
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective adhesive cell capture without molecular specificity: new surfaces exploiting nanoscopic polycationic features as discrete adhesive units†
S. Kalasina,
E. P. Browneb,
K. F. Arcarob and
M. M. Santore*a
aDepartment of Polymer Science and Engineering, 120 Governors Drive, Amherst, MA 01003, USA. E-mail: Santore@mail.pse.umass.edu
bDepartment of Veterinary and Animal Science, 240 Thatcher Road, Amherst, MA 01003, USA
First published on 28th February 2017
Abstract
This work explored how molecularly non-specific polycationic nanoscale features on a collecting surface control kinetic and selectivity aspects of mammalian cell capture. Key principles for selective collector design were demonstrated by comparing the capture of two closely related breast cancer cell lines: MCF-7 and TMX2-28. TMX2-28 is a tamoxifen-selected clone of MCF-7. The collector was a silica surface, negatively-charged at pH 7.4, containing isolated molecules (∼8 nm diameter) of the cationic polymer, poly(dimethyl-aminoethylmethacrylate), pDMAEMA. Important in this work is the non-selective nature of the pDMAEMA interactions with cells: pDMAEMA generally adheres negatively charged particles and cells in solution. We show here that selectivity towards cells results from collector design: this includes competition between repulsive interactions involving the negative silica and attractions to the immobilized pDMAEMA molecules, the random pDMAEMA arrangement on the surface, and the concentration of positive charge in the vicinity of the adsorbed pDMAEMA chains. The latter act as nanoscopic cationic surface patches, each weakly attracted to negatively-charged cells. Collecting surfaces engineered with an appropriate amount pDMAEMA, exposed to mixtures of MCF-7 and TMX2-28 cells preferentially captured TMX2-28 with a selectivity of 2.5. (This means that the ratio of TMX2-28 to MCF cells on the surface was 2.5 times their compositional ratio in free solution.) The ionic strength-dependence of cell capture was shown to be similar to that of silica microparticles on the same surfaces. This suggests that the mechanism of selective cell capture involves nanoscopic differences in the contact areas of the cells with the collector, allowing discrimination of closely related cell line-based small scale features of the cell surface. This work demonstrated that even without molecular specificity, selectivity for physical cell attributes produces adhesive discrimination.
Introduction
Advances in the capture of rare circulating tumor cells, relevant to metastasis and treatment evaluation,1–5 have sparked broader interest in the enrichment of various types of targeted cells.6,7 Cell capture/enrichment schemes often bring cells to interfaces and rely on immobilized antibodies or other specialized molecules (e.g. aptamers) for selectivity. Compared with approaches employing solution-phase antibodies, immobilization of targeting molecules introduces complexity by restricting their access and adding non-specific interactions involving the particle or the microfluidic surface.8,9 Immunomagnetic beads, as an example, achieve purities of 97% at best10 and 50% at worst (in the cases we found).9,11 Further, not all targeted cells present surface markers such as EpCam.12,13 Additionally, other markers are shared by tumor and healthy cells but presented at different levels.14,15 The current paper explores how interfacial interactions, which are molecularly non-specific, can be exploited to distinguish cells by features other than surface markers and how the background interactions, which sometimes give rise to undesired adhesion, can be exploited in accomplishing targeted capture.Established fractionation methods producing cell cuts based on size, dielectric properties, and/or density, are powerful when used upstream of other methods, and also show promise on their own for discriminating circulating epithelial cells.16–19 Accordingly, we expect proliferation of methods that exploit physical differences in cells, as the cells themselves become better characterized in terms of their mechanical and interfacial physical–chemical properties. One example is the well-known enhanced binding and selectivity associated with the clustering of targeted groups rather than their uniform distribution.20–22 This multivalency effect could be refined and put into practice if the targets, i.e., the arrangements of markers on the cell surfaces, were better understood.
Electrostatic interactions, because they are often longer range than some interactions driving molecular recognition (for example hydrogen bonding), could be exploited to enhance or shift molecular targeting. Studies with non-biological particles suggest that electrostatic interactions could, on their own23,24 or combined with molecular recognition molecules,25,26 facilitate selective capture of cells. For instance, on solid surfaces that lack specific targeting molecules, nanoscale distributions of surface charge such as cationic regions or “patches”, facilitate adhesion explained only by the charge distribution itself and not the average surface charge or zeta potential.27,28 (The term “patch” was previously employed to describe a nanoscopic region containing multiple charges. The abstraction of adsorbed pDMAEMA to surface patches, in this work, emphasizes that the details of the adsorbed chain configuration are unimportant.) It has been further shown that collecting surfaces can be engineered with precisely-controlled regions of positive or negative charge to produce sharp selectivity of target particles, based on the local curvature of the particles at the point of contact.27,29
The current study explores the translation of the “nanoscale polycationic region” or “cationic patch” mechanism from silica particles28 to mammalian cells. In this study, we employ collecting surfaces with nanoscale regions of cationic charge, previously well-characterized and well-understood in terms of their physical makeup and interactions with silica particles.28 These collecting surfaces are microscope slides, whose negatively charged silica surfaces (at pH ∼ 7) are modified with extremely small amounts of a cationic polymer, pDMAEMA [poly(dimethylaminomethylmethacrylate)] so that individual isolated chains present islands (∼8 nm) of cationic charge in a sea of negative charge from the silica.29 These polymer chains have been shown to be immobilized30 and flat to the surface,31 and arranged in a random distribution within the plane.32 They are completely retained over a broad ionic strength range and also in a moderate pH range bracketing the buffered pH 7.4 conditions studied here. Suspended cells, whose surfaces are net negative in charge, are flowed gently over these collectors, as shown in Fig. 1, and their capture behavior recorded. The capture behavior of two similar breast cancer cell lines, MCF-7, which is often used as a model of a circulating tumor cell,8,33–35 and TMX2-28, a tamoxifen-selected clone of MCF-7, is compared and used to guide the design of surfaces that selectively capture one of the two cell lines. In order to establish mechanism, the influence of salt is examined and cell capture is benchmarked against a model system of silica particles.
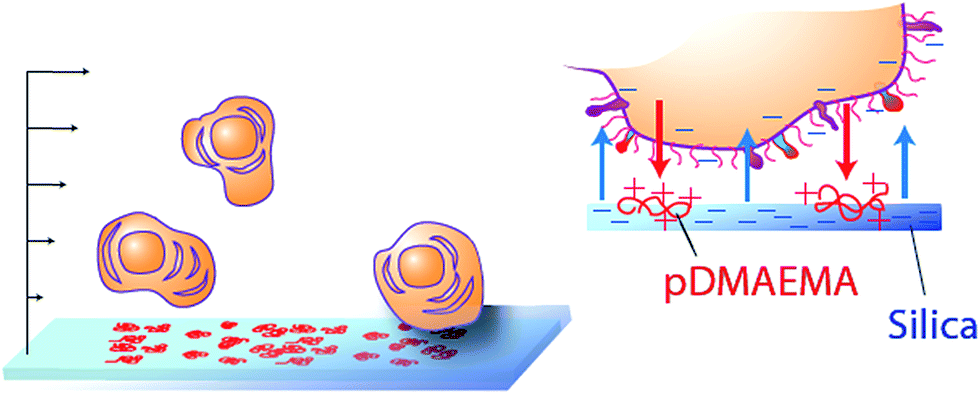 | ||
| Fig. 1 Schematic of cell capture platform. Gently flowing cells encounter a negative surface containing a low level of randomly arranged, immobilized, nanoscale (8–10 nm) cationic patches, here made of coils of pDMAEMA adsorbed on silica. The main negative surface repels the cells while the cationic patches are electrostatically attractive. | ||
Results
Key features
The first series of cell-capture studies were conducted using cell suspensions that each contained a single cell line. Buffered suspensions of cells (∼106/mL, pH 7.4) were flowed gently through a slit shear chamber, with a wall shear rate of 22 s−1, corresponding to a wall stress of 23 mPa. Cell accumulation on the engineered surface, comprising one wall of the flow chamber, was tracked in real time using optical video microscopy. Cell accumulation rates are compared in Fig. 2, for TMX2-28 or MCF-7 cells in single-cell line suspensions. Here the osmolarity and pH were fixed but the ionic strength was varied. Cells in standard phosphate buffer (osmolarity 280 mOsm, ionic strength, I = 0.176 M, and Debye length κ−1 = 0.74 nm) are compared with low ionic strength buffer containing the same concentrations of buffering salts, Na2HPO4 and KH2PO4, but achieving a lower ionic strength of I = 0.005 M because NaCl is not included. Sucrose was added, near a concentration of 0.25 M, to maintain the same osmotic pressure (280 mOsm).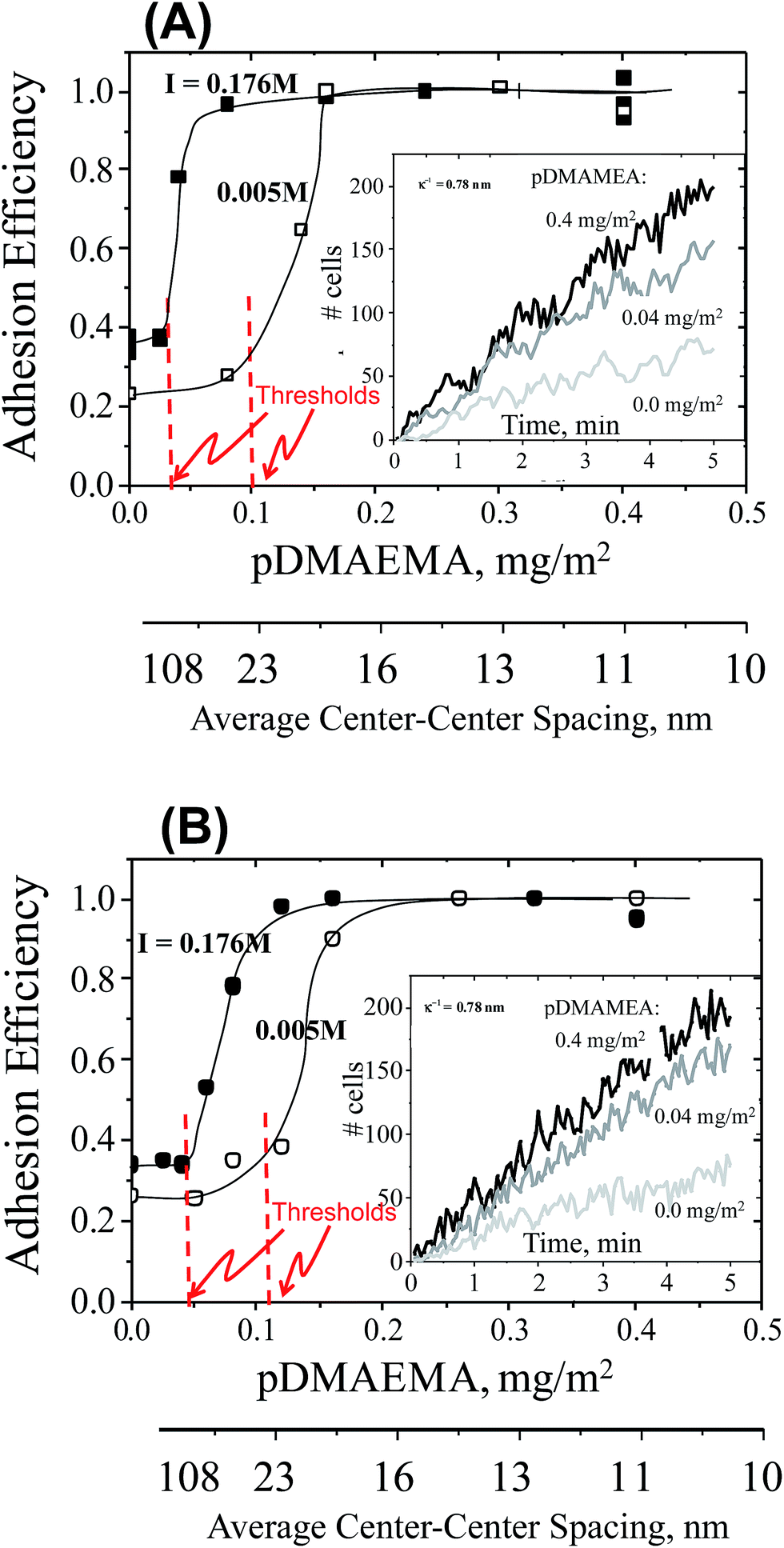 | ||
| Fig. 2 Main: adhesion efficiencies (measured accumulation rate normalized on the transport-limited rate) of cells in two different ionic strengths buffers (I = 0.176 M filled symbols, and 0.005 M open symbols) on collectors of varied pDMAEMA content (on the x-axis) and a wall shear rate of 22 s−1. Lines guide the eye. Adhesion thresholds sketched in. Inset: 3 example data sets for the accumulation of cells on collectors with different pDMAEMA loadings. The slopes of these data each contribute a datum in the main figure. Part (A): all cells are TMX2-28. Part (B): all cells are MCF-7. | ||
The main parts of Fig. 2 summarize the initial cell capture rates, for TMX2-28 in part A and MCF-7 in part B, as a function of the amount of pDMAEMA on the collecting wall. The capture rates are represented as capture efficiencies. Capture efficiency is the observed capture rate normalized on the maximum or transport-limited rate, which occurs on a strongly adhesive surface. Since the capture rates on test surfaces are proportional to the cell concentration (which varied somewhat from cell batch to cell batch, made fresh for each day of experiments) each was datum calibrated by the rate measured on the highly-adhesive saturated pDMAEMA surface. In this way, batch-to-batch variations in cell concentration were eliminated. Data points in Fig. 2 represent the average of 3 runs on each type of surface, with data points always falling within 10% of each other. Since pDMAEMA loading is sparse on test surfaces, a second x-axis in Fig. 2 provides physical perspective by showing the average center–center spacing between pDMAEMA molecules or “patches”, calculated from the known pDMAEMA loading.
The initial cell capture rates of Fig. 2 are dominated, by design, by cell–collector pairwise interactions: the cell density on the collector is sufficiently low that captured cells do not interfere with the approach of additional ones. The example data in the insets of Fig. 2 confirm the linearity of the initial cell accumulation on the surfaces, proving that the captured cells do not hinder subsequent cell capture. The data in the main figure are determined by slopes of data like those in the inset. Of note, viability 1 minute before each run and 5 minutes after capture was confirmed by trypan blue staining: initial cell viability was always in excess of 98% and there was no significant difference between free and captured cell viability.
Key qualitative features in Fig. 2 are common to all data sets. First without any pDMAEMA on the glass, cells are captured at a low rate that depends slightly on ionic strength. Next, within a given series that varies the pDMAEMA loading using many test surfaces, a low pDMAEMA-independent cell capture rate is evident at modest pDMAEMA loadings on the collector. Only above a threshold in the pDMAEMA loading does the pDMAEMA influence cell capture, and then cell capture increases substantially. The effect of additional pDMAEMA terminates when the cell capture rate reaches an efficiency of unity. Here the cells are captured at the transport-limited rate, the maximum possible, equal to the arrival rate of the cells to the interface and dependent only on the cell concentration, chamber geometry and flow.
Significant for both cell types in Fig. 2, and discussed below, is a shifting of the adhesion threshold to higher pDMAEMA loadings at lower ionic strength. There is an additional small but important difference in the exact threshold values of the two cell lines.
The appearance of a threshold has been previously reported for the capture of well-characterized particles, such as 1 μm silica microspheres, on collecting surfaces similar to ones in this study.28,30 Accordingly as a benchmark, for the pH and ionic conditions of the cell studies of Fig. 2 and 3 summarizes silica microparticle capture on the same series of collecting surfaces used in the cell capture study, with each point the average of two runs for surfaces of a targeted composition (and error less than 10%). Like the cells, the silica particles exhibit an ionic strength-dependent threshold in the pDMAEMA needed for capture, and approach an efficiency of unity at high pDMAEMA loadings. The main qualitative difference between the capture of microspheres and cells is the lack of silica microsphere capture on collectors containing no pDMAEMA. This is a result of the strong negative charge of the silica microspheres, which produces a strong electrostatic repulsion towards silica collecting surfaces containing no pDMAEMA.27,28 The similarities between the cell and microparticle capture, despite the substantial differences between cells and particles, is the key to understanding the mechanism of capture, discussed below.
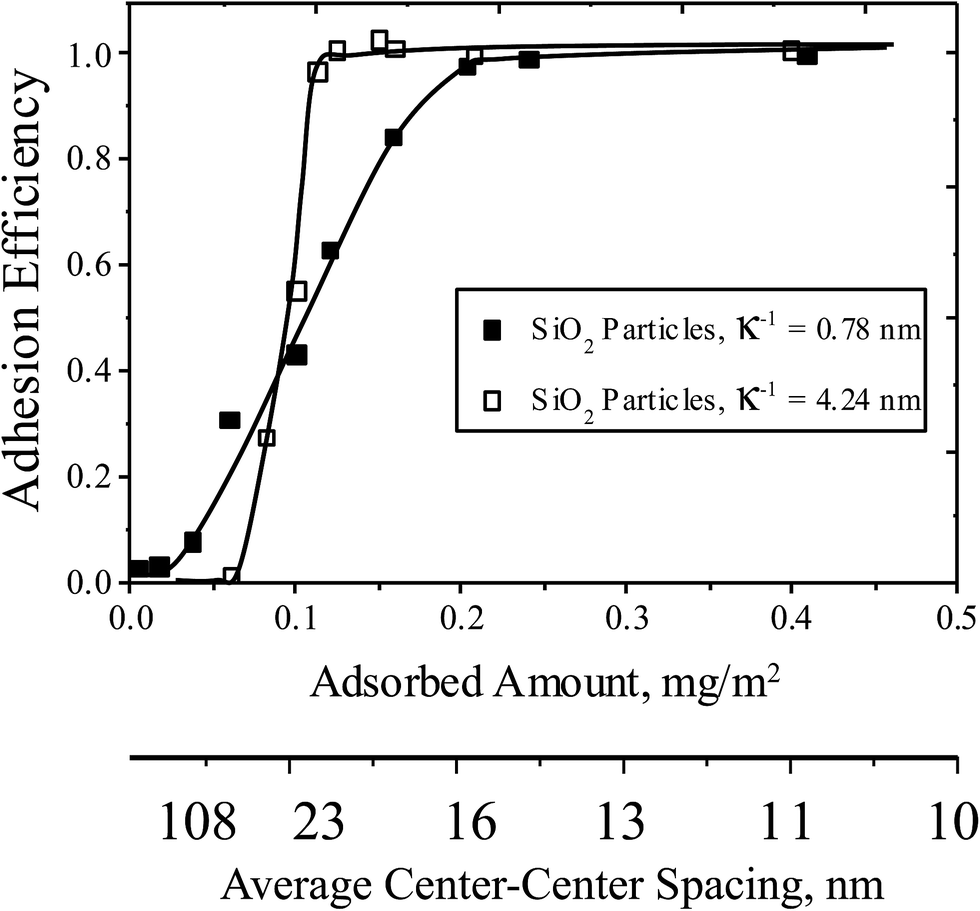 | ||
| Fig. 3 Adhesion efficiencies (measured accumulation rates normalized on the transport-limited rate), of silica microspheres on surfaces with different amounts of pDMAEMA, at a wall shear rate 22 s−1 and ionic strengths 5 mM (hollow points) and 176 mM (solid points). The pH is 7.4. Curves are drawn to guide the eye. | ||
Selective capture
The difference between the adhesion thresholds for MCF-7 and TMX2-28 cells, though small, was thought to be sufficient to facilitate selective cell capture. Engineering a single collecting surface between the thresholds of two competing particulate species has been shown to allow one of the two to be selectively captured with relatively high purity, for instance with selectivity S > 0.99.27,29 Here, selectivity S, between two competing species is defined as the ratio of their free solution concentration ratio to the surface concentration ratio. For example if a 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :65 (number ratio) of A and B in solution produces a surface ratio of 70:30, then the selectivity S = (7/3)/(35/65) = 4.33. Notably this definition implies selectivity itself is independent of the solution composition, appropriate for dilute suspensions. A given selectivity value predicts a surface composition for a particular proportion of competing species in solution.
:65 (number ratio) of A and B in solution produces a surface ratio of 70:30, then the selectivity S = (7/3)/(35/65) = 4.33. Notably this definition implies selectivity itself is independent of the solution composition, appropriate for dilute suspensions. A given selectivity value predicts a surface composition for a particular proportion of competing species in solution.
Using F-TMX2-28, a TMX2-28 line modified to express GFP (green fluorescent protein), we compared in Fig. 4, cell capture from 50–50 mixtures of F-TMX2-28 and MCF-7 to those of individual cell lines in suspension. We first established, in the ESI,† that the capture rates of TMX2-28 and F-TMX2-28 are identical over the full range of collector compositions. For the mixture experiments in Fig. 4, key collector compositions are considered: bare glass, a saturated pDMAEMA layer, and compositions between the thresholds for the two cell lines. For each mixture datum in Fig. 4, 3 runs are averaged. Data points were within 10%, with typical spread shown at 0 and 0.4 mg m−2 pDMAEMA on the x-axis. The inset of Fig. 4 summarizes the calculated selectivity values at key pDMAEMA surface loadings. The selectivity values were averaged from 3 runs of each collector composition and 10 areas (470 μm by 350 μm at a magnification of 10×) of each surface imaged. Example images of the surfaces with their captured cells are included in Fig. 5. In Fig. 5, all cells are visible in bright field, but only F-TMX2-28 are visible in fluorescence. The rightmost panels are bright field images with dots manually placed on the F-TMX2-28 cells, based on the fluorescence image.
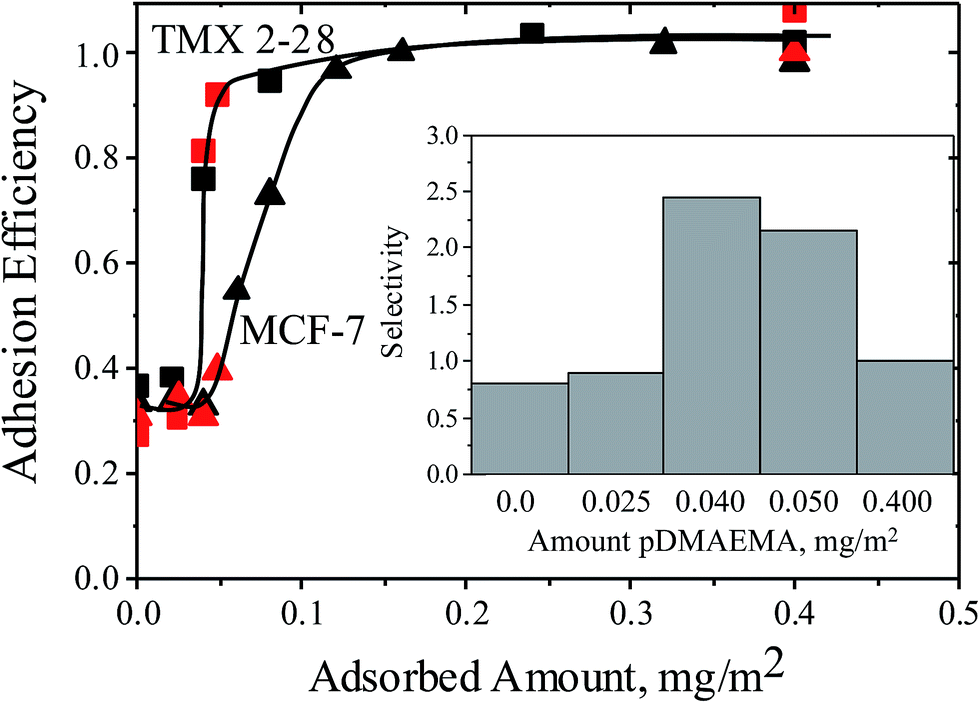 | ||
| Fig. 4 Capture rates for TMX2-28 (squares) and MCF-7 (triangles) cells, comparing data for single cell lines (black data from Fig. 2), to data obtained using 1:1 mixtures of the two cell lines (red). Curves are drawn to guide the eye. The flow rate is rate 22 s−1 and the buffer has an ionic strength of 0.176 M. The inset shows the selectivities measured from the mixture runs at specific collector compositions. | ||
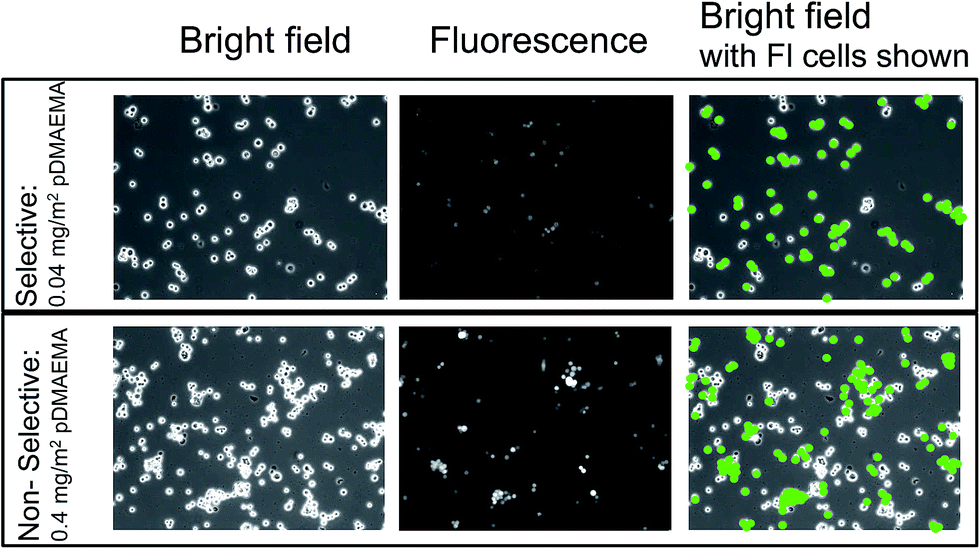 | ||
| Fig. 5 Typical bright field and fluorescence micrographs (indicated) showing cells on selective and nonselective collectors containing 0.04 and 0.004 mg m−2 of pDMAEMA. MCF-7 and F-TMX2-28 have been captured from 1:1 mixtures. The two rightmost panels identify the F-TMX2-28 cells based on a comparison of the bright field and fluorescent images on the left and middle. Images are 470 × 350 μm. | ||
Fig. 4 and 5 establish two important observations. First, preferential capture of F-TMX2-28 occurs from 50–50 mixtures of F-TMX2-28 and MCF-7 on collectors that fall between the thresholds compositions for the two cell types. Selectivity vanishes (approaches unity, indicating no preference) when the pDMAEMA loading of the collector is above both thresholds and it is also insignificant below both thresholds when cell capture is uninfluenced by the polycations. The second point borne out in Fig. 4 is that cell capture from a 50–50 mixture occurs with the same efficiency as a function of collector composition, as seen for the suspensions of single cell types. This result has implications beyond the utility of suspensions of single cell types to guide the design of surfaces that will contact cell mixtures. The observation argues that cells of different types do not influence each other during the capture process and therefore, the same selectivity values would be obtained in studies where the two cells types were present in different, in fact any, proportion in bulk solution. Said differently, Fig. 4 compares a 50:50 mixture of two cell types to the extreme compositions of 0:100 and 100:0 and finds perfect agreement in the cell capture efficiencies. The data in Fig. 4 therefore bracket the full possible range of bulk solution cell compositions for this pair of cell types. This taken together with the linearity of cell capture with time, for instance in Fig. 2, argue further that at the cell concentration of 106/mL and below, the cell capture behavior is well-described by Fig. 4.
Discussion
This work demonstrates the ability of a negatively charged surface containing randomly-arranged flat nanoscale regions of positive charge, to adhesively discriminate closely-related cell lines with selectivity S = 2.5. This selectivity might, naively, be considered low compared with the antibody-based targeting.9 It is important to remember, however, that the adhesive pDMAEMA patches are cationic homopolymers that are not, themselves fundamentally selective: polycations such as pDMAEMA are well established to adhere indiscriminately to negative targets, for instance in solution.36,37 Further, the two cell types studied here are both breast epithelial cancer lines and one, TMX2-28, is a tamoxifen-selected clone of the MCF-7 line. Worth noting, we did not happen to attempt this work with other cell lines and are publishing the current results, which represent a substantial body of work. Because we had no “throw away data” with other cells, the result are all the more compelling. Put in this perspective, then, the findings here are exciting and unanticipated: adhesive discrimination of closely related cells using simple materials without biomolecular recognition is an enticing prospect.Separation of particulates by physical means, for instance sedimentation, exclusion chromatography, or electrophoresis, requires a difference in size, charge, or density. MCF-7 and TMX2-28 cells do not noticeably exhibit morphological differences, such as size, shape, or appearance.38,39 Further, the two cell lines possess identical zeta potentials of −10 to −12 mV. Their differences lie primarily in resistance to the commonly-used endocrine treatment for estrogen-receptor-positive breast cancer, tamoxifen. This tamoxifen-resistance has rendered TMX2-28 cells estrogen receptor-negative, with increased proliferation and invasion capabilities that are supported by changes in gene expression.38–46 Thus hydrodynamic and other physical methods such as dielectrophoresis would not be expected to distinguish these two cell types. While the selectivity values reported in Fig. 4 are remarkable on their own, the background cell capture on the bare microscope slides and slides containing low pDMAEMA loadings is the sole reason why better selectivity was not obtained. Such background adhesion can also plague technologies that employ immobilized antibodies. In the current study no attempt was made to eliminate background adhesion. Future refinements to eliminate adhesion on the base-substrate will be guided by an understanding of the mechanism for the selective capture.
Mechanism of capture and discrimination: thresholds and dependence on pDMAEMA
The fate of particles and cells, to be captured or to flow past a collector, is determined by particle–surface interactions occurring in the instants when the free particle or cell is within nanometers of the surface. This capture behavior is distinct from subsequent cell response, rearrangement, and deformation.Evidence for the mechanism of cell capture and selectivity is found in the features of the capture curves of Fig. 2 and 3: the adhesion thresholds and the shift of cell capture curves with ionic strength. Indeed, the strong influence of ionic strength argues that capture is dominated by electrostatic interactions. The similarities between cell and silica microsphere capture, where the latter is well understood, provide a basis for understanding cell capture.
The adhesion thresholds, which guide the design of collectors that discriminate cells, are among the key features of cell and microparticle capture on surfaces containing cationic patches. We define an adhesion threshold to be a distinct cut off in the loading of pDMAEMA on the collector, below which no impact of pDMAEMA on cell or particle capture is observed.
Silica particles are electrostatically repelled from negatively charged silica flats in the absence of pDMAEMA. Indeed silica collectors have a net negative charge, even with pDMAEMA loadings up to 0.2 mg m−2. The collectors are substantially negative on average, having a zeta potential of −40 to −50 mV (ref. 32) when pDMAEMA loadings are 0.02–0.1 mg m−2, near the adhesion thresholds. It is the randomly placed pDMAEMA patches that are responsible for microparticle capture despite this net repulsion. However, individual surface-immobilized pDMAEMA molecules are incapable, at the conditions in Fig. 3, of capturing and holding silica microspheres, because their attractions are individually too weak to overcome the background electrostatic repulsion. The argument for this is as follows: if single pDMAEMA molecules were sufficiently attractive towards silica microspheres, then it would be possible to create collectors with extremely low pDMAEMA loadings that captured the microparticles, and the capture efficiency curve would trend towards the origin in plots like Fig. 3. Instead of trending towards the origin, particle capture as a function of pDMAEMA loading exhibits an adhesion threshold. Therefore adhesion thresholds are a signature of weak interactions between microparticles and immobilized cationic patches, and capture occurs only when particles encounter multiple patches. This is a form of multivalency, here facilitated by the random arrangement of the patches on the collector. Such upward-curving binding curves, reported by us for particle capture28,30 are established for multivalent molecular interactions.47–49
Cell capture is similar to silica microparticle capture in key ways. Both MCF-7 and TMX2-28 cells have overall negative charge, with zeta potentials of −10 to −12 mV, albeit weaker than the silica particles, whose zeta potentials are −35 to −40 mV at κ−1 = 1 nm. The cells, therefore experience some electrostatic repulsion towards the bare silica collectors. However, a low level of cell capture is observed on bare silica, perhaps by hydrogen bonding or by the attractions of cationic groups on the cells with the negative silica collector. (Both would involve cell surface functionality not present on the silica microparticles.) It is an important finding that low loadings of pDMAEMA on the collector in Fig. 2 do not influence the low level of cell capture resulting from cell–silica interactions. Instead, pDMAEMA visibly drives capture only above a cell-line-specific pDMAEMA loading on the collector. This suggests that, as with the case of silica microspheres, single sparsely situated pDMAEMA patches on the silica collector are individually too weak to capture and hold MCF-7 or TMX2-28 cells. For the pDMAEMA patches to influence cell capture, MCF-7 or TMX2-28 cells must encounter a region of surface that has a sufficiently high number of pDMAEMA patches to overcome electrostatic repulsions. If individual pDMAEMA molecules could capture and hold cells, data would trend immediately upward from the y-intercept in Fig. 2, instead of the observed zero slope near the y-intercept.
How salt influences the adhesion threshold
The parallel influence of ionic strength on particle and cell capture, especially on the thresholds in Fig. 2 and 3, is important because it establishes that cells and particles interact through a similar mechanism with the collector. Our prior treatment to predict the adhesion thresholds of singlet28 and aggregate particles,27 as a function of surface charge and local curvature, thus provides a foundation for qualitative understanding and future predictive treatments of cell–collector interactions, including the adhesion thresholds and, ultimately, selectivity.Ionic strength couples, through the Debye length κ−1, with local particle or cell curvature of radius a, to determine the effective area of interaction, of radius Rzoi ∼ (aκ−1)1/2 on the collector,27,50 surrounding a given contact point, shown in Fig. 6. For negatively charged microspheres or cells on collectors such as ours, which comprise negatively charged flats containing small randomly-distributed small cationic features, the interaction area in turn determines whether the overall interaction at a given contact point is attractive or repulsive. For instance, with small contact areas, a particle will tend to experience interactions that are either overall repulsive or attractive, depending on where it touches the spatially heterogeneous substrate. Particles will pass over net repulsive regions and eventually adhere on attractive regions, which carry a locally above average density of cationic features. When the contact area is large due to gradual local curvature or low ionic strength, the particle or cell will experience a more nearly-average surface character which, in our work, is substantially negative. Hence it will not find an opportunity to adhere at any point on the collector. This mechanism, which also produces shifts of the adhesion threshold with ionic strength, is well-quantified for spheres.28 As suggested by the parallel impact of ionic strength on cell and particle capture in Fig. 2 and 3, the mechanism must also govern the interaction of cells with the same collectors.
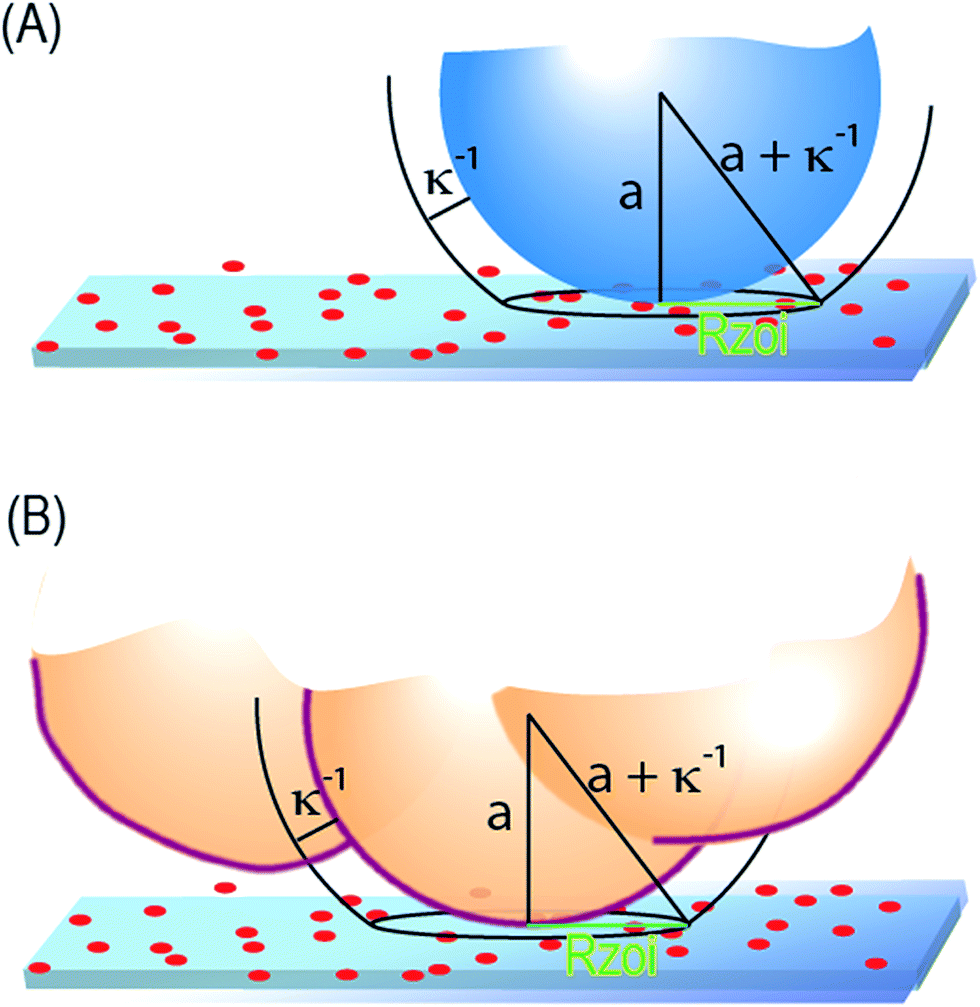 | ||
| Fig. 6 (A) Illustration of the zone of influence for a spherical particle interacting electrostatically with a flat, where a is the sphere radius. (B) The zone of influence for the electrostatic interaction of an irregularly shaped cell with a flat collector. Here a is the curvature of the cell where it contacts the surface. | ||
The presence of a threshold and its dependence on ionic strength are distinct signatures of the interaction of a curved object with a repulsive surface containing randomly distributed discrete weakly-adhesive stickers. Curvature based adhesive-selectivity has been established for both spheres and irregular silica aggregates.27,28,50 The differences in adhesion thresholds were translated to adhesive selectivity, in excess of 99, in the case of particle mixtures differing in size by only a factor of 2. Large aggregates could be separated from uniform spheres of the same overall size, with the same selectivity.27 This implies that similarly sized cells of identical surface chemistry but different submicron and nanometric curvature and protrusions could be adhesively discriminated, with preference given to the cells with more sharply-curved objects. A similar principle was recently borne out in shape-sensitivity of delivery package interactions with other breast cells.51
Translating the mechanism to cell separation
The presence of ionic-strength-dependent adhesion thresholds for pDMAEMA-driven cell-capture, and the similarities of cell capture to particle capture on the same surfaces at the same ionic conditions, argues for a strong role of curvature in cell capture. In Fig. 6, and in the geometrical calculation of the effective contact radius, Rzoi, Debye length and particle curvature are equivalent. Debye length is shown to shift the adhesion thresholds with particles and cell, and curvature is known to shift the thresholds for particles. Thus, slight differences in local curvature of similarly surface charged TMX2-28 and MCF-7 cells, for instance arising from cellular protrusions, could well explain their different adhesion thresholds. Engineering collectors between the adhesion thresholds of two cell types allows selectivity to be engineered into cell capture.Given the complexity of cell surfaces, however, it is worth considering other potential contributions to observed differences in the capture of the two cell lines. The curvature-sensitive mechanism of cell interactions acts through the size of the interactive zone, Rzoi: cells having smaller interactive areas with the collector area more readily captured. Thus if the cell surface or its protrusions are soft enough to be hydrodynamically deformed (flattened) at nanoscopic length scales during approach to the surface, Rzoi will be increased and the threshold will shift to the right. Thus, if MCF-7 were more nanoscopically compliant than TMX2-28 but otherwise identical, the threshold of MCF-7 would appear to the right of TMX2-28, as was observed. Non-uniform distributions of surface charge on the cell, not detectible for instance by zeta potential, might also produce differences in cell adhesion. Thus, besides the microscopic cell shape in the absence of force, nanoscale cell mechanics and charge heterogeneity on the cell will influence cell capture and could enhance, detract from, or dominate selectivity.
It remains a grand challenge to physically and mechanically characterize cells with sufficient precision to enable rational design of collectors such as ours. While studies have revealed differing stiffnesses in immobilized cells52–55 and differing deformabilities in suspended cells,56–58 for instance comparing healthy and cancerous cells or different cancer cells, the particular length and time scales of the deformation is often not clear and is critical. Specific separations strategies distinguish only a particular range in cell properties: the deformations relevant to our method could be as small as a few nanometers, evidenced by the sensitivity to ionic strength. Thus care must be taken when interpreting literature reports of cell deformation to ensure that the reported differences in cell mechanics are truly relevant. We did not find a report comparing TMX2-28 and MCF-7 cell mechanics; however, it is noted that the studies referenced here report greater deformability in cancerous cells compared with non-tumorigenic lines and the greatest deformabilities in more metastatic lines.
A final point regarding the mechanism of capture and discrimination is worth mentioning. It is critical to the particle-specific or cell-specific thresholds that there is substantial background (here electrostatic) repulsion which is overcome by the randomly distributed stickers.50 A more neutral background surface or one made only mildly non-adhesive by the placement of lubricating polymer films, for instance, may not necessarily provide sufficient background repulsion to produce target-sensitive adhesion thresholds. Therefore the appropriate type of surface passivation to eliminate the background adhesion is challenging and must preserve the competition between weak discrete attractions and uniform repulsion.
Prospects
The creation of inexpensive materials with adhesive selectivity for targeted cells or the ability to adhesively cut or fractionate cells is a translational goal with potential impact in lab-scale analytics and patient-scale cell-based therapeutics. Materials that exploit commodity-scale compounds [such as acrylics, including (pDMAEMA)] rather than purified biomolecular fragments or compounds synthesized sequentially (such as peptides), benefit from lower cost, greater robustness, and longer storage life, opening the door to wide use. Our ability to exploit inexpensive materials in a physical mechanism that adhesively distinguishes closely-related cell lines is encouraging, as is the observation of cell viability over the (admittedly short) experimental timescale.The selectivity of S = 2.5 reported here is remarkable considering the relationship of the two cell lines. An important point is that this selectivity is a property of the surface which applies strictly to a single pass operation and does not account for transport. Separations technologies, for instance chromatography and microfluidic methods, incorporate functional surfaces, but also rely on other features such as staging or extended contact with a surface. A column or device output can exceed the intrinsic surface selectivity to obtain a purer product. Thus incorporation of the current surfaces into devices and columns with appropriate transport and hydrodynamic designs would yield a device capable of purification beyond that achieved on a single contact. For instance, in order to capture all the TMX2-28 cells from a mixture, one requires a device that enables all cells to encounter the active surface.59 Further, if future generations of surfaces such as ours can achieve selectivities in excess of 98–99, then a single pass becomes adequate and the material becomes competitive with immobilized antibodies. Elimination of the nonspecific adhesion (onto the non-pDMAEMA surface regions) is key to achieving this goal and a target we believe to be realizable.
A few other points are worth mentioning. First, the concepts demonstrated here employ acrylic polymers as polycationic units. The principles explained above, using weakly adhesive randomly distributed stickers against a background repulsive field, producing adhesion thresholds, do not place specific molecular requirements on the patches beyond an attraction to the target. This means the principles demonstrated here for pDMAEMA are likely to apply for other polycations. Indeed we have demonstrated similar thresholds and highly-selective (in excess of 99) for surfaces containing poly-L-lysine and gold-core nanoparticulate stickers that manipulate of silica particles27 and proteins.60 The findings reported here with pDMAEMA are therefore expected to be general, though interfacial compositions may differ quantitatively if other polycations are used. Also the physical mechanism suggests further generalization to other cells. MCF-7 and TMX2-28 are representative of epithelial cells in terms of physical properties, for instance size and negative charge. The parallels between cell behavior and microparticles make a strong argument for generalizability. Also, the current data are the only cells we studied on these surfaces, before shifting efforts to improved surfaces in a separate project. No data were eliminated (for instance failures with other cell lines) in developing this paper.
Conclusions
This work demonstrates the design of surfaces, made from adhesive components which are individually non-selective in their cellular interactions but which, when immobilized in a random configuration on a cell-repelling surface, adhesively discriminate similar cell lines. In this study, a selectivity of 2.5 is reported for the preferential capture of TMX2-28 cells from mixtures containing TMX2-28 and MCF-7 lines, when collecting surfaces are designed with the appropriate amount of weakly-binding randomly-distributed pDMAEMA coils immobilized on a negatively-charged slide.Cell capture as a function of collector design parameters was quantified in detail and the impact of ionic strength was additionally probed. The influence of collector composition and ionic strength on cell capture was further benchmarked against the capture of microparticles. Both cell and microparticle systems exhibited similar features and trends, supporting a similar mechanism for capture. The presence of thresholds in the pDMAEMA content on the collector, needed to facilitate the polycation-driven capture of cell or microparticle targets, is an indicator that multiple pDMAEMA molecules engage each particle or cell in the moments leading to its capture. Substantial capture does not occur when a particle or cell encounters a single immobilized pDMAEMA molecule. The numbers of pDMAEMA molecules needed, at the point of cell or particle contact with the collector, depends on the area of the collector that exerts force on the particle or cell. This area is sensitive not only to ionic strength but also to the particle or cell curvature at the point of contact. For rigid objects this area is fixed, but for soft objects it is sensitive to mechanics: nanometer-scale deformations as a cell approaches the surface change curvature and can influence selectivity. The mechanism is also sensitive to the local charge in the region of contact and is, accordingly, sensitive to heterogeneous charge distributions on the particles or cells. These factors lead to different adhesion thresholds for different objects, allowing surfaces to be engineered to adhesively discriminate objects in suspension.
In summary, this work has demonstrated how weak pairwise discrete attractions between a collecting surface and suspended cells can produce adhesive selectivity towards a particular cell type that would be difficult to discriminate by methods relying on biomolecular recognition, size, or net electrostatic charge.
Experimental description
Preparation of collector surfaces, in pH 7.4 phosphate buffer (0.002 M KH2PO4 and 0.008 M Na2HPO4), used Fisherfinest (Fisher Scientific) microscope slides, and poly(dimethylaminoethyl methacrylate), pDMAEMA, MW 31300 a gift from DuPont. Slides were soaked in concentrated sulfuric acid overnight and rinsed in DI water before being mounted in the flow chamber in which their surfaces were modified. The flow chamber itself is a standard laminar slit flow chamber measuring 25 × 10 × 1 mm. Functional surfaces were prepared by first flowing buffer, then flowing a buffered 5 ppm pDMAEMA solution for a controlled period of time, and then re-introducing the flowing buffer, all at a wall shear rate of 5 s−1. The amount of pDMAEMA adsorbed to the surface was previously studied in detail using near-Brewster reflectometry27,61 and calibration curves for the adsorbed amounts were checked with each new stock solution of pDMAEMA, as needed to ensure precision control, within 0.015 mg m−2 of deposited pDMAEMA. Due to the strong electrostatic attractions between the polycation and the negative silica surface, the pDMAEMA is immobilized on the timescales and conditions in this study.62,63
The MCF-7 cells were purchased from American Type Culture Collection (Manassas, VA 20108 USA) and the TMX2-28 cells were obtained from Dr John Gierthy (New York State Department of Health). MCF-7, TMX2-28, and F-TMX2-28 were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% calf serum, 10 μg mL−1 insulin, 100× non-essential amino acids, 10000 μg mL−1 penicillin–streptomycin, and 200 mM L-glutamine as previously described.41 Cells preserved in liquid nitrogen were passaged twice before being utilized for experiments. To dissociate the adherent cells from the flask, media was removed, the cells were rinsed with phosphate buffered saline and 2 mL of 0.25% (w/v) trypsin/0.53 mM EDTA was added to each flask. The cells were then incubated at 37 °C/5% CO2 for 5 minutes. The trypsin was neutralized with DMEM + 5% calf serum, and a cell pellet was obtained by spinning the neutralized cell suspension for 5 minutes at 200 × g. The cells were counted using a hemocytometer, and the cell pellet was brought to a concentration of 106 cells per mL of PBS. The cell solution was then used within 1 hour for subsequent capture experiments. Cells were always pulse vortexed briefly within minutes of experiments to ensure single cell suspension. Cell viability was assessed using a trypan blue stain.
Cells were studied at a concentration of ∼106/mL in phosphate buffered saline (0.002 M KH2PO4, 0.008 M Na2HPO4, and 0.15 M NaCl) or at lower ionic strength but with the identical osmotic pressure (0.002 M KH2PO4, 0.008 M Na2HPO4, and 0.25 M sucrose). The studies were conducted on a Nikon Diaphot 300 microscope in the same chambers in which the collecting surfaces were prepared. In particular, the 10× objective produced a field of view 470 μm by 350 μm in size. In each cell capture run, the buffer of interest was first flowed through the chamber, then the cell suspension in the same buffer was flowed, then finally, the buffer was reintroduced, typically after 10 minutes. For each separate batch of cells, cell capture was measured on a saturated layer of pDMAEMA on the microscope slide, used to normalize cell concentration effects on the capture rates observed on test surfaces. Reporting capture efficiencies in this fashion, rather than the un-normalized capture rates, eliminated variations due to bulk solution cell concentration. A saturated layer of pDMAEMA was employed as the surface for the normalization because it is positive charged and highly adhesive to negative particles and cells. In our flow chamber it has always produced the quantitative transport-limited capture rate for bacteria and microparticles,28 and gave consistently rapid capture of all cells studied here.
Cell capture was monitored on video, and subsequently analyzed to determine the numbers of captured cells as a function of time. In studies distinguishing fluorescent F-TMX2-28 cells from others on the surface, fluorescent optics (a fluorescein cube) was employed.
Acknowledgements
This work was supported by the Center for Excellence in Apoptosis Research and the NIH (5 R21CA159109-02).References
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235 CrossRef CAS PubMed.
- S. L. Stott, C. H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd, A. J. Gilman, J. B. Lord, D. Winokur, S. Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392–18397 CrossRef CAS PubMed.
- S. Riethdorf, H. Wikman and K. Pantel, Int. J. Cancer, 2008, 123, 1991–2006 CrossRef CAS PubMed.
- I. Baccelli, A. Schneeweiss, S. Riethdorf, A. Stenzinger, A. Schillert, V. Vogel, C. Klein, M. Saini, T. Bauerle, M. Wallwiener, T. Holland-Letz, T. Hofner, M. Sprick, M. Scharpff, F. Marme, H. P. Sinn, K. Pantel, W. Weichert and A. Trumpp, Nat. Biotechnol., 2013, 31, 539 CrossRef CAS PubMed.
- A. J. Armstrong, M. S. Marengo, S. Oltean, G. Kemeny, R. L. Bitting, J. D. Turnbull, C. I. Herold, P. K. Marcom, D. J. George and M. A. Garcia-Blanco, Mol. Cancer Res., 2011, 9, 997–1007 CrossRef CAS PubMed.
- T. M. Cao, M. J. Mitchell, J. Liesveld and M. R. King, Sensors, 2013, 13, 12516–12526 CrossRef PubMed.
- N. Charles, J. L. Liesveld and M. R. King, Geometry Optimization of a Flow-Based Device to Maximize Selectin-Mediated Hematopoietic Stem Cell Enrichment, Asme, 2008, pp. 1609–1614 Search PubMed.
- Z. Svobodova, J. Kucerova, J. Autebert, D. Horak, L. Bruckova, J. L. Viovy and Z. Bilkova, Electrophoresis, 2014, 35, 323–329 CrossRef CAS PubMed.
- Y. Chen, P. Li, P.-H. Huang, Y. Xie, J. D. Mai, L. Wang, N. Nam-Trung and T. J. Huang, Lab Chip, 2014, 14, 626–645 RSC.
- S. Kim, S. I. Han, M. J. Park, C. W. Jeon, Y. D. Joo, I. H. Choi and K. H. Han, Anal. Chem., 2013, 85, 2779–2786 CrossRef CAS PubMed.
- C. L. Chen, K. C. Chen, Y. C. Pan, T. P. Lee, L. C. Hsiung, C. M. Lin, C. Y. Chen, C. H. Lin, B. L. Chiang and A. M. Wo, Lab Chip, 2011, 11, 474–483 RSC.
- L. X. Zhang, L. D. Ridgway, M. D. Wetzel, J. Ngo, W. Yin, D. Kumar, J. C. Goodman, M. D. Groves and D. Marchetti, Sci. Transl. Med., 2013, 5, 128er5 Search PubMed.
- G. Bertolini, L. D'Amico, M. Moro, E. Landoni, P. Perego, R. Miceli, L. Gatti, F. Andriani, D. Wong, R. Caserini, M. Tortoreto, M. Milione, R. Ferracini, L. Mariani, U. Pastorino, I. Roato, G. Sozzi and L. Roz, Cancer Res., 2015, 75, 3636–3649 CrossRef CAS PubMed.
- V. Plaks, C. D. Koopman and Z. Werb, Science, 2013, 341, 1186–1188 CrossRef CAS PubMed.
- J. T. Kaifi, M. Kunkel, A. Das, R. A. Harouaka, D. T. Dicker, G. F. Li, J. J. Zhu, G. A. Clawson, Z. H. Yang, M. F. Reed, N. J. Gusani, E. T. Kimchi, K. F. Staveley-O'Carroll, S. Y. Zheng and W. S. El-Deiry, Cancer Biol. Ther., 2015, 16, 699–708 CrossRef CAS PubMed.
- S. Zheng, H. Lin, J. Q. Liu, M. Balic, R. Datar, R. J. Cote and Y. C. Tai, J. Chromatogr. A, 2007, 1162, 154–161 CrossRef CAS PubMed.
- S. Y. Zheng, H. K. Lin, B. Lu, A. Williams, R. Datar, R. J. Cote and Y. C. Tai, Biomed. Microdevices, 2011, 13, 203–213 CrossRef PubMed.
- M. Hosokawa, T. Hayata, Y. Fukuda, A. Arakaki, T. Yoshino, T. Tanaka and T. Matsunaga, Anal. Chem., 2010, 82, 6629–6635 CrossRef CAS PubMed.
- W. J. Zhang, K. Kai, D. S. Choi, T. Iwamoto, Y. H. Nguyen, H. L. Wong, M. D. Landis, N. T. Ueno, J. Chang and L. D. Qin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18707–18712 CrossRef CAS PubMed.
- E. Kokkoli, S. E. Ochsenhirt and M. Tirrell, Langmuir, 2004, 20, 2397–2404 CrossRef CAS PubMed.
- G. Maheshwari, G. Brown, D. A. Lauffenburger, A. Wells and L. G. Griffith, J. Cell Sci., 2000, 113, 1677–1686 CAS.
- S. P. Massia and J. A. Hubbell, J. Cell Biol., 1991, 114, 1089–1100 CrossRef CAS PubMed.
- A. Ahmad, H. M. Evans, K. Ewert, C. X. George, C. E. Samuel and C. R. Safinya, J. Gene Med., 2005, 7, 739–748 CrossRef CAS PubMed.
- Y. Rao, X. F. Wu, P. Yip, J. Gariepy and C. H. Siu, J. Biol. Chem., 1993, 268, 20630–20638 CAS.
- H. E. Davis, M. Rosinski, J. R. Morgan and M. L. Yarmush, Biophys. J., 2004, 86, 1234–1242 CrossRef CAS PubMed.
- P. T. Wong, J. A. Schauerte, K. C. Wisser, H. Ding, E. L. Lee, D. G. Steel and A. Gafni, J. Mol. Biol., 2009, 386, 81–96 CrossRef CAS PubMed.
- M. M. Santore and N. Kozlova, Langmuir, 2007, 23, 4782–4791 CrossRef CAS PubMed.
- R. Duffadar, S. Kalasin, J. M. Davis and M. M. Santore, J. Colloid Interface Sci., 2009, 337, 396–407 CrossRef CAS PubMed.
- M. M. Santore, J. Zhang, S. Srivastava and V. M. Rotello, Langmuir, 2009, 25, 84–96 CrossRef CAS PubMed.
- N. Kozlova and M. M. Santore, Langmuir, 2006, 22, 1135–1142 CrossRef CAS PubMed.
- Y. Shin, J. E. Roberts and M. M. Santore, Macromolecules, 2002, 35, 4090–4095 CrossRef CAS.
- S. Kalasin, S. Martwiset, E. B. Coughlin and M. M. Santore, Langmuir, 2010, 26, 16865–16870 CrossRef CAS PubMed.
- S. C. Hur, N. K. Henderson-MacLennan, E. R. B. McCabe and D. Di Carlo, Lab Chip, 2011, 11, 912–920 RSC.
- S. T. Wang, K. Liu, J. A. Liu, Z. T. F. Yu, X. W. Xu, L. B. Zhao, T. Lee, E. K. Lee, J. Reiss, Y. K. Lee, L. W. K. Chung, J. T. Huang, M. Rettig, D. Seligson, K. N. Duraiswamy, C. K. F. Shen and H. R. Tseng, Angew. Chem., Int. Ed., 2011, 50, 3084–3088 CrossRef CAS PubMed.
- H. J. Yoon, T. H. Kim, Z. Zhang, E. Azizi, T. M. Pham, C. Paoletti, J. Lin, N. Ramnath, M. S. Wicha, D. F. Hayes, D. M. Simeone and S. Nagrath, Nat. Nanotechnol., 2013, 8, 735–741 CrossRef CAS PubMed.
- Y. Lvov, K. Ariga, I. Ichinose and T. Kunitake, J. Am. Chem. Soc., 1995, 117, 6117–6123 CrossRef CAS.
- S. A. Sukhishvili and S. Granick, J. Chem. Phys., 1998, 109, 6861–6868 CrossRef CAS.
- J. M. Gozgit, B. T. Pentecost, S. A. Marconi, C. N. Otis, C. Wu and K. F. Arcaro, Mol. Cancer Res., 2006, 4, 905–913 CrossRef CAS PubMed.
- J. M. Gozgit, B. T. Pentecost, S. A. Marconi, R. S. J. Ricketts-Loriaux, C. N. Otis and K. F. Arcaro, Br. J. Cancer, 2007, 97, 809–817 CrossRef CAS PubMed.
- K. D. Fagan-Solis, B. T. Pentecost, J. M. Gozgit, B. A. Bentley, S. M. Marconi, C. N. Otis, D. L. Anderton, S. S. Schneider and K. F. Arcaro, J. Cell. Physiol., 2014, 229, 1160–1169 CrossRef CAS PubMed.
- K. D. Fagan-Solis, S. S. Schneider, B. T. Pentecost, B. A. Bentley, C. N. Otis, J. F. Gierthy and K. F. Arcaro, J. Cell. Biochem., 2013, 114, 1385–1394 CrossRef CAS PubMed.
- A. Hammer, S. Laghate and M. Diakonova, Biochem. Biophys. Res. Commun., 2015, 463, 644–649 CrossRef CAS PubMed.
- L. Rider, P. Oladimeji and M. Diakonova, Mol. Endocrinol., 2013, 27, 1048–1064 CrossRef CAS PubMed.
- C. M. Turk, K. D. Fagan-Solis, K. E. Williams, J. M. Gozgit, S. Smith-Schneider, S. A. Marconi, C. N. Otis, G. M. Crisi, D. L. Anderton, M. W. Kilimann and K. F. Arcaro, Cancer Cell Int., 2012, 12, 17 CrossRef CAS PubMed.
- K. E. Williams, D. L. Anderton, M. P. Lee, B. T. Pentecost and K. F. Arcaro, Epigenetics, 2014, 9, 297–307 CrossRef CAS PubMed.
- M. J. Fasco, A. Amin, B. T. Pentecost, Y. Yang and J. F. Gierthy, Mol. Cell. Endocrinol., 2003, 206, 33–47 CrossRef CAS PubMed.
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Curr. Opin. Chem. Biol., 2000, 4, 696–703 CrossRef CAS PubMed.
- J. E. Gestwicki, C. W. Cairo, L. E. Strong, K. A. Oetjen and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 14922–14933 CrossRef CAS PubMed.
- W. Sheng, T. Chen, W. Tan and Z. H. Fan, ACS Nano, 2013, 7, 7067–7076 CrossRef CAS PubMed.
- R. D. Duffadar and J. M. Davis, J. Colloid Interface Sci., 2007, 308, 20–29 CrossRef CAS PubMed.
- J. F. Alexander, V. Kozlovskaya, J. Chen, T. Kuncewicz, E. Kharlampieva and B. Godin, Adv. Healthcare Mater., 2015, 4, 2657–2666 CrossRef CAS PubMed.
- Q. S. Li, G. Y. H. Lee, C. N. Ong and C. T. Lim, Biochem. Biophys. Res. Commun., 2008, 374, 609–613 CrossRef CAS PubMed.
- M. Prabhune, G. Belge, A. Dotzauer, J. Bullerdiek and M. Radmacher, Micron, 2012, 43, 1267–1272 CrossRef PubMed.
- A. Geltmeier, B. Rinner, D. Bade, K. Meditz, R. Witt, U. Bicker, C. Bludszuweit-Philipp and P. Maier, PLoS One, 2015, 10, e0134999 Search PubMed.
- E. A. Corbin, F. Kong, C. T. Lim, W. P. King and R. Bashir, Lab Chip, 2015, 15, 839–847 RSC.
- K. Sliogeryte, S. D. Thorpe, Z. Wang, C. L. Thompson, N. Gavara and M. M. Knight, J. Biomech., 2016, 49, 310–317 CrossRef PubMed.
- R. M. Vazquez, G. Nava, M. Veglione, T. Yang, F. Bragheri, P. Minzioni, E. Bianchi, M. Di Tano, I. Chiodi, R. Osellame, C. Mondello and I. Cristiani, Integr. Biol., 2015, 7, 477–484 RSC.
- T. Yang, F. Bragheri, G. Nava, I. Chiodi, C. Mondello, R. Osellame, K. Berg-Sorensen, I. Cristiani and P. Minzioni, Sci. Rep., 2016, 6, 23946 CrossRef CAS PubMed.
- J. P. Smith, A. C. Barbati, S. M. Santana, J. P. Gleghorn and B. J. Kirby, Electrophoresis, 2012, 33, 3133–3142 CrossRef CAS PubMed.
- S. Gon and M. M. Santore, Langmuir, 2011, 27, 1487–1493 CrossRef CAS PubMed.
- Z. G. Fu and M. M. Santore, Colloids Surf., A, 1998, 135, 63–75 CrossRef CAS.
- N. Hansupalak and M. M. Santore, Langmuir, 2003, 19, 7423–7426 CrossRef CAS.
- N. Hansupalak and M. M. Santore, Macromolecules, 2004, 37, 1621–1629 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01217a |
| This journal is © The Royal Society of Chemistry 2017 |