
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Challenging cyclopropanation reactions on non-activated double bonds of fatty esters†
B. Angulo ,
J. M. Fraile*,
C. I. Herrerías* and
J. A. Mayoral
,
J. M. Fraile*,
C. I. Herrerías* and
J. A. Mayoral
Departamento de Catálisis y Procesos Catalíticos, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, Facultad de Ciencias, C/Pedro Cerbuna 12, 50009 Zaragoza, Spain. E-mail: jmfraile@unizar.es; clarah@unizar.es
First published on 31st March 2017
Abstract
Monounsaturated fatty esters with non-activated double bonds have been made to react with two different diazocompounds, ethyl diazoacetate (EDA) and dimethyl diazomalonate (DDM), in copper and rhodium catalyzed cyclopropanation reactions. The corresponding cyclopropanes have been obtained with quantitative or nearly quantitative yields (>99%) in all the cases, with only the exception of the reaction of methyl elaidate, an E alkene, with the less reactive DDM. For the first time, the two stereoisomers of ethyl (2R*,3S*)-2-(7-methoxycarbonyl)heptyl-3-octylcyclopropane-1-carboxylate, obtained from the reaction of methyl oleate and EDA, have been separated and fully characterized. Finally, the diesters and triesters with cyclopropane structure have been reduced to diols and triols, and hydrolyzed to the corresponding diacids and triacids, to demonstrate the potential usefulness of this methodology to prepare monomers from renewable sources.
Introduction
Vegetal oils and animal fats are among the most important renewable feedstocks in the chemical industry. Although classical oleochemical transformations occur preferentially at the ester functionality of the triglycerides,1 modern synthetic methods have been recently applied to modify the alkyl chain.2 In this regard, catalytic C–C bond forming reactions on unsaturated compounds have been used to modify the size and the structure of the chains of natural fatty derivatives.3 Cyclopropane fatty acids are present in nature,4 and their synthesis has been usually tackled by construction of the alkyl chains from the pre-formed cyclopropane ring.5,6 The Simmons–Smith reaction has been also described for this purpose,7 but surprisingly, the catalytic cyclopropanation of carbon–carbon double bonds has been scarcely explored. Only in one case unsaturated fatty esters were made react with diazomethane in a Pd catalyzed cyclopropanation,8 whereas one single example was described for the copper catalyzed cyclopropanation with ethyl diazoacetate9 and another one using diazoketones.10 In fact, the resulting difunctional compounds of the latter reactions may open the way to further modifications for different uses. However, the articles describing the preparation of these functionalised cyclopropanes9,10 were published 50 years ago and hence both the methods for determination of results and, specially, the characterization of the products were limited or very deficient.On the other hand, vegetable oils have been used as such, or after suitable modifications, to produce an impressive variety of macromolecular materials through different alternative polymerization mechanisms, leading to a great number of structures with different properties and promising applications.11–13 With this aim, several methods for the synthesis of diacids from fatty acid derivatives have been described in the literature.14,15 Generally these monomers, due to their longer alkyl chains, impart certain properties in the resulting polymers, such as elasticity, flexibility, high impact strength, hydrolytic stability, hydrophobicity or lower transition temperatures.15
Given the extended use of elastomers, it is interesting to develop bio-based materials; regarding polymer properties, the inherent internal position of the functionalities has great interest in the preparation of soft materials because the alkyl dangling chains act as internal plasticizers. With this aim castor oil, containing 85–90% of easily polymerizable ricinoleic acid, has attracted much attention.16,17 The scarcely explored cyclopropanation with diazoesters would allow the straightforward synthesis of di- or polyesters with well-defined geometries and bearing dangling alkyl chains. Furthermore, the selective transformation of the ester functionalities opens the way to a plethora of new compounds with different geometries and functionalities.
Our research group has wide experience in the cyclopropanation reaction between olefins and diazoesters promoted by different kind of copper catalytic systems.18–23 Therefore, we decided to apply this knowledge in carrying out the cyclopropanation reaction over non-activated double bonds of different unsaturated fatty esters using copper and rhodium salts as catalysts.
In this manuscript, we describe for the first time, the synthesis, characterization and isolation of several di- and tri-esters resulting from the reaction between diazoesters and fatty acid methyl esters, as well as some simple transformations of the ester groups.
Results and discussion
Copper catalysed cyclopropanation of methyl oleate with ethyl diazoacetate (EDA)
The cyclopropanation of methyl oleate (1) with EDA (Scheme 1) was chosen as test reaction to optimize the conditions, namely solvent, type of catalyst, amount of catalyst, EDA amount and addition method, temperature and time.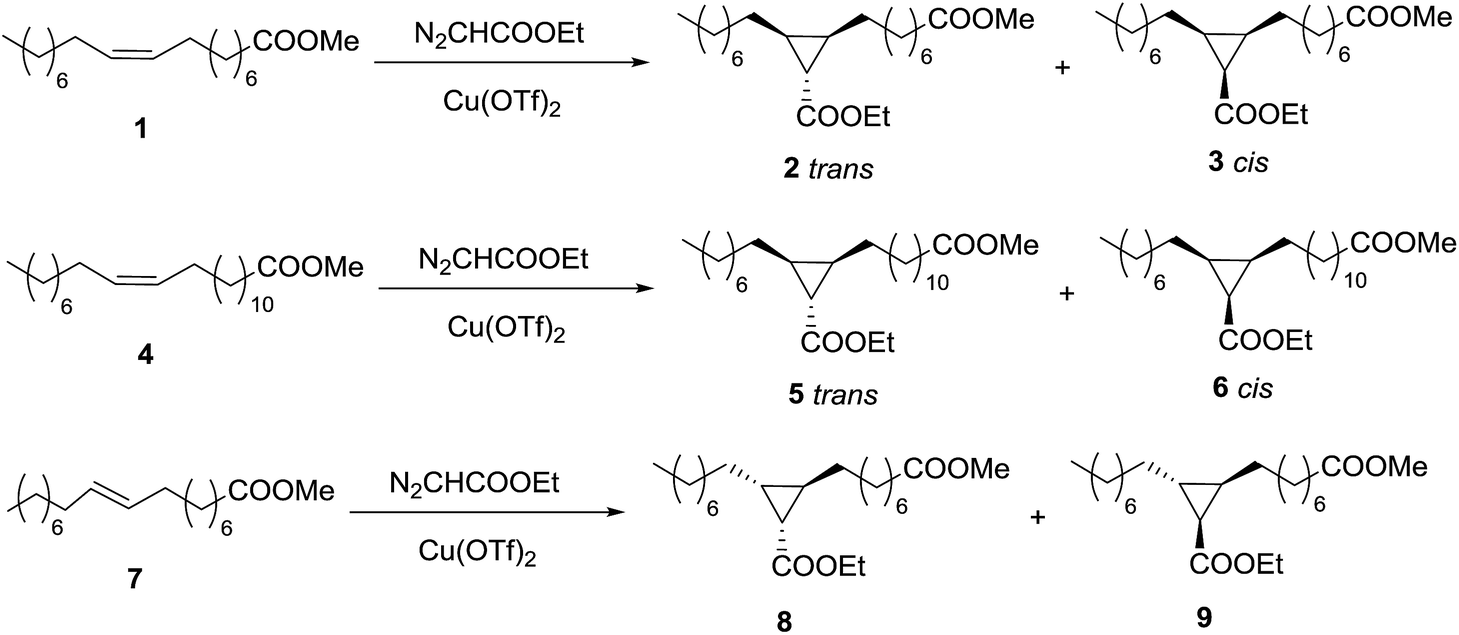 | ||
| Scheme 1 Cyclopropanation reactions of methyl oleate (1), methyl erucate (4), and methyl elaidate (7) with EDA. | ||
The results obtained with Cu(OTf)2 are gathered in Table 1. Initially the stoichiometric amount of EDA was used with 1 mol% Cu(OTf)2 as catalyst, and different solvents were tested in a 24 h period (entries 1–5). At room temperature, the best result (23% yield) was obtained with dichloromethane (entry 1), whereas toluene and THF (entries 2–3) led to lower yields with nearly the same trans/cis selectivity (2/3 = 68/32), typical for this kind of reaction. The increase in reaction temperature (entries 4–5) up to 110 °C did not improve the results. A larger amount of catalyst (5 mol%, entry 6) did not produce any positive effect either. Under the best conditions, the reaction was shown to proceed at longer reaction times, with a maximum of 36% yield after 48 h (entry 7).
| Entry | EDA/oleate | EDA addition (h) | mol% Cu(OTf)2 | T (°C) | Solvent | Timeb (h) | Yieldc (%) | trans/cisc |
|---|---|---|---|---|---|---|---|---|
| a Reagents and conditions: methyl oleate (2.5 mmol), ethyl diazoacetate (EDA), Cu(OTf)2, solvent (2 mL).b Total reaction time (including addition time).c Determined by gas chromatography.d 1,2-DCE = 1,2-dichloroethane. | ||||||||
| 1 | 1/1 | 0 | 1 | 25 | CH2Cl2 | 24 | 23 | 68/32 |
| 2 | 1/1 | 0 | 1 | 25 | Toluene | 24 | 8 | 66/34 |
| 3 | 1/1 | 0 | 1 | 25 | THF | 24 | 5 | 69/31 |
| 4 | 1/1 | 0 | 1 | 60 | 1,2-DCEd | 24 | 16 | 67/33 |
| 5 | 1/1 | 0 | 1 | 110 | Toluene | 24 | 13 | 66/34 |
| 6 | 1/1 | 0 | 5 | 25 | CH2Cl2 | 24 | 18 | 70/30 |
| 7 | 1/1 | 0 | 1 | 25 | CH2Cl2 | 48 | 36 | 67/33 |
| 8 | 1/1 | 10 | 1 | 25 | CH2Cl2 | 24 | 34 | 62/38 |
| 9 | 2/1 | 10 | 1 | 25 | CH2Cl2 | 24 | 71 | 67/33 |
| 10 | 3/1 | 6 | 1 | 25 | CH2Cl2 | 24 | 98 | 65/35 |
| 11 | 3/1 | 10 | 1 | 25 | CH2Cl2 | 15 | >99 | 68/32 |
| 12 | 3/1 | 10 | 1 | 25 | None | 24 | 71 | 64/36 |
| 13 | 4/1 | 20 | 1 | 25 | None | 24 | 91 | 64/36 |
| 14 | 6/1 | 20 | 1 | 25 | None | 24 | >99 | 64/36 |
Given the total conversion of EDA, this value represents the chemoselectivity of the reaction with respect to the competitive dimerization of EDA to diethyl fumarate and maleate. The typical method to optimize this chemoselectivity is the slow addition of the diazocompound. When EDA was slowly added during 10 h, the same maximum yield was obtained at shorter reaction time (entry 8). Considering the importance of EDA dimerization, the amount of diazocompound was increased (entries 9–11), and quantitative yield of cyclopropanes 2 + 3 was obtained with an EDA/oleate molar ratio of 3.
As dichloromethane is not a good solvent from the point of view of sustainability, the reaction was also tested in the absence of solvent. Under the same conditions, the yield was only 71%, with similar diastereoselectivity (entry 12). The further addition of EDA led to a progress in the reaction, indicating that the catalyst was still active and the lower yield was a problem of chemoselectivity. In fact, when the EDA/oleate molar ratio was increased to 4/1 (entry 13), over 90% yield was obtained, whereas the quantitative conversion of oleate was reached with 6 equivalents of EDA (entry 14). This lower chemoselectivity in the absence of solvent is probably due to a poor solubility of the copper catalyst in the non-polar methyl oleate.
Given the possible advantages of heterogeneous catalysts in easy separation and reuse, a solid catalyst was prepared by cation exchange of Cu(OTf)2 in LAPONITE®.24 This synthetic clay has sodium cations in the interlayer space that are exchangeable in a polar medium. The copper content of LAPONITE®-Cu was 0.48 mmol g−1, and this solid was tested in the optimal conditions found in homogeneous phase (Table 2). As can be seen, the first run of the catalyst led to analogous results to the homogeneous counterpart (93% yield, trans/cis 68/32), but after filtration and washing with dichloromethane, the catalyst showed very low catalytic activity (entry 2). The side reactions of EDA, dimerization and oligomerization, produce coordinating by-products able to poison the copper sites of LAPONITE®. Thus, the solid recovered in another run, with reproducible results (entry 3 vs. entry 1), was thoroughly washed with acetonitrile. The catalytic activity was recovered in larger extension (entry 4), but the catalyst is not fully recoverable. In previous works, we have observed an effect of LAPONITE® on the stereoselectivity of the cyclopropanation reactions,25 favouring the less stable cis-isomer, and this effect was enhanced by the use of a non-polar solvent. However, when the reaction was carried out in hexane (entry 5), yield was very low and no changes in the diastereoselectivity were observed. It was possible to reach a quantitative yield under reflux, but the trans/cis selectivity remained unchanged. This is probably due to the highly flexible hydrocarbon chains, able to accommodate in the close proximity of the clay surface, in contrast with the rigid structure of styrenes or cycloalkenes tested until now.26 Finally, the solventless conditions were also tested, with the expected negative effect on the catalytic activity. In view of the lack of advantages in using LAPONITE®-Cu, the rest of cyclopropanation reactions were only tested with Cu(OTf)2.
| Entry | Run | Solvent | Yieldb (%) | trans/cisb |
|---|---|---|---|---|
| a Reagents and conditions: methyl oleate (2.5 mmol), ethyl diazoacetate (EDA) (7.5 mmol, 10 h addition), LAPONITE®-Cu (1 mol%), solvent (2 mL), room temperature, 24 h.b Determined by gas chromatography.c Catalyst recovered by centrifugation and washing with CH3CN prior to use.d Reaction carried out under reflux. | ||||
| 1 | 1 | CH2Cl2 | 93 | 68/32 |
| 2 | 2 | CH2Cl2 | 8 | 68/32 |
| 3 | 1 | CH2Cl2 | 91 | 69/31 |
| 4 | 2c | CH2Cl2 | 34 | 69/31 |
| 5 | 1 | Hexane | 5 | 65/35 |
| 6 | 1d | Hexane | 99 | 69/31 |
| 7 | 1 | None | 31 | 69/31 |
Initially, the two diastereomers were assigned as trans-2 and cis-3 from mechanistic considerations. However, it was necessary to confirm the stereochemistry of both compounds. In order to carry out their characterization, firstly, they were separated by flash-chromatography. The only signal that enabled the structural elucidation was the CH–COOEt in the cyclopropane, which would be coupled to the other two protons of the ring with different coupling constant depending on the relative position. The CH groups in both molecules were identified on the basis of the HSQC NMR spectra in CDCl3, but the signal was overlapped with those of the alkyl chain in one of the isomers, and the triplet in the other one was quite large and poorly defined (see ESI†). Thus, other deuterated solvents were tried in an attempt to distinguish those signals and benzene-d6 was shown to be the most suitable solvent. The key signal (Fig. 1) was clearly separated in the cis-3 isomer, whereas it was overlapped in the trans-2 isomer, although the three peaks were easily detectable and the coupling constant measurable. In that way, it was possible to confirm the stereochemistry of the two compounds, as the J coupling constant was 8.7 Hz in the case of cis-3 and 4.5 Hz in the trans-2.27,28
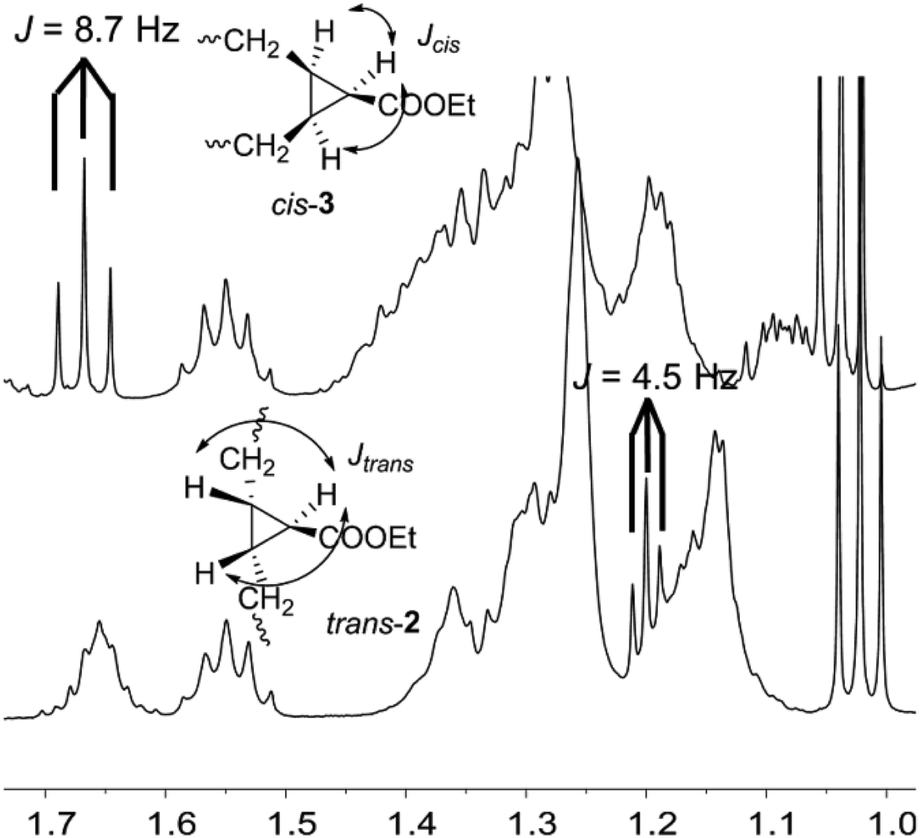 | ||
| Fig. 1 1H NMR spectra (benzene-d6) in the range of 1.8–0.9 ppm of cis-3 and trans-2 cyclopropanes. | ||
Copper catalyzed cyclopropanation of methyl erucate with ethyl diazoacetate (EDA)
Given the good result obtained with methyl oleate, we decided to apply this methodology to the cyclopropanation of different unsaturated fatty esters. As first step, a larger cis monounsaturated fatty ester, methyl erucate (4) (C22:1 cis-Δ13) was tested (Scheme 1).Under the optimized conditions, EDA/erucate molar ratio = 3, EDA slowly added during 10 h, 1 mol% Cu(OTf)2, CH2Cl2 as solvent, room temperature, 15 h total reaction time, total conversion of methyl erucate to the trans-5 and cis-6 cyclopropanes was obtained, with a trans/cis selectivity of 69/31. Both diastereomers were separated and characterized by mass spectrometry and NMR using CDCl3 and bezene-d6 as solvents (see ESI†).
Copper catalysed cyclopropanation of methyl elaidate with ethyl diazoacetate (EDA)
The next variation in the substrate was the stereochemistry of the C–C double bond. Methyl elaidate (7) (C18:1 trans-Δ9) was made react with EDA (Scheme 1) under the optimized conditions found for methyl oleate. As expected, two products (8 and 9) were detected in a 50/50 ratio, but yield was only 84% after 15 h. The relative cis position of one of the long chains with the ester group of the copper–carbene in every transition state explains the lower reactivity of this alkene and the lack of selectivity. The increase of the EDA/elaidate molar ratio to 4 led to the desired quantitative yield. However, it was not possible to separate the diastereomers and furthermore, the 1H NMR signal used for identification in the case of 2 and 3 would not be useful in this case. In both isomers, 8 and 9, the CH–COOEt has one H in relative position cis and another one in relative position trans, showing then both a doublet–doublet with the same J coupling constants. Therefore, it was not possible to identify the two compounds separately but we did the characterization of the equimolecular mixture.Cyclopropanation of methyl fatty esters with EDA catalysed by Rh2(OAc)4
Rhodium is the most commonly used catalyst for cyclopropanation reactions,29,30 and hence Rh2(OAc)4 was tested as catalyst in the three reactions described above. The results are summarized in Table 3. The reactions were first carried out under the optimized conditions found for Cu(OTf)2 (entries 1, 3 and 4). In the case of the Z alkenes (oleate and erucate), quantitative yields were obtained with slightly lower selectivity to the trans cyclopropane. With the E alkene (elaidate, entry 4), a slight preference for one of the unidentified diastereomers was observed. The excess of EDA could be decreased up to 2/1 with methyl oleate and 3/1 with methyl elaidate to obtain the same final yields, showing that rhodium leads to a slightly better chemoselectivity for cyclopropanation with respect to the side reactions of EDA (dimerization and oligomerization). As the advantages of rhodium are marginal in comparison with the higher price compared with copper, the rest of the work was carried out with Cu(OTf)2 as catalyst.| Entry | Fatty ester | EDA/fatty ester | Timeb (h) | Yieldc (%) | trans/cisc |
|---|---|---|---|---|---|
| a Reagents and conditions: fatty ester (2.5 mmol), ethyl diazoacetate (EDA) slowly added (10 h), Rh2(OAc)4 (1 mol%), CH2Cl2 (2 mL), room temperature.b Total reaction time.c Determined by gas chromatography.d Diastereomers (unidentified) ratio. | |||||
| 1 | Methyl oleate (1) | 3/1 | 15 | >99 | 60/40 |
| 2 | Methyl oleate (1) | 2/1 | 15 | 97 | 60/40 |
| 3 | Methyl erucate (4) | 3/1 | 24 | >99 | 65/35 |
| 4 | Methyl elaidate (7) | 4/1 | 15 | 93 | 54/46d |
| 5 | Methyl elaidate (7) | 3/1 | 15 | 93 | 55/45d |
Cyclopropanation of methyl oleate (1) with dimethyl diazomalonate (DDM)
In the cyclopropanations described above, the final products are diesters with different geometry in the cyclopropane ring or different chain length, which can be envisaged as monomers for linear polymers. However, the use of diazomalonate would lead to the formation of triesters that could play the role of cross-linkers. Thus, dimethyl diazomalonate (DDM) was used as carbenoid source in the cyclopropanation of methyl oleate (Scheme 2). The results are gathered in Table 4.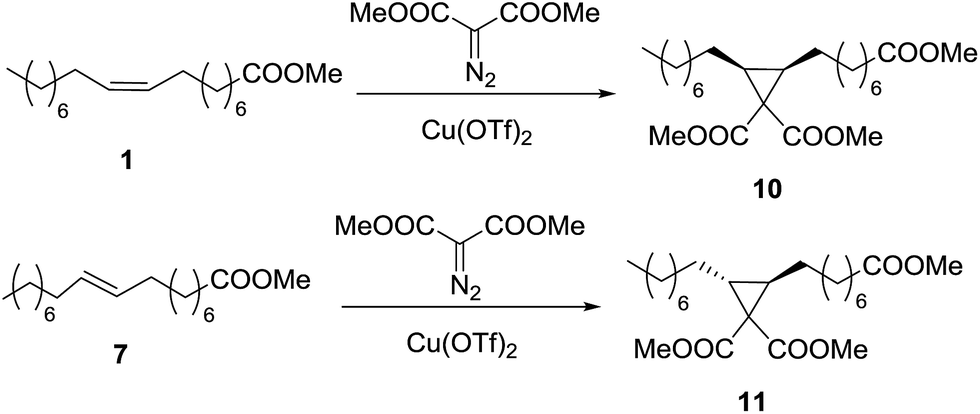 | ||
| Scheme 2 Cyclopropanation reactions of methyl oleate (1) and methyl elaidate (7) with DDM. | ||
| Reaction # | T (°C) | DDM/oleate | % mol Cu(OTf)2 | Time (h) | Yieldb (%) |
|---|---|---|---|---|---|
| a Reagents and conditions: methyl oleate (2.5 mmol), dimethyl diazomalonate (DDM) in a single addition, Cu(OTf)2, 1,2-dichloroethane (2 mL).b Determined by gas chromatography.c Using CH2Cl2 as solvent. | |||||
| 1c | 25 | 3/1 | 1 | 24 | 1 |
| 2 | 65 | 3/1 | 1 | 24 | 50 |
| 96 | 67 | ||||
| 144 | 67 | ||||
| 2 | 168 | 80 | |||
| 192 | 86 | ||||
| 3 | 264 | 86 | |||
| 3 | 65 | 3/1 | 1 | 7 | 43 |
| 2 | 9 | 72 | |||
| 3 | 24 | 72 | |||
| 4/1 | 3 | 30 | 72 | ||
| 4 | 48 | 85 | |||
| 4 | 85 | 4/1 | 1 | 5 | 59 |
| 2 | 24 | 77 | |||
| 3 | 30 | 77 | |||
| 5 | 85 | 5/1 | 1 | 6 | 80 |
| 6/1 | 2 | 24 | >99 | ||
| 6 | 85 | 6/1 | 1 | 4 | 47 |
| 6 | 56 | ||||
| 7/1 | 2 | 24 | 81 | ||
The presence of two electron withdrawing groups in DDM makes this diazocompound less reactive than EDA,31 and the reaction does not take place at room temperature. On the contrary, when the reaction is carried out at 65 °C (reaction 2), it proceeds with moderate yield, up to 67% with 3 equivalents of DDM, in a quite long time. The addition of another 1 mol% of catalyst increased the yield up to 86%, showing that there was a problem of catalyst deactivation. The reaction can be speeded up with successive additions of catalyst and one more equivalent of DDM, reaching 85% yield in only 48 h (reaction 3).
However, the result is not improved by increasing the reaction temperature (reaction 4). The yield limit is then a combination of catalyst deactivation, probably due to the chelating character of both DDM and cyclopropane 10, and the low yield of the reaction, due to the low reactivity of DDM. Quantitative yield can be obtained with 6/1 DDM/oleate molar ratio and 2 mol% of Cu(OTf)2 (reaction 5), but from results in reaction 6 it can be seen that it is better to introduce both diazocompound and catalyst in portions.
Cyclopropanation of methyl elaidate (7) with dimethyl diazomalonate (DDM)
As expected from the lower reactivity of methyl elaidate (7) observed with EDA, the cyclopropanation with DDM (Scheme 2) proceeds with much lower yield than the analogous reaction of methyl oleate described above. Starting from a DDM/elaidate molar ratio of 5 and 1 mol% of Cu(OTf)2 at 85 °C in 1,2-dichloroethane, the amounts of DDM and catalyst were increased up to 7 equivalents and 4 mol% respectively, reaching a moderate 55% yield of cyclopropane 11.Reduction of the cyclopropanic esters to primary alcohols
The possibility to convert the obtained cyclopropanes into valuable monomers was tested first by reduction of the ester groups to primary alcohols using LiAlH4 (Scheme 3). The reaction with the trans cyclopropane 2 was very clean and fast, leading to the expected diol 12 with total GC conversion and 73% of isolated yield (Table 5, entry 1). The reaction of the cis cyclopropane 3 led diol 13 with 93% of isolated yield (entry 2).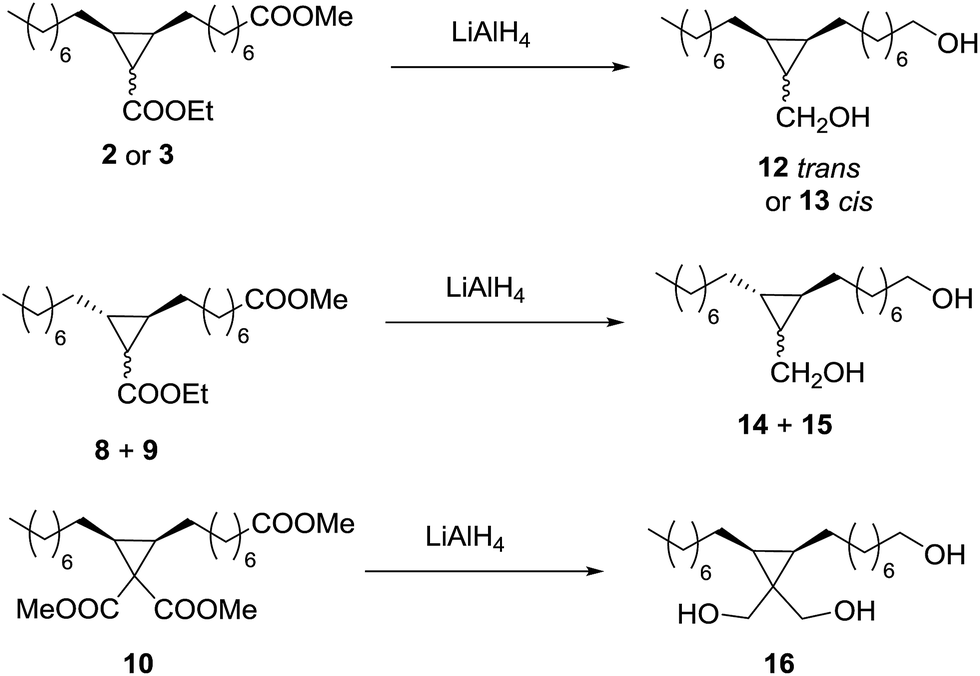 | ||
| Scheme 3 Reduction of the cyclopropanic esters to diols or triols. | ||
| Entry | Ester | Alcohol | Conv.b (%) | Isolated yield (%) |
|---|---|---|---|---|
| a Reagents and conditions: ester (100 mg), LiAlH4 (2.1 eq.), anhydrous Et2O (15 mL), room temperature, 2 h.b Determined by gas chromatography.c Crude of reaction only washed with brine. | ||||
| 1 | 2 | 12 | >99 | 73 |
| 2 | 3 | 13 | >99 | 93 |
| 3c | 2 | 12 | >99 | 92 |
| 4 | 8 + 9 | 14 + 15 | >99 | 92 |
| 5 | 10 | 16 | >99 | 70 |
The problem with diol 12 was supposed to be the solubility in the aqueous phase used for washing the organic phase, and the optimization of this step allowed obtaining 12 with 92% isolated yield (entry 3). The 50/50 mixture of the cyclopropanes 8 and 9, coming from methyl elaidate and EDA, was reduced to an equimolecular mixture of the diols 14 and 15 with also 92% of isolated yield (entry 4). Finally, the triester 10 gave the triol 16 with 70% of isolated yield (entry 5), probably due to an increase in the aqueous solubility produced by the third hydroxyl group.
Hydrolysis of the cyclopropanic esters
As a proof of concept, the diesters 2 and 3, and the triester 10 were hydrolysed with KOH/tBuOH and then neutralized to obtain the corresponding diacids 17 and 18, and the triacid 19 (Scheme 4). The diesters 2 and 3, as well as the triester 10, gave quantitative conversions (Table 6) to the corresponding diacids and triacid, but the partial solubility in the aqueous phase decreases the isolated yields to 83% in the case of 17 and 18, and more significantly in the case of the triacid 19 up to 59%.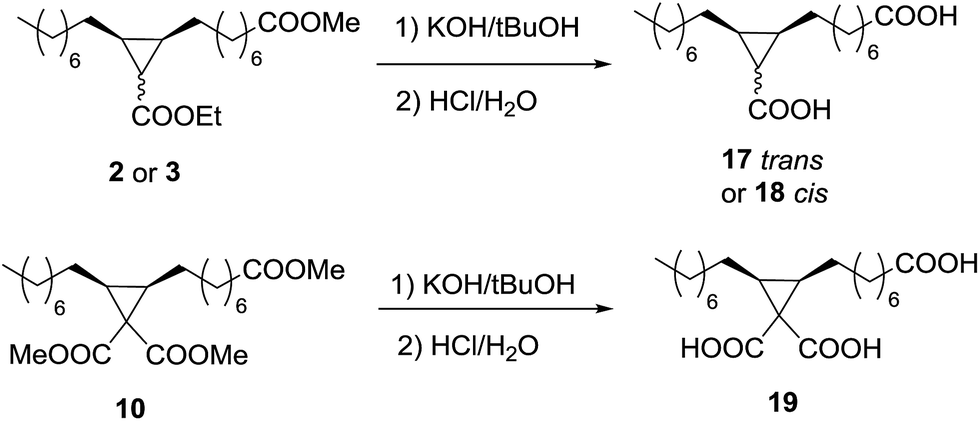 | ||
| Scheme 4 Hydrolysis of the cyclopropanic esters. | ||
Conclusions
The cyclopropanation reaction between different fatty esters and ethyl diazoacetate (EDA) has been carried out in a single step with quantitative yields, using 1 mol% Cu(OTf)2 as catalyst under mild conditions. For the first time, both diastereomers (trans and cis) were separated by flash-chromatography and unambiguously identified by NMR. Harsher reaction conditions were required for the cyclopropanation with dimethyl diazomalonate (DDM) because of the lower reactivity of this diazocompound. Even so, yields from moderate to excellent were reached. The products obtained in the cyclopropanation reaction have been easily transformed into diacids or triacids by hydrolysis, and into diols and triols by reduction. These compounds have the potential to be used as monomers or co-monomers in polymerization reactions for the obtaining of new materials.Experimental
General considerations
Methyl fatty esters and ethyl diazoacetate were purchased from different commercial sources, all of them were used as received without further purification. Dichloromethane was dried in an SPS-Device. Dimethyl 2-diazomalonate32 was prepared by reaction of dimethyl malonate with 4-methylbenzenesulfonyl azide.33 Reaction products were purified from the reaction crude by column chromatography on silica gel, using a mixture of hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate (9:1) as eluent, or by medium pressure liquid chromatography with the same mixture of solvents at 60 mL min. 1H- and 13C-NMR spectra of the products were recorded in a 400 MHz NMR spectrometer. CDCl3, C6D6 and CD3OD were used as NMR solvents. Spectral assignments were achieved by 1H–COSY, 1H–13C HSQC and 1H–13C HMBC experiments. HRMS spectra were measured in a Quadrupole Time of Flight (QTOF) mass spectrometer microTOF-Q equipped with an electrospray ionization (ESI) source.
:ethyl acetate (9:1) as eluent, or by medium pressure liquid chromatography with the same mixture of solvents at 60 mL min. 1H- and 13C-NMR spectra of the products were recorded in a 400 MHz NMR spectrometer. CDCl3, C6D6 and CD3OD were used as NMR solvents. Spectral assignments were achieved by 1H–COSY, 1H–13C HSQC and 1H–13C HMBC experiments. HRMS spectra were measured in a Quadrupole Time of Flight (QTOF) mass spectrometer microTOF-Q equipped with an electrospray ionization (ESI) source.
Representative procedure: Cu(OTf)2 catalysed cyclopropanation reaction of methyl oleate with EDA
A mixture of Cu(OTf)2 (9 mg, 0.025 mmol, 1 mol%) and eicosane (125 mg, 0.44 mmol; internal standard) in 2 mL of anhydrous dichloromethane was stirred for 20 min at room temperature under argon atmosphere. Then, methyl oleate (741.2 mg, 2.5 mmol) was added to the resulting solution. Finally, ethyl diazoacetate (7.5 mmol) was slowly added via syringe pump during 10 hours. The reaction was monitored by gas chromatography until total consumption of the diazoacetate. After the reaction was finished, the crude was filtered through a silica pad to adsorb the catalyst. The results were determined by gas chromatography, the products were separated by flash chromatography using a mixture of hexane:ethyl acetate (9:1) as eluent and they were identified by NMR and HRMS. All the cyclopropanation reactions were carried out in a similar way under the conditions detailed in tables.
Preparation of Cu-exchanged LAPONITE®
This method is a modification of the one previously described.24 To a solution of Cu(OTf)2 (0.7 mmol) in acetonitrile (6 mL) was added 1 g of LAPONITE® and the resulting suspension was stirred at room temperature for 24 h. The solid was separated by filtration, thoroughly washed with 10 mL of methanol and then with 5 mL of dichloromethane, and dried under vacuum before use. Analysis of the solid showed that the copper content was 0.48 mmol g−1 of dry solid.Representative procedure: LAPONITE®-Cu catalysed cyclopropanation reaction of methyl oleate with EDA
To a suspension of LAPONITE®-Cu (52.1 mg, 1 mol%) and eicosane (125 mg, 0.44 mmol; internal standard) in 2 mL of anhydrous dichloromethane was added methyl oleate (741.2 mg, 2.5 mmol) at room temperature and under argon atmosphere. Finally, the corresponding amount of ethyl diazoacetate was slowly added via syringe pump during 10 hours. The reaction was monitored by gas chromatography until total consumption of the diazoacetate. After the reaction was finished, the catalyst was filtered off, washed with dichloromethane and dried under vacuum. The recovered catalyst was reused following the same method.Representative procedure for the reduction of the ester groups in products (2) (trans) or (3) (cis) from methyl oleate
A suspension of LiAlH4 (42.9 mg, 1.13 mmol) in dry diethyl ether (15 mL) was stirred for 1 hour at room temperature. Afterwards, 100 mg (0.26 mmol) of (2), (3) or a mixture of both products was added to the previous solution and stirred for 2 hours. After this time, it was added, sequentially, 3 mL of ethyl acetate, 3 mL of EtOH, 10 mL of H2O and 0.8 mL of HCl 1 N. Aqueous and organic layers were separated and the aqueous one was extracted with ethyl acetate (2 × 15 mL). Organic layers were combined and washed with water (2 × 10 mL) and brine (10 mL). Then, the organic solution was dried with anhydrous MgSO4 and concentrated under vacuum.Representative procedure for the hydrolysis of the ester groups in products (2) (trans) or (3) (cis) from methyl oleate
100 mg (0.26 mmol) of (2), (3) or a mixture of both products was dissolved in 2 mL of tBuOH. Then KOH (117.2 mg, 2.09 mmol) was added and the solution was warmed up to 70 °C and stirred for 2 hours. After that time, 10 mL of H2O were added and HCl 6 N until pH = 2. The resulting solution was extracted with diethyl ether (3 × 10 mL). Organic layers were combined, dried with anhydrous MgSO4 and concentrated under vacuum.Acknowledgements
Financial support from the Spanish Ministerio de Economía y Competitividad (MINECO) (project number CTQ2014-52367-R) and Gobierno de Aragón (E11 Group co-financed by the European Regional Development Funds) is gratefully acknowledged.Notes and references
- H. Baumann, M. Bühler, H. Fochem, F. Hirsinger, H. Zoebelein and J. Falbe, Angew. Chem., Int. Ed. Engl., 1988, 27, 41–62 CrossRef.
- U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 CrossRef CAS PubMed.
- U. Biermann and J. O. Metzger, Top. Catal., 2004, 27, 119–130 CrossRef CAS.
- G. Knothe, Lipids, 2006, 41, 393–396 CrossRef CAS PubMed.
- S. Hartmann, D. E. Minnikin, H.-J. Römming, M. S. Baird, C. Ratledge and P. R. Wheeler, Chem. Phys. Lipids, 1994, 71, 99–108 CrossRef CAS PubMed.
- S. Shah, J. M. White and S. J. Williams, Org. Biomol. Chem., 2014, 12, 9427–9438 CAS.
- F. D. Gunstone and B. S. Perera, Chem. Phys. Lipids, 1973, 10, 303–308 CrossRef CAS PubMed.
- A. Gangadhar, R. Subbarao and G. Lakshminarayana, J. Am. Oil Chem. Soc., 1988, 65, 601–606 CrossRef CAS.
- K. U. M. Prasad, S. N. Balasubrahmanyam and B. H. Iyer, Indian J. Chem., 1969, 7, 460–462 CAS.
- D. Lefort, J. Sorba and A. Pourchez, Bull. Soc. Chim. Fr., 1966, 2223–2232 CAS.
- L. Maisonneuve, T. Lebarbé, E. Grau and H. Cramail, Polym. Chem., 2013, 4, 5472–5517 RSC.
- M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788–1802 RSC.
- Y. Lu and R. C. Larock, ChemSusChem, 2009, 2, 136–147 CrossRef CAS PubMed.
- U. Biermann, W. Friedt, S. Lang, W. Lühs, G. Machmüller, J. O. Metzger, M. R. gen. Klaas, H. J. Schäfer and M. P. Schneider, Angew. Chem., Int. Ed., 2000, 39, 2206–2224 CrossRef CAS PubMed.
- K. Hill, Pure Appl. Chem., 2000, 72, 1255–1264 CrossRef CAS.
- H. Mutlu and M. A. R. Meier, Eur. J. Lipid Sci. Technol., 2010, 112, 10–30 CrossRef CAS.
- D. Ogunniyi, Bioresour. Technol., 2006, 97, 1086–1091 CrossRef CAS PubMed.
- J. M. Fraile, J. I. García, J. A. Mayoral and T. Tarnai, Tetrahedron: Asymmetry, 1998, 9, 3997–4008 CrossRef CAS.
- J. M. Fraile, J. I. García, C. I. Herrerías, J. A. Mayoral, O. Reiser and M. Vaultier, Tetrahedron Lett., 2004, 45, 6765–6768 CrossRef CAS.
- J. M. Fraile, J. I. García, C. I. Herrerías and J. A. Mayoral, Chem. Commun., 2005, 4669–4671 RSC.
- H. Werner, C. I. Herrerías, M. Glos, A. Gissibl, J. M. Fraile, I. Perez, J. A. Mayoral and O. Reiser, Adv. Synth. Catal., 2006, 348, 125–132 CrossRef CAS.
- J. M. Fraile, J. I. Garcia, C. I. Herrerias, J. A. Mayoral, E. Pires and L. Salvatella, Catal. Today, 2009, 140, 44–50 CrossRef CAS.
- J. I. García, C. I. Herrerías, B. López-Sánchez, J. A. Mayoral and O. Reiser, Adv. Synth. Catal., 2011, 353, 2691–2700 CrossRef.
- J. M. Fraile, B. García, J. I. García, J. A. Mayoral and F. Figueras, in Heterogeneous Catalysis and Fine Chemicals IV, ed. H. U. Blaser, A. Baiker and R. Prins, 1997, vol. 108, pp. 571–578 Search PubMed.
- J. M. Fraile, J. I. García, M. A. Harmer, C. I. Herrerías, J. A. Mayoral, O. Reiser and H. Werner, J. Mater. Chem., 2002, 12, 3290–3295 RSC.
- J. M. Fraile, J. I. García and J. A. Mayoral, Chem. Commun., 1996, 1319–1320 RSC.
- J. D. Graham and M. T. Rogers, J. Am. Chem. Soc., 1962, 84, 2249–2252 CrossRef CAS.
- A. Solladié-Cavallo and T. Isarno, Tetrahedron Lett., 1999, 40, 1579–1582 CrossRef.
- A. Caballero, A. Prieto, M. M. Díaz-Requejo and P. J. Pérez, Eur. J. Inorg. Chem., 2009, 1137–1144 CrossRef CAS.
- J. I. García, L. Salvatella, E. Pires, J. M. Fraile and J. A. Mayoral, in Comprehensive Organic Synthesis II, Elsevier, Amsterdam, 2014, pp. 1081–1280 Search PubMed.
- H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977–1050 CrossRef CAS PubMed.
- F. De Nanteuil, E. Serrano, D. Perrotta and J. Waser, J. Am. Chem. Soc., 2014, 136, 6239–6242 CrossRef CAS PubMed.
- T. Kang, Y. Kim, D. Lee, Z. Wang and S. Chang, J. Am. Chem. Soc., 2014, 136, 4141–4144 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of products and GC chromatograms. See DOI: 10.1039/c7ra01017f |
| This journal is © The Royal Society of Chemistry 2017 |