
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Prenylated phenylpropanoids with unprecedented skeletons from Illicium burmanicum†
Xinhui
Tian‡
 a,
Xin
Guo‡
a,
Zhiguo
Zhuo
b,
Rentao
Zeng
b,
Xin
Fang
b,
Xike
Xu
b,
Huiliang
Li
b,
Yunheng
Shen
b and
Weidong
Zhang
*abc
a,
Xin
Guo‡
a,
Zhiguo
Zhuo
b,
Rentao
Zeng
b,
Xin
Fang
b,
Xike
Xu
b,
Huiliang
Li
b,
Yunheng
Shen
b and
Weidong
Zhang
*abc
aInterdisciplinary Science Research Institute, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, P. R. China. E-mail: wdzhangy@hotmail.com; Web: http://www.shutcm.edu.cn/
bDepartment of Phytochemistry, School of Pharmacy, Second Military Medical University, Shanghai 200433, P. R. China
cInnovative Research Center of Traditional Chinese Medicine, Shanghai Institute of Pharmaceutical Industry, Shanghai 201204, P. R. China
First published on 9th February 2017
Abstract
A phytochemical investigation on the branches and leaves of Illicium burmanicum led to the isolation of two unique allo-thujane-phenylpropane and lavandulane-phenylpropane hetero-adducts (1 and 2), two new prenylated phenylpropanoids (3 and 4), and one new neolignan (5). Their structures were established by comprehensive NMR and CD spectroscopic analysis. Burmaniols A (1) and B (2) showed appreciable cytotoxicity against A549 and HCT116 cells with IC50 values of 6.40–7.76 μM.
Introduction
There are approximately 50 species in the genus Illicium, and 28 of which occur exclusively in China.1Illicium plants are rich sources of structurally diverse sesquiterpenoids, lignans and prenylated phenylpropanoids.2–9 Plants of the Illicium genus have attracted considerable attention due to their diverse bioactivities such as neurotrophic,4,10–22 neurotoxic,23 cytotoxic,24 cancer chemopreventive,25 anti-inflammatory9,26–27 and anti-depressant activities.28Illicium burmanicum E. H. Wilson (Schisandraceae) is an evergreen tree or shrub distributed in Burma and Yunnan province of China. Its roots, leaves, and fruit have been locally used as folk medicine for preventing vomiting, relieving pain, promoting tissue regeneration and setting a broken bone in the southwest of China.29 Previously, several new sesquiterpene lactones and prenylated phenylpropanoids with anti-inflammatory activity have been isolated from the stem bark of I. burmanicum.9,26 In this study further investigation on the branches and leaves of I. burmanicum was performed to search for more compounds with novel structures and potent bioactivities. As a result, five new (1–5) and six known compounds (6–11) were isolated from this plant. Herein, the isolation and structural elucidation of compounds 1–11, and their cytotoxic activities against human tumor cell lines are described (Fig. 1).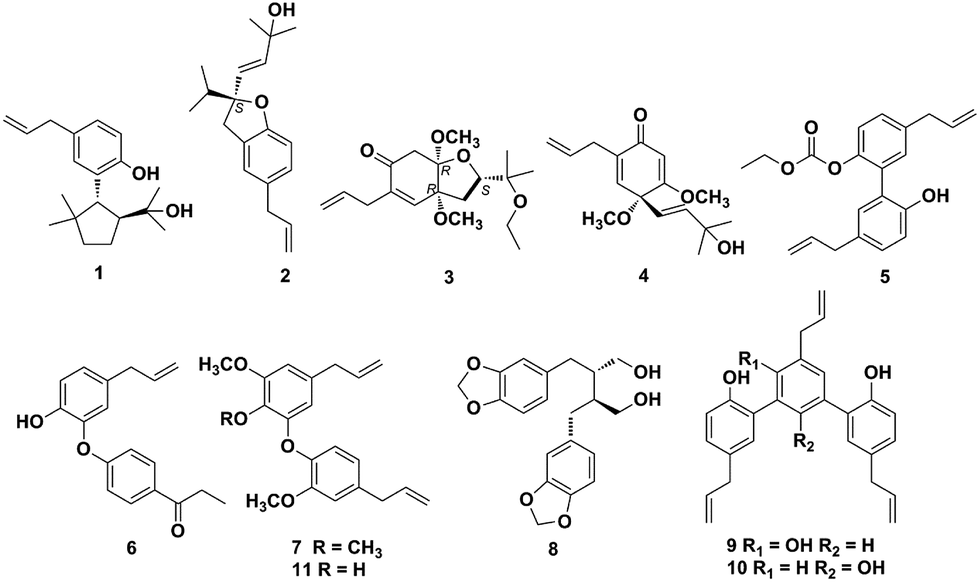 | ||
| Fig. 1 Chemical structures of compounds 1–11. | ||
Results and discussion
Compound 1 was isolated as white wax. The pseudo-molecular ion peak at m/z = 287.2022 [M − H]− (calcd 287.2011) established the molecular formula of C19H28O2. The 1H NMR spectrum showed signals attributed to four methyl groups at δH = 0.74 (3H, s), 0.97 (3H, s), 0.99 (3H, s), 1.13 (3H, s), an allyl group at δH = 3.29 (2H, d, J = 6.5 Hz), 5.00 (1H, dd, J = 17.0, 2.0 Hz), 5.02 (1H, dd, J = 11.0, 2.0 Hz), 5.95 (1H, ddt, J = 17.0, 11.0, 6.5 Hz), and a 1,3,4-substituted phenyl group at δH = 6.70 (1H, d, J = 8.0 Hz), 6.85 (1H, dd, J = 8.0, 1.5 Hz), 6.99 (1H, br s). More detailed information about the structure of 1 came from the interpretation of 1H–1H COSY, HSQC, HMBC, and NOESY spectra. In the 1H–1H COSY spectrum of 1, correlations of 8-H (δH = 5.95) with 7-H2 (δH = 3.29) and 9-H2 (δH = 5.00, 5.02) were observed, whereas 5-H (δH = 6.70) displayed correlation with 6-H (δH = 6.85). The above evidence, together with the key HMBC correlations from 7-H2 to C-1 (δC = 131.2), C-2 (δC = 129.6), C-6 (δC = 126.8), C-8 (δC = 138.1), from 5-H to C-3 (δC = 128.9) and C-4 (δC = 151.9), and from 2-H (δH = 6.99) to C-4 established the established the phenylpropene moiety in 1. Additionally, the spin coupling system 1′-H/2′-H/3′-H2/4′-H2 was also established on the basis of their mutual 1H–1H COSY correlations. Observation of HMBC correlations from 9′-H3 (δH = 0.97) and 10′-H3 (δH = 0.74) to C-1′ (δC = 47.4), C-4′ (δC = 41.6), C-5′ (δC = 43.9), and 7′-H3 (δH = 1.13) and 8′-H3 (δH = 0.99) to C-2′ (δC = 54.3), C-6′ (δC = 73.7) established an allo-thujane monoterpenoid moiety. A connection between C-3 and C-1′ was proved on the basis of the key HMBC correlations of 2-H with C-1′, and 2′-H (δH = 2.47) with C-3. Considering the molecular formula, there were two additional hydroxyls in 1, and they were supposed to locate at C-4 and C-6′ positions based on the 13C NMR chemical shift. Strong NOESY correlations of 10′-H3 with 2-H, and 1′-H (δH = 3.19) with 7′-H3, 8′-H3, 9′-H3 established the relative configuration of 1. Therefore, the structure of 1 was established, and named burmaniol A (Fig. 2).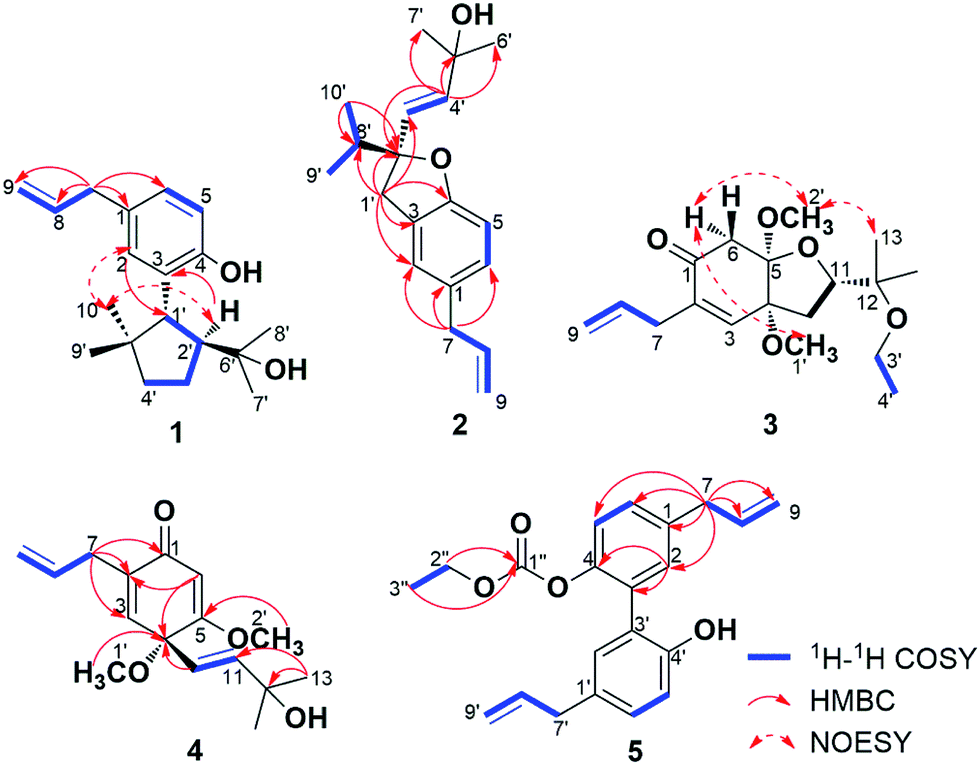 | ||
| Fig. 2 Key 1H–1H COSY, HMBC and NOESY correlations of compounds 1–5. | ||
Compound 2 was isolated as colorless oil. The HREIMS data at m/z 286.1931 [M]+˙ (calcd 286.1933) established its molecular formula of C19H26O2, suggesting seven degrees of unsaturation. The 1H NMR spectrum displayed the presence of two singlet methyl groups at δH = 1.27 (3H, s), 1.30 (3H, s), an isopropyl group at δH = 0.96 (6H, d, J = 6.5 Hz), 2.00 (1H, sept, J = 6.5 Hz), an allyl group at δH = 3.29 (2H, d, J = 6.5 Hz), 5.03 (1H, dd, J = 10.0, 2.0 Hz), 5.06 (1H, dd, J = 17.0, 2.0 Hz), 5.95 (1H, ddt, J = 17.0, 10.0, 6.5 Hz), a trans-double bond at δH = 5.75 (1H, d, J = 15.5 Hz), 5.88 (1H, d, J = 15.5 Hz), and a 1,2,4-trisubstituted phenyl group at δH = 6.69 (1H, d, J = 8.0 Hz), 6.91 (1H, d, J = 8.0 Hz), 6.93 (1H, s). More detailed information about the structure of 2 came from the interpretation of 1H–1H COSY, HSQC, and HMBC spectra. Part of the NMR spectra of 2 strongly resembled to those of 1, indicating that it also has a propen-2-ylphenyl. Additionally, two spin coupling systems of 3′-H/4′-H and 9′-H3/8′-H/10′-H3 were also established on the basis of their mutual 1H–1H COSY correlations. Further observation of HMBC correlations from 9′-H3 (δH = 0.96), 10′-H3 (δH = 0.96) to C-2′ (δC = 92.3) and C-8′ (δC = 37.1), from 1′-H2 (δH = 3.00, 3.18) to C-2′, C-3′ (δC = 128.0) and C-8′, as well as the correlations from 4′-H (δH = 5.88) to C-2′, C-5′ (δC = 70.8), C-6′, 7′ (δC = 29.9) established a lavandulane monoterpenoid fragment. The monoterpenoid fragment was supposed to link to C-3 on the basis of the key HMBC correlations of 1′-H2 with C-2 (δC = 124.9), C-3 (δC = 126.5), C-4 (δC = 157.7), and 2-H (δH = 6.93) with C-1′ (δC = 38.4). Deducting six degrees of unsaturation accounted for one phenyl group and two double bonds, the remaining one degree of unsaturation suggested that an additional ring was required. On the basis of the chemical shift, C-2′ was supposed to link with C-4 through an oxygen atom, forming a furan ring. Thus the planar structure of 2 was established as depict, and named burmaniol B. The absolute configuration of the sole C-2′ chiral carbon was determined to be S by comparison of its specific rotation value with (S)-2-ethenyl-2-methyl-2,3-dihydrobenzofuran reported in literature.30
Compound 3 was obtained as colorless oil, and its molecular formula of C18H28O5 was indicated by HRESIMS at m/z = 347.1854 [M + Na]+ (calcd 347.1834). The 1H NMR spectrum showed signals attributed to four singlet methyls at δH = 1.09 (3H, s), 1.12 (3H, s), 3.37 (3H, s), 3.39 (3H, s), an ethoxyl group at δH = 1.11 (3H, t, J = 7.0 Hz), 3.40 (2H, q, J = 7.0 Hz), an isolated methylene group at δH = 2.57 (1H, d, J = 16.5 Hz), 3.21 (1H, d, J = 16.5 Hz), an allyl group at δH = 3.04 (1H, dd, J = 7.0, 1.0 Hz), 3.05 (1H, dd, J = 7.0, 1.0 Hz), 5.09 (1H, dd, J = 17.0, 1.0 Hz), 5.11 (1H, dd, J = 10.0, 1.0 Hz), 5.82 (1H, ddt, J = 17.0, 10.0, 7.0 Hz), and a tri-substituted double bond at δH = 6.39 (1H, s). The above spectroscopic data strongly resembled to those of 2,3-dehydro-4,5-di-O-methyl-illifunone E isolated from Illicium anisatum,5 except for an additional ethoxyl group (δH = 1.11, 3.40; δC = 16.2, 57.0) in 3. The ethoxyl group was supposed to attach at C-12 on the basis of the HMBC correlations of 3′-H2 (δH = 3.40) and 4′-H3 (δH = 1.11) with C-12 (δH = 75.4). The relative configuration of 3 was established based on the NOESY correlations of 6α-H (δH = 2.57) with 1′-H3 (δH = 3.37) and 2′-H3 (δH = 3.39), and 2′-H3 with 13-H3 (δH = 1.12). The absolute configuration of 3 was determined to be 4R,5R,11S when compared its CD spectrum with illicinone E and 4-epi-illicinone E-12-shikimate at 320 nm (Fig. S23†).2,7 Thus, the structure of 3 was identified as (4R,5R,11S)-2,3-dehydro-4,5-di-O-methyl-12-O-ethyl-illifunone E.
Compound 4 was obtained as colorless oil, and its molecular formula of C16H22O4 was indicated by HRESIMS at m/z = 279.1597 [M + H]+ (calcd 279.1596). The 1H NMR spectrum showed signals attributed to four singlet methyls at δH = 1.27 (6H, s), 3.13 (3H, s), 3.74 (3H, s), an allyl group at δH = 3.02 (1H, ddd, J = 16.0, 7.0, 1.0 Hz), 3.09 (1H, ddd, J = 16.0, 7.0, 1.0 Hz), 5.07 (2H, m), 5.81 (1H, ddt, J = 18.0, 10.0, 7.0 Hz), a trans double bond at δH = 5.58 (1H, d, J = 16.0 Hz), 5.94 (1H, d, J = 16.0 Hz), and two tri-substituted double bonds at δH = 5.64 (1H, s), 6.15 (1H, br s). The above spectroscopic data exhibited great similarity to illicinone G isolated from Illicium tashiroi.3 The differences between them was the absence of the methylenedioxy (δH = 5.41, 5.60; δC = 97.7), and these signals were replaced by two new emerged methoxyl groups (δH = 3.13, 3.74; δC = 52.4, 56.1) in 4. The HMBC correlations of 1′-H3 (δH = 3.13) with C-4 (δC = 76.5) and 2′-H3 (δH = 3.74) with C-5 (δC = 172.8) attributed the positions of the two methoxyl groups to be at C-4 and C-5, respectively. Thus, the structure of 4 was identified as 4,5-dimethoxyillicinone G.
Compound 5 was isolated as yellow oil. Its molecular formula was established as C21H22O4 on the basis of its positive HRESIMS peak at m/z = 356.1862 [M + NH4]+ (calcd 356.1862). The 1H NMR spectrum of 5 showed signals attributed to an ethoxyl group at δH = 1.16 (3H, t, J = 7.0 Hz), 4.11 (2H, q, J = 7.0 Hz), two allyl units at δH = 3.33 (2H, d, J = 7.0 Hz), 3.42 (2H, d, J = 7.0 Hz), 5.09 (4H, m), 5.95 (2H, m), and two 1,3,4-substituted phenyl groups at δH = 6.91 (1H, d, J = 8.5 Hz), 6.96 (1H, br d, J = 2.5 Hz), 7.07 (1H, dd, J = 8.5, 2.5 Hz) and δH = 7.18 (1H, d, J = 8.5 Hz), 7.19 (1H, br d, J = 2.5 Hz), 7.25 (1H, dd, J = 8.5, 2.5 Hz). The above spectroscopic data exhibited great similarities to the known compound magnolol,31 except that there was an additional ethoxycarboxyl group in 5. The existence of ethoxycarboxyl group was confirmed by the 1H–1H COSY correlation of 2′′-H2 (δH = 4.11) with 3′′-H3 (δH = 1.16) and HMBC correlation of 2′′-H2 and 3′′-H3 with C-1′′ (δC = 153.7). According to the chemical shift of C-4 and C-4′, the ethoxycarboxyl group was supposed to locate at C-4 position. Thus the structure of 5 was established and named 4-[(ethoxycarbonyl)oxy]magnolol.
The known compounds (6–11) were identified as isomagnolone (6),32 1-(8-propenyl)-3-[3′-methoxy-1′-(8-propenyl)phenoxy]-4,5-dimethoxybenzene (7, Fig. S48–S54†),33,34 dihydrocubebin (8),35 macranthol (9),36 dunnianol (10),37 dehydrodieugenol B (11)38 by comparison of their spectroscopic data with those reported previously. The isolated compounds (1–11) were evaluated for cytotoxic activity against human tumor cell lines A549 (non-small-cell lung cancer cells), HCT116 (human colon cancer cells), MDA-MB-231 (human breast cancer cells), and HepG2 (human hepatocellular carcinoma cells) in vitro (Table 3).39 The results showed that compounds 1 and 2 displayed appreciable cytotoxic activity against A549 and HCT116 cell lines with IC50 values of 6.40–7.76 μM, whereas compounds 9 and 10 only exhibited potent inhibition against A549, HCT116 and HepG2 cells with IC50 values of 9.46–15.07 μM. Doxorubicin was employed as the positive control and its IC50 values against A549, HCT116, MDA-MB-231 and HepG2 cells were 0.18 ± 0.004, 0.07 ± 0.001, 0.59 ± 0.010 and 0.06 ± 0.001 μM, respectively.
Conclusions
The eleven compounds (1–11) isolated from I. burmanicum indicated that Illicium plants are rich in structurally diverse lignans, neolignans and prenylated phenylpropanoids. Among the five new compounds (1–5), burmaniols A (1) and B (2) were unique allo-thujane-phenylpropane and lavandulane-phenylpropane hetero-adducts, and compound 1 contains a new furan ring formed through C-2′ and C-4. Compound 5 had an ethoxycarboxyl group at C-4 position, and the organic carbonates was encountered very rarely. These new compounds add to the current list of miscellaneous constituents isolated from the Illicium genus. MTT assay indicated that compounds 1 and 2 have appreciable cytotoxicity against A549 and HCT 116 cells, while initial evaluation of the anti-tumor efficacy of these compounds is not enough, further studies are necessary for understanding their cytotoxic mechanisms.Experimental section
General
1D and 2D NMR spectral data were obtained on a Bruker Avance III 500 MHz NMR spectrometer (Bruker, Fallanden, Switzerland) with TMS as internal standard. HRESIMS spectra were measured on an Agilent 6520 Accurate-MS Q-TOF LC/MS system (Agilent Technologies, Santa Clara, CA, USA). HREIMS spectra were measured on Autospec-Ultima ETOF MS spectrometer (Micromass Ltd., Wythenshawe, Manchester, UK). Optical rotations were recorded on a Perkin-Elmer 341 digital polarimeter and CD spectra were recorded on a JASCO-J-810 spectrometer (JASCO Perkin-Elmer, Maryland, USA). Reversed phase medium pressure liquid chromatography (RP-MPLC) was performed on a Büchi Sepacore system (Büchi Labortechnik AG, Flawil, Switzerland). Materials for column chromatography were silica gel (100–200, 200–300 mesh, Huiyou Silica Gel Development Co. Ltd, Yantai, P. R. China), YMC-GEL ODS-A (50 μm, Milford, Massachusetts, USA) and Sephadex LH-20 (40–70 μm, Amersham Pharmacia Biotech AB, Uppsala, Sweden). All chemicals and solvents were analytical or high-performance liquid chromatography grade.Plant material
The branches and leaves of Illicium burmanicum were collected in Gongshan county, Yunnan province, P. R. China, in September 2011, and authenticated by Prof. Han-Ming Zhang from Second Military Medical University. A voucher specimen (no. 20110925) is deposited in School of Pharmacy, Second Military Medical University.Extraction and isolation
Air-dried branches and leaves of Illicium burmanicum (20.0 kg) were powdered and extracted with 95% EtOH (80 L) three times (1 h) under condition of reflux. The solvent was removed under low pressure to afford a crude extract (1.2 kg), which was then suspended in water and extracted with petroleum ether (442 g), CH2Cl2 (188 g), EtOAc (120 g) and n-BuOH (155 g) successively. The petroleum ether (PE) extract was subjected to silica gel column chromatography (CC) (ϕ 10 × 120 cm, 100–200 mesh, 2.6 kg) and eluted with PE/EtOAc (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–0:1) to give seven fractions [Fr. 1 (102 g), Fr. 2 (31 g), Fr. 3 (53 g), Fr. 4 (48 g), Fr. 5 (46 g), Fr. 6 (42 g), Fr. 7 (54 g)] based on TLC analysis. Fraction 3 was subjected to silica gel CC (ϕ 4.5 × 100 cm, 200–300 mesh, 1.0 kg) and eluted with PE/EtOAc (20:1–0:1) to afford eleven subfractions (Fr. 3-1–Fr. 3-11). Subfraction 3-4 (6.5 g) was subjected to Sephadex LH-20 CC (ϕ 5.0 × 120 cm) using CH2Cl2/MeOH (1:1) elution to give compounds 4 (12.7 mg), 8 (15.3 mg), and 9 (7.0 mg). Subfraction 3-9 (3.2 g) was also passed over Sephadex LH-20 CC (ϕ 5.0 × 120 cm) using CH2Cl2/MeOH (1:1) to give compounds 7 (31.0 mg) and 10 (6.3 mg). Fraction 5 was subjected to RP-MPLC (ϕ 6.0 × 50 cm, MeOH/H2O, 40–100%, 15 mL min−1) to give eight subfractions (Fr. 5-1–Fr. 5-8). Subfraction 5-2 (8.1 g) was applied to silica gel CC (ϕ 4.5 × 100 cm, 200–300 mesh, 800 g) and eluted with PE/EtOAc (10:1–0:1) to give compounds 1 (4.2 mg) and 2 (18.5 mg). Subfraction 5-5 (2.1 g) was passed over Sephadex LH-20 CC (ϕ 3.0 × 150 cm) using CH2Cl2/MeOH (1:1) elution to give compounds 3 (28.0 mg) and 11 (7.2 mg). Compounds 5 (34 mg) and 6 (3.6 mg) were isolated from subfraction 5-8 (2.6 g) by applying to RP-MPLC (ϕ 3.5 × 50 cm) and eluted with a gradient of MeOH/H2O (40–100%, 15 mL min−1).
:1–0:1) to give seven fractions [Fr. 1 (102 g), Fr. 2 (31 g), Fr. 3 (53 g), Fr. 4 (48 g), Fr. 5 (46 g), Fr. 6 (42 g), Fr. 7 (54 g)] based on TLC analysis. Fraction 3 was subjected to silica gel CC (ϕ 4.5 × 100 cm, 200–300 mesh, 1.0 kg) and eluted with PE/EtOAc (20:1–0:1) to afford eleven subfractions (Fr. 3-1–Fr. 3-11). Subfraction 3-4 (6.5 g) was subjected to Sephadex LH-20 CC (ϕ 5.0 × 120 cm) using CH2Cl2/MeOH (1:1) elution to give compounds 4 (12.7 mg), 8 (15.3 mg), and 9 (7.0 mg). Subfraction 3-9 (3.2 g) was also passed over Sephadex LH-20 CC (ϕ 5.0 × 120 cm) using CH2Cl2/MeOH (1:1) to give compounds 7 (31.0 mg) and 10 (6.3 mg). Fraction 5 was subjected to RP-MPLC (ϕ 6.0 × 50 cm, MeOH/H2O, 40–100%, 15 mL min−1) to give eight subfractions (Fr. 5-1–Fr. 5-8). Subfraction 5-2 (8.1 g) was applied to silica gel CC (ϕ 4.5 × 100 cm, 200–300 mesh, 800 g) and eluted with PE/EtOAc (10:1–0:1) to give compounds 1 (4.2 mg) and 2 (18.5 mg). Subfraction 5-5 (2.1 g) was passed over Sephadex LH-20 CC (ϕ 3.0 × 150 cm) using CH2Cl2/MeOH (1:1) elution to give compounds 3 (28.0 mg) and 11 (7.2 mg). Compounds 5 (34 mg) and 6 (3.6 mg) were isolated from subfraction 5-8 (2.6 g) by applying to RP-MPLC (ϕ 3.5 × 50 cm) and eluted with a gradient of MeOH/H2O (40–100%, 15 mL min−1).
| No. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 | |||||
| 2 | 6.99 br s | 6.93 s | 6.96 br d (2.5) | ||
| 3 | 6.39 br s | 6.15 br s | |||
| 4 | |||||
| 5 | 6.70 d (8.0) | 6.69 d (8.0) | 6.91 d (8.5) | ||
| 6 | 6.85 dd (8.0, 1.5) | 6.91 d (8.0) | 2.57 d (16.5) 3.21 d (16.5) | 5.64 s | 7.07 dd (8.5, 2.5) |
| 7 | 3.29 d (6.5) | 3.29 d (6.5) | 3.04 dd (7.0, 1.0) | 3.02 ddd (16.0, 7.0, 1.0) | 3.33 d (7.0) |
| 3.05 dd (7.0, 1.0) | 3.09 ddd (16.0, 7.0, 1.0) | ||||
| 8 | 5.95 ddt (17.0, 11.0, 6.5) | 5.95 ddt (17.0, 10.0, 6.5) | 5.82 ddt (17.0, 10.0, 7.0) | 5.81 ddt (18.0, 10.0, 7.0) | 5.95 m |
| 9 | 5.00 dd (17.0, 2.0) | 5.03 dd (10.0, 2.0) | 5.09 dd (17.0, 1.0) | 5.07 m | 5.09 m |
| 5.02 dd (11.0, 2.0) | 5.06 dd (17.0, 2.0) | 5.11 dd (10.0, 1.0) | |||
| 10 | 1.99 dd (12.0, 6.0) | 5.58 d (16.0) | |||
| 2.53 dd (12.0, 11.0) | |||||
| 11 | 3.59 dd (11.0, 6.0) | 5.94 d (16.0) | |||
| 12 | |||||
| 13 | 1.12 s | 1.27 s | |||
| 14 | 1.09 s | 1.27 s | |||
| 1′ | 3.19 d (10.0) | 3.00 d (15.5), 3.18 d (15.5) | 3.37 s | 3.13 s | |
| 2′ | 2.47 m | 3.39 s | 3.74 s | 7.19 br d (2.5) | |
| 3′ | 1.69 m, 1.95 m | 5.75 d (15.5) | 3.40 q (7.0) | ||
| 4′ | 1.59 m | 5.88 d (15.5) | 1.11 t (7.0) | ||
| 5′ | 7.18 d (8.5) | ||||
| 6′ | 1.27 s | 7.25 dd (8.5, 2.5) | |||
| 7′ | 1.13 s | 1.30 s | 3.42 d (7.0) | ||
| 8′ | 0.99 s | 2.00 sept (6.5) | 5.95 m | ||
| 9′ | 0.97 s | 0.96 d (6.5) | 5.09 m | ||
| 10′ | 0.74 s | 0.96 d (6.5) | |||
| 2′′ | 4.11 q (7.0) | ||||
| 3′′ | 1.16 t (7.0) |
| No. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 | 131.2 s | 131.5 s | 194.9 s | 186.6 s | 131.9 s |
| 2 | 129.6 d | 124.9 d | 141.5 s | 138.7 s | 130.6 d |
| 3 | 128.9 s | 126.5 s | 142.2 d | 140.7 d | 123.6 s |
| 4 | 151.9 s | 157.7 s | 80.9 s | 76.5 s | 151.3 s |
| 5 | 115.6 d | 108.7 d | 102.0 s | 172.8 s | 116.4 d |
| 6 | 126.8 d | 127.9 d | 42.6 t | 103.9 d | 129.7 d |
| 7 | 39.6 t | 39.7 t | 33.1 t | 32.7 t | 39.3 t |
| 8 | 138.1 d | 138.2 d | 134.7 d | 134.9 d | 137.7 d |
| 9 | 115.2 t | 115.2 t | 117.5 t | 117.1 t | 115.5 t |
| 10 | 37.1 t | 125.3 d | |||
| 11 | 83.0 d | 140.3 d | |||
| 12 | 75.4 s | 70.7 s | |||
| 13 | 21.6 q | 29.7 q | |||
| 14 | 20.9 q | 29.7 q | |||
| 1′ | 47.4 d | 38.4 t | 52.7 q | 52.4 q | 130.0 s |
| 2′ | 54.3 d | 92.3 s | 48.8 q | 56.1 q | 131.8 d |
| 3′ | 25.2 t | 128.0 d | 57.0 t | 138.9 s | |
| 4′ | 41.6 t | 137.4 d | 16.2 q | 147.2 s | |
| 5′ | 43.9 s | 70.8 s | 122.4 d | ||
| 6′ | 73.7 s | 29.9 q | 129.6 d | ||
| 7′ | 27.9 q | 29.9 q | 39.5 t | ||
| 8′ | 28.5 q | 37.1 d | 136.7 d | ||
| 9′ | 28.6 q | 17.0 q | 116.5 t | ||
| 10′ | 23.9 q | 17.5 q | |||
| 1′′ | 153.7 s | ||||
| 2′′ | 64.9 t | ||||
| 3′′ | 13.9 q |
| Compounds | IC50 (μM) | |||
|---|---|---|---|---|
| A549 | HCT116 | MDA-MB-231 | HepG2 | |
| a n = 3, means ± SD. b Positive control. | ||||
| 1 | 6.42 ± 0.13 | 7.76 ± 0.11 | 17.08 ± 0.09 | 84.10 ± 1.15 |
| 2 | 6.40 ± 0.12 | 7.18 ± 0.08 | 28.66 ± 0.27 | 12.88 ± 0.17 |
| 9 | 9.46 ± 0.18 | 10.86 ± 0.15 | 17.30 ± 0.31 | 13.64 ± 0.13 |
| 10 | 12.91 ± 0.24 | 15.07 ± 0.29 | 30.72 ± 0.66 | 13.49 ± 0.25 |
| Doxorubicinb | 0.18 ± 0.004 | 0.07 ± 0.001 | 0.59 ± 0.010 | 0.06 ± 0.001 |
Chemicals and reagents for biological activities
The human non-small-cell lung carcinoma cells (A549), human colon cancer cells (HCT116), human breast cancer cells (MDA-MB-231), and human hepatocellular carcinoma cells (HepG2) were obtained from the Cell Bank of Shanghai Institute of Biochemistry & Cell Biology, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences; Dimethylsulfoxide (DMSO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and doxorubicin were purchased from Sigma Chemical Co. (St. Louis, MO, USA).MTT assay
The cytotoxicity of compounds against human non-small-cell lung carcinoma cells (A549), human colon cancer cells (HCT116), human breast cancer cells (MDA-MB-231), and human hepatocellular carcinoma cells (HepG2) was determined by MTT assay.39 The assay was performed in triplicate. All cells were seeded in 96-well plate at a density of 104 cells per well and incubated in a humidified 5% CO2 atmosphere at 37 °C for 24 h. Then the medium was removed, and each well was treated with 50 μL of medium containing 0.2% DMSO (control group), or 1–100 μM tested compounds, or the positive control doxorubicin. After 24 h of treatment, the medium was removed, and 20 μL of MTT solution (5 mg mL−1; Sigma; St. Louis, MO) was added to each well, and the cultures were incubated for another 3 h at 37 °C. Upon removal of MTT medium, 100 μL of DMSO was added to each well and agitated at 60 rpm for 5 min to dissolve the precipitate. The absorbance was measured at 570 nm by a SYNERGY microplate reader (Bio Tek, Winooski, VT). Doxorubicin was employed as the positive control and its IC50 values against A549, HCT116, MDA-MB-231, and HepG2 cells were 0.18 ± 0.004, 0.07 ± 0.001, 0.59 ± 0.010 and 0.06 ± 0.001 μM, respectively.Acknowledgements
The work was supported by Professor of Chang Jiang Scholars Program, NSFC (81230090, 81520108030, 81573318, 81373301, 1302658), Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products (10DZ2251300), the Scientific Foundation of Shanghai China (12401900801, 13401900101), National Major Project of China (2011ZX09307-002-03) and the National Key Technology R & D Program of China (2012BAI29B06).References
- Y. H. Liu, X. R. Luo, R. F. Wu and B. N. Zhang, Flora Republicae Popularis Sinicae, Science Press, Beijing, 1996, vol. 30, pp. 199–231 Search PubMed.
- Y. Fukuyama, N. Shida, T. Sakurai and M. Kodama, Phytochemistry, 1992, 31, 3975–3979 CrossRef CAS.
- Y. Fukuyama, N. Shida, Y. Hata and M. Kodama, Phytochemistry, 1994, 36, 1497–1503 CrossRef CAS.
- Y. Fukuyama, K. Okamoto, Y. Kubo, N. Shida and M. Kodama, Chem. Pharm. Bull., 1994, 42, 2199–2201 CrossRef CAS PubMed.
- I. Kouno, S. Shimamoto, Z. H. Jiang and T. Tanaka, Phytochemistry, 1997, 46, 1389–1392 CrossRef CAS.
- Y. N. Liu, X. H. Su, C. H. Huo, X. P. Zhang, Q. W. Shi and Y. C. Gu, Chem. Biodiversity, 2009, 6, 963–989 CrossRef CAS PubMed.
- X. F. Wu, Y. Li, H. N. Lu, S. S. Yu, S. G. Ma and J. Liu, J. Asian Nat. Prod. Res., 2009, 12, 1056–1061 CrossRef PubMed.
- Y. Z. He, E. K. Osoro, S. I. Palmer, L. N. Wang and N. S. Aboud, Chin. Herb. Med., 2014, 6, 76–79 CrossRef CAS.
- D. D. Huang, H. Y. Zhu, Y. H. Chen, W. S. Chen, D. Xue and L. N. Sun, Fitoterapia, 2015, 107, 22–28 CrossRef CAS PubMed.
- Y. Fukuyama, N. Shida, M. Kodama, H. Chaki and T. Yugami, Chem. Pharm. Bull., 1995, 43, 2270–2272 CrossRef CAS PubMed.
- T. Nakamura, M. Okuyama and M. Yamazaki, Chem. Pharm. Bull., 1996, 44, 1908–1914 CrossRef CAS PubMed.
- Y. Fukuyama, Y. Hata and M. Kodama, Planta Med., 1997, 63, 275–277 CrossRef CAS PubMed.
- J. M. Huang, R. Yokoyama, C. S. Yang and Y. Fukuyama, Tetrahedron Lett., 2000, 41, 6111–6114 CrossRef CAS.
- J. M. Huang, R. Yokoyama, C. S. Yang and Y. Fukuyama, J. Nat. Prod., 2001, 64, 428–431 CrossRef CAS PubMed.
- R. Yokoyama, J. M. Huang, C. S. Yang and R. Fukuyama, J. Nat. Prod., 2002, 65, 527–531 CrossRef CAS PubMed.
- M. Moriyama, J. M. Huang, C. S. Yang, H. Hioki, M. Kubo, K. Harada and Y. Fukuyama, Tetrahedron, 2007, 63, 4243–4249 CrossRef CAS.
- M. Moriyama, J. M. Huang, C. S. Yang, M. Kubo, K. Harada, H. Hioki and Y. Fukuyama, Chem. Pharm. Bull., 2008, 56, 1201–1204 CrossRef CAS PubMed.
- M. Kubo, C. Okada, J. M. Huang, K. Harada, H. Hioki and Y. Fukuyama, Org. Lett., 2009, 22, 5190–5193 CrossRef PubMed.
- M. Kubo, K. Kobayashi, J. M. Huang, K. Harada and Y. Fukuyama, Tetrahedron Lett., 2012, 53, 1231–1235 CrossRef CAS.
- S. Takaoka, N. Takaoka, Y. Minoshima, J. M. Huang, M. Kubo, K. Harada, H. Hioki and Y. Fukuyama, Tetrahedron, 2009, 65, 8354–8361 CrossRef CAS.
- J. Xu, M. H. Lacoske and E. A. Theodorakis, Angew. Chem., Int. Ed., 2014, 53, 956–987 CrossRef CAS PubMed.
- X. H. Tian, R. C. Yue, H. W. Zeng, H. L. Li, L. Shan, W. W. He, Y. H. Shen and W. D. Zhang, Sci. Rep., 2015, 5, 16982 CrossRef CAS PubMed.
- Y. Kudo, J. I. Oka and K. Yamada, Neurosci. Lett., 1981, 25, 83–88 CrossRef CAS PubMed.
- X. H. Tian, L. Li, J. P. Pei, R. C. Yue, X. Fang, J. P. Zhang, W. W. He, L. Shan, Y. H. Shen and W. D. Zhang, RSC Adv., 2015, 5, 75857–75862 RSC.
- M. Itoigawa, C. Ito, H. Tokuda, F. Enjo, H. Nishino and H. Furukawa, Cancer Lett., 2004, 214, 165–169 CrossRef CAS PubMed.
- D. D. Huang, H. P. Deng, W. S. Chen, G. H. Huang, C. Chen and L. N. Sun, Fitoterapia, 2014, 92, 194–199 CrossRef CAS PubMed.
- T. Matsui, C. Ito, M. Itoigawa, T. Okada and H. Furukawa, Planta Med., 2007, 73, 662–665 CrossRef CAS PubMed.
- J. Li, D. Geng, J. Xu, L. J. Weng, Q. Liu and L. T. Yi, Eur. J. Pharmacol., 2013, 707, 112–119 CrossRef CAS PubMed.
- J. P. Wang, Z. Y. Guan, C. F. Dong, L. Gao, S. D. Luo and Y. F. Wang, China J. Chin. Mater. Med., 2014, 39, 2526–2530 CAS.
- Y. Uozumi, H. Kyota, K. Kato, M. Ogasawara and T. Hayashi, J. Org. Chem., 1999, 64, 1620–1625 CrossRef CAS PubMed.
- C. Q. Liang, X. L. Zhou, Z. Wang, X. J. Su and Q. Xu, Chin. Tradit. Pat. Med., 2010, 32, 824–827 CAS.
- I. Kouno, C. Iwamoto, Y. Kameda, T. Tanaka and C. S. Yang, Chem. Pharm. Bull., 1994, 42, 112–114 CrossRef.
- N. I. Baek, H. Kim, Y. H. Lee, J. D. Park, K. S. Kang and S. I. Kim, Planta Med., 1992, 58, 566–568 CrossRef CAS PubMed.
- T. A. D. Costa-Silva, S. S. Grecco, F. S. De Sousa, J. H. G. Lago, E. G. A. Martins, C. A. Terrazas, S. Varikuti, K. L. Owens, S. M. Beverley, A. R. Satoskar and A. G. Tempone, J. Nat. Prod., 2015, 78, 653–657 CrossRef PubMed.
- B. J. Cabanillas, A. Le Lamer, D. Castillo, J. Arevalo, R. Rojas, G. Odonne, G. Bourdy, B. Moukarzel, M. Sauvain and N. Fabre, J. Nat. Prod., 2010, 73, 1884–1890 CrossRef CAS PubMed.
- P. J. Yin, J. S. Wang, P. R. Wang and L. Y. Kong, Chin. J. Nat. Med., 2012, 10, 383–387 CAS.
- S. Yahara, T. Nishiyori, A. Kohda, T. Nohara and I. Nishioka, Chem. Pharm. Bull., 1991, 39, 2024–2036 CrossRef CAS.
- A. M. P. De Diaz, H. E. Gottlieb and O. R. Gottlieb, Phytochemistry, 1980, 19, 681–682 CrossRef CAS.
- X. H. Tian, R. C. Yue, S. D. Zhang, Y. H. Shen, J. Ye, L. Shan, H. L. Li, B. Wen, X. K. Xu and W. D. Zhang, Eur. J. Org. Chem., 2014, 4753–4758 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01143a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |