
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile synthesis of a α-MoO3 nanoplate/TiO2 nanotube composite for high electrochemical performance†
Shupei Sun,
Xiaoming Liao *,
Yu Sun,
Guangfu Yin,
Yadong Yao,
Zhongbing Huang and
Ximing Pu
*,
Yu Sun,
Guangfu Yin,
Yadong Yao,
Zhongbing Huang and
Ximing Pu
College of Materials Science and Engineering, Sichuan University, Chengdu, Sichuan 610065, China. E-mail: sherman_xm@163.com; Fax: +86 28 85413003
First published on 26th April 2017
Abstract
A novel α-MoO3/TiO2 composite electrode material for high performance supercapacitor applications was synthesized using a facile electrodeposition technique. The surface morphology, microstructure, chemical composition and chemical states of the electrode material were analyzed using scanning electron microscopy, X-ray diffraction, energy dispersive X-ray spectroscopy, and X-ray photoelectron spectroscopy. Cyclic voltammetry tests, galvanostatic charge–discharge measurements, and electrochemical impedance spectroscopy were employed to analyze electrochemical behavior. The results demonstrate that a very high capacitance (43.42 mF cm−2) can be achieved at a scan rate of 20 mV s−1 in 1 M aqueous Na2SO4 solution and good capacitance behavior even after 3000 continuous charge–discharge cycles, indicating potential applications as an electrode material for supercapacitors.
1. Introduction
Over the past few years, extensive studies have focused on the development of new electrode materials for advanced energy storage devices,1–6 due to the ever-increasing power and energy requirements in the modern electronics industry. Among the various energy-storage devices, electrochemical capacitors (EC), also called supercapacitors, are one of the most promising electrochemical energy-storage systems and have attracted tremendous attention. Generally, according to the charge/discharge mechanism, supercapacitors can be classified into two categories: electrical double-layer capacitors (EDLCs), which commonly use carbon-active materials (activated carbon,7,8 carbon nanotubes,9,10 and graphene11,12), and pseudocapacitors, which use transition metal oxides/sulfides (RuO2,13,14 NiOx,15,16 MnOx,17,18 CoS,19 ZnS,20 etc.) and conducting polymers (e.g. polypyrrole,21–23 polyaniline24,25). Among the different transition metal oxides, titanium dioxide (TiO2)26,27 and molybdenum oxide (MoO3−x)28–35 have received immense interest for applications in electrode materials, due to their low cost, low toxicity, natural abundance, and environmentally friendly nature.Self-assembled titania nanotubes (TNTs) can be easily fabricated by one-step anodization of Ti sheets in a two-electrode system in fluoride-containing electrolytes.15–20 The highly ordered structure and large surface area of TNTs increase reaction sites as well as promote charge and ions transfer, enhancing supercapacitor performance of composite electrode. Therefore, many researches employ TNTs as a substrate and construct composite electrode material. Zhou et al. achieved MnO2–TiO2 composite electrode via a sequential chemical bath deposition.18 Ray et al. fabricated titania nanotube/cobalt sulfide composite electrode by electrochemical technique.19 Du et al. prepared polypyrrole–titania nanotube hybrid through a normal pulse voltammetry deposition process.22 Xie et al. prepared polypyrrole/titania/polyaniline coaxial nanotube hybrid through an electrodeposition process.21 In conclusion, transition metal oxide or conducting polymer incorporated with TNTs exhibited superior electrochemical performance resulted from their synergistic effect.
MoO3 has attracted considerable attention as a promising transition metal oxide widely used in the fields of photochromic and electrochromic devices,33 gas sensors,34 and energy storage.28–30 There are three basic polytypes of MoO3, orthorhombic MoO3 (α-MoO3), monoclinic MoO3 (β-MoO3), and hexagonal MoO3 (h-MoO3). Among them, α-MoO3 has received immense attention because of its unique layer structure.28,30,32 The unique structure allows atoms, ions or molecules to be introduced into the layers through intercalation. α-MoO3 also has a high theoretical capacity of 1111 mA h g−1,36 almost three times that of the graphite.37 Li et al. studied the electrochemical behavior of α-MoO3 nanobelts deposited on a foam nickel plate, whose specific capacitance was as high as 280 F g−1.28 Sarfraz et al. reported the electrochemical performance of MoO3 nanowires coated onto a conductive carbon cloth, which exhibited a high specific capacitance of 288 F g−1.29 Compared to foam nickel plate or carbon cloth, the well-aligned nanotubes vertically oriented from the surface of Ti substrate, as current collector, do not need polymer binder and conductive additives. Additionally, Liu et al.38–40 pointed out that 3D architecture nanocomposites contributed to achieving high-performance, low-cost and safe energy storage devices. Hence, we fabricated α-MoO3 nanoplates/TiO2 nanotubes composite via a facile electrochemical deposition on nanotube surface and extensively studied its application for supercapacitor.
In this work, anatase TNTs were prepared by anodization and subsequent thermal treatment. Electrodeposition in molybdenum salt solutions was performed on as-prepared nanotube arrays and bare Ti sheets to obtain amorphous coatings, which then were transformed into the crystalline phase upon annealing in air. We compared the electrochemical behavior of molybdenum trioxide coated on TNTs and Ti metal. Our results demonstrate that these TNTs could be an excellent substrate and MoO3/TiO2 is a promising electrode material.
2. Experimental procedure
2.1. Materials
The titanium sheet (TA2, purity > 99.6%, thickness 1 mm) was purchased from LuoKe titanium Ltd. Ethylene glycerol (C2H6O2, purity > 99.5%), ammonium fluoride (NH4F, purity > 96%) and hexaammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O, purity > 99.0%) were purchased from KeLong Chemical Ltd. Other chemical reagents used in these experiments were of analytical grade and were used directly without further purification. All aqueous solutions were prepared using deionized water.2.2. Fabrication of MoO3/TiO2
Highly ordered titania nanotubes (TNTs) were fabricated by one-step anodization of Ti sheets in ethylene glycerol containing 0.3 wt% NH4F and 6 vol% deionized H2O at 50 V for 30 min. A calcination treatment at 450 °C for 1 h was used to obtain crystalline TNTs, which are referred as TNT. Then electrochemical deposition of MoO3 onto TNTs was carried out with a three-electrode system composed of annealed TNTs as the working electrode, a Pt foil as the counter electrode and an Ag/AgCl as the reference electrode. The deposition was performed in 0.05 M (NH4)6Mo7O24·4H2O aqueous solution by cycling from −0.6 to 0 V with a scan rate of 20 mV s−1 for 17 cycles. The as-deposited TNTs are designated as AMO-TNT, which were then annealed in air at 450 °C for 1 h to obtain crystalline MoO3-coated nanotubes (CMO-TNT). For comparison, a MoO3 film was directly grown by electrodeposition on Ti sheets via 17 cycles and then heat-treated at 450 °C for 1 h, which is referred as CMO-T.2.3. Materials characterization and electrochemical measurements
The surface morphology and microstructure of the fabricated samples were investigated by scanning electron microscopy (SEM, Hitachi S4800, Japan), X-ray diffraction (XRD, X'Pert Pro MPD, Holland), respectively. Chemical composition of prepared samples was characterized by an energy dispersive X-ray spectroscope (EDS, Oxford) attached on SEM. An X-ray photoelectron spectroscope (XPS, Escalab 250Xi, USA) was employed to analyze chemical states of the coating film. Electrochemical measurements were performed in a 1 M Na2SO4 aqueous solution with an electrochemical workstation (CHI660E, China). Cyclic voltammetry (CV) tests were conducted over a potential voltage range from −0.9 to −0.2 V (vs. Ag/AgCl) at different scan rates (from 10 to 200 mV s−1). The galvanostatic charge–discharge (CD) testing was performed under different current densities (from 0.4 to 1 mA cm−2). Electrochemical impedance spectroscopy (EIS) measurements were conducted over a frequency range of 0.01 Hz to 100 kHz at an AC voltage amplitude of 5 mV. The cycling stability of the samples was tested by galvanostatic CD measurement performed up to 3000 cycles at a current density of 0.6 mA cm−2.3. Results and discussion
3.1. Morphology and structure analyses
Fig. 1 shows the XRD patterns obtained over 2θ range of 10° to 80° from TNT, CMO-T, AMO-TNT, and CMO-TNT. The peaks centered at ∼25.2° and ∼47.8° are the characteristic peaks of anatase TiO2 (JCPDF 21-1272) while peaks at ∼38.3° and ∼40.1° are assigned to Ti (JCPDF 44-1294).18 After deposition and thermal treatment, the pattern exhibits new peaks. The peaks with 2θ of ∼12.6°, ∼25.5° and ∼27.2° are indexed to orthorhombic α-MoO3 (JCPDF 05-0508).28 However, there is no diffraction peak of α-MoO3 from the pattern of AMO-TNT, indicating deposition of amorphous molybdenum oxides onto anatase TiO2 nanotubes.41 We can observe the peaks of MoO3 from the patterns of CMO-T and CMO-TNT, suggesting the formation of crystalline α-MoO3 phase after annealing at 450 °C for 1 h.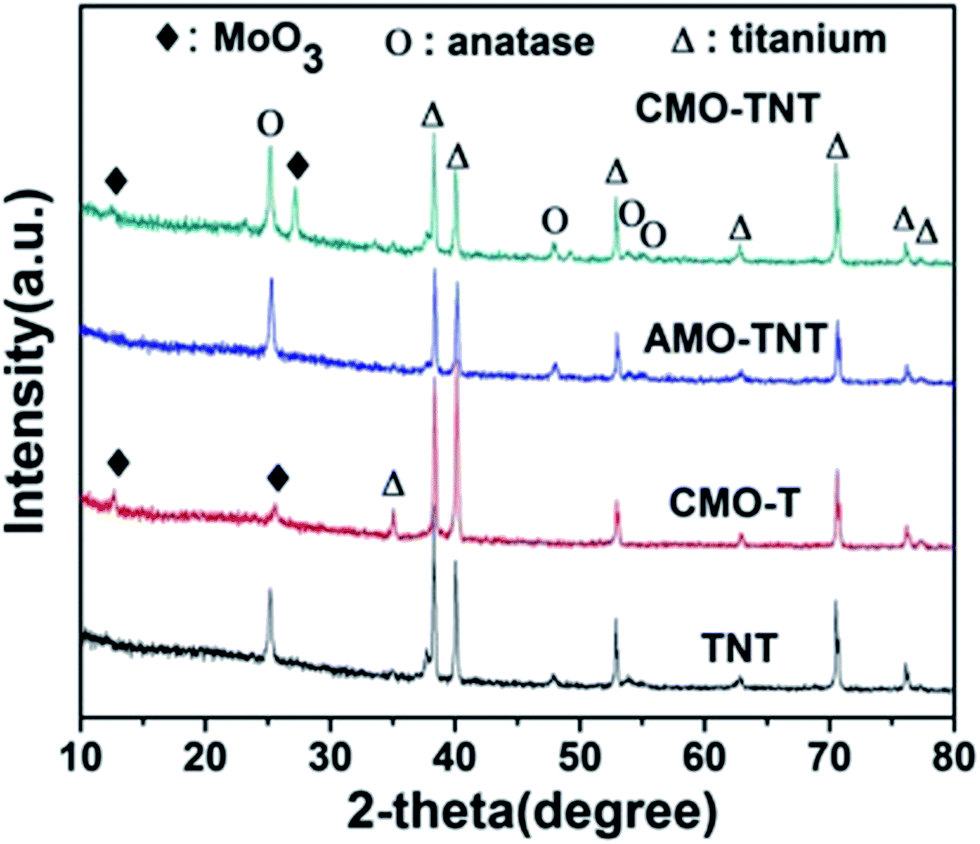 | ||
| Fig. 1 XRD patterns of the samples TNT, AMO-TNT, CMO-TNT and CMO-T. | ||
An EDS analysis was also made to claim the presence of Mo elements. Fig. 2 shows the EDS spectra in which Mo, Ti and O elements are detected, suggesting formation of molybdenum oxide layer. In addition, there is no significant change in the amount of Mo from CMO-TNT when compared to AMO-TNT. The results confirm that annealing in the ambient environment after electrochemical deposition does not decrease the amount of MoO3. However, the Mo amount of CMO-T is lower than that of AMO-TNT and CMO-TNT, indicating that TNTs substrate can contribute to the deposition of Mo.
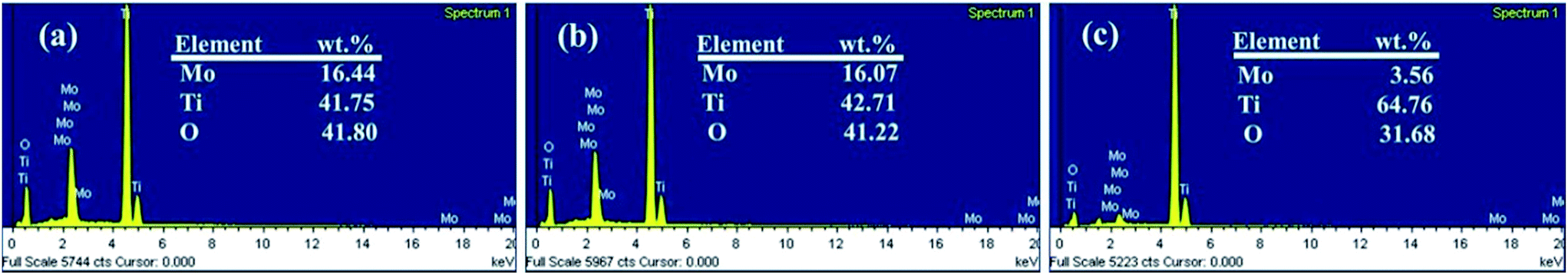 | ||
| Fig. 2 EDS spectra of the samples CMO-TNT (a), AMO-TNT (b), and CMO-T (c). | ||
Fig. 3 shows the surface morphology of TNT, CMO-T, AMO-TNT and CMO-TNT. The anodized TNTs (Fig. 3a) have open-top and uninterrupted nanotube structure which contribute to accessible surface area and promote the speedy permeation of electrolyte ions. Fig. 3b presents SEM image of AMO-TNT. Compared to TNT, there is a coating layer on the top of nanotubes and the precipitates are confirmed to be molybdenum oxides by EDS spectra. After heat treatment, the coating layer (Fig. 3c) becomes roughened. It is evident to see a large number of nanoplates aggregate with each other. From the results of XRD patterns, the coated layer is confirmed to be crystalline MoO3. For a comparative study, planar Ti substrate was treated with electrochemical process to deposit MoO3 species. The dense film is confirmed to be orthorhombic molybdenum trioxide. It should be noted that morphological difference is appeared between the CMO-TNT and CMO-T, i.e., the smaller MoO3 nanoplates and more pores could be seen in the former samples compared to the latter prepared with the same method.
 | ||
| Fig. 3 SEM images of TNT (a), AMO-TNT (b), CMO-TNT (c) and CMO-T (d). | ||
In our study, we discover the amount and morphological difference of MoO3 layer between CMO-TNT and CMO-T, which is attributed to the effect of nanotube arrays. On the one hand, the unique nanotube structure of anodized TNTs provide large surface area for absorbing more precursors and providing more nucleation points for MoO3 nanoplates compared to the planar Ti substrate.18 On the other hand, the rugged tubular opening of the TNTs substrate inhibits growth of MoO3 layer in two-dimensional direction, beneficial to achieving small nanoplates. Thus TNTs play an important role in the quality of coating material.16 As a result, it is understood that the hollow nanotubes of TNTs not only provide unique electron transport pathways for electrochemical reactions, especially for the rapid transfer of Na+ and H+ in a fast charge–discharge process, but offer large surface area for deposition of MoO3 to obtain composite electrode material. We can anticipate that TNTs coated with MoO3 exhibit excellent electrochemical behavior.
Furthermore, XPS was employed to analyze the chemical states of the coating layer. Fig. 4a shows the typical XPS survey spectra of AMO-TNT electrode. The Mo 3d5/2 peak at 233.1 eV and the Mo 3d3/2 peak at 236.3 eV correspond to MoVI, and the Mo 3d5/2 peak at 232.3 eV and the Mo 3d3/2 peak at 235.6 eV correspond to MoV.41,42 As shown in Fig. 4b, the XPS spectrum shifting to high energies (Mo 3d5/2 peak at 233.3 eV and the Mo 3d3/2 peak at 236.5 eV) is assigned to α-MoO3. As for the O 1s peak, a slight shift (0.2 eV) to a lower energy is observed, which is resulted from elimination of –OH groups and oxidation of MoV ions during annealing process.43 In summary, the composition of amorphous coating layer after electrochemical deposition is MoV oxides and MoVI oxides. Annealing in ambient environment leads to transition of amorphous molybdenum oxide to crystalline α-MoO3. All in all, we successfully prepared α-MoO3/TiO2 composite electrode material through a facile electrodeposition method in molybdenum salt solutions.
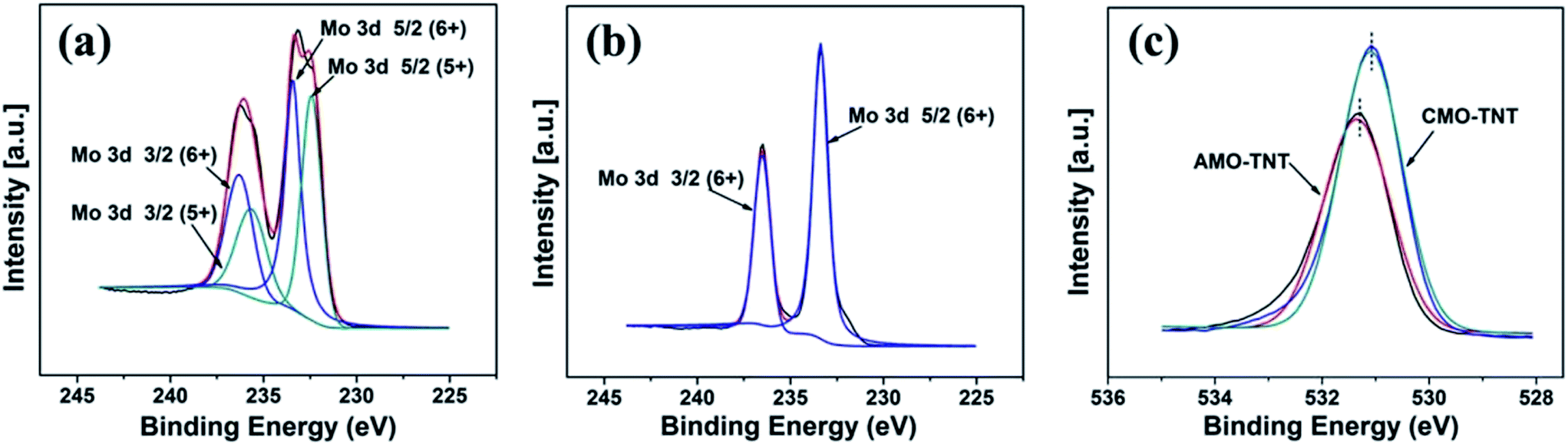 | ||
| Fig. 4 XPS spectra in the Mo 3d binding energy region of AMO-TNT (a), CMO-TNT (b), and O 1s XPS spectra taken from samples AMO-TNT and CMO-TNT (c). | ||
3.2. Electrochemical characterizations
Electrochemical measurements were performed with a three-electrode system in 1 M Na2SO4 electrolyte to evaluate the electrochemical performance of the MoO3/TiO2 nanocomposite. For a comparison, the pristine TiO2 nanotubes and MoO3/Ti substrate were also intensively studied. The CV curves for the TNT, CMO-T, AMO-TNT and CMO-TNT samples collected at a scan rate of 20 mV s−1 are shown in Fig. 5a. The increased current of the CV curves for AMO-TNT and CMO-TNT clearly demonstrates improved electrochemical behavior after coating the nanotubes with MoO3. The Ti substrate coated MoO3 film also exhibits low current response, resulting from its dense structure which is not beneficial to electrolyte ions diffusion. Compared to CMO-TNT, higher resistance at higher potentials of AMO-TNT indicates lower electroactivity, suggesting poorer capacitive behavior.44 Furthermore, the area under CV curve for CMO-TNT is larger than that of other samples, implying enhanced electrochemical properties. The effect of different scan rates on the electrochemical behavior of CMO-TNT has been studied and the results are presented in Fig. 5b. All plots are close to an ideal rectangular shape without evident redox peaks, which is a typical character of double-layer capacitance. Additionally, the shapes of CV curves do not significantly change as the scan rate is increased to 0.2 V s−1, indicating good capacitive behavior and high-rate capability. Fig. 5c shows the calculated areal capacitance of these electrodes as a function of scan rate. The specific capacitances are calculated by the following equation:| Cs = C/S = I/[(dV/dt) × S] | (1) |
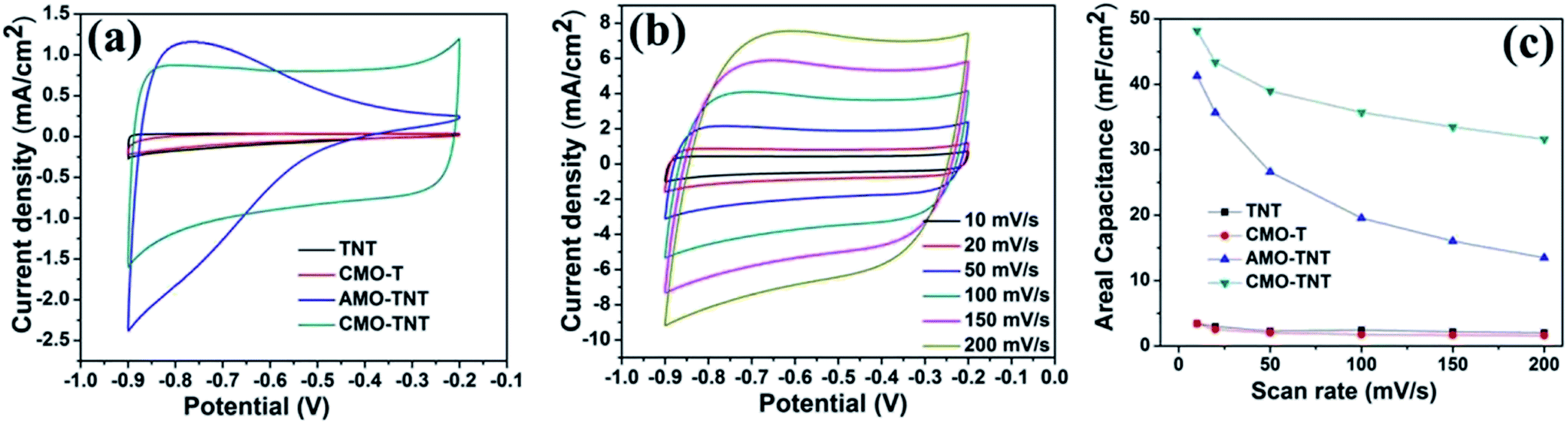 | ||
| Fig. 5 The CV plots at a scan rate of 20 mV s−1 (a) for TNT, CMO-T, AMO-TNT, and CMO-TNT, the CV plots at different scan rates for CMO-TNT (b), and areal capacitances of the samples measured as a function of scan rate (c). | ||
The areal capacitances of TNT, CMO-T, AMO-TNT, and CMO-TNT are 2.97 mF cm−2, 2.55 mF cm−2, 35.67 mF cm−2 and 43.42 mF cm−2 at a scan rate of 20 mV s−1, respectively. For all of the samples, the specific capacitance decreases with increasing scan rate. Notably, the specific capacitance of the CMO-TNT remains as high as 31.60 mF cm−2 at a high scan rate (0.2 V s−1), demonstrating better rate capability and ion transportation performance than the other investigated samples. The decay of the specific capacitance is attributed to the charge not being able to completely accumulate in the materials at high scan rates.
α-MoO3 is known as a layered structure of [MoO6] octahedron. These layers are connected by weak van der Waals force containing van der Waals gaps which are favorable for intercalation of electrolyte ions and accommodate large quantities of positive ions.28,45,46 Dunn and his coworkers47 systematically study capacitance contribution of porous electrode. When the grain size decreases to nanoscale dimensions, there is some amount of charge stored from the insertion process. Thus, we propose that nanosize α-MoO3 plates with distinct layer structure are favorable of intercalation and deintercalation of electrolyte ions, and thus enhancing the electrochemical behavior. Additionally, the layered structure also offers superior charge transfer behavior.48 The difference between AMO-TNT and CMO-TNT is attributed to the crystal structure and chemical states of molybdenum oxides, capacitive factors and electrochemical kinetics.49,50 Therefore, if crystalline α-MoO3 is used as coating material, the hybrid electrode material will exhibit excellent electrochemical performance. The highly ordered and uninterrupted structure of TNTs not only provides large surface area and direct pathways, but also helps to make best utilization of nanosized α-MoO3 and promote charge transfer inside a porous electrode. There is no doubt that TNTs have a progressive and positive influence on the capacitive behavior of the coated layer compared to the planar Ti substrate. In conclusion, it is the combined effect of TNTs and MoO3 layer that improves electrochemical properties of CMO-TNT.
Galvanostatic CD tests, which were performed in the potential range from −0.9 to −0.2 V, were also used to characterize the electrochemical properties and specific capacitance of the composite electrodes. From Fig. 6a, AMO-TNT and CMO-TNT exhibit longer charge/discharge time compared to TNT, indicating enhanced electrochemical performance. The results also indicate that the combination of TNTs and α-MoO3 can enhance the electrochemical performance and increase specific capacitance. Fig. 6b presents the galvanostatic CD plots of CMO-TNT. The curves keep linear and symmetric at different current densities, which typically are the characteristic of double layer capacitance. Areal capacitances of the electrodes can be calculated from the galvanostatic CD curves using the following equation:
| Cs = [I × Δt]/[S × ΔV] | (2) |
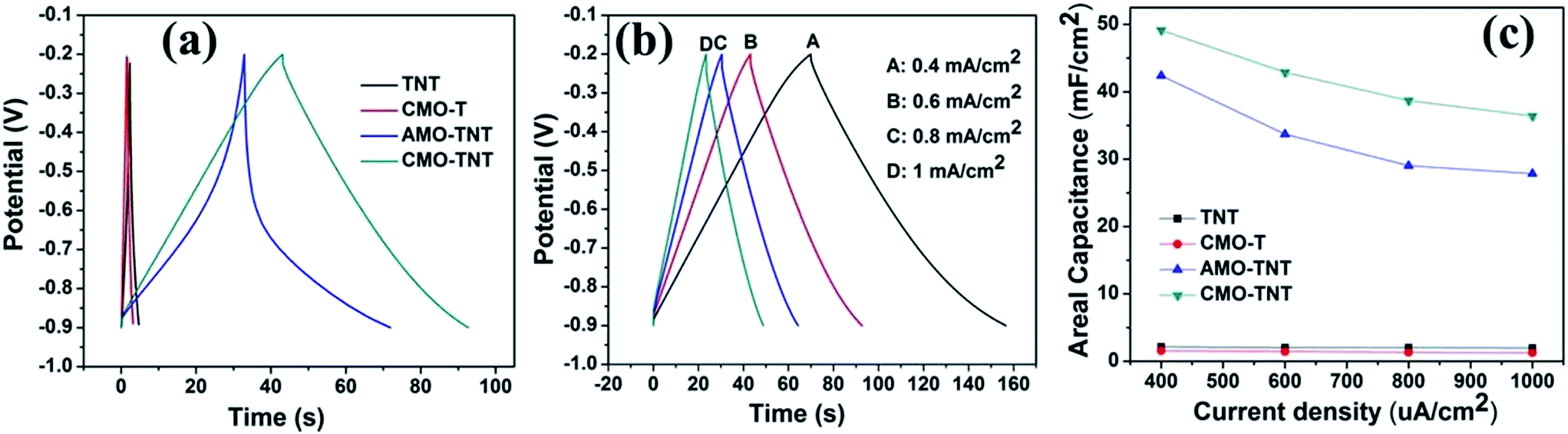 | ||
| Fig. 6 The galvanostatic charge–discharge curves at a current density of 0.6 mA cm−2 for TNT, CMO-T, AMO-TNT, and CMO-TNT (a), galvanostatic charge–discharge curves for CMO-TNT achieved from 0.4 mA cm−2 to 1 mA cm−2 (b), the average areal capacitance at different current densities (c). | ||
To further understand electrochemical behavior of composite electrode materials, EIS measurements were carried out over a frequency range of 0.01 Hz to 100 kHz. Fig. 7 shows the Nyquist plots of TNT, CMO-T, AMO-TNT, and CMO-TNT. The high frequency region of the spectra is shown as the inset. Semicircular arcs are observed in the samples of TNT, CMO-T, and AMO-TNT, indicating poor charge transfer performance due to the interfacial transfer resistance between the electrode and electrolyte systems. However, no such a semicircular arc for CMO-TNT is observed, indicating good charge transport capability. The high surface area and unique structure of the CMO-TNT electrode could explain the decrease in charge transfer resistance. These results indicate better capacitance performance of the CMO-TNT electrode.
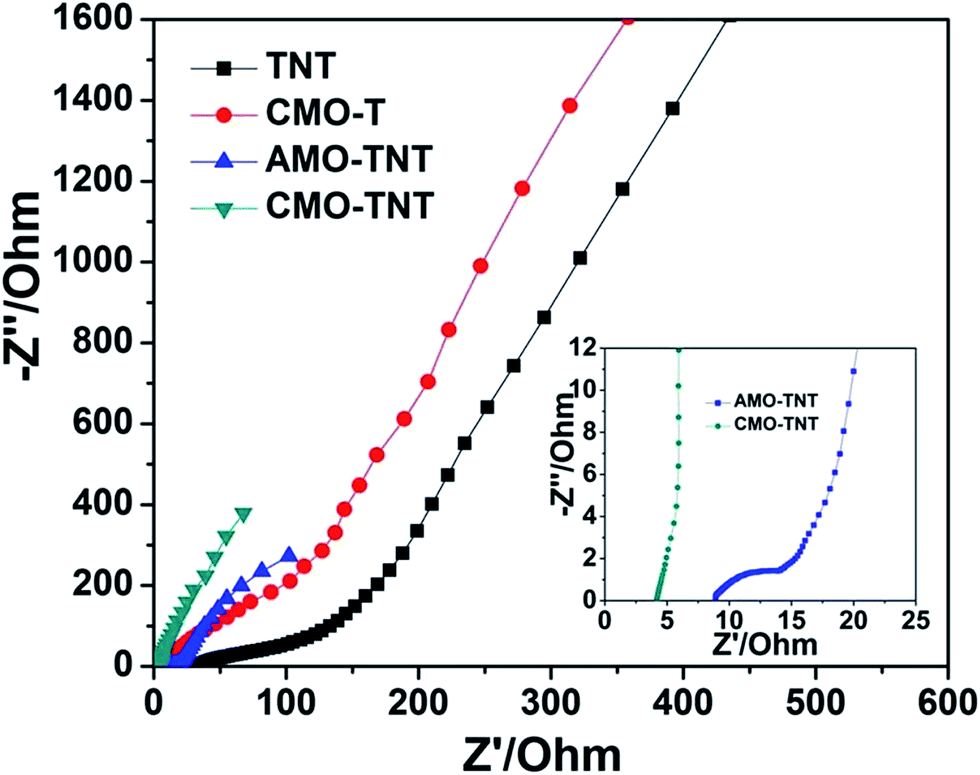 | ||
| Fig. 7 Nyquist plots of TNT, CMO-T, AMO-TNT, and CMO-TNT. | ||
The CMO-TNT electrode was measured at the current density of 0.6 mA cm−2 for 3000 cycles, to study its long-term capability. As shown in Fig. 8, the overall capacitance retention of CMO-TNT is about 51.7% after 3000 cycles. Interestingly, there is significant capacitance reduction (∼12%) in first 50 cycles. However, the capacitance fading rate of later cycle numbers becomes much lower than that of first 50 cycles. Fig. S5a and S5b† show SEM image and EDS spectrum of the sample achieved by galvanostatic CD testing of CMO-TNT for 50 cycles, respectively. A large number of pores are appeared, which is different from the top view of CMO-TNT (Fig. 3c). The EDS spectrum shows that the amount of Mo element after galvanostatic CD testing for 50 cycles is just 8.9%, which is lower than that of CMO-TNT (16.44%). These results indicate poor adhesion of MoO3 nanoplates to nanotube substrate, and thus detachment to a certain degree from nanotube surface over cycling. The XPS survey spectrum confirms the formation of MoO2 after galvanostatic CD testing for 50 cycles (Fig. S6†). Wang et al.30 reported that the presence of MoO2 reasonably reduces the electrochemical reactivity of molybdenum oxides, leading to a rapid capacity decay in the initial cycles. Taking these results into consideration, the rapid capacity decay may be ascribed to the detachment of MoO3 nanoplates from nanotube surface and the formation of MoO2 in the first 50 cycles. Additionally, above electrochemical analysis suggests the existence of intercalation and deintercalation of cations. On the one hand, the intercalated Na ion could occupy some active sites, leading to the decrease of capacitance.51 On the other hand, the rapid capacity reduction of composite electrode is caused by the irreversible structure destruction resulting from the Na+ insertion.42,51,52 With the increasing cycle numbers, some Na ions are introduced to Mo–O interlayers, which effectively inhibits the irreversible phase transition and stabilize the layer structure.51 Thus, capacitance fading is greatly alleviated. Although our composite electrode has somewhat large capacitance loss, the final capacitance (21.72 mF cm−2) after 3000 continuous charge–discharge cycles is still higher than the initial capacitance (2.50–20.08 mF cm−2) reported in literatures.53–57
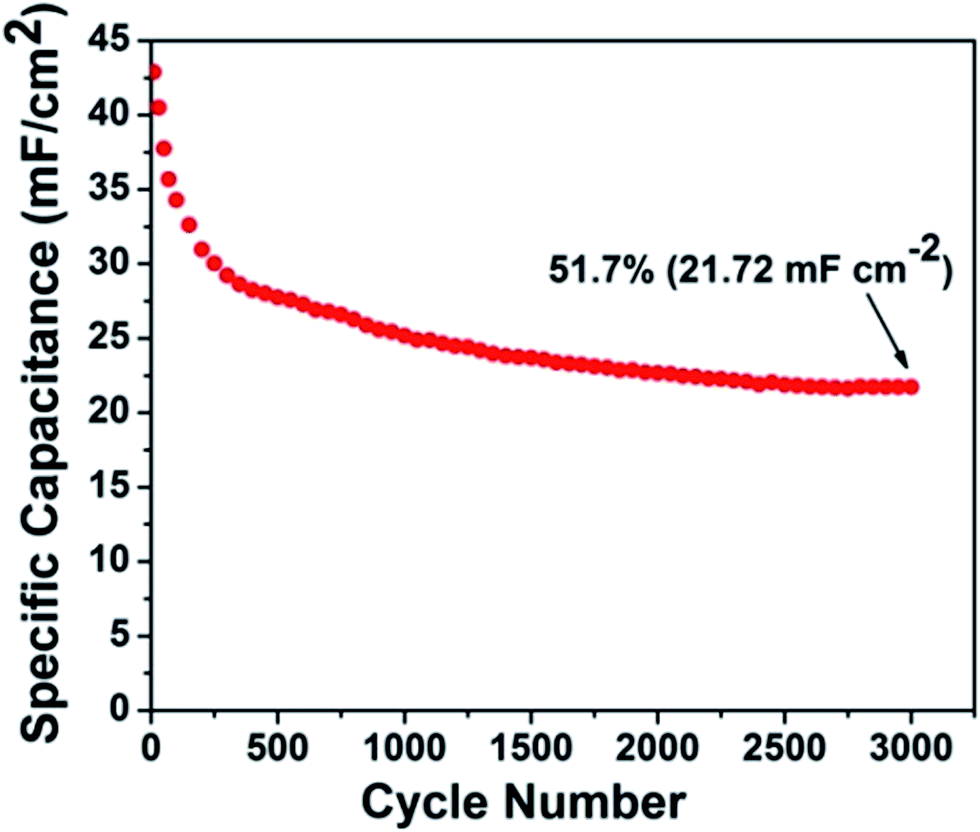 | ||
| Fig. 8 Cyclic performance of CMO-TNT composite electrode at a current density of 0.6 mA cm−2. | ||
4. Conclusion
In our study, highly ordered TiO2 nanotube arrays were synthesized by one-step anodization and then annealed at 450 °C in air to achieve crystalline phase, which worked as active surface coated with MoO3 through electrochemical deposition. The resulting CMO-TNT electrode shows a large area capacitance of 43.42 mF cm−2 at a scan rate of 20 mV s−1. The enhanced electrochemical properties are attributed to combined effect of the TiO2 nanotube arrays and MoO3 layer. The results suggest that TiO2 nanotube coated with crystalline α-MoO3 film can be used as a promising electrode material.Acknowledgements
The authors are very much grateful to the National Engineering Research Center for Biomaterials, Sichuan University for the assistance with the microscopy work. S. P. Sun thanks to Mr Yan for his encouragement and support during the research.References
- Y. Liu, X. Y. Cai, J. H. Jiang, M. Yan and W. D. Shi, Appl. Surf. Sci., 2017, 396, 774–779 CrossRef CAS.
- P. Bhojane, S. Sen and P. M. Shirage, Appl. Surf. Sci., 2016, 377, 376–384 CrossRef CAS.
- C. N. He, S. Wu, N. Q. Zhao, C. S. Shi, E. Z. Liu and J. J. Li, ACS Nano, 2013, 7, 4459–4469 CrossRef CAS PubMed.
- G. Q. Zhang, L. Yu, H. B. Wu, H. E. Hoster and X. W. Lou, Adv. Mater., 2012, 24, 4609–4613 CrossRef CAS PubMed.
- M. P. A. Muthalif, Y.-S. Lee, C. D. Sunesh, H.-J. Kim and Y. Choe, Appl. Surf. Sci., 2017, 396, 582–589 CrossRef CAS.
- B. Filanovsky, E. Granot, R. Dirawi, I. Presman, I. Kuras and F. Patolsky, Nano Lett., 2011, 11, 1727–1732 CrossRef CAS PubMed.
- C. Kim, J. Power Sources, 2005, 142, 382–388 CrossRef CAS.
- C. Merino, P. Soto, E. Vilaplana-Ortego, J. M. G. d. Salazar, F. Pico and J. M. Rojo, Carbon, 2005, 43, 551–557 CrossRef CAS.
- A. Izadi-Najafabadi, T. Yamada, D. N. Futaba, M. Yudasaka, H. Takagi, H. Hatori, S. Iijima and K. Hata, ACS Nano, 2011, 5, 811–819 CrossRef CAS PubMed.
- P. Chen, H. T. Chen, J. Qiu and C. W. Zhou, Nano Res., 2010, 3, 594–603 CrossRef CAS.
- Z. J. Fan, J. Yan, L. J. Zhi, Q. Zhang, T. Wei, J. Feng, M. L. Zhang, W. Z. Qian and F. Wei, Adv. Mater., 2010, 22, 3723–3728 CrossRef CAS PubMed.
- L. L. Zhang, R. Zhou and X. S. Zhao, J. Mater. Chem., 2010, 20, 5983–5992 RSC.
- C.-C. Hu, K.-H. Chang, M.-C. Lin and Y.-T. Wu, Nano Lett., 2006, 6, 2690–2695 CrossRef CAS PubMed.
- K.-M. Lin, K.-H. Chang, C.-C. Hu and Y.-Y. Li, Electrochim. Acta, 2009, 54, 4574–4581 CrossRef CAS.
- L. H. Cui, Y. Wang, X. Shu, J. F. Zhang, C. P. Yu, J. W. Cui, H. M. Zheng, Y. Zhang and Y. C. Wu, RSC Adv., 2016, 6, 12185–12192 RSC.
- F. Gobal and M. Faraji, Electrochim. Acta, 2013, 100, 133–139 CrossRef CAS.
- S. S. Zhu, P. P. Zhang, L. Chang, Y. Zhong, K. Wang, H. B. Shao, J. M. Wang, J. Q. Zhang and C.-N. Cao, Phys. Chem. Chem. Phys., 2016, 18, 8529–8536 RSC.
- H. Zhou and Y. R. Zhang, J. Power Sources, 2014, 272, 866–879 CrossRef CAS.
- R. S. Ray, B. Sarma, A. L. Jurovitzki and M. Misra, Chem. Eng. J., 2015, 260, 671–683 CrossRef CAS.
- B. Sarma, R. S. Ray and M. Misra, Mater. Lett., 2015, 139, 77–80 CrossRef CAS.
- Y. B. Xie, D. Wang, Y. Z. Zhou, H. X. Du and C. Xia, Synth. Met., 2014, 198, 59–66 CrossRef CAS.
- H. X. Du, Y. B. Xie, C. Xia, W. Wang and F. Tian, New J. Chem., 2014, 38, 1284–1293 RSC.
- Y. Gao, Y. Z. Wang, X. Xu, K. Ding and D. M. Yu, RSC Adv., 2014, 4, 63719–63724 RSC.
- K. Y. Xie, J. Li, Y. Q. Lai, Z. A. Zhang, Y. X. Liu, G. G. Zhang and H. T. Huang, Nanoscale, 2011, 3, 2202–2207 RSC.
- H. F. Su, T. Wang, S. Y. Zhang, J. M. Song, C. J. Mao, H. L. Niu, B. K. Jin, J. Y. Wu and Y. P. Tian, Solid State Sci., 2012, 14, 677–681 CrossRef CAS.
- R. Ren, Z. H. Wen, S. M. Cui, Y. Hou, X. R. Guo and J. H. Chen, Sci. Rep., 2015, 5, 10714 CrossRef CAS PubMed.
- T. Close, G. Tulsyan, C. A. Diaz, S. J. Weinstein and C. Richter, Nat. Nanotechnol., 2015, 10, 418–422 CrossRef CAS PubMed.
- J. Li and X. H. Liu, Mater. Lett., 2013, 112, 39–42 CrossRef CAS.
- M. Sarfraz, M. F. A. Aboud and I. Shakir, J. Alloys Compd., 2015, 650, 123–126 CrossRef CAS.
- Z. Y. Wang, S. Madhavi and X. W. Lou, J. Phys. Chem. C, 2012, 116, 12508–12513 CAS.
- P. Meduri, E. Clark, J. H. Kim, E. Dayalan, G. U. Sumanasekera and M. K. Sunkara, Nano Lett., 2012, 12, 1784–1788 CrossRef CAS PubMed.
- L. L. Cai, P. M. Rao and X. L. Zheng, Nano Lett., 2011, 11, 872–877 CrossRef CAS PubMed.
- D. Yao, M. R. Field, A. P. O'Mullane, K. Kalantar-zadeh and J. Z. Ou, Nanoscale, 2013, 5, 10353–10359 RSC.
- L. Q. Wang, P. Gao, D. Bao, Y. Wang, Y. J. Chen, C. Chang, G. B. Li and P. P. Yang, Cryst. Growth Des., 2014, 14, 569–575 CAS.
- J. C. Park and H. Song, Chem. Mater., 2007, 19, 2706–2708 CrossRef CAS.
- M. F. Hassan, Z. P. Guo, Z. Chen and H. K. Liu, J. Power Sources, 2010, 195, 2372–2376 CrossRef CAS.
- H. Li, P. Balaya and J. Maier, J. Electrochem. Soc., 2004, 151, A1878–A1885 CrossRef CAS.
- W. H. Zuo, W. H. Zhu, D. F. Zhao, Y. F. Sun, Y. Y. Li, J. P. Liu and X. W. (David) Lou, Energy Environ. Sci., 2016, 9, 2881–2891 CAS.
- J. P. Liu, C. Guan, C. Zhou, Z. Fan, Q. Q. Ke, G. Z. Zhang, C. Liu and J. Wang, Adv. Mater., 2016, 28, 8732–8739 CrossRef CAS PubMed.
- C. Wang, L. X. Wu, H. Wang, W. H. Zuo, Y. Y. Li and J. P. Liu, Adv. Funct. Mater., 2015, 25, 3524–3533 CrossRef CAS.
- D. S. Guan, J. Y. Li, X. F. Gao and C. Yuan, RSC Adv., 2014, 4, 4055–4062 RSC.
- B. Ahmed, M. Shahid, D. H. Nagaraju, D. H. Anjum, M. N. Hedhili and H. N. Alshareef, ACS Appl. Mater. Interfaces, 2015, 7, 13154–13163 CAS.
- M. L. Petrova, M. S. Bojinov and I. H. Gadjov, Bulg. Chem. Commun., 2011, 43, 60–63 CAS.
- Y. B. Xie, Y. Wang and H. X. Du, J. Mater. Sci. Eng. B, 2013, 178, 1443–1451 CrossRef CAS.
- R. B. Pujari, V. C. Lokhande, V. S. Kumbhar, N. R. Chodankar and C. D. Lokhande, J. Mater. Sci.: Mater. Electron., 2016, 27, 3312–3317 CrossRef CAS.
- I. Shakir and M. Sarfraz, Electrochim. Acta, 2014, 147, 380–384 CrossRef CAS.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CAS.
- S. Balendhran, J. K. Deng, J. Z. Ou, S. Walia, J. Scott, J. S. Tang, K. L. Wang, M. R. Field, S. Russo, S. Zhuiykov, M. S. Strano, N. Medhekar, S. Sriram, M. Bhaskaran and K. Kalantar-zadeh, Adv. Mater., 2013, 25, 109–114 CrossRef CAS PubMed.
- C. V. Ramana, V. V. Atuchin, H. Groult and C. M. Julien, J. Vac. Sci. Technol., A, 2012, 30, 04D105 Search PubMed.
- T. M. McEvoy and K. J. Stevenson, Langmuir, 2003, 19, 4316–4326 CrossRef CAS.
- Y. F. Dong, X. M. Xu, S. Li, C. H. Han, K. N. Zhao, L. Zhang, C. J. Niu, Z. Huang and L. Q. Mai, Nano Energy, 2015, 15, 145–152 CrossRef.
- T. Tao, Q. Y. Chen, H. P. Hu and Y. Chen, Mater. Lett., 2012, 66, 102–105 CrossRef CAS.
- S. P. Sun, X. M. Liao, G. F. Yin, Y. D. Yao, Z. B. Huang and X. M. Pu, J. Alloys Compd., 2016, 680, 538–543 CrossRef CAS.
- H. Wu, D. D. Li, X. F. Zhu, C. Y. Yang, D. F. Liu, X. Y. Chen, Y. Song and L. F. Lu, Electrochim. Acta, 2014, 116, 129–136 CrossRef CAS.
- L. X. Zheng, C. D. Wang, Y. C. Dong, H. D. Bian, T. F. Hung, J. Lu and Y. Y. Li, Appl. Surf. Sci., 2016, 362, 399–405 CrossRef CAS.
- Z. Li, Y. T. Ding, W. J. Kang, C. Li, D. Lin, X. Y. Wang, Z. W. Chen, M. H. Wu and D. Y. Pan, Electrochim. Acta, 2015, 161, 40–47 CrossRef CAS.
- H. Zhou and Y. R. Zhang, J. Phys. Chem. C, 2014, 118, 5626–5636 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01164d |
| This journal is © The Royal Society of Chemistry 2017 |