
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of including electron-withdrawing atoms on the physical and photovoltaic properties of indacenodithieno[3,2-b]thiophene-based donor–acceptor polymers: towards an acceptor design for efficient polymer solar cells
Ping Cai *a,
Xiaofeng Xu*b,
Jiangman Sunc,
Junwu Chen*c and
Yong Caoc
*a,
Xiaofeng Xu*b,
Jiangman Sunc,
Junwu Chen*c and
Yong Caoc
aSchool of Materials Science and Engineering, Guangxi Key Laboratory of Information Materials, Guilin University of Electronic Technology, Guilin 541004, P. R. China. E-mail: caiping@guet.edu.cn
bDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-412 96, Göteborg, Sweden. E-mail: xixu@chalmers.se
cInstitute of Polymer Optoelectronic Materials & Devices, State Key Laboratory of Luminescent Materials & Devices, South China University of Technology, Guangzhou 510640, P. R. China. E-mail: psjwchen@scut.edu.cn
First published on 7th April 2017
Abstract
Three new D–A polymers PIDTT-DTBO, PIDTT-DTBT and PIDTT-DTFBT, using indacenodithieno[3,2-b]thiophene (IDTT) as the electron-rich unit and benzoxadiazole (BO), benzodiathiazole (BT) or difluorobenzothiadiazole (FBT) as the electron-deficient unit, were synthesized via a Pd-catalyzed Stille polymerization. The included electron-withdrawing atoms of the acceptor portion were varied between O, S, and F for tailoring the optical and electrochemical properties and the geometry of structures. Their effects on the film topography, photovoltaic and hole-transporting properties of the polymers were thoroughly investigated via a range of techniques. As expected, the stronger electron-withdrawing BO unit affords red-shifted absorption, low-lying HOMO and LUMO levels for the polymer PIDTT-DTBO. However, it depicts lower hole mobility and a less efficient charge collection in the active layer compared to the polymer PIDTT-DTBT. In addition, degradation of the solubility is observed in the fluorinated polymer PIDTT-DTFBT. As a result, a BHJ PSC (ITO/PEDOT:PSS/polymer:PC71BM/interlayer/Al) fabricated with PIDTT-DTBT attains the best power conversion efficiency (PCE) of 4.91%. These results thus demonstrate the potential effects of electronegative atoms on IDTT-based polymers and the structure–function correlations of such electron-donor materials for efficient PSCs.
Introduction
As a novel renewable energy source, polymer solar cells (PSCs) have attracted growing attention from academic and industry communities as a potential solar energy harvesting medium by virtue of their intriguing lightweight, flexible and large-area features at low manufacturing costs.1,2 During the past decade, bulk-heterojunction polymer solar cells (BHJ PSCs), using a solution-processed active layer composed of an electron-donor and an electron-acceptor, sandwiched between an ITO anode and a low work-function metal cathode, have been intensively investigated.3,4 To date, significant progress has been made in polymer/fullerene based BHJ PSCs with varying degrees of success.5–18 Since the development of BHJ PSCs largely relies on the rational design and synthesis of narrow band-gap materials, conjugated polymers based on alternating electron-donating (D) and electron-withdrawing (A) moieties can offer us an opportunity for tuning the D–A strength and nature of π-electron delocalization, in order to match the requirements of band gaps and energy levels (HOMO and LUMO) for ideal electron-donor materials.19–21 Up to now, a strategy toward using weak donor and strong acceptor units for polymer skeleton construction is especially effective to improve device performance.22 The weak donor moiety would anchor a low-lying HOMO level, meanwhile, strong acceptor moiety would optimize the value of a favorable LUMO level and narrow band-gap.23–25Recently, some electron-rich arenes with high level of fusion have been reported.26 Their rigid backbones with horizontally or vertically extended π-conjugation were formed by fastening or fusing adjacent rings. Especially, a class of ladder-type frameworks with forced planarization structures has been regarded as appealing electron-donating moieties.27–38 First, the appropriate planarization of aromatic rings can easily afford a red-shifted absorption spectrum, and also positively impact the intermolecular interactions between conjugated main chains. In addition, the relatively higher level of coplanar backbones are conducive to intermolecular charge carrier hopping and suppress the interannular rotation, which is an effective way to improve the intrinsic charge mobility. Recently, we reported a class of ladder-type D–A polymers composed of a indacenodithieno[3,2-b]thiophene-based arene (IDTT; weak donor) as an electron-rich unit and diphenylquinoxaline (Qx; strong acceptor) as an electron-deficient unit with the best power conversion efficiency of 6.8% for BHJ PSCs.39,40 Generally, indacenodithiophene (IDT)-based polymers demonstrate low-lying HOMO levels, efficient solar energy harvest and high hole mobility.41–44 In order to further extend linear π-conjugation of IDT unit, the central phenyl ring was covalently fastened with two thieno[3,2-b]thiophenes (TT) to form the IDTT skeleton, which can afford an enhanced planarization via increasing the pentacyclic rings to heptacyclic rings. Moreover, four branched and aliphatic side chains can afford sufficient solubility for post-processing and device fabrication. The appealing optoelectronic characteristics of IDTT make it promising building blocks for D–A conjugated polymers.45–47 Very recently, there are evidences indicating that IDTT unit is also a promising building block to synthesize small molecular acceptors used for non-fullerene PSCs.48,49 Open questions remain regarding further structural modifications for IDTT based polymers, tailoring the deep HOMO level without sacrificing much of narrow band-gap, and its relationship to the trade-off between obtained Voc, Jsc and FF for more efficient device performance.
In an effort to provide more insight to the effect of electron-withdrawing moieties on IDTT-based polymers, in this study we incorporate three different electron acceptors (BO, BT and FBT) for comparison (Scheme 1). First, we choose benzodiathiazole (BT) as the building block due to its easy synthesis, strong electron-withdrawing property and excellent stability. Compared with BT unit, benzoxadiazole (BO) unit would have enhanced electron-withdrawing ability due to the stronger electronegativity of oxygen than that of sulfur.50 Another effective method is covalently addition of electronegative groups on the BT ring.23,51 Since the most electronegative element is fluorine, two fluorine atoms are fastened on 5, 6 position of BT core to form the difluorobenzothiadiazole (FBT) unit. Moreover, the electron-withdrawing (BO, BT, FBT) and electron-donating (IDTT) moieties are coupled via bis-thiophene linkages to form D–π–A conjugation, which is often employed to improve the planarity of conjugated backbones. On the basis of the above consideration, three new conjugated polymers PIDTT-DTBO, PIDTT-DTBT and PIDTT-DTFBT were successfully prepared (Scheme 2). The solubility, thermal stability, UV-vis absorption, electrochemical properties and dipole moments were systematically investigated to understand the structure–property correlation. BHJ PSCs using PC71BM as the electron acceptor were fabricated for evaluation. The hole mobility and film morphology of the blend layers were also discussed.
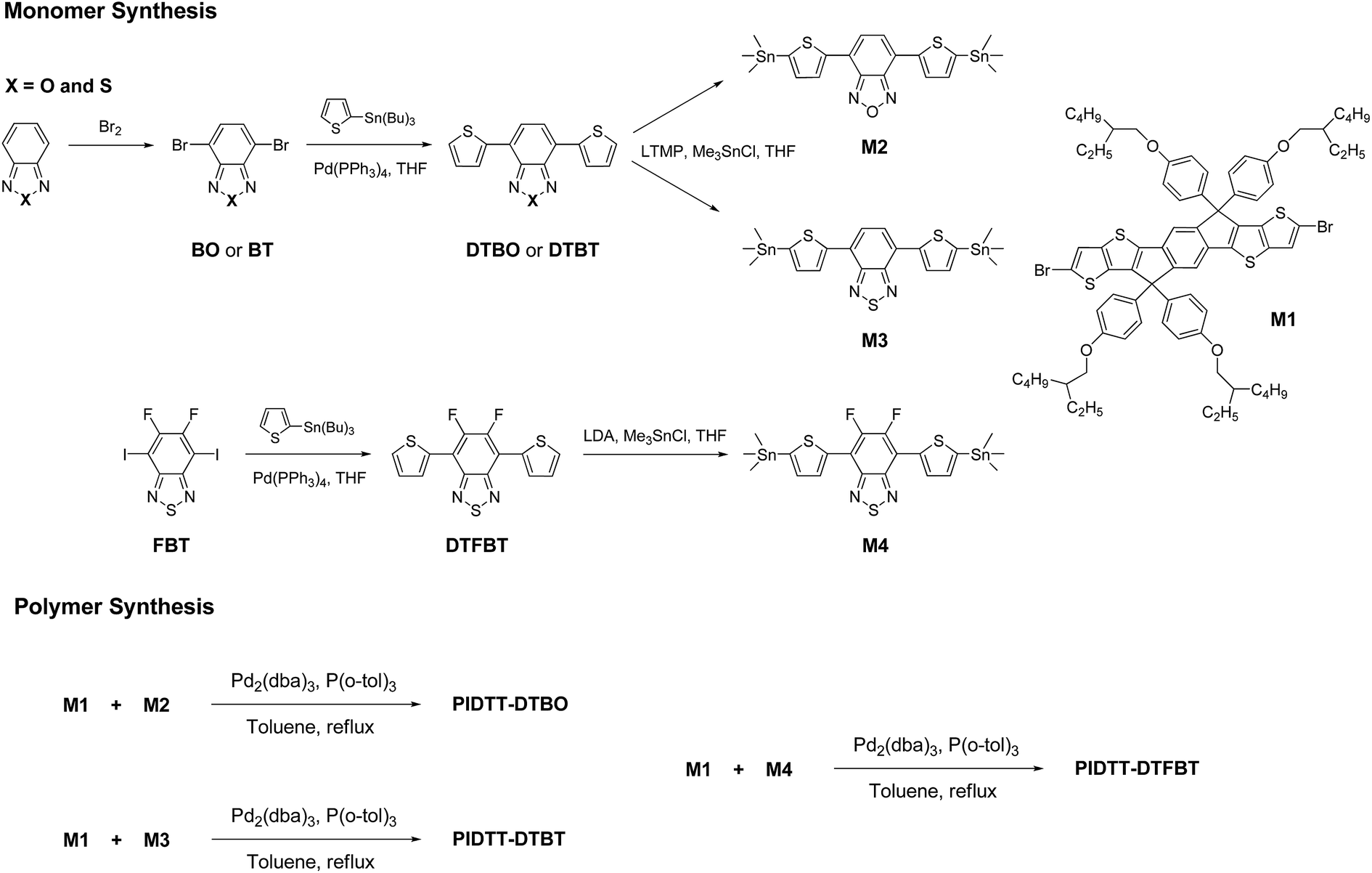 | ||
| Scheme 1 Monomer and polymer synthesis. | ||
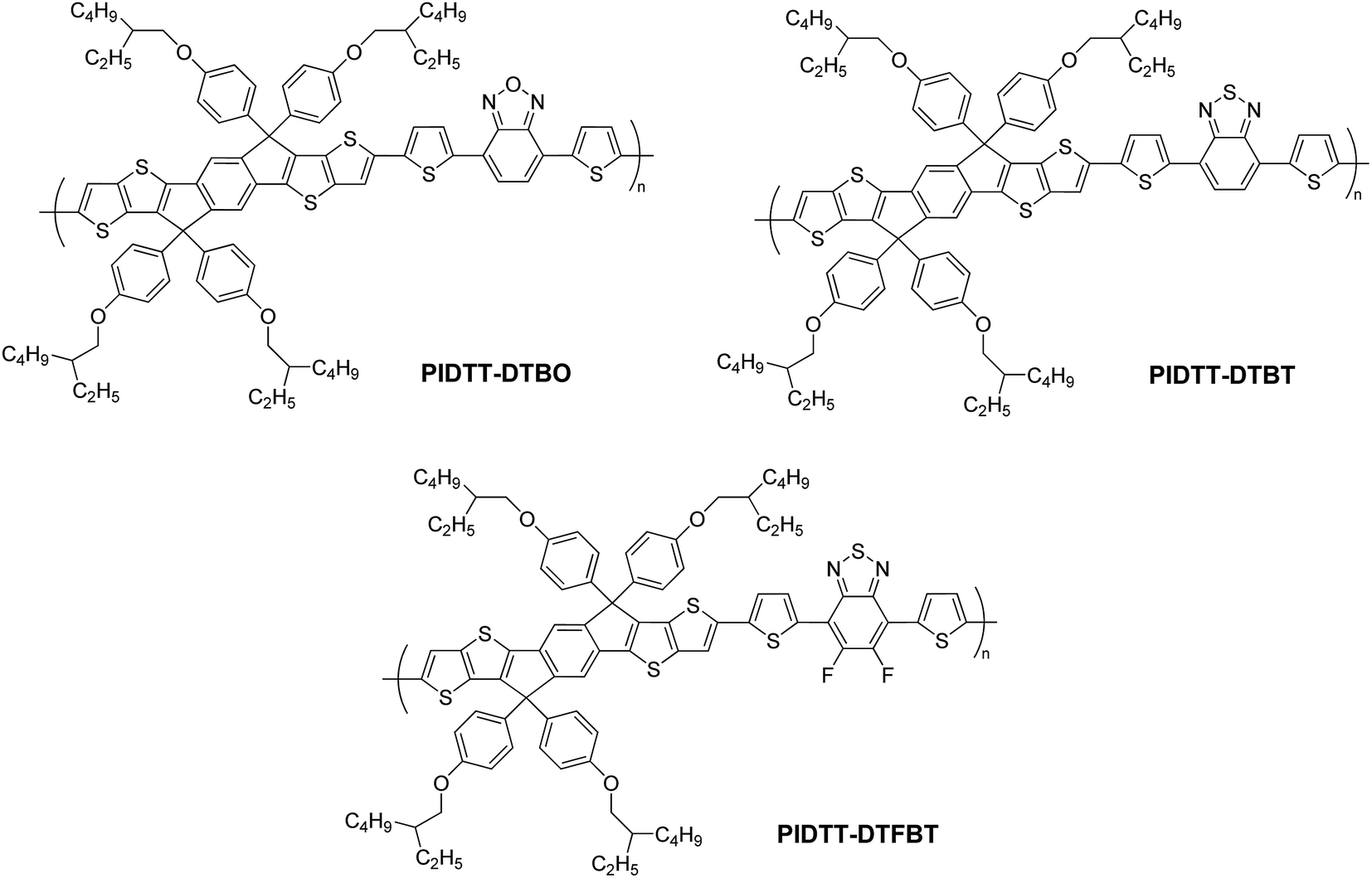 | ||
| Scheme 2 Polymer structures. | ||
Experimental section
Materials
All reagents and solvents were obtained from Aldrich, Alfa Aesar, and TCI Chemical Co. Anhydrous tetrahydrofuran(THF) and chlorobenzene were distilled over sodium/benzophenone under N2 prior to use. All manipulations involving air-sensitive reagents were performed under an atmosphere of argon.Characterization
1H and 13C NMR spectra were recorded on a Bruker AV 300 spectrometer with tetramethylsilane (TMS) as the internal reference. Elemental analyses were performed on a Vario EL elemental analysis instrument (Elementar Co.). The molecular weight of polymers were obtained on a Polymer Laboratories PL 220 Chromatograph using linear polystyrene as references, with 150 °C 1,2,4-trichlorobenzene as the eluent. UV-vis absorption spectra were recorded on a Shimadzu UV 2450 spectrophotometer. Thermogravimetric analysis (TGA) was carried out on a PerkinElmer Diamond TGA/DTA, at a heating rate of 10 °C min−1 from 50 °C to 650 °C under a nitrogen atmosphere. Differential scanning calorimeter (DSC) measurement was carried out on a PerkinElmer Diamond DSC, at a heating rate of 10 °C min−1 from 25 °C to 200 °C under a nitrogen atmosphere. Cyclic voltammetry was carried out on a CHI660A electrochemical workstation with platinum electrodes at a scan rate of 50 mV s−1 against the Ag/Ag+ reference electrode with an argon-saturated solution of 0.1 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) in acetonitrile (CH3CN). The deposition of a polymer on the electrode was done by the evaporation of its dilute chloroform solution. Tapping mode atomic force microscopy (AFM) images were obtained using a NanoScope NS3A system (Digital Instrument).Synthesis
The synthetic routes of monomers and polymers are shown in Schemes 1 and 2. Monomer M1 was synthesized according to our previous work. Monomer 4,7-bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole (M3) and 5,6-difluoro-4,7-bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole (M4) were prepared according to the literatures.7,52 The detailed synthetic processes of 4,7-bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]oxadiazole (M2) are as follows.4,7-Di(thiophen-2-yl)benzo[c][1,2,5]oxadiazole (DTBO)
4,7-Dibromobenzo[c][1,2,5]oxadiazole (1.39 g, 5.0 mmol), 2-tributylstannylthiophene (4.66 g, 12.5 mmol) and Pd(PPh3)2Cl2 (0.18 g, 0.25 mmol) were dissolved in anhydrous THF (50 mL). The reaction mixture was heated to 75 °C for 12 hours under an argon atmosphere. After cooling to room temperature, the organic layer was extracted with dichloromethane (100 mL), washed successively with water and then dried over anhydrous MgSO4. The residue was purified by column chromatography (dichloromethane/hexane = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 as the eluent) and recrystallized from THF\ethanol to give the final compound as a red solid (1.18 g, yield: 83%). 1H NMR (300 MHz, CDCl3), δ (ppm): 8.11 (d, J = 4.8 Hz, 2H), 7.62 (s, 2H), 7.44 (d, J = 6.0 Hz, 2H), 7.20 (m, 2H). 13C NMR (75 MHz, CDCl3), δ (ppm): 147.9, 137.9, 128.7, 128.6, 127.0, 126.4, 122.1. Anal. calcd (%) for C14H8N2OS2: C, 59.13; H, 2.84; N, 9.85; S, 22.55. Found: C, 58.14; H, 2.30; N, 9.60; S, 21.99.
:5 as the eluent) and recrystallized from THF\ethanol to give the final compound as a red solid (1.18 g, yield: 83%). 1H NMR (300 MHz, CDCl3), δ (ppm): 8.11 (d, J = 4.8 Hz, 2H), 7.62 (s, 2H), 7.44 (d, J = 6.0 Hz, 2H), 7.20 (m, 2H). 13C NMR (75 MHz, CDCl3), δ (ppm): 147.9, 137.9, 128.7, 128.6, 127.0, 126.4, 122.1. Anal. calcd (%) for C14H8N2OS2: C, 59.13; H, 2.84; N, 9.85; S, 22.55. Found: C, 58.14; H, 2.30; N, 9.60; S, 21.99.
4,7-Bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]oxadiazole (M2)
To a solution of 2,2,6,6-tetramethylpiperidine (TMP) (0.64 g, 4.5 mmol) in anhydrous THF (50 mL) was added a 2.2 M solution of n-BuLi in hexane (2.05 mL, 4.5 mmol) at −78 °C under an argon atmosphere. The reaction mixture was warmed to room temperature, stirred for another 30 min and cooled to −78 °C again. A solution of 4,7-di(thiophen-2-yl)benzo[c][1,2,5]oxadiazole (0.43 g, 1.5 mmol) in anhydrous THF (10 mL) was added dropwise. The solution turned deep purple immediately and was stirred at −78 °C for 1 hour. Then Me3SnCl (1.20 g, 6.0 mmol) was added in one portion. The reaction mixture was allowed to warm to room temperature and stirred overnight. The resulting mixture was poured into water, extracted with diethyl ether (100 mL) and dried over anhydrous MgSO4. After the solvent was removed, the residue was recrystallized from THF\ethanol to give the final compound as a red solid (0.72 g, yield: 79%). 1H NMR (300 MHz, CDCl3), δ (ppm): 8.17 (d, J = 3.6 Hz, 2H), 7.62 (s, 2H), 7.28 (d, J = 3.6 Hz, 2H), 0.43 (s, 18H). 13C NMR (75 MHz, CDCl3), δ (ppm): 147.9, 143.5, 141.0, 136.6, 129.4, 126.4, 121.7, −8.1. Anal. calcd (%) for C20H24N2OS2Sn2: C, 39.38; H, 3.97; N, 4.59; S, 10.51. Found: C, 39.72; H, 3.47; N, 4.74; S, 10.47.General procedure for polymerization
The three polymers were synthesized by the palladium(0)-catalyzed Stille coupling reaction with a dibromide monomer and a bis(trimethylstannyl)-substituted monomer under an argon atmosphere.0.25 mmol of dibromide monomer, 0.25 mmol of bis(trimethylstannyl)-substituted monomer, tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3) (7 mg) and tri(o-tolyl)phosphine (P(o-Tol)3) (9 mg) were dissolved in anhydrous chlorobenzene (15 mL). The mixture was refluxed with vigorous stirring for 72 h under an argon atmosphere. After the mixture was cooled to room temperature, it was poured into 250 mL of methanol. The precipitated material was dissolved and filtrated through a funnel and precipitated again. The polymer was washed in a Soxhlet extractor with acetone (12 h), hexane (12 h), and chloroform (12 h). The chloroform fraction was concentrated and poured into methanol (250 mL). Finally, the precipitate was collected and dried in vacuum to afford the title polymer as a dark fiber.
PSC fabrication and characterization
Patterned indium tin oxide (ITO) coated glass with a sheet resistance of 15–20 ohm per square was cleaned by a surfactant scrub and then underwent a wet-cleaning process inside an ultrasonic bath, beginning with deionized water followed by acetone and isopropanol. After oxygen plasma cleaning for 5 min, a 40 nm thick poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) (Bayer Baytron 4083) anode buffer layer was spin-cast onto the ITO substrate and then dried by baking in a vacuum oven at 80 °C for overnight. The active layer, with a thickness in the range of 80–90 nm, was then deposited on top of the PEDOT:PSS layer by spin-coating from a chlorobenzene or dichlorobenzene solution. The interlayer solution in methanol was spin-coated on the top of the active layer to form a thin interlayer of 3–5 nm. The thickness of the PEDOT:PSS and active layer was verified by a surface profilometer (Tencor, Alpha-500). At last, a 100 nm aluminium layer was thermally evaporated with a shadow mask at a base pressure of 3 × 10−4 Pa. The overlapping area between the cathode and anode defined a pixel size of 0.15 cm2. The thickness of the evaporated cathodes was monitored by a quartz crystal thickness/ratio monitor (model: STM-100/MF, Sycon). Except the deposition of the PEDOT:PSS layers, all the fabrication processes were carried out inside a controlled atmosphere of nitrogen drybox (Vacuum Atmosphere Co.) containing less than 10 ppm oxygen and moisture. The power conversion efficiencies of the resulting polymer solar cells were measured under 1 sun, AM 1.5 G (air mass 1.5 global) spectrum from a solar simulator (Oriel model 91192) (100 mW cm−2). The current density–voltage (J–V) characteristics were recorded with a Keithley 236 source unit. The external quantum efficiencies of PSCs were measured with a commercial photomodulation spectroscopic setup (DSR 100UV-B) including a xenon lamp, an optical chopper, a monochromator, and a lock-in amplifier operated by a PC computer (Zolix Instruments), and a calibrated Si photodiode was used as a standard.Hole mobility measurement
The SCLC mobility was determined by fitting the dark current to the model of a single carrier SCLC, which is described by the equation:where J is the current, μh is the charge mobility at zero field, ε0 is the free-space permittivity, εr is the relative permittivity of the material, d is the thickness of the active layer, and V is the effective voltage. V = Vappl − Vbi − Vs, Vappl is the applied potential, Vbi is the built-in potential and Vs is the voltage drop due to the substrate series resistance. The hole mobility can be calculated from the slope of the J1/2–V curve.
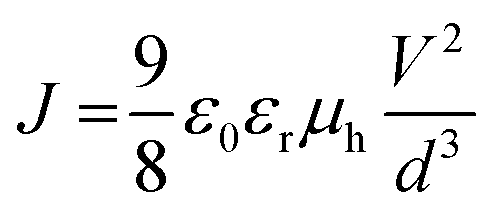
Results and discussion
Design and synthesis
The synthetic routes for monomers and polymers are shown in Scheme 1. Initial reactions started from respective bromination of benzo[c][1,2,5]oxadiazole and benzo[c][1,2,5]thiadiazole in the presence of excess bromide. Two obtained dibromo compounds 4,7-dibromobenzo[c][1,2,5]oxadiazole and 4,7-dibromobenzo[c][1,2,5]thiadiazole were flanked by two thiophene rings via Pd-catalyzed Stille coupling reaction, respectively. Finally, respective reactions of 4,7-di(thiophen-2-yl)benzo[c][1,2,5]oxadiazole (DTBO) and 4,7-di(thiophen-2-yl)benzo[c][1,2,5]thiadiazole (DTBT) with freshly made 2,2,6,6-tetramethylpiperidyl lithium (LTMP) and then trimethylchlorostannane, afforded monomer M2 and M3 in good yield. Here, LTMP is used as a non-nucleophilic base due to its strong steric hindrance coming from the tetramethylpiperidide ring. Monomer M1 was synthesized according to our previous work.39 Monomer M4 was prepared for comparison according to the literature.52 The chemical structure of all the compounds were confirmed using 1H NMR, 13C NMR and elemental analysis. (see Experiment section for details).The preparations of three polymers PIDTT-DTBO, PIDTT-DTBT and PIDTT-DTFBT were accomplished via Pd2(dba)3-catalyzed Stille coupling reaction. After polymerization, the crude polymers were purified by filtration and continuously extracting with acetone, hexane and chloroform in the Soxhlet extractor. The chemical structure of polymer PIDTT-DTBO, PIDTT-DTBT and PIDTT-DTFBT (Scheme 2) were confirmed by 1H NMR and elemental analysis. Polymer PIDTT-DTBO and PIDTT-DTBT were readily soluble in tetrahydrofuran (THF), chloroform, toluene and chlorobenzene (CB). However, after polymerization and precipitation, the crude polymer PIDTT-DTFBT was only partially soluble in warm CB and dichlorobenzene (DCB). Its poor solubility may be ascribed to enhanced intermolecular non-covalent interactions of the fluorinated polymer backbones. No clear 1H NMR spectrum of PIDTT-DTFBT was observed presumably due to strong aggregation of polymer main chains in solution. We failed to obtain a thick and smooth film from the solution of polymer PIDTT-DTFBT, thus hampering investigation of its absorption spectrum, electrochemical properties and device fabrication. The molecular weight of three polymers were evaluated by high temperature gel permeation chromatography (GPC) at 150 °C with 1,2,4-trichlorobenzene as eluent. The number-average molecular weight (Mn) of polymer PIDTT-DTBO and PIDTT-DTBT was 33.5 and 30.3 kDa with polydispersity index (PDI) of 2.2 and 2.4, respectively. For polymer PIDTT-DTFBT, a rather lower Mn of 8.3 kDa and board PDI up to 3.6 was obtained. It is noted that once the limit of solubility was reached in hot chlorobenzene, the length of polymer skeletons would stop increasing and precipitate may appear during polymerization.
Thermal properties
Thermal stability of polymers was investigated using thermogravimetric analysis (TGA), as shown in Fig. 1(a). The TGA curves exhibit onset temperatures with 5% weight loss (Td) of polymer PIDTT-DTBO, PIDTT-DTBT and PIDTT-DTFBT are between 380 and 430 °C, indicating these polymers have sufficient stability for optoelectronic applications. In addition, DSC measurement was carried out to investigate the phase transitions and crystallization properties of the polymers. As shown in Fig. 1(b), neither melting point (Tm) nor crystallization temperature (Tc) was observed from two cooling and heating cycles between 25 °C and 230 °C.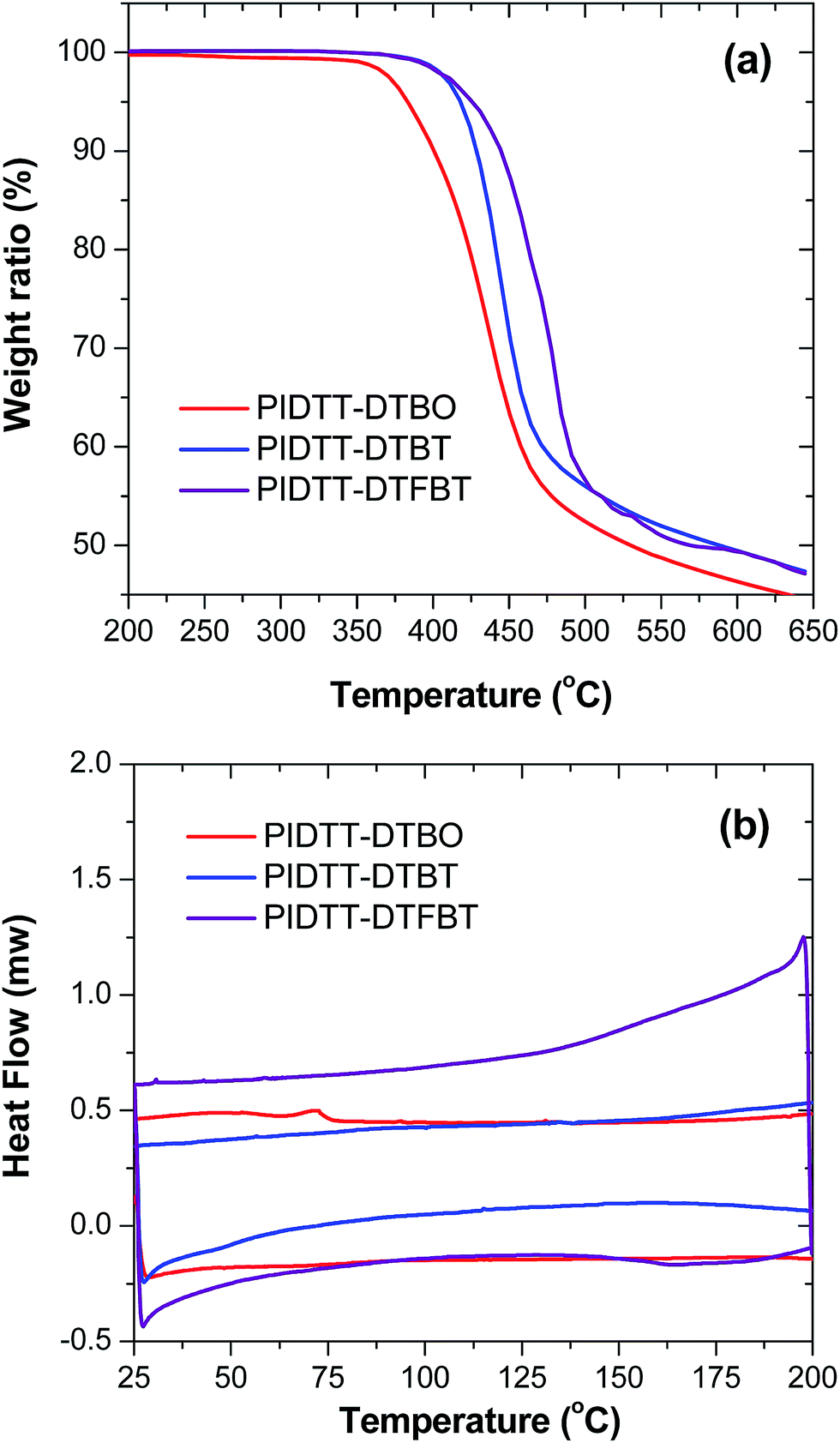 | ||
| Fig. 1 TGA and DSC plot of polymers with a heating rate of 10 °C min−1 under a nitrogen atmosphere. | ||
Absorption spectra
In order to determine the effects of electron-withdrawing atoms on the optoelectronic properties of IDTT-based polymers, UV-vis absorption spectra were obtained for each polymer in solution and film (Fig. 2, Table 1). The absorption spectra of polymer PIDTT-DTBO and PIDTT-DTBT show two distinct absorption bands in the wavelength range of 350–450 and 500–700 nm, corresponding to the π–π* transition and intramolecular charge transfer (ICT) between the donor and acceptor units. The absorption maximum (λmax) of polymer PIDTT-DTBO and PIDTT-DTBT in dilute chloroform solutions are at 610 and 585 nm, respectively. In thin films, the absorption λmax of polymer PIDTT-DTBO and PIDTT-DTBT are red-shifted to 622 and 607 nm, owing to closer π–π stacking of polymer main chains in the solid state. The absorption edges of the film spectra are located at 732 and 721 nm, respectively. Compared to the absorption spectrum of PIDTT-DTBT, polymer PIDTT-DTBO exhibits a bathochromic shift (10 nm) in the long wavelength regime, implying stronger intramolecular charge transfer between the IDTT donor and BO acceptor units. Since the oxygen atom has relatively stronger electronegativity, the stronger D–A interactions within polymer PIDTT-DTBO would be originated from more electron-withdrawing ability of BO unit than that of BT unit. Generally, enhancement of light harvest is beneficial to higher Jsc achievement of PSCs. As shown in Fig. 2(b), we also investigate the effects of thermal annealing on the absorption spectra in solid films. For each polymer, no spectral red-shifts are recorded after thermal annealing at 100 °C for 10 min. The result appear to suggest that the polymer backbones can attain desirable π–π stacking in the solid state without any thermal treatment. In addition, as compared with the previously reported polymer PIDTT-Qx incorporating IDTT and Qx units, very similar absorption spectra are observed in the neat polymer films.39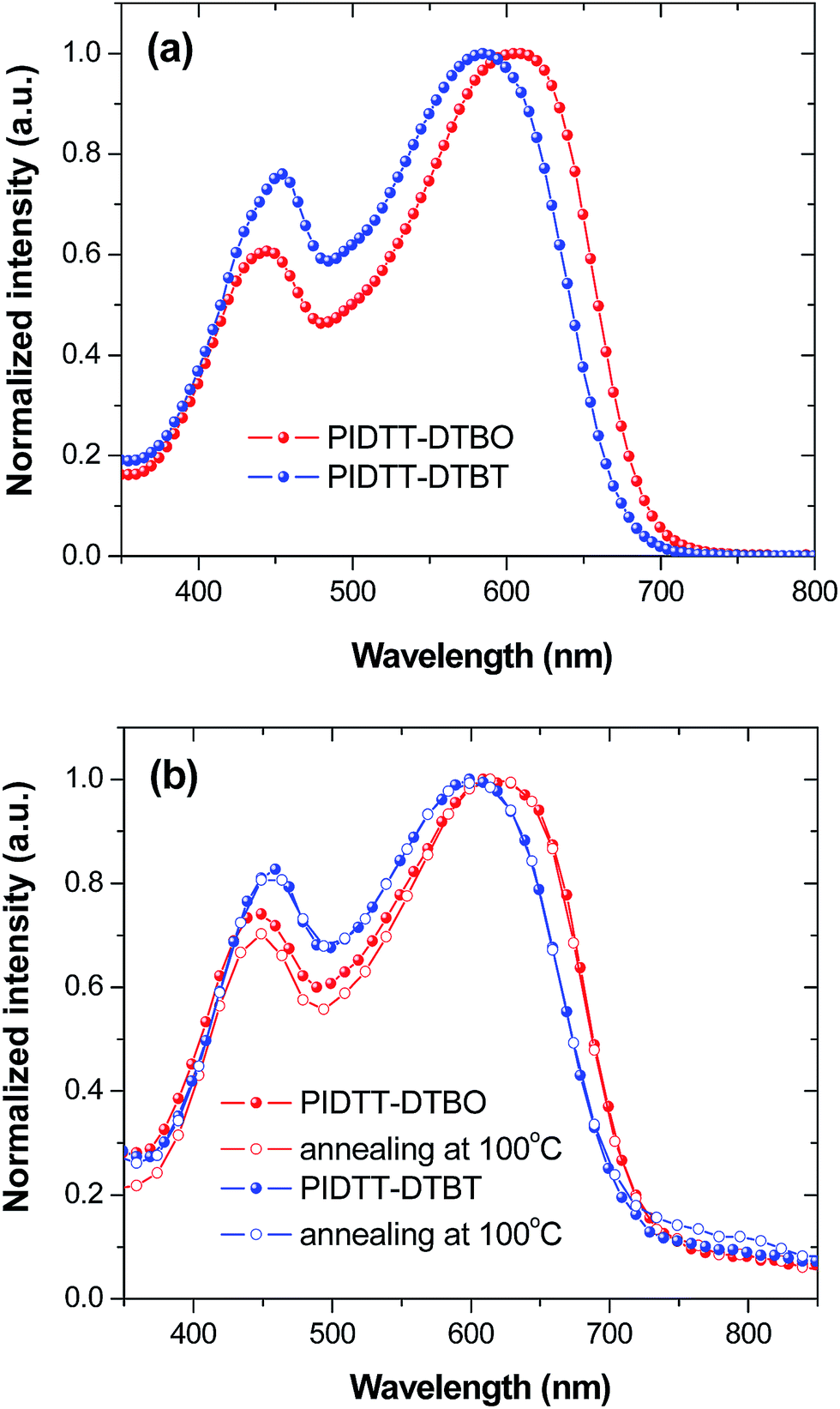 | ||
| Fig. 2 (a) Normalized UV-vis-NIR absorption spectra of polymers in chloroform. (b) Normalized UV-vis-NIR absorption spectra of polymer films before and after annealing at 100 °C for 10 min. | ||
| Polymer | UV-vis absorption spectra | Cyclic voltammetry | |||||
|---|---|---|---|---|---|---|---|
| Solution | Film | p-Doping | n-Doping | ||||
| λabsa (nm) | λabsb (nm) | λonset (nm) | Eoptg (eV)c | φoxd/HOMOe (V)/(eV) | φred/LUMOf (V)/(eV) | Eecgg (eV) | |
| a Absorption peak in chloroform solution.b Absorption peak in film.c Optical band gap.d Onset voltage of oxidation process during cyclic voltammetry with Ag/Ag+ reference electrode.e Calculated according to HOMO = −e(Eox + 4.49) (eV).f LUMO = −e(Ered + 4.49) (eV).g Electrochemical band gap. | |||||||
| PIDTT-DTBO | 610 | 622 | 732 | 1.69 | 0.87/−5.36 | −0.80/−3.69 | 1.67 |
| PIDTT-DTBT | 585 | 607 | 721 | 1.72 | 0.82/−5.31 | −0.82/−3.67 | 1.64 |
Electrochemical properties
Fig. 3 shows the cyclic voltammograms of polymer films on Pt disk electrode in a 0.1 M Bu4NPF6 acetonitrile solution. The highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO) energy levels and electrochemical band-gaps of polymers were obtained from the onsets of oxidation and reduction potentials with an Ag/Ag+ electrode as reference. HOMO level of ferrocene (−4.80 eV) was used as the internal standard. The onset oxidation potentials (φox) of polymer PIDTT-DTBO and PIDTT-DTBT are 0.87, 0.82 V vs. Ag/Ag+ electrode, respectively. The onset reduction potentials (φred) are −0.82 and −0.80 V vs. Ag/Ag+ electrode, respectively. The HOMO and LUMO energy levels were calculated according to the equations, HOMO = −e(Eox + 4.49) (eV), LUMO = −e(Ered + 4.49) (eV). The results of electrochemical measurements are summarized in Table 1. The HOMO level of polymer PIDTT-DTBO is around −5.36 eV, which is 0.05 eV lower than that of polymer PIDTT-DTBT (−5.31 eV). The HOMO shift would be ascribed to stronger electronegativity of oxygen than sulfur atom in acceptor part. Since the Voc of BHJ PSCs is positively correlated to the energy difference between HOMO level of electron donor and LUMO level of electron acceptor, the low-lying HOMO level of polymer PIDTT-DTBO is the precondition for a high Voc achievement. After the isolated acceptors are coupled by thiophene units on both sides, a little higher HOMO energy (up-lying HOMO level) are recorded compared to the direct D–A structure of our previous work.30 It is ascribed to the increased planarity and enhanced π-delocalization brought by the thiophene rings, which is common in many D–π–A conjugated polymers. The LUMO levels of polymer PIDTT-DTBO and PIDTT-DTBT are −3.69 and −3.67 eV, respectively. Similarly, the larger electron-withdrawing effect from oxygen results in a little stable LUMO level of BO compared to BT. According to the previous report, the HOMO and LUMO levels of PIDTT-Qx are −5.39 and −3.73 eV respectively, which are relatively higher than those of PIDTT-DTBO and PIDTT-DTBT.39 As shown from the Fig. 4, the energy difference between the LUMO levels of two polymers and that of PC71BM (−4.2 eV) are large enough for efficient exciton dissociation. According to the equation, Eecg = e(Eox − Ered) (eV), the electrochemical band-gaps of polymer PIDTT-DTBO and PIDTT-DTBT are calculated to be 1.67 and 1.64 eV respectively, which are similar with that of PIDTT-Qx (1.66 eV).39 The coupling of bis-thiophenes to acceptor unit shows minimal impact of the band-gaps. The optical band-gaps of polymer PIDTT-DTBO and PIDTT-DTBT estimated from the corresponding absorption edges of solid films are 1.69 and 1.72 eV, respectively, which match well with their electrochemical band-gaps within the experimental error.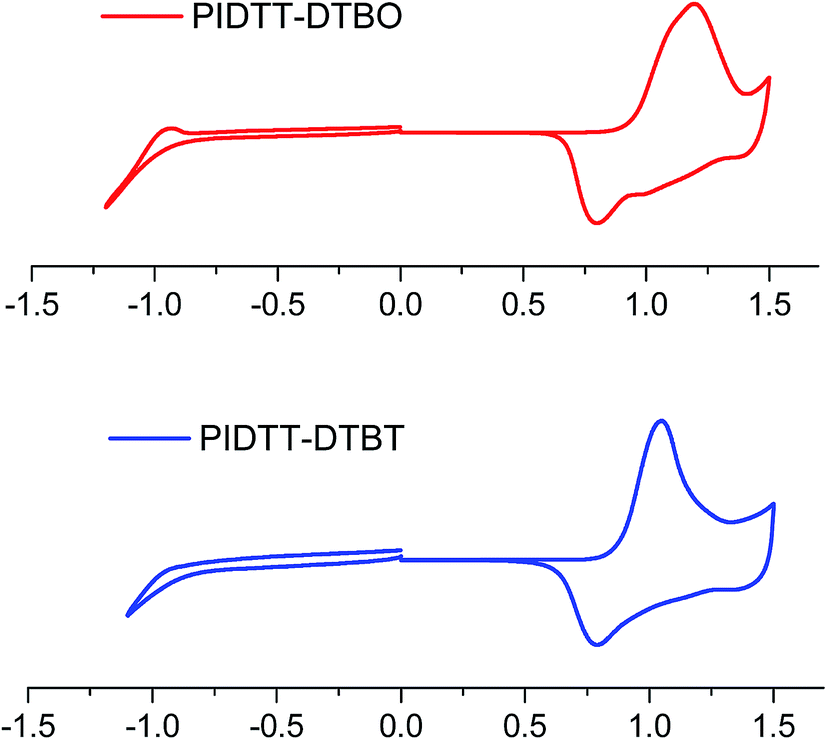 | ||
| Fig. 3 Cyclic voltammogram of the polymer film on a platinum electrode measured in a 0.1 M Bu4NPF6 acetonitrile solution at a scan rate of 100 mV s−1. | ||
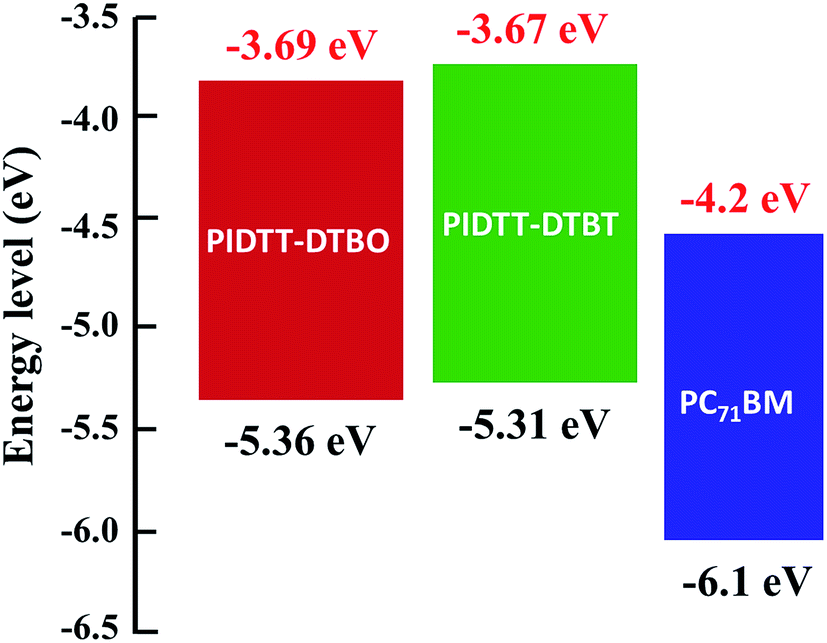 | ||
| Fig. 4 HOMO and LUMO energy levels of the polymers and PC71BM. | ||
Recently, theoretical calculations were used to study the effect of dipole moments on performance of conjugated molecules in PSCs.53,54 Here, the additional dipole moment of 4,7-di(thiophen-2-yl)benzooxadiazole (DTBO) unit was performed with hybrid DFT B3LYP/6-31g*. In comparison with 4,7-di(thiophen-2-yl)benzothiadiazole (DTBT) (0.13 Debye),55 DTBO unit exhibits a codirectional but higher dipole moment of 2.32 Debye, which would be ascribed to the inclusion of more electronegative oxygen atom. Compared to DTBO and DTBT units, 5,6-difluoro-4,7-di(thiophen-2-yl)benzothiadiazole (DTFBT) has an opposite dipole moment of 1.64 Debye due to the 5,6-difluorinated BT core.56 The natural differences of dipole moment would affect the molecular conformations on acceptor portion during thin film fabrication. When using a polar solvent like chlorobenzene, the conformation with a higher dipole moment may appear dominantly, thus affecting intermolecular interactions in the solid film. As a result, it is noted that the replacement of BT by FBT unit can decrease the nonplanar conformation and promote the π–π stacking of polymer main chains,52 which can also explain the noticeable degradation of solubility for our polymer PIDTT-DTFBT.
Photovoltaic properties
To investigate the photovoltaic properties of polymer PIDTT-DTBO and PIDTT-DTBT, bulk-heterojunction polymer solar cells (BHJ PSCs) with the device architecture of ITO/PEDOT:PSS (40 nm)/polymer:PC71BM (85 nm)/PFN interlayer (3 nm)/Al (100 nm) were fabricated. An alcohol-soluble polymer PFN was used as an interfacial modifier between the active layer and Al cathode, which can intensely promote the interfacial contact and electron extraction between the active layer and metallic cathode.57 Polymer and PC71BM was dissolved in o-dichlorobenzene for spin-coating. The measurements of photovoltaic performances were carried out under illumination of AM 1.5 G simulated solar light at 100 mW cm−2. Different weight ratios of polymer:PC71BM were carefully optimized. The corresponding PSC parameters (short-circuit current density Jsc, open circuit voltage Voc, and fill factor FF) are summarized in Table 2, and the J–V curves are shown in Fig. 5(a) and (b). From the device based on polymer PIDTT-DTBO with an optimum D/A ratio (1:4), a PCE of 4.37% with Voc = 0.88 V, Jsc = 9.97 mA cm−2 and FF = 49.84% was obtained, while the device based on polymer PIDTT-DTBT shows a higher PCE of 4.82%, with Voc = 0.86 V, Jsc = 10.05 mA cm−2, and FF = 55.80%. The slightly increased Voc of PIDTT-DTBO-base device is in accordance with its low-lying HOMO level compared to polymer PIDTT-DTBO. In addition, thermal annealing was employed to improve the photovoltaic performances of the PSCs. After annealing at 100 °C for 10 min, the Voc value of each polymer doesn't alter and little changes of FF are observed. The Jsc of PIDTT-DTBO-based devices increase from 9.97 to 10.50 mA cm−2, its PCE thus increase from 4.37 to 4.60%. Compared to PIDTT-DTBT, the PIDTT-DTBO-based device displays a little larger Jsc value, which is in accordance with its 10 nm bathochromic absorption. For polymer PIDTT-DTBT, the measured Jsc exhibits relatively limited increase, the corresponding PCE displays a little increase from 4.88 to 4.91%.
| Polymer | Ratioa | Annealing temperature | Voc (V) | Jsc (mA cm−2) | FF (%) | PCEc (%) |
|---|---|---|---|---|---|---|
| a Polymer:PC71BM weight ratio.b Room temperature without annealing.c The values in the parentheses are from the best devices, and the statistics are from twelve devices. |
||||||
| PIDTT-DTBO | 1:1 |
r.t.b | 0.88 | 5.02 | 32.34 | 1.35 ± 0.07 (1.43) |
| 1:2 |
r.t. | 0.88 | 8.24 | 37.48 | 2.64 ± 0.08 (2.72) | |
| 1:3 |
r.t. | 0.88 | 9.19 | 43.40 | 3.42 ± 0.08 (3.51) | |
| 1:4 |
r.t. | 0.88 | 9.97 | 49.84 | 4.24 ± 0.09 (4.37) | |
| 1:4 |
100 °C | 0.88 | 10.50 | 49.74 | 4.47 ± 0.10 (4.60) | |
| PIDTT-DTBT | 1:1 |
r.t. | 0.84 | 4.15 | 30.15 | 0.93 ± 0.10 (1.05) |
| 1:2 |
r.t. | 0.86 | 6.93 | 34.89 | 1.95 ± 0.10 (2.08) | |
| 1:3 |
r.t. | 0.86 | 8.87 | 51.04 | 3.78 ± 0.08 (3.89) | |
| 1:4 |
r.t. | 0.86 | 10.05 | 55.80 | 4.70 ± 0.10 (4.82) | |
| 1:4 |
100 °C | 0.86 | 10.20 | 55.99 | 4.79 ± 0.09 (4.91) | |
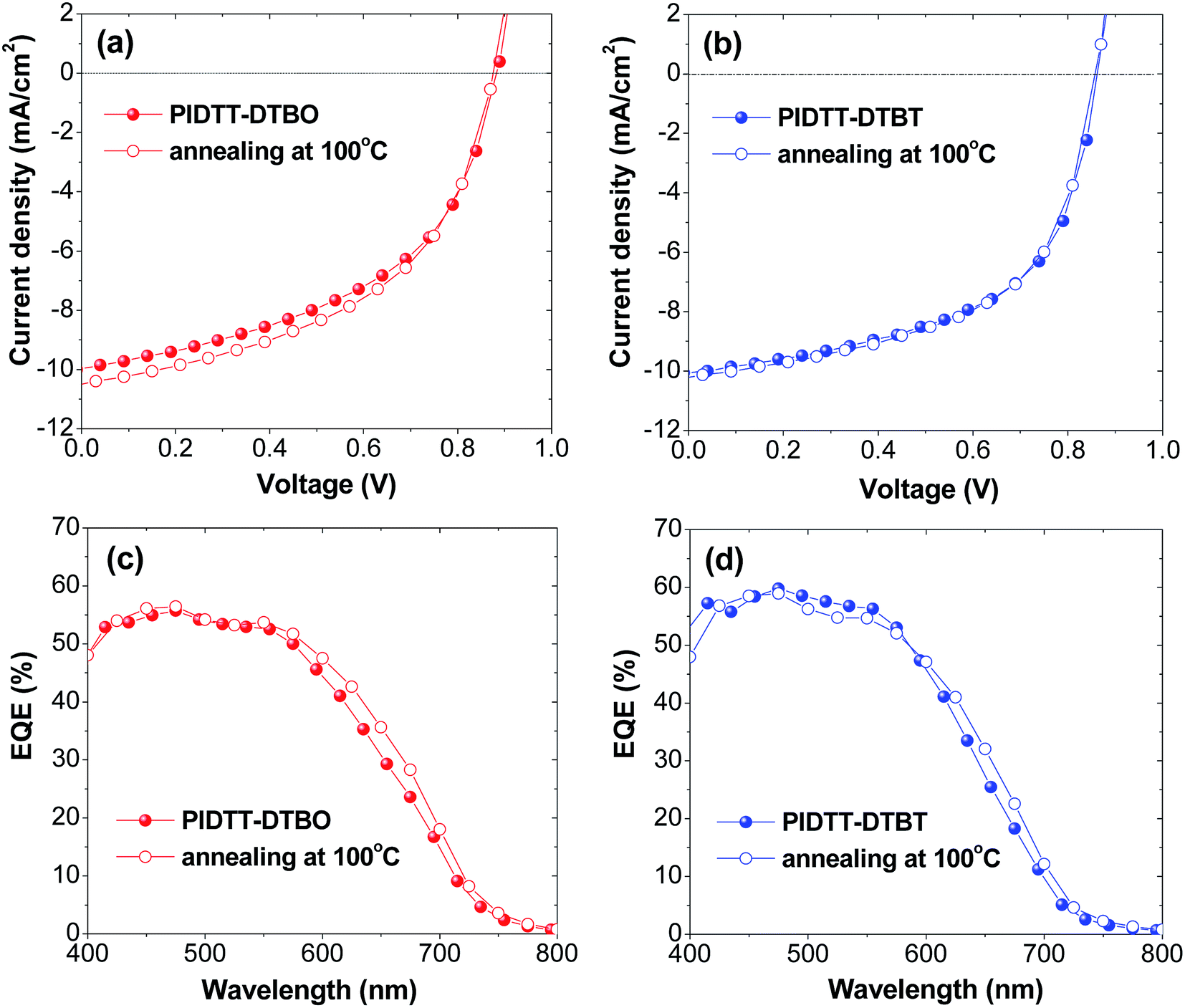 | ||
| Fig. 5 (a, b) Current density–voltage (J–V) characteristics of PSCs before and after annealing at 100 °C for 10 min (c, d) EQE curves of PSCs before and after annealing at 100 °C for 10 min. | ||
To evaluate the photoresponse of PSCs, external quantum efficiencies (EQE) before and after annealing at 100 °C for 10 min were measured. As shown in Fig. 5(c) and (d), the EQE curves generally correspond with UV-vis absorption spectra of polymer PIDTT-DTBO and PIDTT-DTBT in the range of 400 to 750 nm. The enhanced quantum efficiency in the region of 400–500 nm is attributable to the absorption of PC71BM in the visible region. Compared to the devices without annealing, both devices after annealing can demonstrate a higher EQE in the long wavelength region (550 to 750 nm). It's coincident with the J–V results, indicating the solar absorption and conversion becomes more efficiently in the relative strong region (500 to 750 nm) of solar irradiance. The devices based on polymer PIDTT-DTBT respond to a higher photo conversion efficiency over the whole visible region, implying more efficient charge collection and less charge recombination at the junction between polymer PIDTT-DTBT and PC71BM.
In order to determine the effects of thermal annealing on film topography, the surface morphology of polymer/PC71BM blend films before and after post-thermal treatment were studied via atom force microscopy (AFM). As shown in Fig. 6(a–c), without any thermal annealing, the blend films of polymer PIDTT-DTBO and PIDTT-DTBT exhibit smooth morphology with low root mean-square roughness (Rq) of 0.32 and 0.37 nm, respectively. Generally, a smooth film with uniform nanoscale network is desirable in PSCs. Any large domain and aggregate would hinder exciton dissociation, since its size may exceed the average diffusion lengths of exciton. As shown in Fig. 6(b–d), after thermal annealing at 100 °C for 10 min, the Rq is raised to 0.42 nm for polymer PIDTT-DTBO. Some tiny visible domains appears in its blend film, suggesting the polymer affords better intermixing with PC71BM, which corresponds to the obvious improvement (5%) of device efficiency. And the post-thermal treatment slightly increase the Rq to 0.40 nm for polymer PIDTT-DTBT, which may attribute to the slightly stronger aggregation and thus leading to the improved device performance.
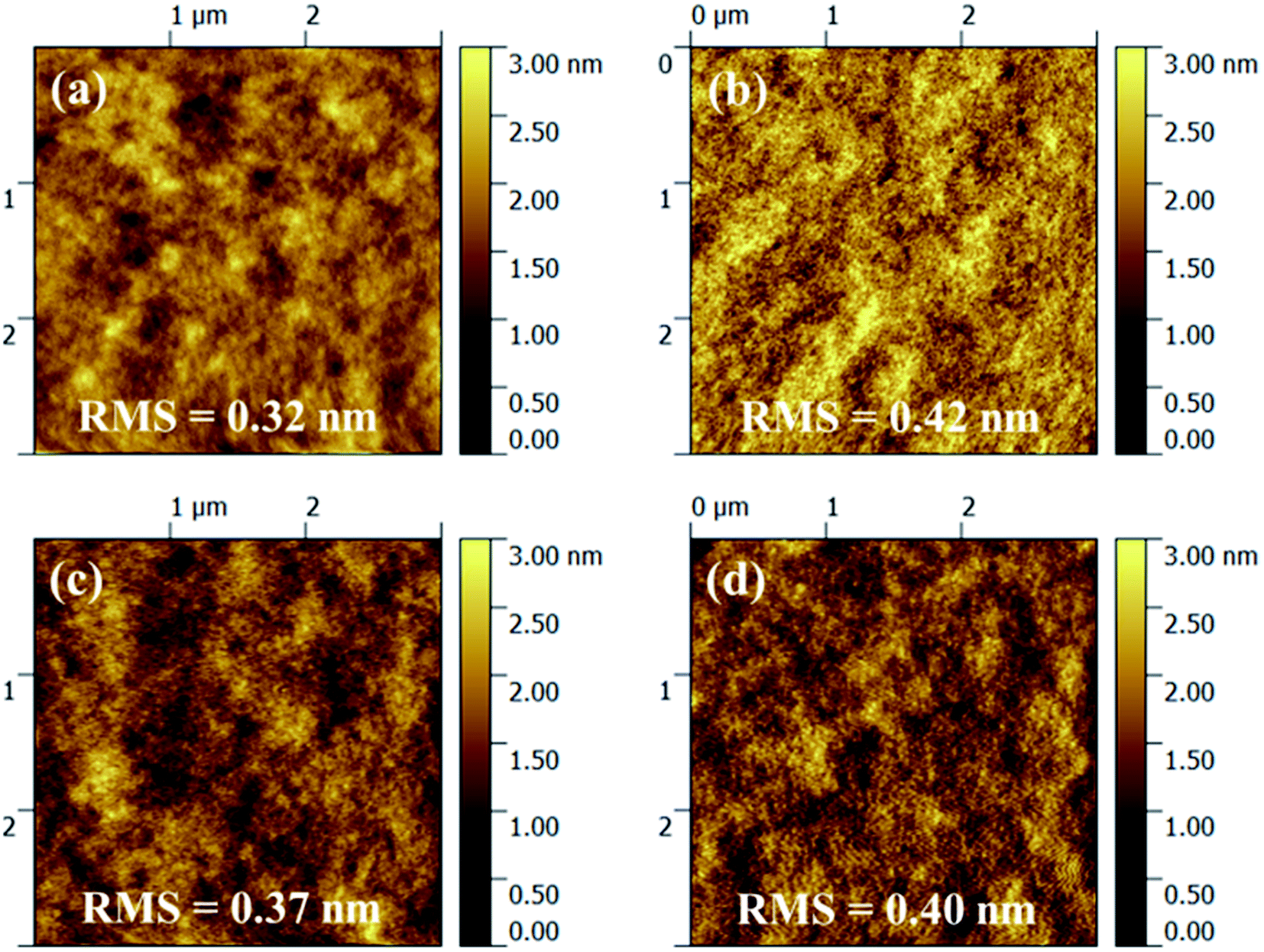 | ||
| Fig. 6 Tapping mode AFM topography images (3 × 3 μm2) of polymer\PC71BM blend films, (a) PIDTT-DTBO\PC71BM before annealing. (b) PIDTT-DTBO\PC71BM after annealing at 100 °C for 10 min (c) PIDTT-DTBT\PC71BM before annealing. (d) PIDTT-DTBT\PC71BM after annealing at 100 °C for 10 min. | ||
Hole mobility
As shown in Fig. 7, the hole mobility of polymer/PC71BM blend films were investigated using the space charge limited current (SCLC) method. The structure of hole-only device is ITO/PEDOT:PSS/polymer:PC71BM (85 nm)/MoO3/Al. After thermal annealing at 100 °C for 10 min, the calculated mobility of polymer PIDTT-DTBO-based device is 9.2 × 10−5 cm2 V−1 s−1, while a higher hole mobility of 1.1 × 10−4 cm2 V−1 s−1 is recorded for polymer PIDTT-DTBT-based device. The better hole-transporting property of polymer PIDTT-DTBT would benefit more balanced hole and electron transport in the polymer/PC71BM blend layer. Since the FF value of PSC is positively correlated to the balance of hole and electron transport, the calculated mobility are consistent with the device results, from which polymer PIDTT-DTBT-based devices show higher FF in comparison with those based on polymer PIDTT-DTBO. In addition, the hole mobilities of PIDTT-DTBO and PIDTT-DTBT are lower than that of PIDTT-Qx (2.9 × 10−4 cm2 V−1 s−1) from the previous report, which can attribute to the relatively inferior PCEs of the solar cells based on PIDTT-DTBO and PIDTT-DTBT.39 Among the high-performing donor polymers in the current reports, the hole mobilities of PIDTT-DTBO and PIDTT-DTBT are on the low side.13,14,18,58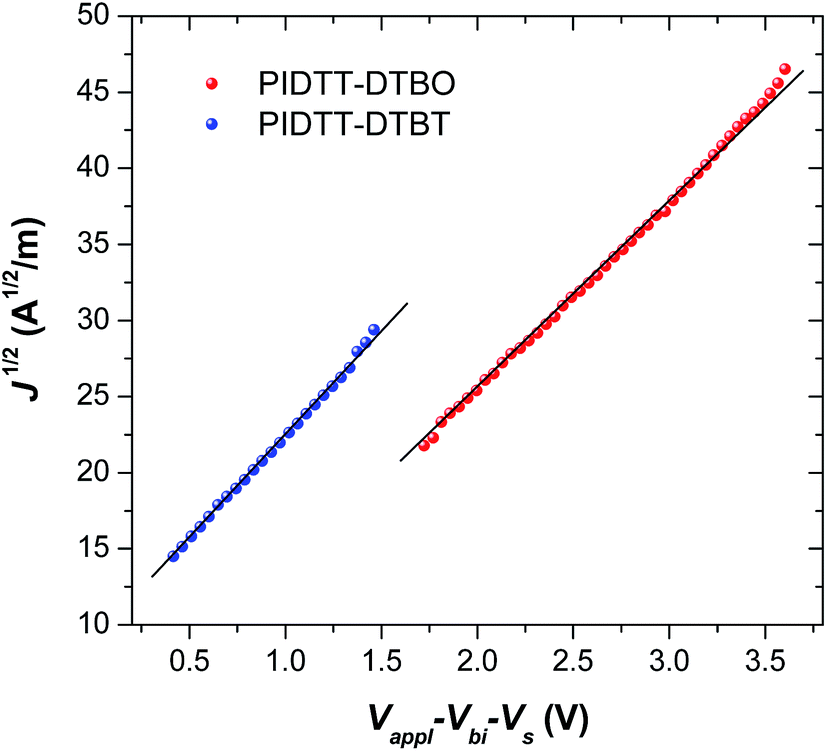 | ||
| Fig. 7 The J1/2–V characteristics of polymer PIDTT-DTBO and PIDTT-DTBT-based hole-only devices. | ||
Conclusions
In summary, three new ladder-type conjugated polymers PIDTT-DTBO, PIDTT-DTBT and PIDTT-DTFBT have been synthesized and well characterized to understand the effects of inclusive electronegative atoms (O, S and F) on properties of IDTT-based polymers. The extended π-conjugation of polymer backbones were formed with electron-rich IDTT unit and electron-poor BO, BT and FBT units. The donor and acceptor portion coupled via two bis-thiophenes appear to afford a high level of coplanar D–π–A conjugation. As expected, the strong electron-withdrawing atoms on acceptor moieties can affect the processing, optical, electrochemical properties and dipole moments of IDTT-based polymers in different ways. After introduction of the most electronegative fluorine atoms onto BT unit, intensely decreased solubility of polymer PIDTT-DTFBT is observed. On the contrary, polymer PIDTT-DTBO and PIDTT-DTBT display good solubility, sufficient thermal stability, low band-gaps and deep HOMO levels. Compared to BT unit, the stronger electron-withdrawing BO unit can afford net red-shifted absorption, more stable HOMO and LUMO levels for polymer PIDTT-DTBO. In contrast, after thermal annealing, polymer PIDTT-DTBT depicts a slight ascent of surface roughness, more efficient charge collection and higher hole mobility to maintain the balance of charge transport. As a result, BHJ PSCs incorporating blends of polymer PIDTT-DTBT with PC71BM have provided reasonably good PCE of 4.9%, while a PCE of 4.6% is noted in polymer PIDTT-DTBO-based devices. The results attest the inclusive electronegative atoms (O, S and F) in acceptor portion can tailor the properties of IDTT-based polymers in many different ways, thus affect the final device performances of PSCs. Through judicial selection of electronegative atoms and structural optimization in acceptor moieties, rational synthesis of more performing conjugated polymers is underway to drive the photovoltaic efficiency to economically viable levels.Acknowledgements
The authors gratefully acknowledge the financial support of the Guangxi Natural Science Foundation (2016GXNSFBA380129), the Department of Education of Guangxi (ky2016YB146), the PhD research startup foundation of Guilin university of Electronic Technology (UF15011Y), and the EU projects SUNFLOWER (FP7-ICT-2011-7, Grant number: 287594).References
- F. C. Krebs, S. A. Gevorgyan and J. Alstrup, J. Mater. Chem., 2009, 19, 5442–5451 RSC.
- N. Espinosa, M. Hösel, D. Angmob and F. C. Krebs, Energy Environ. Sci., 2012, 5, 5117–5132 CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS.
- M. Wang, X. W. Hu, P. Liu, W. Li, X. Gong, F. Huang and Y. Cao, J. Am. Chem. Soc., 2011, 133, 9638–9641 CrossRef CAS PubMed.
- E. G. Wang, Z. F. Ma, Z. Zhang, K. Vandewal, P. Henriksson, O. Inganäs, F. L. Zhang and M. R. Andersson, J. Am. Chem. Soc., 2011, 133, 14244–14247 CrossRef CAS PubMed.
- K. H. Ong, S. L. Lim, H. S. Tan, H. K. Wong, J. Li, Z. Ma, L. C. H. Moh, S. H. Lim, J. C. de Mello and Z. K. Chen, Adv. Mater., 2011, 23, 1409–1413 CrossRef CAS PubMed.
- C. E. Small, S. Chen, J. Subbiah, C. M. Amb, S. W. Tsang, T. H. Lai, J. R. Reynolds and F. So, Nat. Photonics, 2012, 6, 115–120 CrossRef CAS.
- Z. C. He, C. M. Zhong, S. J. Su, M. Xu, H. B. Wu and Y. Cao, Nat. Photonics, 2012, 6, 591–595 Search PubMed.
- A. C. Stuart, J. R. Tumbleston, H. X. Zhou, W. T. Li, S. B. Liu, H. Ade and W. You, J. Am. Chem. Soc., 2013, 135, 1806–1815 CrossRef CAS PubMed.
- H. L. Zhong, Z. Li, F. Deledalle, E. C. Fregoso, M. Shahid, Z. P. Fei, C. B. Nielsen, N. Yaacobi-Gross, S. Rossbauer, T. D. Anthopoulos, J. R. Durrant and M. Heeney, J. Am. Chem. Soc., 2013, 135, 2040–2043 CrossRef CAS PubMed.
- Y. Wu, Z. J. Li, W. Ma, Y. Huang, L. J. Huo, X. Guo, M. J. Zhang, H. Ade and J. H. Hou, Adv. Mater., 2013, 25, 3449–3455 CrossRef CAS PubMed.
- Z. H. Chen, P. Cai, J. W. Chen, X. C. Liu, L. J. Zhang, L. F. Lan, J. B. Peng, Y. G. Ma and Y. Cao, Adv. Mater., 2014, 26, 2586–2591 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- J. D. Chen, C. H. Cui, Y. Q. Li, L. Zhou, Q. D. Ou, C. Li, Y. F. Li and J. X. Tang, Adv. Mater., 2015, 27, 1035–1041 CrossRef CAS PubMed.
- S. Zhang, L. Ye and J. Hou, Adv. Energy Mater., 2016, 1502529 CrossRef.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- J. W. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709–1718 CrossRef CAS PubMed.
- Y. J. Cheng, S. H. Yang and C. S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- Y. F. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed.
- H. X. Zhou, L. Q. Yang and W. You, Macromolecules, 2012, 45, 607–632 CrossRef CAS.
- H. X. Zhou, L. Q. Yang, A. C. Stuart, S. C. Price, S. B. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- S. C. Price, A. C. Stuart, L. Q. Yang, H. X. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625–4631 CrossRef CAS PubMed.
- A. C. Stuart, J. R. Tumbleston, H. X. Zhou, W. T. Li, S. B. Liu, H. Ade and W. You, J. Am. Chem. Soc., 2013, 135, 1806–1815 CrossRef CAS PubMed.
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed.
- C. P. Chen, S. H. Chan, T. C. Chao, C. Ting and B. T. Ko, J. Am. Chem. Soc., 2008, 130, 12828–12833 CrossRef CAS PubMed.
- T. W. Lee, N. S. Kang, J. W. Yu, M. H. Hoang, K. H. Kim, J. I. Jin and D. H. Cho, J. Polym. Sci. Part A: Polym. Chem., 2010, 48, 5921–5929 CrossRef CAS.
- C. Y. Chang, Y. J. Cheng, S. H. Hung, J. S. Wu, W. S. Kao, C. H. Lee and C. S. Hsu, Adv. Mater., 2012, 24, 549–553 CrossRef CAS PubMed.
- H. Bronstein, R. S. Ashraf, Y. Kim, A. J. P. White, T. Anthopoulos, K. Song, D. James, W. Zhang and I. McCulloch, Macromol. Rapid Commun., 2011, 32, 1664–1668 CrossRef CAS PubMed.
- J. S. Wu, Y. J. Cheng, T. Y. Lin, C. Y. Chang, P. I. Shih and C. S. Hsu, Adv. Funct. Mater., 2012, 8, 1711–1722 CrossRef.
- Y. J. Cheng, S. W. Cheng, C. Y. Chang, W. S. Kao, M. H. Liao and C. S. Hsu, Chem. Commun., 2012, 48, 3203–3205 RSC.
- Y. L. Chen, C. Y. Chang, Y. J. Cheng and C. S. Hsu, Chem. Mater., 2012, 24, 3964–3971 CrossRef CAS.
- C. P. Chen and H. L. Hsu, Macromol. Rapid Commun., 2013, 34, 1623–1628 CrossRef CAS PubMed.
- X. Guo, M. J. Zhang, J. H. Tan, S. Q. Zhang, L. J. Huo, W. P. Hu, Y. F. Li and J. H. Hou, Adv. Mater., 2012, 24, 6536–6541 CrossRef CAS PubMed.
- Q. P. Fan, Y. Liu, H. X. Jiang, W. Y. Su, L. R. Duan, H. Tan, Y. Y. Li, J. Y. Deng, R. Q. Yang and W. G. Zhu, Organic. Electron., 2016, 33, 128–134 CrossRef CAS.
- Q. P. Fan, W. Y. Su, X. Guo, X. Zhang, Z. Xu, B. Guo, L. Jiang, M. J. Zhang and Y. F. Li, J. Mater. Chem. A, 2017, 5, 5106–5114 CAS.
- M. J. Zhang, X. Guo, X. C. Wang, H. Q. Wang and Y. F. Li, Chem. Mater., 2011, 23, 4264–4270 CrossRef CAS.
- X. F. Xu, P. Cai, Y. Lu, S. C. Ng, J. W. Chen, B. S. Ong and X. Hu, Macromol. Rapid Commun., 2013, 34, 681–688 CrossRef CAS PubMed.
- X. F. Xu, Z. J. Li, O. Bäcke, K. Bini, D. I. James, E. Olsson, M. R. Andersson and E. G. Wang, J. Mater. Chem. A, 2014, 2, 18988–18997 CAS.
- Y. Zhang, J. Y. Zou, H. L. Yip, K. S. Chen, D. F. Zeigler, Y. Sun and A. K. Y. Jen, Chem. Mater., 2011, 23, 2289–2291 CrossRef CAS.
- M. Wang, X. W. Hu, L. Q. Liu, C. H. Duan, P. Liu, L. Ying, F. Huang and Y. Cao, Macromolecules, 2013, 46, 3950–3958 CrossRef CAS.
- R. S. Ashraf, B. C. Schroeder, H. A. Bronstein, Z. G. Huang, S. Thomas, R. J. Kline, C. J. Brabec, P. Rannou, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Adv. Mater., 2013, 25, 2029–2034 CrossRef CAS PubMed.
- W. M. Zhang, J. Smith, S. E. Watkins, R. Gysel, M. McGehee, A. Salleo, J. Kirkpatrick, S. Ashraf, T. Anthopoulos, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2010, 132, 11437–11439 CrossRef CAS PubMed.
- H. H. Chang, C. E. Tsai, Y. Y. Lai, D. Y. Chiou, S. L. Hsu, C. S. Hsu and Y. J. Cheng, Macromolecules, 2012, 45, 9282–9291 CrossRef CAS.
- Y. X. Xu, C. C. Chueh, H. L. Yip, F. Z. Ding, Y. X. Li, C. Z. Li, X. S. Li, W. C. Chen and A. K. Y. Jen, Adv. Mater., 2012, 24, 6356–6361 CrossRef CAS PubMed.
- Y. X. Xu, C. C. Chueh, H. L. Yip, C. Y. Chang, P. W. Liang, J. J. Intemann, W. C. Chen and A. K. Y. Jen, Polym. Chem., 2013, 4, 5220–5223 RSC.
- Y. Z. Lin, J. Y. Wang, Z.-G. Zhang, H. T. Bai, Y. F. Li, D. B. Zhu and X. W. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- Y. K. Yang, Z.-G. Zhang, H. J. Bin, S. S. Chen, L. Gao, L. W. Xue, C. D. Yang and Y. F. Li, J. Am. Chem. Soc., 2016, 138, 15011–15018 CrossRef CAS PubMed.
- L. Pandey, C. Risko, J. E. Norton and J. L. Brédas, Macromolecules, 2012, 45, 6405–6414 CrossRef CAS.
- Z. Li, J. P. Lu, S. C. Tse, J. Y. Zhou, X. M. Du, Y. Tao and J. F. Ding, J. Mater. Chem., 2011, 21, 3226–3233 RSC.
- J. F. Jheng, Y. Y. Lai, J. S. Wu, Y. H. Chao, C. L. Wang and C. S. Hsu, Adv. Mater., 2013, 25, 2445–2451 CrossRef CAS PubMed.
- B. Carsten, J. M. Szarko, H. J. Son, W. Wang, L. Y. Lu, F. He, B. S. Rolczynski, S. J. Lou, L. X. Chen and L. P Yu, J. Am. Chem. Soc., 2011, 133, 20468–20475 CrossRef CAS PubMed.
- C. J. Takacs, Y. M. Sun, G. C. Welch, L. A. Perez, X. F. Liu, W. Wen, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2012, 134, 16597–16606 CrossRef CAS PubMed.
- C. Risko, M. D. McGeheeb and J. L. Brédas, Chem. Sci., 2011, 2, 1200–1218 RSC.
- H. Bronstein, J. M. Frost, A. Hadipour, Y. J. Kim, C. B. Nielsen, R. S. Ashraf, B. P. Rand, S. Watkins and I. McCulloch, Chem. Mater., 2013, 25, 277–285 CrossRef CAS.
- Z. C. He, C. Zhang, X. F. Xu, L. J. Zhang, L. Huang, J. W. Chen, H. B. Wu and Y. Cao, Adv. Mater., 2011, 23, 3086–3089 CrossRef CAS PubMed.
- P. Cai, Z. H. Chen, L. J. Zhang, J. W. Chen and Y. Cao, J. Mater. Chem. C, 2017, 5, 2786–2793 RSC.
| This journal is © The Royal Society of Chemistry 2017 |