstenhaupt, C. J. Rose and R. A. Marriott, Langmuir, 2016, 32, 9197–9205 CrossRef CAS PubMed.
- L. Sigot, G. Ducom and P. Germain, Chem. Eng. J., 2016, 287, 47–53 CrossRef CAS.
- E. Colombo, F. Cavalieri and M. Ashokkumar, ACS Appl. Mater. Interfaces, 2015, 7, 12972–12980 CAS.
- Q. Xiang, J. Yu and P. K. Wong, J. Colloid Interface Sci., 2011, 357, 163–167 CrossRef CAS PubMed.
- M. Zhou, B. Babgi, S. Gupta, F. Cavalieri, Y. Alghamdi, M. Aksu and M. Ashokkumar, RSC Adv., 2015, 5, 20265–20269 RSC.
- A. Corma, P. Concepción, I. Domínguez, V. Forné and M. J. Sabater, J. Catal., 2007, 251, 39–47 CrossRef CAS.
- X. Zhou, Z.-X. Xie, Z.-Y. Jiang, Q. Kuang, S.-H. Zhang, T. Xu, R.-B. Huang and L.-S. Zheng, Chem. Commun., 2005, 5572–5574 RSC.
- H. Pan, J. Yi, L. Shen, R. Wu, J. Yang, J. Lin, Y. Feng, J. Ding, L. Van and J. Yin, Phys. Rev. Lett., 2007, 99, 127201 CrossRef CAS PubMed.
- M. Atashbar, H. Sun, B. Gong, W. Wlodarski and R. Lamb, Thin Solid Films, 1998, 326, 238–244 CrossRef CAS.
- J. Yu, X. Zhao, J. Du and W. Chen, J. Sol-Gel Sci. Technol., 2000, 17, 163–171 CrossRef CAS.
- S. Södergren, H. Siegbahn, H. Rensmo, H. Lindström, A. Hagfeldt and S.-E. Lindquist, J. Phys. Chem. B, 1997, 101, 3087–3090 CrossRef.
- H. Irie, Y. Watanabe and K. Hashimoto, Chem. Lett., 2003, 32, 772–773 CrossRef CAS.
- R. Asahi, Y. Taga, W. Mannstadt and A. J. Freeman, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 7459 CrossRef CAS.
- V. Srikant and D. R. Clarke, J. Appl. Phys., 1998, 83, 5447–5451 CrossRef CAS.
- I. Dimitrov, A. O. Dikovska, P. Atanasov, T. Stoyanchov and T. Vasilev, J. Phys.: Conf. Ser., 2008, 012044 CrossRef.
- L. Kuang and W. Zhang, RSC Adv., 2016, 6, 2479–2488 RSC.
- P. Wang, B. Huang, Y. Dai and M.-H. Whangbo, Phys. Chem. Chem. Phys., 2012, 14, 9813–9825 RSC.
- C. C. Huang, Z. Yang, K. H. Lee and H. T. Chang, Angew. Chem., 2007, 119, 6948–6952 CrossRef.
- A. Degen and M. Kosec, J. Eur. Ceram. Soc., 2000, 20, 667–673 CrossRef CAS.
- Y. Paz, Z. Luo, L. Rabenberg and A. Heller, J. Mater. Res., 1995, 10, 2842–2848 CrossRef CAS.
- T. Kimura, Y. Yamauchi and N. Miyamoto, Chem.–Eur. J., 2010, 16, 12069–12073 CrossRef CAS PubMed.
- R. Su, R. Tiruvalam, Q. He, N. Dimitratos, L. Kesavan, C. Hammond, J. A. Lopez-Sanchez, R. Bechstein, C. J. Kiely and G. J. Hutchings, ACS Nano, 2012, 6, 6284–6292 CrossRef CAS PubMed.
- D. Beck, J. White and C. Ratcliffe, J. Phys. Chem., 1986, 90, 3123–3131 CrossRef CAS.
- A. Bouzaza, S. Marsteau, A. Laplanche and B. Garrot, http://lodel.irevues.inist.fr/pollution-atmospherique/index.php?id=1964, 2003, 2268–3798.
- X.-R. Dai and V. Blanes-Vidal, J. Environ. Manage., 2013, 115, 147–154 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01227f |
| This journal is © The Royal Society of Chemistry 2017 |