
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot homopolymerization of thiophene-fused isoindigo for ambient-stable ambipolar organic field-effect transistors†
Huanrui Zhang‡
a,
Zhiyuan Zhao‡bc,
Na Zhaoa,
Yu Xiea,
Mian Caiad,
Xiao Wanga,
Yunqi Liu b,
Zhenggang Lana and
Xiaobo Wan*ad
b,
Zhenggang Lana and
Xiaobo Wan*ad
aCAS Key Laboratory of Bio-based Materials, Qingdao Institute of Bioenergy & Bioprocess Technology, Chinese Academy of Sciences, 189 Songling Road, Qingdao 266101, People's Republic of China. E-mail: wanxb@qibebt.ac.cn
bInstitute of Chemistry, Chinese Academy of Sciences, Beijing 100080, People's Republic of China
cNortheast Normal University, Changchun 130024, People's Republic of China
dUniversity of Chinese Academy of Sciences, Beijing 100049, People's Republic of China
First published on 10th May 2017
Abstract
Donor–acceptor (D–A) type conjugated polymers have been extensively studied for organic field-effect transistors (OFETs), while the homopolymers of acceptors were much less investigated. However, conjugated homopolymers are structurally simpler, so their solid-phase behavior such as packing pattern, long-range order and crystallinity are more predictable, so are their charge-transport properties. Here we report the synthesis of the homopolymer of thiophene-fused isoindigo (TII), which was directly obtained using a “one-pot” polymerization method either via Suzuki condensation polymerization (denoted as PTII-1) or Stille condensation polymerization (denoted as PTII-2), respectively. To further elucidate the structure–property relationship of the PTII, TII was also copolymerized with II to give P(TII-co-II) for comparison. Encouragingly, solution-processed OFETs based on PTII-2 exhibited ambipolar transport behavior with balanced hole and electron mobilities (0.065 and 0.15 cm2 V−1 s−1, respectively) after annealing, which is two orders higher than that of P(TII-co-II). 2D grazing incidence X-ray diffraction (2D-GIXRD) analysis explicitly showed that PTII films have much better well-ordered structure and crystallinity than P(TII-co-II), which accounted for their better OFET performance. To the best of our knowledge, PTII-2 is among the best homopolymers of A units with high ambipolar charge-carrier mobility, indicating that the design of such homopolymers might be an important strategy toward high performance polymer OFETs. In addition, the “one-pot” homopolymerization of dibromo-monomers via Suzuki or Stille condensation also provides a more convenient way to construct conjugated homopolymers, which deserves a closer study.
Introduction
Organic field-effect transistors (OFETs) have aroused great attention in the past decades owing to their potential applications in a wide variety of electronic devices, such as electronic papers, displays, radio frequency identification tags, smart cards, and sensors.1–5 Depending on the energy level of the frontier molecular orbitals of the conjugated polymers, both p-type and n-type OFET devices could be obtained. To date, compared with the massive high-mobility unipolar polymer semiconductors, much fewer high-mobility ambipolar polymer semiconductors have been uncovered, especially those ambient-stable ones.6,7 Due to its important role in logic circuits, the development of ambient-stable ambipolar polymer materials remains a critical issue in OFETs.8The design of donor–acceptor (D–A) type conjugated polymers is the major route toward ambipolar or n-type OFET materials with improved ambient stability. Many amide/imide-containing structures, such as naphthalene diimide (NDI), diketopyrrolopyrrole (DPP), and isoindigo (II), are important building blocks for such applications. Especially, the modification on the II core structure has become a very important pathway to modulate the HOMO/LUMO energy level of the resulting D–A type conjugated polymers for better ambipolar properties. For example, Pei et al. demonstrated that charge transport properties of the polymers could evolve from solely p-type to ambipolar by using fluorine or chlorine substituted II as the A unit.9,10 Later on, inspired by II structure, they developed a novel acceptor unit BDOPV (benzodifurandione-based oligo(p-phenylene vinylene)) with an extended conjugated length and a further lowered HOMO/LUMO energy level, which endows the resultant polymer with high electron mobility and ambient stability.11–13 Very recently, Geng et al. showed that isoindigo[7,6-g]isoindigo-based D–A conjugated polymers exhibited balanced hole and electron mobilities.14
The design of acceptor–acceptor (A–A′) type conjugated polymers, as another important strategy, however, is less studied for the ambipolar and n-type OFET devices. Only a few cases were found in the literatures. For instance, Meager and coworkers reported the copolymer of thieno[3,2-b]-thiophene flanked isoindigo (iITT) with benzothiadiazole, which exhibited ambipolar characteristics in thin film OFET devices with hole mobility up to 0.4 cm2 V−1 s−1 and electron mobility up to 0.7 cm2 V−1 s−1.15 Using a similar method, a copolymer designed by Zaumseil consisting of thieno-isoindigo and benzothiadiazole units showed hole and electron mobilities in the order of 10−2 cm2 V−1 s−1.16 Recently, Liu and coworkers copolymerized II with bithiazole, which exhibited ambipolar transistor behavior with the balanced hole and electron mobilities of 0.030 and 0.022 cm2 V−1 s−1, respectively.17 Although less studied, theoretical studies indicate that, unlike most D–A type conjugated polymers with LUMO localized on the A units, A–A′ type conjugated polymers have well delocalized LUMO and HOMO over the entire polymeric backbone,15 which may facilitate both electron and hole transportation. Thus, it is a promising strategy that needs a further study for ambipolar OFETs.
Homopolymer of A units could be considered as a subgroup of A–A′ type conjugated polymer, however, draws even less attention. Compared to conjugated copolymers, conjugated homopolymers are structurally simple, so their solid-phase behaviors such as packing pattern, long-range order and crystallinity are more predictable, so are their charge-transport properties. Actually, the long-range packing and crystal structures of homopolymers such as poly(3-hexylthiophene) (P3HT) have been well-studied,18 and P3HT has been investigated as a p-type OFET material.19,20 Unfortunately, the solid-phase behaviors of A–A′ type conjugated copolymers are complicated and difficult to predict due to the presence of at least two structurally different comonomers in the polymeric backbone. The existence of two different components in D–A type conjugated polymer makes it even more difficult to predict its solid-state structures, since more complicated D–A interactions should be taken into account besides the size difference between D and A units. Such microscopic disorders affect the effective conjugation length, and consequently, their electronic and charge transport properties,21,22 which remains a sophisticated topic for investigation.23 So it will be an interesting topic to investigate the solid-phase behavior and charge transport properties of a homopolymer consisting of only one kind of A units with simplified backbone. To the best of our knowledge, however, only a few homopolymers for ambipolar or n-type OFETs were reported in literature.24–30 For instance, the homopolymer of N-alkyl-2,2′-bithiophene-3,3′-di-carboximide exhibited a high degree of crystallinity and an electron mobility up to 0.011 cm2 V−1 s−1 after annealing.25 Swager and coworkers synthesized a promising class of water and/or methanol soluble poly(pyridinium phenylene)s via post-polymerization modification, which exhibited high electron mobilities.27 The homopolymerization of diketopyrrolopyrrole–bithiophene (PDBT) was also reported, and the OFET devices based on this polymer exhibited a typical ambipolar characteristic with balanced high hole and electron mobilities (μh = 0.024 cm2 V−1 s−1 and μe = 0.056 cm2 V−1 s−1).30 However, due to the existence of both the DPP unit and the bithiophene unit, it was actually a copolymer. In 2011, the homopolymer of isoindigo (i.e. polyisoindigos, PII) was reported by Reynolds' group as a potential n-type polymer for both OFETs and organic solar cells (OSCs), however with a very low electron mobility of 10−7 cm2 V−1 s−1, which was recently improved to 10−4 cm2 V−1 s−1,29,31 much lower than the values reported for other II-based copolymers. We reasoned that the steric hindrance between II units caused by phenyl–phenyl ring repulsion and the slightly twisted structure at the central double bond of II unit32 might largely decrease the coplanarity of the polymeric backbone, which further influenced on the crystallinity, film morphology and microstructures, and thus led to the poor OFET performance.
We thus envisioned that the conjugated homopolymers of isoindigo derivatives could have more predictable crystallinity and exhibit better n-type or ambipolar OFET performance, given that the backbone coplanarity could be well-remained. We have reported the synthesis of thiophene-fused isoindigo (TII), which exhibited better coplanarity than II and its copolymerization with D building blocks led to polymeric materials with balanced ambipolar mobilities.33 We anticipated that compared to the homopolymer of II, the steric hindrance of the adjacent TII unit in the homopolymer of TII (PTII) could be largely alleviated due to the smaller thiophene–thiophene dihedral angle, therefore PTII would be a better n-type or ambipolar OFET material compared to PII.
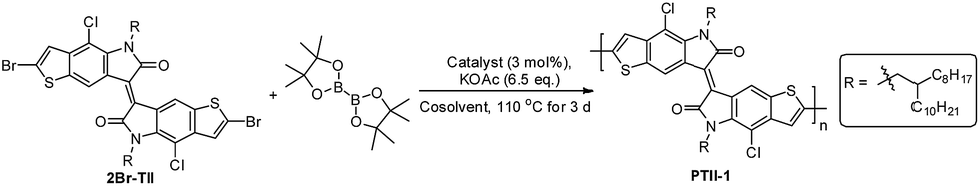
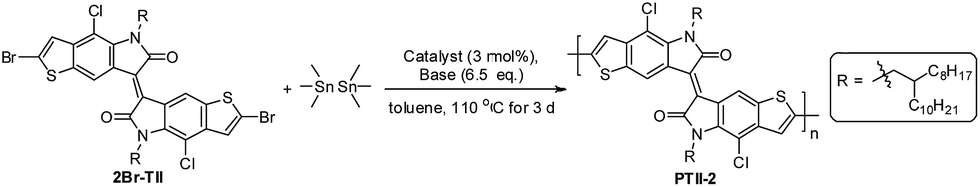
Here we report the synthesis of PTII, which was directly obtained by a one-pot polymerization method using α,α′-dibrominated TII (2Br-TII) as the starting material, either via Suzuki condensation polymerization in the presence of one equivalent of bis(pinacolato)diborane (for PTII-1) or Stille condensation polymerization in the presence of one equivalent of hexamethyldistannane (for PTII-2), respectively. It is worthwhile to mention that such one-pot Suzuki and Stille homopolymerization methodologies are much less studied,34–36 but it provides an easier way to construct conjugated homopolymers, since the tedious and moisture-sensitive preparation and purification of the borylated or stannylated comonomers are avoided. To further elucidate the structure–property relationship of PTII, TII was also copolymerized with II to give P(TII-co-II) as a comparison. All the polymers showed a narrow optical bandgap, very similar to PII. Encouragingly, solution-processed OFETs based on PTII exhibited ambient-stable ambipolar transport behavior with balanced hole and electron mobilities up to 0.065 and 0.15 cm2 V−1 s−1, respectively, which is three orders higher than that of the reported PII, and also two orders higher than P(TII-co-II). Results of density functional theory (DFT) calculation suggested that the better OFET performance of PTII over PII and P(TII-co-II) were attributed to the more planar backbone conformation and more extended HOMO–LUMO orbital overlap. Film morphology and microstructure of the polymers based on TII were analyzed by atomic force microscopy (AFM) and 2D grazing incidence X-ray diffraction (2D-GIXRD). 2D-GIXRD analysis explicitly revealed that PTII showed much better well-ordered structure, as what we expected. To the best of our knowledge, PTII-2 is among the best ambipolar homopolymers of A units with high charge-carrier mobility, indicating that the design of such homopolymers might be an important strategy toward high performance ambipolar polymeric OFETs, which deserves a further study in the future.
Results and discussion
Polymer synthesis
Recently, we presented a novel synthetic route to prepare 2Br-TII, and obtained full-conjugated TII-containing D–A type conjugated copolymers.33 Aiming at the synthesis of PTII, we decided to convert 2Br-TII to the corresponding borylated comonomer. Borylation of 2Br-TII was performed in the presence of two equivalents of bis(pinacolato)diboron in 1,4-dioxane, using Pd (dppf)Cl2 as the catalyst and AcOK as the base. Surprisingly, the color of the reaction solution turned from red to blue in a half hour after the temperature was increased to 80 °C, indicating the formation of oligomers/polymers of TII (Table 1, entry 1).
|
||||
|---|---|---|---|---|
| Entrya | Catalyst | Base | Solvent (toluene/dioxane) | Yieldb (%) |
| a Unless otherwise noted, all reactions were carried out with 2Br-TII (0.05 mmol), bis(pinacolato)diboron (0.055 mmol), catalyst (3 mol%), and base (0.325 mmol) in 1.0 mL of cosolvent at 110 °C for 3 days.b Isolated yield from Soxhlet extraction using CHCl3 as the solvent.c 2.0 eq. of bis(pinacolato)diboron was used and the reaction was performed at 80 °C for 10 h.d 1.0 eq. of bis(pinacolato)diboron was used. | ||||
| 1c | Pd(dppf)Cl2 | AcOK | 10/0 | 10 |
| 2 | Pd(dppf)Cl2 | AcOK | 9/1 | 32 |
| 3 | Pd(dppf)Cl2 | AcOK | 8/2 | 30 |
| 4 | Pd(dppf)Cl2 | AcOK | 7/3 | 40 |
| 5d | Pd(dppf)Cl2 | AcOK | 7/3 | 35 |
| 6 | Pd(OAc)2 | AcOK | 7/3 | 30 |
| 7 | Pd(PPh3)2Cl2 | AcOK | 7/3 | 28 |
| 8 | Pd(PPh3)4 | AcOK | 7/3 | — |
The reaction was then monitored by thin-layer chromatography (TLC), and the results indicated that the oligomer/polymer (which is the immovable blue spot) was formed at the early stage of the reaction, and the formation of the borylated TII could not be clearly recognized from the TLC plate. The reaction was quenched after 10 h, and then the blue-colored product was precipitated in methanol. After Soxhlet extraction with methanol, acetone and hexane to remove low molecular weight oligomers, the polymer with relatively high molecular weight (soluble portion in chloroform) was obtained in around 10% yield.
It is well-known that conjugated polymer with higher molecular weight show higher charge carrier mobility due to the increased conjugation length and transfer integral. With the preliminary result in hand, we then optimized the reaction conditions to increase the molecular weight of the homopolymer, as shown in Table 1, entries 2–8. By using 1.1 equivalent of bis(pinacolato)diboron and a cosolvent system (toluene/1,4-dioxane) to improve the solubility of diboron reagent, the polymer with higher molecular weight (soluble part in CHCl3 Soxhlet extraction) was obtained in 30–40% yield after being heated in a sealed tube at 110 °C for 3 days (entries 2–4). However, further decreasing the amount of that borylation reagent to 1.0 equivalent led to a slightly decreased yield (entry 5), presumably due to the presence of side reactions which consumed some of the diboron reagent. Other catalysts such as Pd(OAc)2, Pd(PPh3)2Cl2 and Ph(PPh3)4 were also screened. It could be seen that the homopolymer was obtained in around 30% yield when using Pd(OAc)2 and Pd(PPh3)2Cl2 as the corresponding catalyst (entries 6 and 7), while the homopolymerization did not occur when Ph(PPh3)4 was used as the catalyst (entry 8). It seems that the Pd catalyst with bidental ligands gave better results. Thus, at this stage the optimal reaction conditions were identified as the following: Pd(dppf)Cl2 (3 mol%), bis(pinacolato)diboron (1.1 eq.), and AcOK (6.5 eq.) in toluene/dioxane (7/3, 0.5 M) at 110 °C for 3 days.
Encouraged by the one-pot Suzuki-coupling polymerization, we assumed that the Stille-coupling polymerization might proceed in a similar manner due to the similar palladium-catalyzed process. We chose hexamethylditin as the stannylation reagent due to its high reactivity,37,38 and AcOK as the base in the model reaction with 2Br-TII (Table 2). An initial evaluation of Pd(dppf)Cl2 showed that the polymerization proceeded well and the homopolymer that could be Soxhlet extracted in chloroform was obtained in 35% yield (entry 1). Gratifyingly, when Pd(PPh3)4 was employed as the catalyst, the polymer with higher molecular weight was obtained, as indicated by Soxhlet extraction: most of the polymer could not be dissolved in chloroform, but only could be Soxhlet extracted with chlorobenzene (PhCl) to give a respectable 50% yield (entry 2). This indicated that the one-pot Stille homopolymerization was more effective to prepare high molecular weight PII than the one-pot Suzuki homopolymerization. The homopolymer was collected in only 38% yield from Soxhlet extraction using CHCl3 as the solvent when AcOK was removed, highlighting that the addition of a base was necessary in maintaining the efficiency of the homopolymerization (entry 3). It seems that the role of base in this one-pot Stille-coupling polymerization might be similar to the role of the base in the one-pot Suzuki-coupling polymerization,39,40 in which the transmetalation process was processed more smoothly in the presence of a base.
|
|||
|---|---|---|---|
| Entrya | Catalyst | Base | Yield (%) |
| a Unless otherwise noted, all reactions were carried out with 2Br-TII (0.05 mmol), hexamethylditin (0.055 mmol), catalyst (3 mol%), and base (0.325 mmol) in 1.0 mL of solvent at 110 °C for 3 days.b Isolated yield from Soxhlet extraction using CHCl3 as the solvent.c Isolated yield from Soxhlet extraction using PhCl as the solvent. | |||
| 1b | Pd(dppf)Cl2 | AcOK | 35 |
| 2c | Pd(PPh3)4 | AcOK | 50 |
| 3b | Pd(PPh3)4 | — | 38 |
For comparison, the copolymer P(TII-co-II) was also prepared through the typical Suzuki coupling reaction between 2Br-TII and 6,6′-(N,N′-2-octyldodecyl)-pinacoldiboron-isoindigo in high yield. The number average molecular weights (Mn) of PTII-1 (chloroform-soluble portion obtained by Suzuki-coupling polymerization), PTII-2 (PhCl-soluble portion obtained by Stille-coupling polymerization under optimized conditions) and P(TII-co-II) are 30, 36 and 41 kDa, respectively, as characterized by the high temperature gel permeation chromatography (HT-GPC) using 1,2,4-trichlorobenzene as eluent at 150 °C (Table 3). The molecular weight of PTII-2 is close to that of P(TII-co-II), indicating that the one-pot homopolymerization could result in polymers with similar molecular weight comparable to the traditional cross-coupling polymerization. It is interesting that PTII-1 and PTII-2 show such a different solubility in chloroform although the molecular weight of PTII-2 is only slightly larger than that of PTII-1, inferring that the solubility of the homopolymer shows a dramatic change beyond certain molecular weight, probably around 31–35 kDa. All the polymers showed excellent thermal stability with decomposition temperature above 300 °C, and no obvious phase transition was observed before decomposition (Fig. S1, ESI†).
| Polymer | Mna (kDa) | PDIa | λmax (nm) solution | λmax (nm) filmb | λedge (nm) | Eoptg (eV) | IPc (eV) |
|---|---|---|---|---|---|---|---|
| a Determined by GPC at 150 °C using TCB as the eluent.b Films were prepared by dropping casted the polymer solution on the piezoid.c Films were prepared by dropping casted the polymer solution on the working electrode. | |||||||
| P(TII-co-II) | 41 | 2.2 | 319, 704 | 328, 697 | 770 | 1.61 | 5.83 |
| PTII-1 | 30 | 2.5 | 356, 581 | 359, 590 | 825 | 1.50 | 5.84 |
| PTII-2 | 36 | 2.1 | 370, 602 | 372, 610 | 837 | 1.48 | 5.84 |
The one-pot Suzuki and Stille coupling homopolymerization using dibrominated compounds as the only monomer was occasionally reported in the literature. Masuda et al. first mentioned the dehalogenative coupling polymerization of dihaloarenes such as dibromobenzene and dibromothiophene using bis(pinacolato)diborane as the condensation reagent.41 This method was later optimized by Reynolds et al. using fluoride salts as the additive to promote the polymerization which yielded in polyfluorenes with higher molecular weight.34 Brouwer and co-workers expanded this methodology to thiophene-terminated monomers with either electron-rich or electron-poor core, and found that the reproducibility and the quality of the resulting conjugated polymers were better than those obtained via traditional Suzuki coupling copolymerization.35 Yang et al. mentioned the homopolymerization of a dibrominated monomer with benzothiadiazole[1,2-b:4,3-b′]-dithiophene core in the presence of hexamethylditin.36 More recently, such a one-pot Stille homopolymerization was applied in the synthesis of an insulated conjugated polymer.42 However, compared to the vast number of the reported cases on the copolymerization between a dibrominated monomer with a diboronated or distannylated monomer, the examples for the one-pot Suzuki or Stille homopolymerization are quite limited. Apparently, not all dibrominated monomers are suitable for such a homopolymerization. For example, we attempted the homopolymerization of 6,6′-dibrominated II but failed. It seems that Suzuki or Stille homopolymerization is highly dependent on the nature of the dibrominated monomer, which is still not clearly understood at this stage.
Scheme 1 outlines the possible reaction mechanism of Suzuki or Stille homopolymerization, given that the Pd(0) catalytic cycle stands for true in all cases. It is obvious that the first step of the Suzuki or Stille reaction of a dibrominated compound includes the insertion of Pd(0) into at least one of the C–Br bonds, followed by the transmetalation reaction with either the diborane or distannyl reagents, which involves the B–B bond or Sn–Sn bond breaking. The reductive elimination gives the mono-borylated or mono-stannylated intermediate. If this process is repeated on the other C–Br bond of the same intermediate, the di-borylated or di-stannylated compound will be obtained, which is commonly observed in the preparation of di-borylated or di-stannylated comonomers (Path 1). For the homopolymerization, however, the mono-borylated or mono-stannylated intermediate either undergoes transmetalation and reductive elimination reactions with another Br–Ar–Pd(II)–Br intermediate (Path 2), or the oxidative insertion occurs on the mono-borylated or mono-stannylated intermediate, which is followed by the transmetalation and elimination with another mono-borylated or mono-stannylated intermediate(Path 3). In both Path 2 and 3, the breaking of C–B or C–Sn bond is necessary for the transmetalation to occur, which is different from Path 1, where B–B or Sn–Sn bond breaking is necessary for the transmetalation step. If we assume that the oxidative insertion of Pd(0) into C–Br bond is only marginally influenced by the functionality on the other end of the intermediate and the reductive elimination is the most facile step, we could safely make the conclusion that the difference between the active energy of breaking a B–B/Sn–Sn bond and that of breaking C–B/C–Sn bond determines whether the di-borylated/di-stannylated compound or the homopolymer would be obtained. In other words, if the C–B/C–Sn bond is easier to break than B–B/Sn–Sn bond, homo-polymer would be preferred. We hypothesize that the C–B/C–Sn bond involved in TII intermediate is weaker than B–B/Sn–Sn bond, which might be influenced by the nature of TII core and accounts for the homopolymer formation.
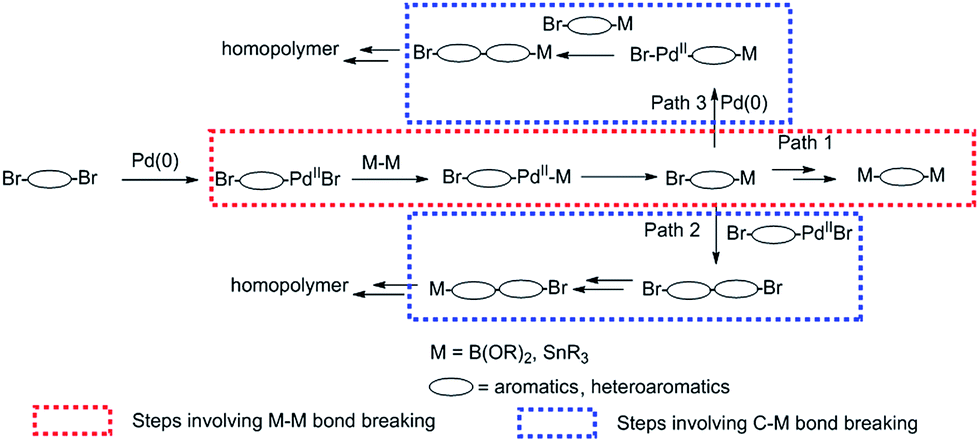 | ||
| Scheme 1 Possible Suzuki or Stille homopolymerization mechanism analysis. | ||
Theoretical calculations
To understand the influence of the steric hindrance between the co-monomers on the polymer backbones and intrinsic electronic structures, density functional theory (DFT) calculations were performed on the molecular geometries and frontier orbitals of PII, P(TII-co-II) and PTII at the B3LYP/6-31G* level. To simplify the calculation, long alkyl chains were replaced by methyl group. The optimized geometries and frontier orbitals of the trimers of PII, P(TII-co-II) and PTII are shown in Fig. 1.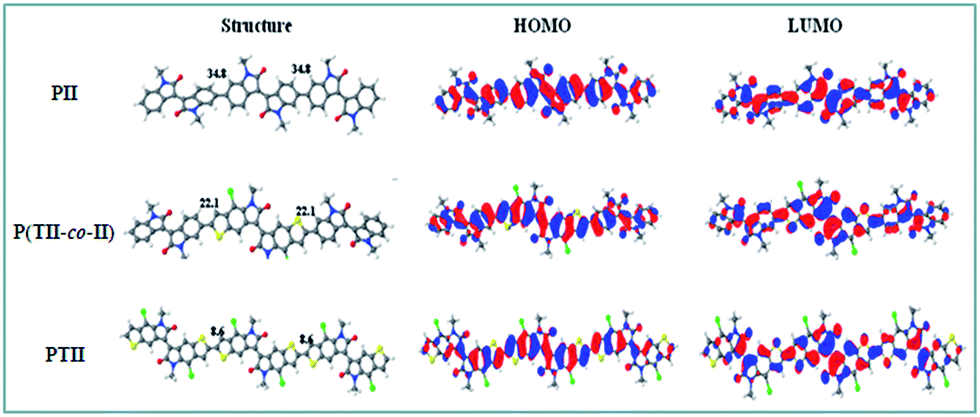 | ||
| Fig. 1 Theoretical calculation of molecular conformation and orbitals of PII and P(TII-co-II), and PTII dimers at ωB97XD functional. | ||
As can be seen from Fig. 1, both the HOMO and LUMO of all the A type and A–A′ type polymers are delocalized along the polymer backbone, which underlines their difference with typical D–A copolymers. It can be seen that the dihedral angles between the adjacent II and TII units in P(TII-co-II) is 22.1°, which is 12.7° smaller than that (34.8°) between the adjacent II units in PII. As we speculated, the backbone of PTII displays the best degree of coplanarity whose dihedral angle between the adjacent TII units is 8.58° owing to the weaker steric hindrance, indicating that charge transport in PTII should be more efficient than the other two polymers. The calculated bandgaps are 2.02 eV for PII, 1.89 eV for P(TII-co-II), and 1.79 eV for PTII, which indicates that PTII shows the narrowest bandgap among the three polymers. Generally, the planar conformation of the polymeric backbone offers expanded π-conjugation and favorable π–π stacking, therefore results in narrower bandgap and higher charge mobility. Accordingly, PTII is expected to exhibit more efficient charge behavior than that of PII and P(TII-co-II).
Photophysical and electrochemical properties
Fig. 2 illustrates a comparison of the normalized UV-vis absorption spectra of the polymers P(TII-co-II), PTII-1 and PTII-2 both in diluted solution state and in thin film state, and the corresponding data are collected in Table 3. The optical band gaps (Eg) of the polymers were estimated from the low-energy band edges in the film state. The solution of P(TII-co-II) exhibits a strong absorption in the 550–750 nm region with a maximum absorption peak at 704 nm. On the other hand, PTII-2 shows a slightly broader absorption region (450–750 nm), however, with a maximum absorption peak at 602 nm. PTII-1, which was synthesized via Suzuki homopolymerization, displays a similar but blue-shifted spectra compared to PTII-2, presumably due to its smaller molecular weight.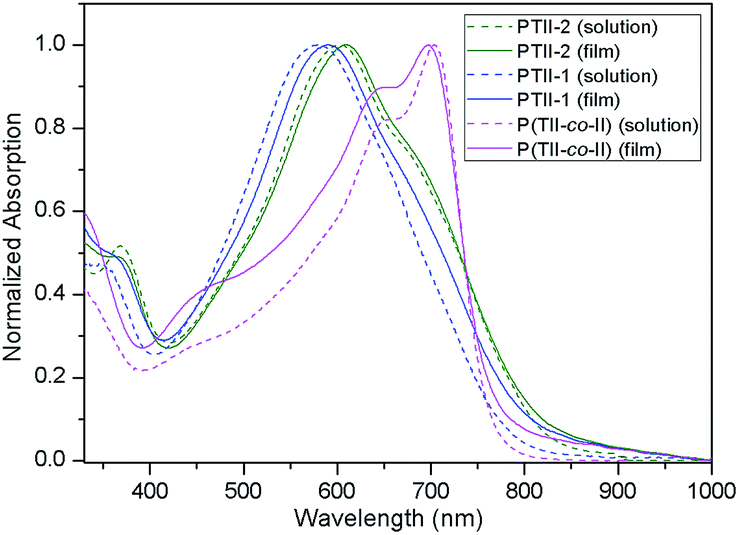 | ||
| Fig. 2 Normalized UV-vis absorption spectra of P(TII-co-II), PTII-1 and PTII-2 both in dichlorobenzene solution (1 × 10−5 M) and in film. | ||
We postulate that the maximum absorption difference between P(TII-co-II) and PTII might be derived from their different electronic structures. In P(TII-co-II), the slight difference in the electron-withdrawing ability between TII and II (II might be a stronger A unit) makes the intramolecular charge transfer between TII and II units still possible, which causes the red-shift of the maximum absorption. On the contrary, there is no such difference between TII units in PTII, so the red-shift of the maximum absorption comparable to P(TII-co-II) should not be expected. In thin film state, PTII-1 and PTII-2 exhibit small redshifts of the maximum absorption peak compared to their solution state, while P(TII-co-II) displays a 7 nm hypsochromic shift, indicating the different aggregation patterns between the homopolymer and copolymer. The redshift of the maximum absorption peak of PTII in the solid state indicates that it might adopt J-aggregate-like stacking, while the hypsochromic shift of the maximum absorption peak of P(TII-co-II) in the solid state indicates that it might adopt H-aggregate-like stacking. It is noted that the thin films of the polymers P(TII-co-II), PTII-1 and PTII-2 exhibit the absorption onset at 770, 825 and 837 nm, respectively, which are redshifted obviously compared to that of PII (λonset = 731).29 The redshifted absorption onset of PTII indicates that the highly planar polymeric backbone of PTII favors longer conjugated length and better intermolecular interaction. The optical bandgap of the polymer films were calculated from the absorption onset. PTII-2 exhibits the narrowest optical bandgap of 1.48 eV comparable to PTII-1 (Eoptg = 1.50 eV), smaller than P(TII-co-II) (Eoptg = 1.61 eV) and PII (Eoptg = 1.70 eV), which is in accordance with the calculated results described above.
The electrochemical cyclic voltammograms (CVs) of P(TII-co-II), PTII-1 and PTII-2 as shown in Fig. S2† were measured to estimate their HOMO and LUMO energy levels, and the data are outlined in Table 3. The actual HOMO/LUMO energy level is hard to be accurately determined, and CVs are used to provide information about the ionization potential (IP) as well as the electron affinity (EA) of the polymers (Fig. S2, ESI†), which could partially reflect the HOMO/LUMO energy level. Similar to PII, all the TII-based polymers show an irreversible oxidation process, with the oxidation onset potential at around 1.43 V. The ionization potential (IP) was calculated according to the equation IP = (Eoxonset − ferrocene) + 4.4 eV, and the results are summarized in Table 3. Unfortunately, no well-defined reduction peak was observed when the potential was scanned to −2.0 V vs. Ag/AgCl, so we could not obtain the EA values of the polymers.
OFETs properties
The potential application of all three polymers for OFETs were investigated using a top-gate/bottom-contact (TG/BC) device configuration. The semiconducting layer was deposited by spin-coating the corresponding polymer solutions (10 mg mL−1 in 1,2-dichlorobenzene) on Au/titanium (source-drain)/SiO2 substrate. A PMMA solution was spin-coated on top of the polymer film as the dielectric layer after the film was annealed at 200 °C for 6 min, and an aluminum layer was thermally evaporated as the gate electrode. All devices were measured on a probe station under ambient conditions, and the results are shown in Table 4. The typical output and transfer characteristics of the devices are shown in Fig. S3, S4† (for P(TII-co-II) and PTII-1) and Fig. 3 (for PTII-2), which were typical for ambipolar OFETs. As shown in Table 4, all the conjugated polymers exhibited ambipolar behavior with better charge mobility than PII. The polymer FET based on P(TII-co-II) showed well balanced hole and electron mobilities around 0.001 cm2 V−1 s−1. Encouragingly, the homopolymer both PTII-1 and PTII-2 exhibited high mobility ambipolar charge transport, with the hole mobility of 0.0059 cm2 V−1 s−1 and electron mobility of 0.058 cm2 V−1 s−1 for PTII-1, and the hole mobility of 0.065 cm2 V−1 s−1 and electron mobility of 0.15 cm2 V−1 s−1 for PTII-2. Compared to PTII-1, PTII-2 exhibited higher and more balanced electron/hole mobility, which is in accordance with its higher molecular weight. From these data we can conclude that the homopolymer PTII possesses superior charge transport than that of P(TII-co-II) and PII, showing that planarity of the polymeric backbone has a significant influence on the carrier transport properties.| Polymer | L (μm) | μh, maxa [cm2 V−1 s−1] | Vth (V) | Ion/Ioff | μe, maxa [cm2 V−1 s−1] | VT (Vth) | Ion/Ioff |
|---|---|---|---|---|---|---|---|
| a Maximum values of hole or electron mobilities measured under ambient conditions. | |||||||
| P(TII-co-II) | 20 | 0.0016 | −55 | 102 | 0.0013 | +70 | 103 |
| PTII-1 | 20 | 0.0059 | −55 | 102 | 0.058 | +59 | 104 |
| PTII-2 | 20 | 0.065 | −75 | 102 | 0.15 | +86 | 102 |
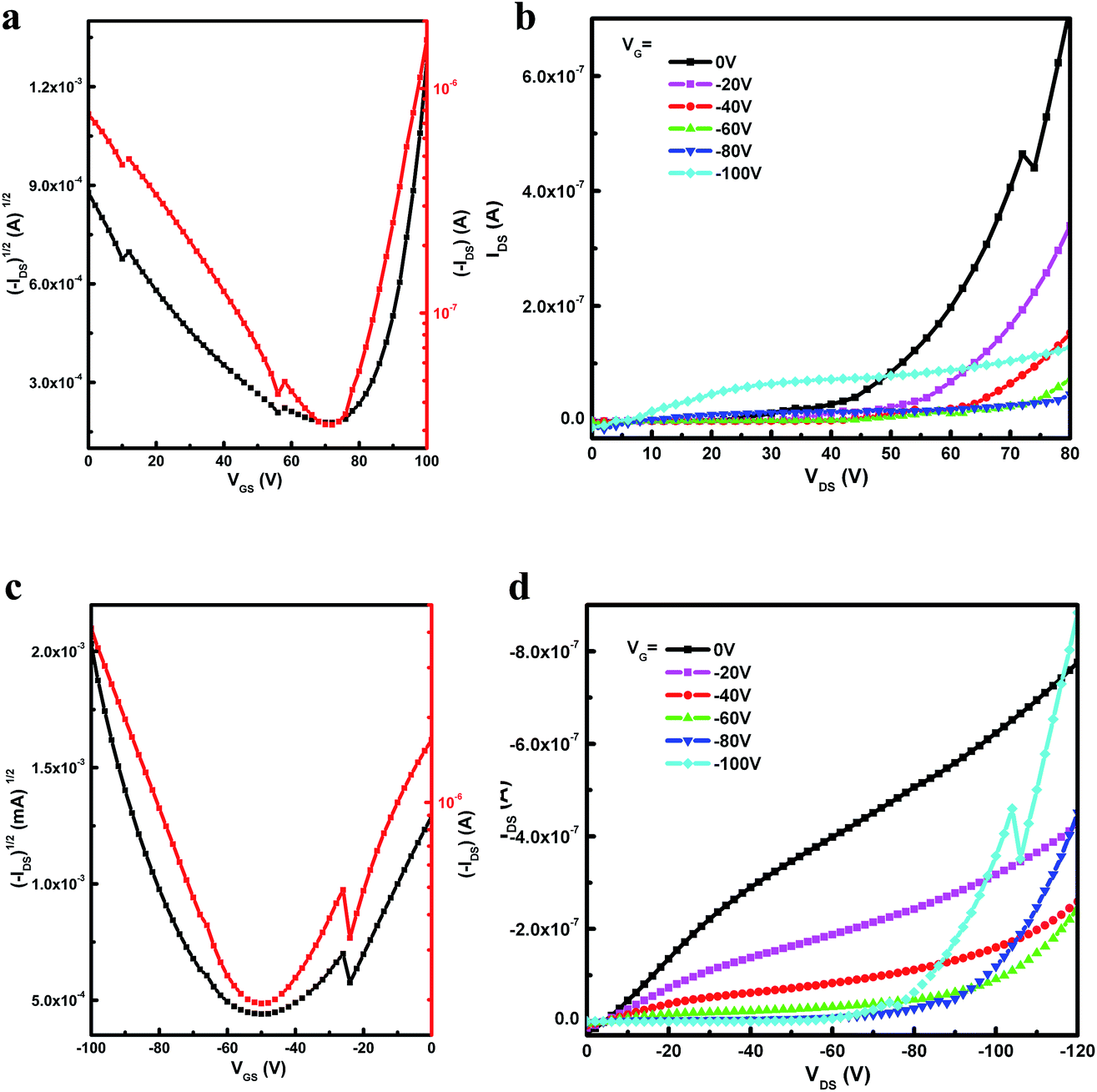 | ||
| Fig. 3 Transfer (a, c) and output (b, d) characteristics of PTII-2 annealed at 200 °C and tested under ambient conditions. | ||
Morphology
Film morphology and microstructure of the polymers based on TII were analyzed by atomic force microscopy (AFM) and 2D grazing incidence X-ray diffraction (2D-GIXRD) to reveal the relationship between the film morphology of the polymers based on TII and device performance, and the results are shown in Fig. 4. AFM image of the film of P(TII-co-II) was featured with a slightly granular morphology (Fig. 4a), with the roughness around 0.88 nm, and only the (100) diffraction peak was observed in the 2D-GIXRD image (Fig. 4d). PTII-1 (the homopolymer with lower molecular weight) showed a similar morphology (Fig. 4b), but with smoother surface, as indicated by the smaller root-mean-square (RMS) value (0.739 nm). Interestingly, the 2D-GIXRD image of PTII-1 showed a drastic difference compared to that of P(TII-co-II): a much stronger (h00) diffraction pattern up to (300) was clearly observed for PTII-1 (Fig. 4e), indicating that PTII-1 has a more well-organized lamellar structure than P(TII-co-II). This is in accordance with the hypothesis that homopolymers should have better crystallinity due to the more uniformed backbone. More strikingly, even better well-ordered structure was observed with the increase of the molecular weight. PTII-2 showed an even stronger (h00) diffraction pattern up to (400) (Fig. 4f), which led to a drastic change in its morphology (Fig. 4c). The AFM image of PTII-2 clearly showed the formation of the fiber-like intercalating networks with an even smoother surface (RMS value 0.731 nm), which is also different from that of PTII-1 and P(TII-co-II). It is worthwhile to mention that such a drastic change in AFM images is the result of a small molecular weight increase of around 6 kDa. Fig. S5† shows the corresponding out-of-plane and in-plane line cuts (see ESI†). P(TII-co-II) showed a lamellar structure with d100 = 20.06 Å. The d values of the lamellar structures of PTII-1 and PTII-2 could also be determined up to (200) level: d100 = 26.74 Å, and d200 = 14.47 Å for PTII-1, and d100 = 26.74 Å, and d200 = 13.58 Å for PTII-2, respectively. Moreover, (010) arcs at 17.03 nm−1 for P(TII-co-II), 17.23 nm−1 for PTII-1, and 17.86 nm−1 for PTII-2 were observed, corresponding to π–π stacking distances of 3.69, 3.64, and 3.52 Å, respectively. It can be seen that the homopolymers exhibited smaller π–π stacking distance than P(TII-co-II) owing to their better coplanarity and crystallinity of the polymeric backbone, which became even shorter with the increase of molecular weight. It is worthwhile to mention that the diffraction patterns indicate the coexistence of both face-on and edge-on polymer backbone orientation in PTII, with no obvious edge-on preference. It is encouraging that high ambipolar mobility was still observed for PTII, since most of the high-performance OFET materials show an edge-on preference. This infers that the charge carrier mobility of homopolymers could be further improved by side-chain engineering such as the introduction of siloxane-terminated side chain which prefers edge-on orientation.43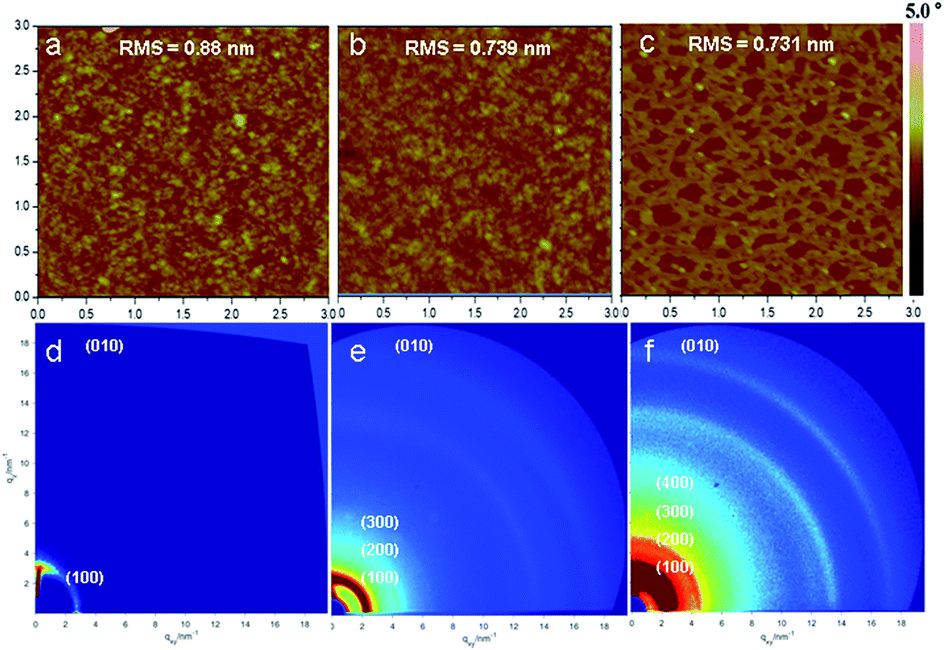 | ||
| Fig. 4 Tapping-mode AFM height images (3.0 × 3.0 μm) of (a) P(TII-co-II), (b) PTII-1 and (c) PTII-2 films. 2D-GIXRD patterns of (d) P(TII-co-II), (e) PTII-1 and (f) PTII-2 films. Films were spin-coated from the DCB solutions of the polymers (5 mg mL−1) and annealed at 200 °C. | ||
Conclusions
In summary, homopolymers of thiophene-fused isoindigo (PTII) were synthesized by a one-pot polymerization method using 2Br-TII as the starting material either via Suzuki condensation polymerization or Stille condensation polymerization, and their ambient-stable ambipolar charge transport behaviors were characterized. Compared to the homopolymer of isoindigo (PII) and P(TII-co-II), the steric hindrance of the repeating TII units was largely alleviated, which resulted in a more planar structure. PTII exhibited better crystallinity, as evidenced by 2D-GIXRD analysis, which could be ascribed to that the repeating units in the homopolymer have the same size and could be more easily fit into the lattice. With the increase of the molecular weight of PTII, better inter-connected polymer networks was observed, as shown in AFM image analysis. As the result, the solution-processed OFET devices based on PTII exhibited ambient-stable ambipolar transport behavior with balanced hole and electron mobilities up to 0.065 and 0.15 cm2 V−1 s−1, respectively, three orders higher than that of the reported PII, and also two orders higher than P(TII-co-II). To the best of our knowledge, PTII is among the best homopolymers consisting of A units with high ambipolar charge-carrier mobility, and such high mobility was obtained with no edge-on orientation preference. This study indicated that the homopolymerization of accepting units is an alternative method supplementary to the traditional D–A type conjugated copolymers for high performance ambipolar OFET materials, given that the high planarity of the polymer backbone could be guaranteed. Moreover, the one-pot homopolymerization of a dibromo-monomer via Suzuki or Stille condensation also deserves a closer study, since it provides a more convenient way to construct conjugated homopolymers. The extension of such a one-pot homopolymerization method to other acceptors and the side-chain engineering of homopolymers are currently undergoing in our laboratory and will be reported in due course.Experimental
General
All reactions were performed under argon atmosphere unless stated otherwise. All glassware was completely dried before use. Chemicals and solvents were purchased from commercial suppliers or purified by standard techniques. 1H NMR spectra were recorded on a Bruker Avance-III 600 MHz with tetramethylsilane (TMS) as an internal standard at 298 K.Cyclic voltammetry (CV) curves were carried out by using an electrochemistry workstation (CHI660D, Chenhua, Shanghai) in an anhydrous argon-saturated chloroform solution (10−3 mol L−1) of 0.1 M tetrabutylammonium hexafluorophosphate (n-Bu4NPF6) at room temperature. We utilized the glassy carbon electrode as the working electrode, Pt as the counter electrode and Ag/AgCl as the reference electrode at a potential scan rate of 0.1 V s−1. Measurements were calibrated with ferrocene as the internal standard. The UV-vis spectra were recorded with a Hitachi U-4100 UV-vis spectrophotometer in an anhydrous chloroform solution (10−5 mol L−1). Gel permeation chromatography (GPC) was performed on a Waters 1151 pump and a UV-vis monitor (700 nm) using 1,2,4-trichlorobenzene (TCB) as the eluent (150 °C). The 2D-GIXRD data was obtained at 1W1A, Beijing Synchrotron Radiation Facility. AFM patterns were obtained with a Digital Instrument NanoScope IIIa. TGA patterns were collected with a TGA Q50 V20.13 Build 39.
Device fabrication
Top-gate/bottom-contact FET devices were fabricated using SiO2 (300 nm) substrates. The gold source and drain bottom electrodes (with Ti as the adhesion layer) were patterned by photolithography on the SiO2 surface. The substrates were treated with ultrasonication in acetone, cleaning agent, deionized water (twice), and isopropanol. Then the substrates were dried under vacuum at 80 °C and used in a glove box. Octadecyltrichlorosilane (OTS) modification was performed on the surface of SiO2 in a vacuum to obtain an OTS-self-assembled monolayer. In a nitrogen glove-box, the hot polymer solution in o-DCB (10 mg mL−1) was spin-coated onto the substrates yielding a polymer thin-film (∼20 nm). Then the film samples were further annealed at 200 °C for 6 min in a N2 glove box. Next, a PMMA (Mw = 996 kDa) solution in anhydrous n-butyl acetate (80 mg mL−1) was spin-coated onto the surface of the polymer thin-film (PMMA thickness ∼1350 nm, capacitance ∼2.17 nF cm−2). The samples were then dried at 90 °C for 30 min in a N2 glove box. Then a layer of Al (50 nm) was evaporated onto the dielectric layer by thermal evaporation as gate electrodes. The OFET devices exhibited a channel ratio of width to length (W/L = 280). The performances of the OFETs were investigated on a probe stage with a Keithley 4200 parameter analyzer under an atmosphere (humidity 50–60%). The carrier mobility, μ, was calculated from the data in the saturated regime according to the equation IDS = (W/2L)Ciμh or e(VG − VT),2 where IDS is the drain current in the saturated regime. W and L are the semiconductor channel width and length, respectively. Ci is the capacitance of PMMA/SiO2 gate-dielectric VG and VT are the gate voltage and threshold voltage, respectively.General procedures for synthesis of P(TII-co-II), PTII-1 and PTII-2
Acknowledgements
This work is supported by “100 Talents” Program from Chinese Academy of Sciences and National Natural Science Foundation of China (NSFC51573204 and 21402220). The authors gratefully acknowledge the assistance of scientists of Diffuse X-ray Scattering Station in Beijing during the experiments.Notes and references
- A. N. Sokolov, B. C. K. Tee, C. J. Bettinger, J. B. H. Tok and Z. Bao, Acc. Chem. Res., 2012, 45, 361–371 CrossRef CAS PubMed.
- L. Torsi, M. Magliulo, K. Manoli and G. Palazzo, Chem. Soc. Rev., 2013, 42, 8612–8628 RSC.
- C. Liao and F. Yan, Polym. Rev., 2013, 53, 352–406 CrossRef CAS.
- B. Kumar, B. K. Kaushik and Y. S. Negi, J. Mater. Sci.: Mater. Electron., 2014, 25, 1–30 CrossRef CAS.
- P. Heremans, A. K. Tripathi, A. de Jamblinne de Meux, E. C. Smits, B. Hou, G. Pourtois and G. H. Gelinck, Adv. Mater., 2016, 28, 4266–4282 CrossRef CAS PubMed.
- H. Dong, X. Fu, J. Liu, Z. Wang and W. Hu, Adv. Mater., 2013, 25, 6158–6183 CrossRef CAS PubMed.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296–1323 CrossRef CAS PubMed.
- T. Lei, J. H. Dou, Z. J. Ma, C. H. Yao, C. J. Liu, J. Y. Wang and J. Pei, J. Am. Chem. Soc., 2012, 134, 20025–20028 CrossRef CAS PubMed.
- T. Lei, J.-H. Dou, Z.-J. Ma, C.-J. Liu, J.-Y. Wang and J. Pei, Chem. Sci., 2013, 4, 2447–2452 RSC.
- T. Lei, J.-H. Dou, X.-Y. Cao, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2013, 135, 12168–12171 CrossRef CAS PubMed.
- T. Lei, J.-H. Dou, X.-Y. Cao, J.-Y. Wang and J. Pei, Adv. Mater., 2013, 25, 6589–6593 CrossRef CAS PubMed.
- T. Lei, X. Xia, J. Y. Wang, C. J. Liu and J. Pei, J. Am. Chem. Soc., 2014, 136, 2135–2141 CrossRef CAS PubMed.
- Y. Jiang, Y. Gao, H. Tian, J. Ding, D. Yan, Y. Geng and F. Wang, Macromolecules, 2016, 49, 2135–2144 CrossRef CAS.
- I. Meager, M. Nikolka, B. C. Schroeder, C. B. Nielsen, M. Planells, H. Bronstein, J. W. Rumer, D. I. James, R. S. Ashraf, A. Sadhanala, P. Hayoz, J.-C. Flores, H. Sirringhaus and I. McCulloch, Adv. Funct. Mater., 2014, 24, 7109–7115 CAS.
- M. Held, Y. Zakharko, M. Wang, F. Jakubka, F. Gannott, J. W. Rumer, R. S. Ashraf, I. McCulloch and J. Zaumseil, Org. Electron., 2016, 32, 220–227 CrossRef CAS.
- P. Li, H. Wang, L. Ma, L. Xu, F. Xiao, Z. Yi, Y. Liu and S. Wang, Sci. China: Chem., 2016, 59, 679–683 CrossRef CAS.
- Y. Olivier, D. Niedzialek, V. Lemaur, W. Pisula, K. Mullen, U. Koldemir, J. R. Reynolds, R. Lazzaroni, J. Cornil and D. Beljonne, Adv. Mater., 2014, 26, 2119–2136 CrossRef CAS PubMed.
- H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. Herwig and D. M. D. Leeuw, Nature, 1999, 401, 685–688 CrossRef CAS.
- Z. Bao, A. Dodabalapur and A. J. Lovinger, Appl. Phys. Lett., 1996, 69, 4108 CrossRef CAS.
- S. Westenhoff, W. J. D. Beenken, A. Yartsev and N. C. Greenham, J. Chem. Phys., 2006, 125, 154903 CrossRef PubMed.
- S. N. Yaliraki and R. J. Silbey, J. Chem. Phys., 1996, 104, 1245–1253 CrossRef CAS.
- C.-F. Huang, S.-L. Wu, Y.-F. Huang, Y.-C. Chen, S.-T. Chang, T.-Y. Wu, K.-Y. Wu, W.-T. Chuang and C.-L. Wang, Chem. Mater., 2016, 28, 5175–5190 CrossRef CAS.
- S. Janietz, J. Barche, A. Wedel and D. Sainova, Macromol. Chem. Phys., 2004, 205, 1916–1922 CrossRef CAS.
- J. A. Letizia, M. R. Salata, C. M. Tribout, A. Facchetti, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 9679–9694 CrossRef CAS PubMed.
- C. R. Hakan Usta, Z. Wang, H. Huang, M. K. Deliomeroglu, A. F. Aleksandr Zhukhovitskiy and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 5586–5608 CrossRef PubMed.
- D. Izuhara and T. M. Swager, J. Am. Chem. Soc., 2009, 131, 17724–17725 CrossRef CAS PubMed.
- M. M. Durban, P. D. Kazarinoff and C. K. Luscombe, Macromolecules, 2010, 43, 6348–6352 CrossRef CAS.
- R. Stalder, J. Mei, J. Subbiah, C. Grand, L. A. Estrada, F. So and J. R. Reynolds, Macromolecules, 2011, 44, 6303–6310 CrossRef CAS.
- Y. Li, B. Sun, P. Sonar and S. P. Singh, Org. Electron., 2012, 13, 1606–1613 CrossRef CAS.
- R. Stalder, S. R. Puniredd, M. R. Hansen, U. Koldemir, C. Grand, W. Zajaczkowski, K. Muellen, W. Pisula and J. R. Reynolds, Chem. Mater., 2016, 28, 1286–1297 CrossRef CAS.
- P. Deng and Q. Zhang, Polym. Chem., 2014, 5, 3298–3305 RSC.
- N. Zhao, N. Ai, M. Cai, X. Wang, J. Peib and X. Wan, Polym. Chem., 2016, 7, 235–243 RSC.
- R. M. Walczak, R. N. Brookins, A. M. Savage, E. M. van der Aa and J. R. Reynolds, Macromolecules, 2009, 42, 1445–1447 CrossRef CAS.
- F. Brouwer, J. Alma, H. Valkenier, T. P. Voortman, J. Hillebrand, R. C. Chiechi and J. C. Hummelen, J. Mater. Chem., 2011, 21, 1582–1592 RSC.
- S. Hu, X. Bao, Z. Liu, T. Wang, Z. Du, S. Wen, N. Wang, L. Han and R. Yang, Org. Electron., 2014, 15, 3601–3608 CrossRef CAS.
- T. Iwaki, A. Yasuhara and T. Sakamoto, J. Chem. Soc., Perkin Trans. 1, 1999, 1505–1510 RSC.
- Y. Zhang, J. W. Hubbard, N. G. Akhmedov, J. L. Petersen and B. C. Soderberg, J. Org. Chem., 2015, 80, 4783–4790 CrossRef CAS PubMed.
- A. A. C. Braga, N. H. Morgon, G. Ujaque and F. Maseras, J. Am. Chem. Soc., 2005, 127, 9298–9307 CrossRef CAS PubMed.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed.
- A. Izumi, R. Nomura and T. Masuda, Chem. Lett., 2000, 728–729 CrossRef CAS.
- T. Hosomi, H. Masai, T. Fujihara, Y. Tsuji and J. Terao, Angew. Chem., Int. Ed., 2016, 55, 13427–13431 CrossRef CAS PubMed.
- S.-F. Yang, Z.-T. Liu, Z.-X. Cai, H.-W. Luo, P.-L. Qi, G.-X. Zhang and D.-Q. Zhang, Macromolecules, 2016, 49, 5857–5865 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: TGA, CV, 1H-NMR data and device characterizations. See DOI: 10.1039/c7ra01139c |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |