
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Analytical method for metabolites involved in biosynthesis of plant volatile compounds†
Xiumin Fu‡
a,
Ying Zhou‡
a,
Lanting Zenga,
Fang Dongb,
Xin Meia,
Yinyin Liaoa,
Naoharu Watanabec and
Ziyin Yang
 *a
*a
aKey Laboratory of South China Agricultural Plant Molecular Analysis and Genetic Improvement, Guangdong Provincial Key Laboratory of Applied Botany, South China Botanical Garden, Chinese Academy of Sciences, Xingke Road 723, Tianhe District, Guangzhou 510650, China. E-mail: zyyang@scbg.ac.cn; Tel: +86-20-38072989
bGuangdong Food and Drug Vocational College, Longdongbei Road 321, Tianhe District, Guangzhou 510520, China
cGraduate School of Science and Technology, Shizuoka University, 3-5-1 Johoku, Naka-ku, Hamamatsu 432-8561, Japan
First published on 3rd April 2017
Abstract
Plants synthesize and emit a large variety of volatile organic compounds, which possess extremely important ecological functions. Rapid progress in the method and instruments available to separate and identify plant metabolites has led to intensive investigations of the biosynthetic pathways that form plant volatiles, which has resulted in insightful and useful discoveries. In this review, we summarize and present the successful techniques currently used for studying metabolites involved in the formation of the major classes of plant volatile compounds, including volatile fatty acid derivatives, volatile isoprenoids, volatile phenylpropanoids/benzenoids, carotenoid-derived aroma compounds, and glycosidically bound volatile compounds. We discuss the extraction, identification, and quantitative analysis of many of these metabolites, which are unstable and occur in plants at trace levels.
 Xiumin Fu | Xiumin Fu is an assistant professor at South China Botanical Garden, Chinese Academy of Science, Guangzhou, China. She received her PhD in Pomology from the Zhejiang University, China. Her research interests focus on the biosynthesis of quality-related metabolites in fruits. |
 Ying Zhou | Ying Zhou is an assistant professor at South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, China. She received her PhD in Environmental Biology from the Zhejiang University, China. Her research interests focus on the formation of aroma compounds in Camellia sinensis. |
 Ziyin Yang | Ziyin Yang is a professor at South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, China. He received his PhD in Tea Science from the Zhejiang University, China. His research interests focus on the biosyntheses and regulation mechanisms of specialized metabolites in economic plants. |
1. Introduction
Plants synthesize and emit a large variety of volatile organic compounds, which are involved in plant interactions with the environment.1 To date, more than 1700 volatiles have been identified from more than 90 plant families, and these contribute approximately 1% of all known plant secondary metabolites.2 From a physiological standpoint, plant volatiles are mainly involved in protection against biotic stress by acting as deterrents to herbivores, attractants to beneficial organisms such as species-specific pollinators, and being involved in plant–plant or within-plant signaling. They can also provide protection against abiotic stress by directly or indirectly improving the in resistance to damage by reactive oxygen species.2–4 There are also increasing reports that plant volatiles can be beneficial to human health and have a range of biological activities that could be exploited by incorporating plant volatiles as lead compounds in drug discovery.5 Because of their particular ecological functions and potential medicinal applications, the biosynthesis of plant volatiles should be rigorously investigated at the molecular and biochemical levels.Plant volatiles are lipophilic molecules, and their synthesis involves the removal of hydrophilic moieties, oxidation/hydroxylation, acylation reactions, methylation, and reduction.6 There are many biochemical pathways leading to the production of a diverse range of plant volatiles.1,5–7 Fig. 1 indicates the main metabolic routes involved in the formation of the major classes of plant volatiles, along with representative precursors of the volatiles from each pathway. Most plant volatiles are derived from the isoprenoid pathways, which include the cytosolic mevalonate and the methylerythritol phosphate pathways. Diverse hemi-, mono-, sesqui-, and diterpene plant volatiles are derived from dimethylallyl pyrophosphate (DMAPP), geranyl diphosphate (GPP), farnesyl pyrophosphate (FPP), and geranylgeranyl diphosphate (GGPP) under the action of terpene synthases. Another important class of volatiles derived from terpenoids are the products produced from carotenoids by carotenoid cleavage dioxygenases. Many of these volatile carotenoid derivatives are key aroma constituents of flowers and have extremely low aroma thresholds. The second most ubiquitous class of plant volatiles consists of volatile phenylpropanoids and benzenoids containing an aromatic ring. Most volatiles from this class are derived from shikimate via L-phenylalanine (L-Phe). The first step in the biosynthesis of many volatile phenylpropanoids and benzenoids is the deamination of L-Phe to trans-cinnamic acid (CA) by the enzyme L-Phe ammonia-lyase.7–9 By contrast the biosynthesis of other volatile phenylpropanoid-related compounds, such as phenylacetaldehyde and 2-phenylethanol, does not occur via CA and competes with L-Phe ammonia lyase for L-Phe utilization.10,11 In addition to this, coenzyme A (CoA)-independent, non-β-oxidation pathway benzenoid compounds can also be formed in plants via a CoA-dependent, β-oxidation pathway.12,13 The β-oxidation pathway involves the activation of CA via the formation of a cinnamoyl-CoA ester, followed by hydration of the CoA ester to 3-hydroxy-3-phenylpropionyl-CoA, and oxidation of the hydroxyl group to a ketone (3-oxo-3-phenylpropionyl-CoA).13 Down regulation of cinnamate: CoA ligase from Petunia hybrida decreased emission of benzylbenzoate, phenylethylbenzoate, and methylbenzoate. Furthermore, cinnamoyl–CoA formation by cinnamate:CoA ligase in peroxisomes is important in the β-oxidation pathway in biosynthesis of benzoic acid from L-Phe.14 Studies on the upstream pathway of volatile phenylpropanoids and benzenoids indicate that L-Phe is synthesized predominantly via arogenate from prephenate by the actions of prephenate aminotransferase and arogenate dehydratase in petunia petals.15,16 Plants can also synthesize L-Phe via the intermediate metabolite phenylpyruvic acid, similarly to many microorganisms.17 Interestingly, Hirata et al. reported that 2-phenylethanol can be biosynthesized from L-Phe via phenylpyruvic acid and phenylacetaldehyde in isolated rose petal protoplasts.18 A third group of plant volatiles is derived by oxidative cleavage and decarboxylation of various fatty acids, resulting in the production of short-chain volatiles with aldehyde and ketone moieties, such as cis-3-hexenol, 1-hexanal, nonanal, and methyl jasmonate. These compounds originate from C18 unsaturated fatty acids (linoleic or linolenic acids), which enter the lipoxygenase pathway. The first step of this pathway is the dioxygenation of unsaturated fatty acids, catalyzed by lipoxygenase enzymes.
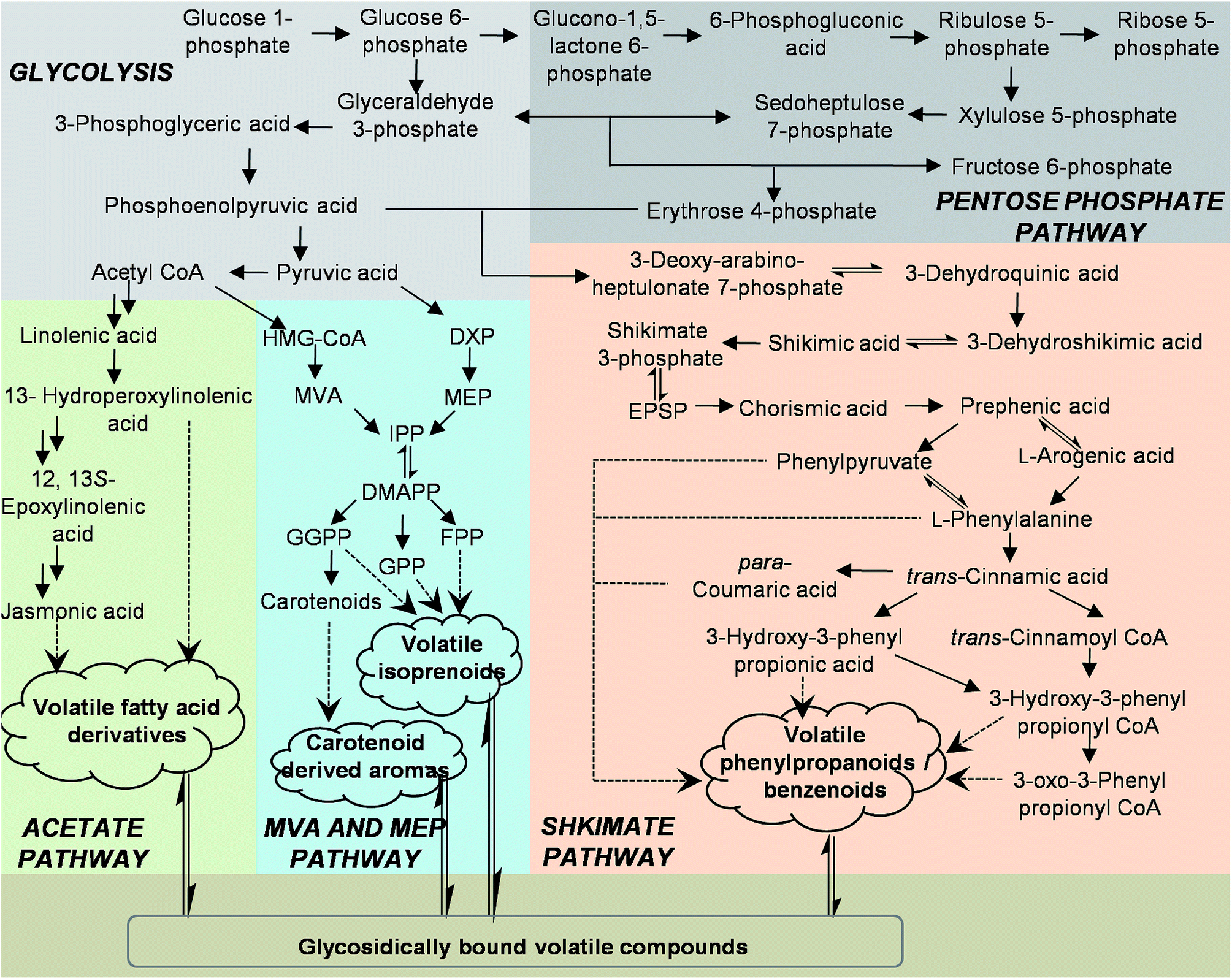 | ||
| Fig. 1 Metabolic routes involved in the formation of the major classes of plant volatiles CoA, coenzyme-A; DMAPP, dimethylallyl diphosphate; DXP, 1-deoxy-D-xylulose 5-phosphate; FPP, farnesyl diphosphate; GGPP, geranylgeranyl diphosphate; GPP, geranyl diphosphate; HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; IPP, isopentenyl diphosphate; MEP, 2-C-methyl-D-erythritol 4-phosphate; MVA, mevalonate. | ||
Studies on the biochemistry and enzymology of plant volatiles have mainly concentrated on the isolation and characterization of enzymes and genes involved in the final steps of the biosynthesis of plant volatiles.13,19 However, the precise biochemical pathways leading to the formation of the volatiles are still unclear. This is because many metabolites involved in the formation of plant volatiles are unstable and occur in plants at trace levels, which means they are difficult to detect. Rapid progress in the methodologies and instruments available for separation and identification of metabolites have allowed for qualitative and quantitative investigations of these metabolites.20–24 Here, we summarize progress in our current understanding of successful techniques used for studying metabolites involved in the metabolic routes of the major classes of plant volatiles (Fig. 1). The advantages and limitations of extraction and analysis protocols for these metabolites are also discussed.
2. Detection and identification of plant volatile compounds
There are many methods available to extract or collect volatiles from plants or foods.25,26 Here, we mostly focus on volatile compounds produced by plants during growth. The volatile compounds can be classified as endogenous or emitted volatiles. For the endogenous volatiles, a direct organic solvent extraction is generally used (Fig. 2A). Many solvents such as hexane, pentane, diethyl ether, dichloromethane, chloroform, ethyl acetate, and solvent mixtures can be used for extraction.27–30 As many enzymes in plants may become active during the preparation of fresh samples, the use of an organic solvent can minimize the activation of enzymes that possibly alter the original composition of volatile compounds. In some cases, a solid-phase extraction (SPE) column has been used to remove some non-volatile compounds from the organic solvent extract of volatile compounds.31 For emitted volatiles, static headspace sampling techniques including solid phase microextraction,32,33 Gerstel Twister,34,35 and Monotrap,36 and dynamic headspace sampling systems have been employed. After the volatiles are extracted or collected, analytical instruments are required for identification of these volatiles. Gas chromatography-mass spectrometry (GC-MS) is a common and powerful technique to analyze volatile compounds, and can give chemical structure information.37,38 For real-time monitoring of the volatile compounds emitted from plants, a zNose technique, which combines a collection device and analytical instrument has been developed.39 The volatile compounds emitted from plants can be simultaneously collected and analyzed by the zNose. The zNose separates compounds by fast gas chromatography (GC) and identifies the compounds with a highly sensitive surface acoustic wave quartz microbalance detector. Because of the high sensitivity of the zNose, volatiles can be collected within intervals as short as 3 min.25 Volatile analysis techniques with high time resolution can accurately monitor volatile emission. For example, using the zNose technique with high time resolution, Arimura et al. found that the nocturnal emission of β-ocimene started slowly and was followed by a very strong emission burst at the beginning of the photophase in MecWorm-treated lima bean leaves.40 By contrast, a technique with low time resolution may omit this important finding (Fig. 2B).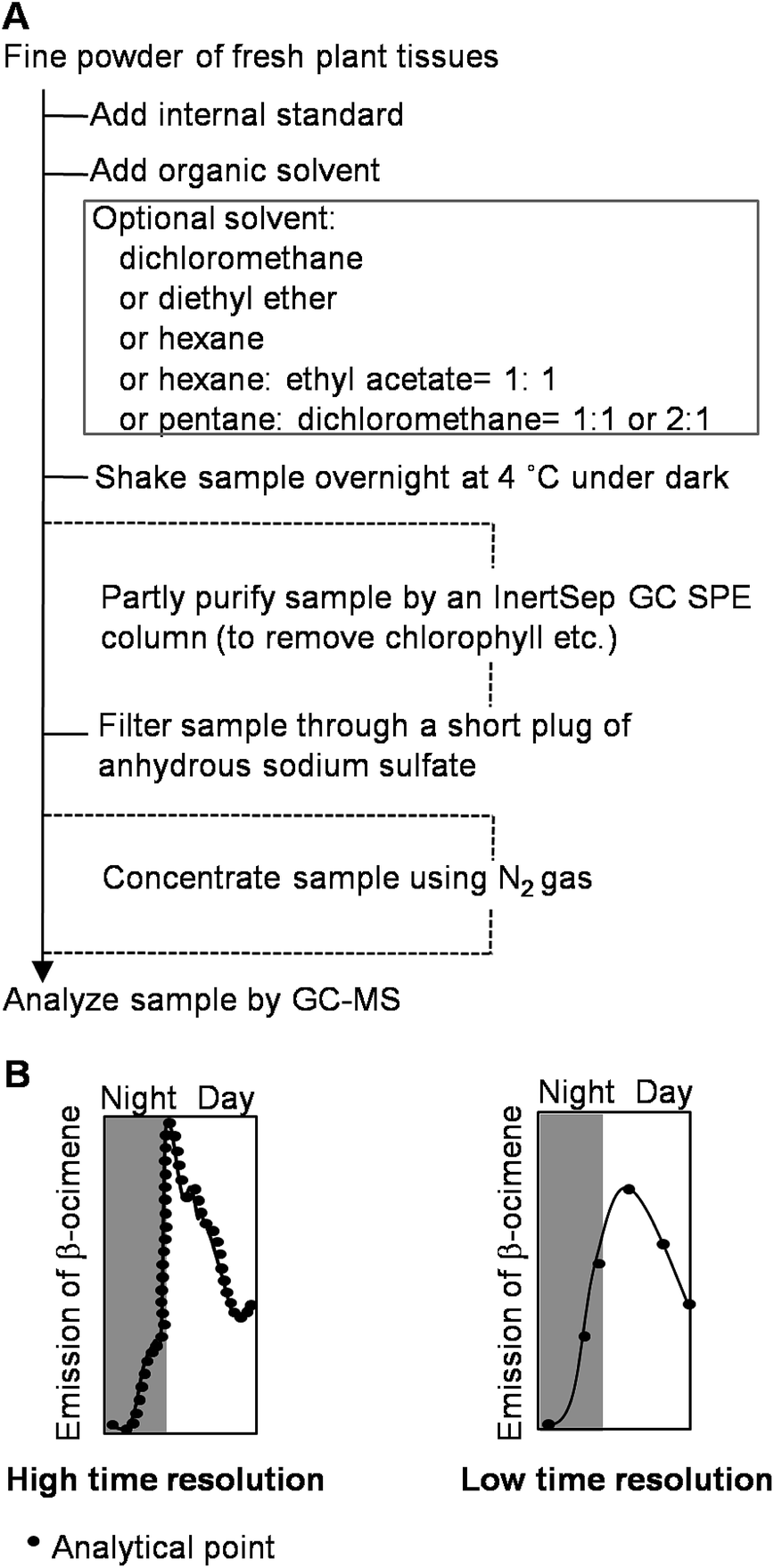 | ||
| Fig. 2 Basic protocol of extraction of endogenous volatiles from plants during the growth of plants using direct solvent organic extraction (A) and importance of high time resolution in analysis of volatile compounds (B). | ||
3. Analysis of metabolites involved in the glycolysis and pentose phosphate pathway
Glycolysis is a primary metabolic route in the central carbon metabolism, playing a very important role in many biological processes and providing precursors for secondary metabolism. The pentose phosphate pathway plays a fundamental role in several cellular processes and is important for biosynthesis and catabolism of five-carbon sugars.41 The concentrations of plant secondary metabolites and changes induced by environmental stresses are of interest for understanding biosynthesis. Phosphoenolpyruvic acid, acetyl CoA, pyruvic acid, erythrose 4-phosphate, and metabolites involved in the glycolysis and pentose phosphate pathways are the main precursors for fatty acids, isoprenoids, carotenoids, and L-Phe, and these lead to the production of a diverse range of volatiles. An indirect method based on spectrophotometric analysis of these compounds has been established for plants.42–45 For simultaneous determination of these metabolites, which have high polarity, low in vivo concentrations, and complex matrices, the preferred technique is mass spectrometry (MS) combined with an isolation technique such as GC, liquid chromatography (LC), or capillary electrophoresis (CE). Two main extraction methods are used to extract these compounds, and the selection of the extraction method depends on the subsequent analytical and quantification methods. If the extracted compounds are to be analyzed by a spectrophotometer, HClO4 is used for the extraction.42–45 Methanol–chloroform–water is used for the extraction if these compounds are to be analyzed by GC-MS,46 CE-MS,47 or LC-MS.48 For GC-MS analysis of these thermo-labile and non-volatile metabolites, pre-column derivatization of the samples is usually required.49 CE-MS analysis does not require a derivatization step, and it is generally suitable for analysis of ionic metabolites, but this technique has poor sensitivity and migration.50 Although LC-MS has mostly been used for analysis of these metabolites in Escherichia coli and yeast and rarely applied to plant samples, it is useful for the determination of sugar phosphates in many plants.51–53 The biggest challenge in LC-MS is the extremely high polarity of these metabolites. Because of the anionic nature of the analytes, ion pair chromatography, which uses a hydrophobic stationary phase and MS compatible solvent to separate ionic compounds on addition of ion pairs with an oppositely charged ion pair reagent, is a promising alternative to ion exchange chromatography. Tetraalkylammonium salts are frequently used as ion pair reagents to separate anionic compounds.54 These salts are non-volatile and therefore incompatible with electrospray ionization (ESI) mass spectrometry. Consequently, the volatile ion pair modifier tributylammonium acetate has been added to the mobile phase for simultaneous separation of sugar phosphates on a common C18 reversed-phase column.514. Analysis of metabolites involved in formation of volatile fatty acid derivatives
Fatty acid derivatives produced from unsaturated fatty acids (linoleic acid and linolenic acid) and saturated fatty acids give plants a “green odor”. Several methods have been reported for extraction of fatty acids. Popular extraction methods for lipids are the Folch extraction,55 the Bligh and Dyer method,56 and Soxhlet extraction.57 Generally, liquid–liquid extractions, such as Bligh and Dyer method, are suitable for lipid extraction from all samples and Soxhlet extraction is convenient only for solid samples.58 In the Folch extraction, a chloroform–methanol mixture (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) is used, and in the Bligh extraction, a chloroform–methanol–water mixture is used. Petroleum ether is the solvent recommended by the AOAC for lipid extraction using the Soxhlet method. However, hexane has also been used for defatting and for analysis of fats, lipids, and fatty acids in oil seeds, cereals, milk, and vegetables.57 GC and LC are the preferred methods for determining these metabolites in plants. GC is the method of choice for characterizing fatty acid profiles of lipids in biological materials, including human food, in the laboratories of various scientific institutions and industrial organizations. In the GC method, lipid derivatization is required to alter the volatility of lipid components and improve peak shape, which provides better separation. There are many derivatization procedures described in the literature, and the majority of these involve conversion of fatty acid components to the corresponding esters, and usually methyl esters. The catalyst used are either acidic (e.g. HCl, H2SO4, and BF3) or alkaline (e.g. sodium methoxide, sodium/potassium hydroxide).59,60 Positional information for double bonds in unsaturated fatty acid methyl esters is considered invaluable. To determine the positions of these double bonds, a method using dimethyl disulfide as an adduct is the most convenient and effective.61–63 Mass spectra of 2-alkenyl-4,4-dimethyloxazoline derivatives of fatty acids are useful for locating double bonds, especially in polyunsaturated and conjugated systems.59,64 Normally, single-column GC is applied. However, for minor compounds with retention times similar to those of large interfering peaks, this technique offers insufficient resolution. Recent studies have shown that comprehensive multidimensional gas chromatography is very useful for unravelling very complex mixtures of analytes, such as the fatty acid mixtures in fish and vegetable oils.65 When GC is not available, high performance liquid chromatography (HPLC) is a useful alternative for accurate quantitative routine analysis.66 For free fatty acids analyzed by HPLC, glacial acetic acid (0.05–0.1%) is normally added to the mobile phase, which is usually 85% methanol in water with a gradient elution to 100% methanol. The glacial acetic acid acts as an ion suppressor and the carboxylic acid remains protonated (non-charged) under acidic conditions (pH < 4).67–69 Sample preparation and derivatization steps for HPLC are time consuming, and methods that do not require derivatization and use MS detection for identification of partially resolved or co-eluting peaks have become popular.
:1, v/v) is used, and in the Bligh extraction, a chloroform–methanol–water mixture is used. Petroleum ether is the solvent recommended by the AOAC for lipid extraction using the Soxhlet method. However, hexane has also been used for defatting and for analysis of fats, lipids, and fatty acids in oil seeds, cereals, milk, and vegetables.57 GC and LC are the preferred methods for determining these metabolites in plants. GC is the method of choice for characterizing fatty acid profiles of lipids in biological materials, including human food, in the laboratories of various scientific institutions and industrial organizations. In the GC method, lipid derivatization is required to alter the volatility of lipid components and improve peak shape, which provides better separation. There are many derivatization procedures described in the literature, and the majority of these involve conversion of fatty acid components to the corresponding esters, and usually methyl esters. The catalyst used are either acidic (e.g. HCl, H2SO4, and BF3) or alkaline (e.g. sodium methoxide, sodium/potassium hydroxide).59,60 Positional information for double bonds in unsaturated fatty acid methyl esters is considered invaluable. To determine the positions of these double bonds, a method using dimethyl disulfide as an adduct is the most convenient and effective.61–63 Mass spectra of 2-alkenyl-4,4-dimethyloxazoline derivatives of fatty acids are useful for locating double bonds, especially in polyunsaturated and conjugated systems.59,64 Normally, single-column GC is applied. However, for minor compounds with retention times similar to those of large interfering peaks, this technique offers insufficient resolution. Recent studies have shown that comprehensive multidimensional gas chromatography is very useful for unravelling very complex mixtures of analytes, such as the fatty acid mixtures in fish and vegetable oils.65 When GC is not available, high performance liquid chromatography (HPLC) is a useful alternative for accurate quantitative routine analysis.66 For free fatty acids analyzed by HPLC, glacial acetic acid (0.05–0.1%) is normally added to the mobile phase, which is usually 85% methanol in water with a gradient elution to 100% methanol. The glacial acetic acid acts as an ion suppressor and the carboxylic acid remains protonated (non-charged) under acidic conditions (pH < 4).67–69 Sample preparation and derivatization steps for HPLC are time consuming, and methods that do not require derivatization and use MS detection for identification of partially resolved or co-eluting peaks have become popular.
13-Hydroperoxylinolenic acid, (12,13S)-epoxylinolenic acid, and octadecanoid 12-oxo-phytodienoic acid (OPDA) are intermediates in the biosynthesis of cis-3-hexenal or jasmonic acid (JA) in plants. Methanol is always used for the extraction of these acids from plants.70 Because of their low content in plants, MS coupled with GC or HPLC is used for detection of these acids.70–72 Commercial internal standards for these compounds are not readily available. Standards can be prepared for 13-hydroperoxylinoleic acid from linoleic acid,73 and heavy isotope-labelled octadecanoid 12-oxo-phytodienoic acid.72
JA could be converted to the volatile ester methyl jasmonate by the enzyme JA carboxyl methyltransferase.74 Methyl esters of secondary metabolites are important components of plant volatiles. Methyl jasmonate is a fragrance compound that was originally identified from the flowers of Jasminum grandiflorum.75 Several quantification methods have been applied to detect endogenous JA, including highly sensitive and reproducible GC-negative ion chemical ionization-MS, which has a limit of detection of −500 fg.71 Currently, normal phase LC-quadrupole time-of-flight MS is used instead of radioimmunoassay and enzyme-linked immunosorbent assay, for the simultaneous discrimination and direct analysis of all four JA stereoisomers without the need for derivatization.76
5. Analysis of metabolites involved in formation of volatile isoprenoids
Although the isoprenoid biosynthetic pathway has been intensively investigated, analytical methodologies for precursors of volatile isoprenoids, for example, DMAPP, GPP, FPP, GGPP, are not well established. To understand the exact biochemical roles of these metabolites and their effects on the formation and emission of volatile isoprenoids, we need to know their levels in plants. However, there are several challenges to overcome when developing methods for determination of these metabolites. Firstly, these compounds are not volatile. Therefore, they cannot be analyzed by GC or GC-MS. Secondly, the ionic nature of the phosphate groups of these compounds makes their isolations using HPLC difficult.77 Thirdly, these compounds do not contain groups with characteristic ultraviolet (UV) adsorption, and are not easily detected by simple UV methods.77 Finally, these compounds have low basal concentrations and are unstable. For example, GPP rapidly degrades in samples, even at 4 °C.78 Several direct and indirect qualitative and quantitative analytical methods have been developed for detection of the precursors of volatile isoprenoids in cultured cells and animal tissues. Detection of these compounds in plants is usually by indirect methods, which typically involve hydrolysis and determination of occurrences and levels of their hydrolyzed products (e.g. linalool and isoprene).Because of the nature of the precursors of volatile isoprenoids, direct analysis of these metabolites is not straightforward.77 Ion-exchange chromatography and CE techniques were used in early attempts at direct analysis of these metabolites.79,80 However, these techniques lack sufficient detection sensitivity and do not allow for analysis of the metabolites at nanomolar or picomolar levels.77 As an alternative, MS has sufficient detection sensitivity and specificity. Recently, HPLC-tandem MS (MS/MS) and ultra HPLC-MS/MS using ion-pair reagents have been used for determination of these metabolites.81,82
6. Analysis of metabolites involved in the formation of carotenoid derived aroma compounds
In addition to their roles in plants as photosynthetic accessory pigments and colorants, carotenoids are thought to be the precursors of terpenoid volatile compounds that contribute to flavor and aroma. Examples include geranyl acetone from phytoene, pseudoionone from lycopene, β-ionone from β-carotene, and α-ionone and pseudoionone from δ-carotene.83,84 Better understanding of these aroma compounds can be achieved by studying their carotenoid precursors. Several methods have been applied for extraction and analysis of carotenoids in plant tissues, and there is no generally accepted standard method for carotenoid extraction in laboratories.85–88 Most extraction methods follow a common path involving release of the desired components from their matrices by disrupting the plant tissue, followed by removal of any unwanted components. Chloroform is more suitable than hexane, light petroleum (boiling point 40–60 °C), diethyl ether, or ethyl acetate as the extraction solvent, and can efficiently extract a broad range of carotenoids.89 Methanol–water–chloroform is particularly effective for extraction of carotenoids from plant materials.90,91 Carotenoids can be divided into two groups, the carotenes group (e.g. lycopene, β-carotene, and α-carotene) and the xanthophylls group (e.g. lutein, β-cryptoxanthin, zeaxanthin, and violaxanthin). Carotenes can be found in their free forms, whereas xanthophylls can be found in their free forms or in a more stable esterified form. Consequently, saponification is required to hydrolyze the esterified forms to free carotenoids before analysis. Saponification performed at 60 °C for 30 min in the dark using 0.02 M or 2.5–6% KOH in methanol provides high yields of free carotenoids.89,92,93A C30 stationary phase provides better separation and selectivity than conventional C18 in HPLC because the longer alkyl chains are better for long-chain analytes. However, the runtime for carotenes and xanthophylls is typically longer (60–100 min) than that with a C18 column (10–25 min) when using a methanol/tert-methyl butyl ether mobile phase. Normally, a photodiode array detector is used to identify carotenoids based on their characteristic spectral properties.89 Recently, MS has been useful for the identification for unknown carotenoids, using the molecular mass and fragmentation patterns.94
7. Analysis of metabolites involved in formation of volatile phenylpropanoids/benzenoids
Several biosynthetic pathways reportedly lead to benzenoid compounds in plants.13 The main pathways are the CoA-dependent, β-oxidation pathway and the CoA-independent, non-β-oxidation pathway. Quantitative analysis of benzenoid/phenylpropanoid-related compounds will help reveal what pathways contribute to the formation of benzenoid compounds in plants. The precursors of volatile phenylpropanoids/benzenoids mainly contain organic acid (e.g. 3-deoxy-arabino-heptulonate 7-phosphate, 3-dehydroquinic acid, 3-dehydroshikimic acid, shikimic acid, shikimate 3-phosphate, 5-enolpyruvylshikimate-3-phosphate, chorismic acid, prephenic acid, phenylpyruvate, trans-cinnamic acid, para-coumaric acid and 3-hydroxy-3-phenylpropionic acid), amino acid (e.g. L-Phe), and phenolic acids conjugated coenzyme A (e.g. trans-cinnamoyl CoA, 3-hydroxy-3-phenylpropionyl CoA, and 3-oxo-3-phenylpropionyl CoA).GC-MS, HPLC, HPLC-MS, CE, and CE-MS have been used for quantitative analysis of organic acids and amino acids in plants. These organic acids are not volatile and can not be detected by GC/MS directly. Derivatization, such as formation of trimethylsilyl derivatives is required and N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) or bis(trimethylsilyl)trifluoroacetamide are commonly used as trimethylsilylation reagents.95 Some organic acids, such as shikimic acid and p-coumaric acid, can be directly detected by HPLC.96–98 Analysis of phenolic acids by HPLC uses UV detection. Preparative sample clean-up using SPE has been applied to improve the resolution between compounds.99 Although no derivatization is needed, the sensitivity may be lower than that obtained with GC-MS. Sometimes insufficient selectivity and efficiency are encountered in HPLC. Among the available methods, HPLC-MS is considered the most sensitive and selective method for organic acids. The development of ionization techniques, such as ESI, has facilitated the analysis of organic acids by HPLC-MS. ESI† provides information on the structures without the need for derivatization. In addition, SPE-LC/MS/MS can be used for simultaneous quantification of short-, medium-, long-chain, and other CoA compounds originating from different biological tissues.100
Because most organic acids lack chromophores, a series of papers has been published reporting on the analysis of phenolic acid by CE.101–103 Recently, CE-MS analysis of sugar phosphates, amino acids, organic acids and CoA compounds has been reported.104 This method offers short analysis times and a high number of theoretical plates. Micellar electrokinetic capillary chromatography has been used for the determination of shikimate and shikimate-3-phosphate in crude plant extracts after extraction with 70% methanol. The use of a polar organic solvent, such as methanol, is acceptable for extraction of highly polar metabolites for CE-MS. Liquid–liquid fractionation following addition of water and chloroform to the methanol extract allows for effective removal of lipid, chlorophyll, and protein contaminants in the sample solution that could interfere with CE-MS analysis.104
For amino acid analysis, the most common methods use amino acid analyzer, HPLC coupled with a diodearray detector105 or a fluorescence detector106 using pre-column derivatization, or HPLC coupled with MS.107,108 Amino acid analyser is typically done by cation-exchange chromatography followed by post-column derivatization with ninhydrin and UV detection. This method lacks throughput and specificity. MS provides high sensitivity and selectivity, but quantitative MS detection often lacks precision. Therefore, isotopically labeled amino acids are required as internal standards for accurate quantitative analysis. Because of its simplicity, sensitivity, and convenience, fluorometry combined with fluorescence derivatization is a popular method for amino acid analysis.
8. Analysis of glycosidically bound volatile compounds
Volatile compounds can be converted from their free forms to glycosidically bound forms, which are more water soluble and less reactive than their free aglycone counterparts. Qualitative and quantitative analyses of glycosidically bound volatile compounds are useful for understanding volatile compounds in plants. However, authentic standards of glycosidically bound volatile compounds are not readily available. In most cases, authentic standards are obtained via syntheses that have several steps.109–112Fig. 3 (part 4) shows a flowchart for the isolation, purification and analysis of glycosidically bound volatile compounds. Methanol–chloroform–water is commonly used as the extraction system.113 The Amberlite XAD-2 column has been widely used for extraction and isolation of glycosidically bound volatile compounds from many plant species, including tea leaves,114 tea flowers,110 lychee,115 pineapple,116 grape,117 ginger,118 and different tissues such as leaves, stems, roots, flowers, and fruits.119 In this method, the Amberlite XAD-2 column is rinsed with distilled water to eliminate sugar, acids, and other water-soluble compounds. Then, the free volatile fraction fixed on the column is eluted using pentane/dichloromethane. Finally, the glycosidically bound fraction is eluted using methanol. The obtained crude extract of glycosidically bound volatile compounds can be analyzed by GC-MS or LC-MS. Because authentic standards are not readily available, enzymatic or acidic hydrolysis of glycosidically bound volatile compounds is used. The free volatiles from the hydrolysis reaction represent the relative contents of glycosidically bound volatile compounds detected by GC-MS. Enzymes used for hydrolysis, such as almond β-glucosidase, can be commercial products or extracted from the target plants.115,120 Hydrochloric acid can be used for hydrolysis but it only gives aglycones and not the sugar residues.115 In addition, the hydrolysis rate is not complete. When authentic standards are available, each glycosidically bound volatile compound can be derivatized and analyzed by GC-MS. MSTFA is widely used as a derivatization reagent. However, the fragmentation patterns are not easy to assign, because the characteristic fragments may be from the cleavage of a sugar moiety. Moreover, the derivatization rate is also not complete. In contrast with indirect analysis by GC-MS combined with enzymatic or acidic hydrolysis and MSTFA derivatization, the direct analysis by LC-MS is more suitable for quantitative analysis.110 There are two points to note for quantitative analysis using LC-MS. Firstly, the crude extracts of glycosidically bound volatile compounds may require multiple purification steps using an ODS-C18 SPE column to obtain a good chromatogram.110 Secondly, the internal standard selection is important. The best internal standard is the stable isotope labeled glycosidically bound volatile compound. However, these labeled compounds are not easy to obtain and their chemical syntheses are complicated. Consequently, p-nitrophenyl (pNP)-glycine, pNP-glutamic acid, and pNP-Pri can be used instead.
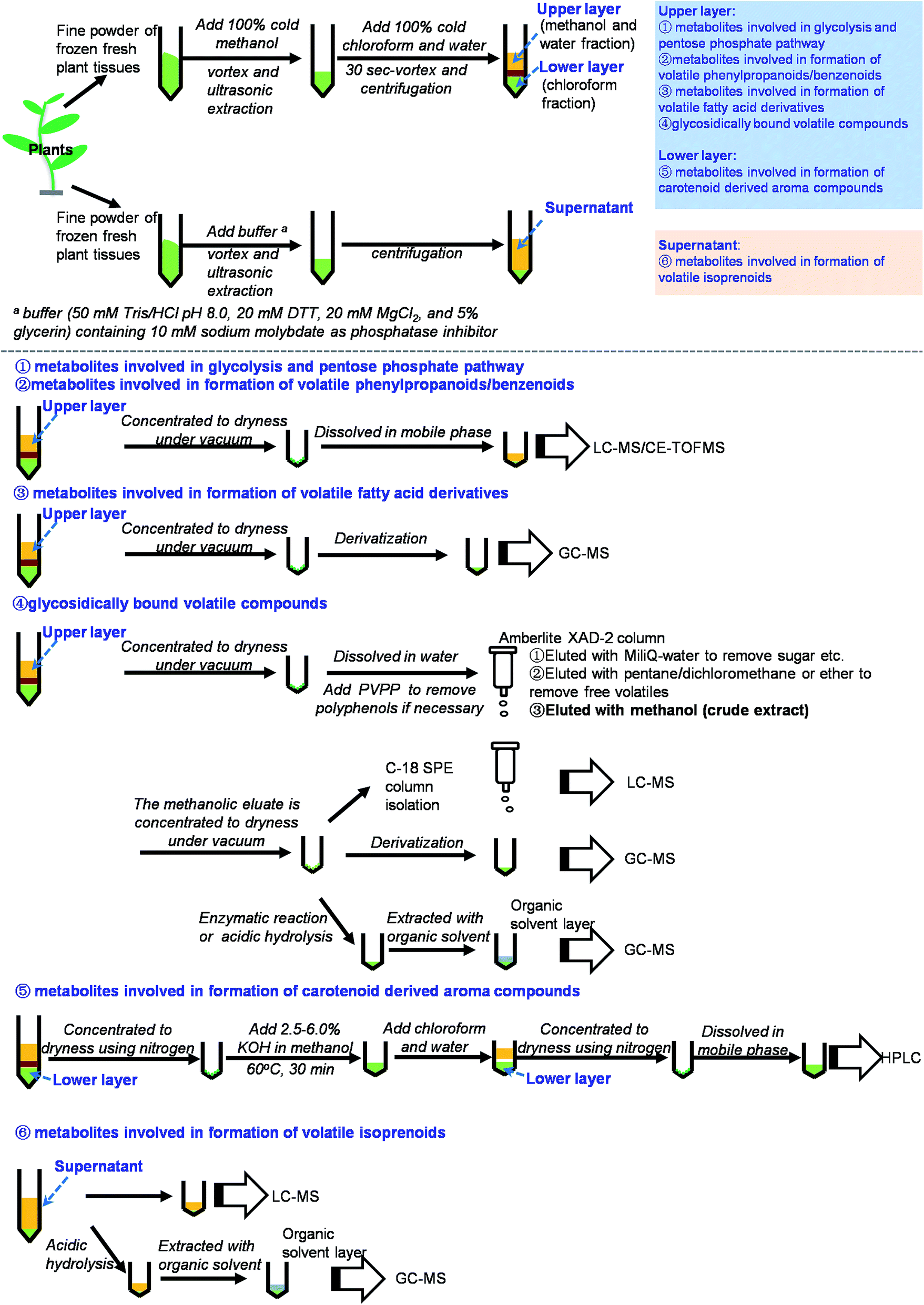 | ||
| Fig. 3 Summary on scheme of extraction and analysis of metabolites involved in biosynthesis of plant volatile compounds. | ||
Enzymatic or acidic hydrolysis and GC-MS analysis is suitable for the aglycone qualitative and relative quantitative analyses of glycosidically bound volatiles when the authentic standards are not available. When the authentic standards are available, the derivatization and GC-MS analysis can be used for qualitative and relative quantitative analyses of glycosidically bound volatiles, and LC-MS can be used for precise quantitative analysis.
9. Concluding remarks and perspectives
This paper provides a comprehensive review of the current knowledge of analytical methodologies for metabolites involved in biosynthesis of plant volatile compounds. Based on previous reports and our own investigations, we have summarized methods for the extraction and analysis of these metabolites involved in biosynthesis of plant volatile compounds (Fig. 3 and Table S1†). Methanol, chloroform, and water are the most useful solvents for extraction of most metabolites involved in biosynthesis of plant volatile compounds. LC-MS is the useful method for detection of these compounds. By comparison, NMR is less sensitive, GC-MS requires derivatization, and LC with UV detection requires the presence of a detectable chromophore in each analyte.20,23 However, several important issues should be addressed in future studies:(1) Methods should be improved for simultaneous analysis of multiple target metabolites.
(2) Because of the differing and complex compositions in samples, some established methodologies that are successful in certain plant species may not work for other plant species. Therefore, methodologies for different plant species or tissues should be improved.
(3) Many authentic standards are unavailable commercially. Establishment of an open access database, including the MS and NMR spectra of the metabolites, will be very helpful for development of this research area.
(4) Some metabolites are very unstable and occur in plants at trace levels. Consequently, more stable and sensitive extraction and analysis protocols should be developed.
(5) Currently methodologies for many metabolites only focused on relative quantitative analyses. Precise quantitative analytical methodologies should be established.
Acknowledgements
A part of the research aspects done by the authors are supported by the financial supports from the National Natural Science Foundation of China (31670690, 31601787, and 31500244), the Guangdong Natural Science Foundation (2016A030306039, 2016A030313652, and 2014A030310131), the Guangdong Innovation Team of Modern Agricultural Industry Technology System (2016LM1143), and the “100 Talents Programme of the Chinese Academy of Sciences” (Y321011001 and 201209). Because of space limitations, we could not cite all publications in the field; we apologize to all colleagues whose work has not been mentioned.References
- F. Negre-Zakharov, M. C. Long and N. Dudareva, in Plant-derived Natural Products, Synthesis, Function, and Application, ed. A. E. Osbourn and V. Lanzotti, Springer, Berlin, Heidelberg, 2009, pp. 405–431 Search PubMed.
- E. Pichersky and J. Gershenzon, Curr. Opin. Plant Biol., 2002, 5, 237 CrossRef CAS PubMed.
- C. E. Vickers, J. Gershenzon, M. T. Lerdau and F. Loreto, Nat. Chem. Biol., 2009, 5, 283 CrossRef CAS PubMed.
- F. Dong, X. M. Fu, N. Watanabe, X. G. Su and Z. Y. Yang, Molecules, 2016, 21, 124 CrossRef PubMed.
- M. E. Maffei, J. Gertsch and G. Appendino, Nat. Prod. Rep., 2011, 28, 1359 RSC.
- E. Pichersky, J. P. Noel and N. Dudareva, Science, 2006, 331, 808 CrossRef PubMed.
- N. Dudareva and E. Pichersky, in Biology of Floral Scent, ed. N. Dudareva and E. Pichersky, CRC Press Taylor & Francis Group, Boca Raton, 2006, pp. 55–78 Search PubMed.
- J. Q. Kong, RSC Adv., 2015, 5, 62587 RSC.
- P. Singh, R. M. Kalunke and A. P. Giri, RSC Adv., 2015, 5, 106886 RSC.
- D. Tieman, M. Taylor, N. Schauer, A. R. Fernie, A. D. Hanson and H. J. Klee, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8287 CrossRef CAS PubMed.
- M. Sakai, H. Hirata, H. Sayama, K. Sekiguchi, H. Itano, T. Asai, H. Dohra, M. Hara and N. Watanabe, Biosci., Biotechnol., Biochem., 2007, 71, 2408 CrossRef CAS PubMed.
- A. P. Jarvis, O. Schaaf and N. J. Oldham, Planta, 2000, 212, 119 CrossRef CAS PubMed.
- J. Boatright, F. Negre, X. L. Chen, C. M. Kish, B. Wood, G. Peel, I. Orlova, D. Gang, D. Rhodes and N. Dudareva, Plant Physiol., 2004, 135, 1 CrossRef PubMed.
- A. Klempien, Y. Kaminaga, A. Qualley, D. A. Nagegowda, J. R. Widhalm, I. Orlova, A. K. Shasany, G. Taguchi, C. M. Kish, B. R. Cooper, J. C. D'Auria, D. Rhodes, E. Pichersky and N. Dudareva, Plant Cell, 2012, 24, 2015 CrossRef CAS PubMed.
- H. Maeda, A. K. Shasany, J. Schnepp, I. Orlova, G. Taguchi, B. R. Cooper, D. Rhodes, E. Pichersky and N. Dudareva, Plant Cell, 2010, 22, 832 CrossRef CAS PubMed.
- H. Maeda, H. Yoo and N. Dudareva, Nat. Chem. Biol., 2011, 7, 19 CrossRef CAS PubMed.
- V. Tzin and G. Galili, Mol. Plant, 2010, 3, 956 CrossRef CAS PubMed.
- H. Hirata, T. Ohnishi, H. Ishida, K. Tomida, M. Sakai, M. Hara and N. Watanabe, J. Plant Physiol., 2012, 169, 444 CrossRef CAS PubMed.
- Z. Y. Yang, M. Sakai, H. Sayama, T. Shimeno, K. Yamaguchi and N. Watanabe, J. Plant Physiol., 2009, 166, 887 CrossRef CAS PubMed.
- K. Dettmer, P. A. Aronov and B. D. Hammock, Mass Spectrom. Rev., 2007, 26, 51 CrossRef CAS PubMed.
- S. U. Kadam, N. N. Misra and N. Zaima, RSC Adv., 2016, 6, 33537 RSC.
- V. Gupta, R. S. Thakur, C. R. K. Reddy and B. Jha, RSC Adv., 2013, 3, 7037 RSC.
- A. C. Martin, A. D. Pawlus, E. M. Jewett, D. L. Wyse, C. K. Angerhofer and A. D. Hegeman, RSC Adv., 2014, 4, 26325 RSC.
- C. E. Rodriguez-Lopez, C. Hernandez-Brenes and R. I. Diaz de la Garza, RSC Adv., 2015, 5, 106019 RSC.
- D. Tholl and U. S. R. Röse, in Biology of Floral Scent, ed. N. Dudareva and E. Pichersky, CRC Press Taylor & Francis Group, Boca Raton, 2006, pp. 3–25 Search PubMed.
- Z. Y. Yang, S. Baldermann and N. Watanabe, Food Res. Int., 2013, 53, 585 CrossRef CAS.
- V. Bergougnoux, J. C. Caissard, F. Jullien, J. L. Magnard, G. Scalliet, J. M. Cock, P. Hugueney and S. Baudino, Planta, 2007, 226, 853 CrossRef CAS PubMed.
- E. Alissandrakis, D. Daferera, P. A. Tarantilis, M. Polissiou and P. C. Harizanis, Food Chem., 2003, 82, 575 CrossRef CAS.
- A. Pérez-Silva, E. Odoux, P. Brat, F. Ribeyre, G. Rodriguez-Jimenes, V. Robles-Olvera, M. A. García-Alvarado and Z. Günata, Food Chem., 2006, 99, 728 CrossRef.
- X. Fu, S. Cheng, Y. Zhang, B. Du, C. Feng, Y. Zhou, X. Mei, Y. Jiang, X. Duan and Z. Yang, Food Chem., 2017, 221, 356 CrossRef CAS PubMed.
- C. F. Poole, TrAC, Trends Anal. Chem., 2003, 22, 362 CrossRef CAS.
- G. Ouyang and J. Pawliszyn, Anal. Bioanal. Chem., 2006, 386, 1059 CrossRef CAS PubMed.
- W. Wang, L. Zhang, Z. Li, S. Zhang, C. Wang and Z. Wang, RSC Adv., 2016, 6, 113951 RSC.
- B. Kolahgar, A. Hoffmann and A. C. Heiden, J. Chromatogr., 2002, 963, 225 CrossRef CAS PubMed.
- D. Bressanello, E. Liberto, C. Cordero, P. Rubiolo, G. Pellegrino, M. R. Ruosi and C. Bicchi, Food Chem., 2017, 214, 218 CrossRef CAS PubMed.
- H. Matsui, T. Inui, K. Oka and N. Fukui, Food Chem., 2016, 202, 15 CrossRef CAS PubMed.
- F. Azimi and M. H. Fatemi, RSC Adv., 2016, 6, 111197 RSC.
- S. Lv, Y. Wu, J. Wei, M. Lian, C. Wang, X. Gao and Q. Meng, RSC Adv., 2015, 5, 87806 RSC.
- J. Lammertyn, E. A. Veraverbeke and J. Irudayaraj, Sens. Actuators, B, 2004, 98, 54 CrossRef CAS.
- G. Arimura, S. Köpke, M. Kunert, V. Volpe, A. David, P. Brand, P. Dabrowska, M. E. Maffei and W. Boland, Plant Physiol., 2008, 146, 965 CrossRef CAS PubMed.
- P. M. Dewick, Medicinal Natural Products: A Biosynthetic Approach, Wiley, 3rd edn, 2009 Search PubMed.
- J. M. Mutuku and A. Nose, Plant Cell Physiol., 2012, 53, 1017 CrossRef CAS PubMed.
- Y. C. Du, A. Nose, K. Wasano and Y. Uchida, Funct. Plant Biol., 1998, 25, 253 CAS.
- R. C. Leegood and R. T. Furbank, Planta, 1984, 162, 450 CrossRef CAS PubMed.
- H. Usuda, M. Ku and G. Edwards, Funct. Plant Biol., 1984, 11, 509 CAS.
- J. Lisec, N. Schauer, J. Kopka, L. Willmitzer and A. R. Fernie, Nat. Protoc., 2006, 1, 387 CrossRef CAS PubMed.
- H. Yamakawa and M. Hakata, Plant Cell Physiol., 2010, 51, 795 CrossRef CAS PubMed.
- B. Luo, K. Groenke, R. Takors, C. Wandrey and M. Oldiges, J. Chromatogr. A, 2007, 1147, 153 CrossRef CAS PubMed.
- U. Roessner, C. Wagner, J. Kopka, R. N. Trethewey and L. Willmitzer, Plant J., 2000, 23, 131 CrossRef CAS PubMed.
- J. Y. Cai and J. Henion, J. Chromatogr. A, 1995, 703, 667 CrossRef CAS.
- B. Luo, K. Groenke, R. Takors, C. Wandrey and M. Oldiges, J. Chromatogr. A, 2007, 1147, 153 CrossRef CAS PubMed.
- S. U. Bajad, W. Lu, E. H. Kimball, J. Yuan, C. Peterson and J. D. Rabinowitz, J. Chromatogr. A, 2006, 1125, 76 CrossRef CAS PubMed.
- A. Jannasch, M. Sedlak and J. Adamec, in Metabolic Profiling, Methods in Molecular Biology, ed. T. O. Metz, Springer Berlin, Heidelberg, 2011, pp. 159–171 Search PubMed.
- P. R. Haddad and P. E. Jackson, Ion Chromatography, Principles and Applications, Elsevier, Amsterdam, 1990, p. 172 Search PubMed.
- J. Folch, M. Lees and G. H. S. Stanley, J. Biol. Chem., 1957, 226, 497 CAS.
- E. G. Bligh and W. J. Dyer, Can. J. Biochem. Physiol., 1959, 37, 911 CrossRef CAS PubMed.
- A. M. Almazan and S. O. Adeyeye, J. Food Compos. Anal., 1998, 11, 375 CrossRef CAS.
- P. Manirakiza, A. Covaci and P. Schepens, J. Food Compos. Anal., 2001, 14, 93 CrossRef CAS.
- B. Qi, T. Fraser, S. Mugford, G. Dobson, O. Sayanova, J. Butler, J. A. Napier, A. K. Stobart and C. M. Lazarus, Nat. Biotechnol., 2004, 22, 739 CrossRef CAS PubMed.
- K. S. Liu, J. Am. Oil Chem. Soc., 1994, 71, 1179 CrossRef CAS.
- S. Shibamoto, T. Murata and K. Yamamoto, Lipids, 2016, 51, 1077 CrossRef CAS PubMed.
- E. Dunkelblum, S. H. Tan and P. J. Silk, J. Chem. Ecol., 1985, 11, 265 CrossRef CAS PubMed.
- G. W. Francis, Chem. Phys. Lipids, 1981, 29, 369 CrossRef CAS.
- L. Fay and U. Richli, J. Chromatogr. A, 1991, 541, 89 CrossRef CAS.
- H. J. de Geus, I. Aidos, J. de Boer, J. B. Luten and U. A. T. Brinkman, J. Chromatogr. A, 2001, 910, 95 CrossRef CAS PubMed.
- J. P. Spychalla and S. L. Desborough, Plant Physiol., 1990, 94, 1207 CrossRef CAS PubMed.
- J. T. Lin, T. A. McKeon and A. E. Stafford, J. Chromatogr. A, 1995, 699, 85 CrossRef CAS.
- B. Marcato and G. Cecchin, J. Chromatogr. A, 1996, 730, 83 CrossRef CAS.
- A. Mehta, A. M. Oeser and M. G. Carlson, J. Chromatogr. B: Biomed. Sci. Appl., 1998, 719, 9 CrossRef CAS.
- A. Müller, P. Düchting and E. W. Weiler, Planta, 2002, 216, 44 CrossRef PubMed.
- M. J. Mueller and W. Brodschelm, Anal. Biochem., 1994, 218, 425 CrossRef CAS PubMed.
- B. A. Stelmach, A. Müller, P. Hennig, D. Laudert, L. Andert and E. W. Weiler, Phytochemistry, 1998, 47, 539 CrossRef CAS PubMed.
- M. O. Funk, R. Isaac and N. A. Porter, Lipids, 1976, 11, 113 CrossRef CAS PubMed.
- M. S. Song, D. G. Kim and S. H. Lee, J. Plant Biol., 2005, 48, 292 CrossRef CAS.
- G. i. Arimura, R. Ozawa, T. Shimoda, T. Nishioka, W. Boland and J. Takabayashi, Nature, 2000, 406, 512 CrossRef CAS PubMed.
- Y. Han, Z. Zhou, H. Wu, H. Nie, R. Lei, Y. Bai and H. Liu, J. Chromatogr. A, 2012, 1235, 125 CrossRef CAS PubMed.
- G. Nürenberg and D. A. Volmer, Anal. Bioanal. Chem., 2012, 402, 671 CrossRef PubMed.
- S. A. Holstein, H. Tong, C. H. Kuder and R. J. Hohl, Lipids, 2009, 44, 1055 CrossRef CAS PubMed.
- R. E. Dugan, E. Rasson and J. W. Porter, Anal. Biochem., 1968, 22, 249 CrossRef CAS PubMed.
- R. K. Keller, P. Bangalore, J. Robert and M. G. Swanson, J. Chromatogr. A, 1996, 737, 325 CrossRef CAS.
- L. Henneman, A. G. van Cruchten, S. W. Denis, M. W. Amolins, A. T. Placzek, R. A. Gibbs, W. Kulik and H. R. Waterham, Anal. Biochem., 2008, 383, 18 CrossRef CAS PubMed.
- L. Henneman, A. G. van Cruchten, W. Kulik and H. R. Waterham, Biochim. Biophys. Acta, 2011, 1811, 227 CrossRef CAS PubMed.
- E. Lewinsohn, Y. Sitrit, E. Bar, Y. Azulay, A. Meir, D. Zamir and Y. Tadmor, J. Agric. Food Chem., 2005, 53, 3142 CrossRef CAS PubMed.
- W. Schwab, R. Davidovich-Rikanati and E. Lewinsohn, Plant J., 2008, 54, 712 CrossRef CAS PubMed.
- S. Rivera and R. Canela, Molecules, 2012, 17, 11255 CrossRef CAS PubMed.
- D. B. Rodriguez-Amaya, J. Food Compos. Anal., 2010, 23, 726 CrossRef CAS.
- A. Singh, S. Ahmad and A. Ahmad, RSC Adv., 2015, 5, 62358 RSC.
- E. Yara-Varon, A. S. Fabiano-Tixier, M. Balcells, R. Canela-Garayoa, A. Bily and F. Chemat, RSC Adv., 2016, 6, 27750 RSC.
- P. D. Fraser, M. E. Pinto, D. E. Holloway and P. M. Bramley, Plant J., 2000, 24, 551 CrossRef CAS PubMed.
- C. J. Xu, P. D. Fraser, W. J. Wang and P. M. Bramley, J. Agric. Food Chem., 2006, 54, 5474–5481 CrossRef CAS PubMed.
- X. M. Fu, W. B. Kong, G. Peng, J. Y. Zhou, M. Azam, C. J. Xu, D. Grierson and K. S. Chen, J. Exp. Bot., 2012, 63, 341 CrossRef CAS PubMed.
- E. García-de Blas, R. Mateo, J. Viñuela and C. Alonso-Álvarez, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 341 CrossRef PubMed.
- M. C. Chan, S. H. Ho, D. J. Lee, C. Y. Chen, C. C. Huang and J. S. Chang, Biochem. Eng. J., 2013, 78, 24 CrossRef CAS.
- B. Y. Hsu, Y. S. Pu, B. S. Inbaraj and B. H. Chen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 899, 36 CrossRef CAS PubMed.
- Z. F. Katona, P. Sass and I. Molnár-Perl, J. Chromatogr. A, 1999, 847, 91 CrossRef CAS.
- N. Ozturk, M. Tuncel and N. B. Tuncel, J. Liq. Chromatogr. Relat. Technol., 2007, 30, 587 CrossRef CAS.
- P. Valentão, G. Lopes, M. Valente, P. Barbosa, P. B. Andrade, B. M. Silva, P. Baptista and R. M. Seabra, J. Agric. Food Chem., 2005, 53, 3626 CrossRef PubMed.
- A. Schieber, P. Keller and R. Carle, J. Chromatogr. A, 2001, 910, 265 CrossRef CAS PubMed.
- P. Oliveira, J. A. Pereira, P. B. Andrade, P. Valentão, R. M. Seabra and B. M. Silva, Food Chem., 2008, 111, 393 CrossRef PubMed.
- C. Magnes, M. Suppan, T. R. Pieber, T. Moustafa, M. Trauner, G. Haemmerle and F. M. Sinner, Anal. Chem., 2008, 80, 5736 CrossRef CAS PubMed.
- C. Mardones, A. Hitschfeld, A. Contreras, K. Lepe, L. Gutiérrez and D. von Baer, J. Chromatogr. A, 2005, 1085, 285 CrossRef CAS PubMed.
- Q. F. Zhang and H. Y. Cheung, Phytochem. Anal., 2011, 22, 18 CrossRef CAS PubMed.
- W. Klampfl, W. Buchberger and P. R. Haddad, J. Chromatogr. A, 2000, 881, 357 CrossRef PubMed.
- K. Harada and E. Fukusaki, Plant Biotechnol., 2009, 26, 47 CrossRef CAS.
- M. Calull, J. Fábregas, R. M. Marcé and F. Borrull, Chromatographia, 1991, 31, 272 CAS.
- N. Tateda, K. Matsuhisa, K. Hasebe, N. Kitajima and T. Miura, J. Chromatogr. B: Biomed. Sci. Appl., 1998, 718, 235 CrossRef CAS.
- R. Rebane, M. L. Oldekop and K. Herodes, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 904, 99 CrossRef CAS PubMed.
- R. Rebane and K. Herodes, J. Chromatogr. A, 2012, 1245, 134 CrossRef CAS PubMed.
- S. J. Ma, M. Mizutani, J. Hiratake, K. Hayashi, K. Yagi, N. Watanabe and K. Sakata, Biosci., Biotechnol., Biochem., 2001, 65, 2719 CrossRef CAS PubMed.
- Y. Zhou, F. Dong, A. Kunimasa, Y. Zhang, S. Cheng, J. Lu, L. Zhang, A. Murata, F. Mayer, P. Fleischmann, N. Watanabe and Z. Yang, J. Agric. Food Chem., 2014, 62, 8042 CrossRef CAS PubMed.
- S. Matsumura, S. Takahashi, M. Nishikitani, K. Kubota and A. Kobayashi, J. Agric. Food Chem., 1997, 45, 2674 CrossRef CAS.
- A. Mulkens and I. Kapetanidis, J. Nat. Prod., 1988, 51, 496 CrossRef CAS PubMed.
- J. Wirth, W. Guo, R. Baumes and Z. Günata, J. Agric. Food Chem., 2001, 49, 2917 CrossRef CAS PubMed.
- D. Wang, T. Yoshimura, K. Kubota and A. Kobayashi, J. Agric. Food Chem., 2000, 48, 5411 CrossRef CAS PubMed.
- C. C. Chyau, P. T. Ko, C. H. Chang and J. L. Mau, Food Chem., 2003, 80, 387 CrossRef CAS.
- P. Wu, M. C. Kuo, T. G. Hartman, R. T. Rosen and C. T. Ho, J. Agric. Food Chem., 1991, 39, 170 CrossRef CAS.
- J. Wirth, W. Guo, R. Baumes and Z. Günata, J. Agric. Food Chem., 2001, 49, 2917 CrossRef CAS PubMed.
- P. Wu, M. C. Kuo and C. T. Ho, J. Agric. Food Chem., 1990, 38, 1553 CrossRef CAS.
- G. Schulz and E. Stahl-Biskup, Flavour Fragrance J., 1991, 6, 69 CrossRef CAS.
- Y. Z. Gunata, C. L. Bayonove, R. L. Baumes and R. E. Cordonnier, J. Chromatogr. A, 1985, 331, 83 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00766c |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2017 |