
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly selective hydrodeoxygenation of anisole, phenol and guaiacol to benzene over nickel phosphide†
Yunhua Li *,
Juan Fu and
Binghui Chen
*,
Juan Fu and
Binghui Chen
Department of Chemical and Biochemical Engineering, College of Chemistry and Chemical Engineering, National Engineering Laboratory for Green Chemical Productions of Alcohols–Ethers–Esters, Xiamen University, Xiamen, 361005, PR China. E-mail: yunhuali@xmu.edu.cn; Fax: +86 0592 2183751; Tel: +86 0592 2183751
First published on 7th March 2017
Abstract
Ni2P supported catalysts have extensively been studied for various hydrodeoxygenation (HDO) reactions. However, the main products are cyclohexane or cyclohexanol for lignin-derived compounds HDO over these catalysts. In this study, we investigate the catalytic conversion of anisole, phenol and guaiacol to benzene over Ni2P/SiO2 by probing the reaction conditions. The results show that a lower reaction temperature and higher H2 pressure favour the hydrogenation of these model chemicals to cyclohexane, whereas a higher reaction temperature and lower H2 pressure aid the generation of benzene. The cyclohexane and benzene yields are 89.8% and 96.0% at 1.5 MPa and 573 K and 0.5 MPa and 673 K, respectively. By eliminating the influence of internal and external diffusion, the low intrinsic activation energy of 58.2 kJ mol−1 is obtained, which explains the high catalytic activity. In addition, although guaiacol HDO has a low conversion due to the space steric effect of its substituents, it presents a similar reaction pathway to obtain anisole and phenol, which is dependent on reaction conditions. The long-run evaluation experiment shows that the activity and selectivity of anisole HDO to benzene changes slightly for 36 h.
Introduction
The concerns of fossil fuel consumption and environmental pollution are driving the development of biomass substitution for fossil fuel resources and chemicals in recent years. However, crude bio-oil from biomass1–3 has some significant disadvantages as vehicle fuel, such as high acid content, viscosity and corrosion, chemical instability and low calorific value. In order to solve these problems, catalytic cracking,4,5 aldol condensation6,7 and catalytic hydrodeoxygenation (HDO) have been adopted.At present, catalysts for crude oil HDO are mainly divided into two types: sulfide and noble metal supported catalysts. The former mainly includes the Ni–MoS2/Al2O3 and Co–MoS2/Al2O3 catalysts.8–11 Although these catalysts have good HDO capability, a certain content of chemical containing sulfur is required to maintain their catalytic performances. Consequently, this brings about not only an increase in operating cost but also inevitable contamination of the bio-oil.12,13 With respect to precious metal catalysts, such as Pd, Pt, Ru, and Rh14–17, despite their excellent HDO activity, their industrial application prospect is restricted due to their resource scarcity and high cost. In contrast, transition metal phosphide,18–22 nitride,23,24 carbide25,26 and Ni27–29 supported catalysts are relatively clean and efficient catalyst systems for the HDO process.
The key point of the hydrodeoxygenation reaction is the selective hydrogenation of certain groups in biomass-derived molecules. This involves two aspects in the HDO process of lignin model chemicals, such as guaiacol, phenol and anisole: selective hydrogenation of Caromatic–O and the benzene ring in oxygen-containing chemicals. Regarding their feasibility as vehicle fuel, higher occurrence of the former is vital because of their higher octane number together with the lower consumption of H2. To the best of our knowledge, the selective hydrogenation process is mainly dependent on the reasonable design of the components and structures of heterogeneous catalysts.30–34
Very recently, Zhao et al. pointed out that for the selective hydrogenolysis of lignin-derived substituted phenols, a higher temperature and lower H2 pressure contribute to the generation of benzene over the Ru/HZSM-5 catalyst.35 In the previous study, we systematically studied phenol HDO over Ni2P and Ni2P@Pd supported catalysts using autoclave reactors.36,37 However, the main products are cyclohexane and cyclohexanol over these catalysts. Herein, the influence of the HDO reaction conditions on the reaction products is investigated over the base metal phosphide in a fixed bed. The reaction pathways of the highly selective HDO of phenol, anisole and guaiacol to benzene are uncovered by the reaction evaluation and physiochemical characterization of the Ni2P catalyst. Compared with the traditional studies, the present study has four advantages: (1) by simply changing the reaction conditions, benzene or cyclohexane is achieved, (2) the intrinsic activation energy is obtained through the elimination of internal and external diffusion to determine the high catalytic performance of the Ni2P catalyst fundamentally, (3) the reaction pathway is elucidated for the efficient conversion of lignin model compounds into benzene over Ni2P supported catalysts, and (4) the phosphide supported catalyst has high stability in the presence of a large amount of benzene.
Experimental
Materials
Guaiacol (GC grade) used for analysis was obtained from Aladdin Chemistry Co., Ltd. All other chemicals, such as benzene, phenol, and anisole, of analytical grade were purchased from Sinopharm Chemical Reagent Co., Ltd, and used as received without any further purification.Catalyst synthesis
Ni2P/SiO2 was prepared according to a similar method in ref. 36. In a typical preparation, 2.62 g Ni (NO3)2·6H2O and 60 g urea were dissolved in 225 mL deionized water and stirred for 30 min. The mixture was slowly added to a tetraethoxysilane (TEOS) precursor solution, which was obtained by dissolving 20.95 g TEOS in 20 mL ethanol. After stirring for 4 h, drying for 12 h at 393 K and calcination for 4 h at 823 K in a muffle furnace, NiO/SiO2 was obtained. Then, the as-obtained NiO/SiO2 was impregnated with a certain amount NH4H2PO4 solution. After reducing for 3 h at 923 K in H2 atmosphere, Ni2P/SiO2 was synthesized after passivation for 2 h in a flow of 0.5% O2/N2 at room temperature.Materials characterization
X-ray powder diffraction (XRD) measurement carried out on a PANalytical X'Pert Pro diffractometer with Cu Kα radiation was used to elucidate the crystallographic properties of the sample. The 2θ scans covered the range of 10–90° at a rate of 10° min−1. The crystallite size of the sample was estimated using the Scherrer equation. The particle sizes of the catalysts were characterized via transmission electron microscopy (TEM, FEI TECNAI F30). Before TEM observation, the Ni2P catalyst was sonicated in ethanol onto a holey-carbon coated copper grid and dried in air for 30 min.The BET (Brunauer–Emmett–Teller) surface area, pore volume and mesopore diameter distribution were measured via N2 adsorption–desorption measurements, which were carried out at 77 K using a Micromeritics ASAP 2020 system. Before the analyses, the samples were outgassed at 523 K for 2 h.
The real Ni loading was tested by X-ray fluorescence (XRF, Bruker S8 TIGER) measurements. Carbon element analysis was carried out with an elemental analyzer (Vario ELIII, Elementar).
X-ray photoelectron spectroscopy (XPS) was conducted using a Quantum 2000 Scanning ESCA Microprobe system with focused monochromatic Al Kα X-rays (1486.6 eV) under ultrahigh vacuum. Prior to the test, the sample was reduced in pure H2 (100 mL min−1) at 723 K for 2 h. Binding energies were calibrated using the C 1s peak at 284.8 eV as a reference.
CO chemisorption was conducted on a Micromeritics ASAP 2020 equipped with a thermal conductivity detector (TCD). Prior to the test, 0.1 g passivated sample was treated at 723 K under an H2 stream (40 mL min−1) for 2 h and evacuated for 2 h at room temperature. The adsorbed CO was determined from the difference between the two isotherms.
Evaluation of HDO on Ni2P catalysts
All reactions were performed using a fixed-bed reactor (I.D. 8 mm) with a thermocouple to monitor the reaction temperature. In a typical operation, 0.2 g catalyst was reduced in pure H2 (100 mL min−1) at 723 K for 1 h and cooled to a certain temperature. The reaction mixture with a ratio of H2/oxygen-containing chemicals, anisole or guaiacol of 45 was introduced into the reactor. In the reaction process, the reaction products were detected online by gas chromatography connected with the fixed-bed.Results and discussion
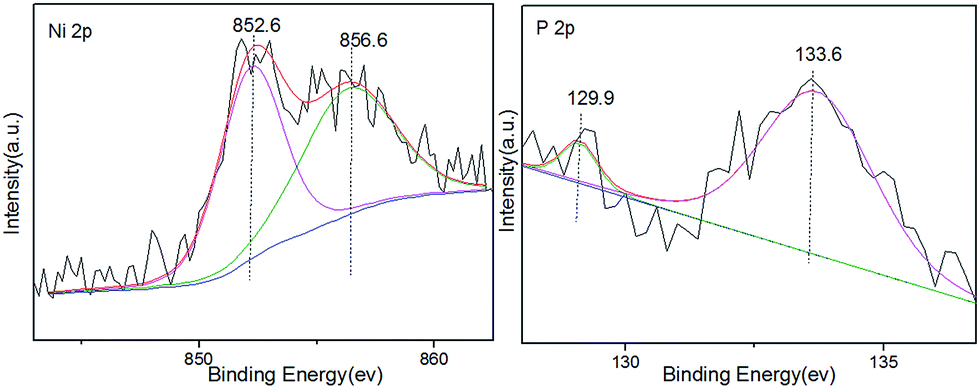
The structure and composition properties of Ni2P/SiO2 were determined by physical characterizations. According to the TEM image shown in Fig. 1, the metal nanoparticles are uniformly distributed on the silica carrier (Fig. 1a–c) and the particle size is determined to be 5.35 nm based on 200 particles (Fig. 1d). This is in agreement with the value of 5.60 nm from the XRD (Fig. S1†) observations. In addition, the HR-TEM images taken of individual particles further present a d-spacing of 0.22 nm, which is consistent with the (111) crystallographic plane of Ni2P.38 Table S1† lists the composition and structure of the Ni2P supported catalyst. The actual load of Ni2P is 7.5% as determined by XRF and the surface area determined by BET is 316 m2 g−1. The amount of CO adsorbed on Ni2P/SiO2 is 114 μmol CO on per gram catalyst. The XPS spectra in Fig. 2 further show that Ni2P/SiO2 includes Niδ+ (0 < δ < 2) and Pδ−(0 < δ < 1) in Ni2P at about 852.6 eV and 129.9 eV (ref. 37) as well as Ni2+ and P5+ from the oxidation of Ni2P at about 856.6 eV and 133.6 eV, respectively. | ||
| Fig. 1 TEM images and particle size distribution for the Ni2P catalyst. | ||
 | ||
| Fig. 2 XPS spectra of Ni 2p and P 2p of the Ni2P/SiO2 catalyst. | ||
In order to make lignin model compounds efficiently convert into benzene by only changing the reaction conditions, the influence of reaction temperature was systematically investigated on the product distributions of anisole HDO.
As can be seen from Table 1, at 523 K, anisole conversion is very low and the main products are cyclohexane and methoxylcyclohexane. When the temperature increases to 573 K, cyclohexane further increases with a decrease in methoxylcyclohexane. In other words, the Caromatic–O bonds cleave and anisole is converted into cyclohexane with an increase in temperature, which indicates the occurrence of the reaction according to pathways I and II in Fig. 3. In contrast, the selectivity of benzene remains almost constant below 573 K. As the temperature further increases up to 673 K, the benzene selectivity prominently increases from 5.8% to 60.6%, which indicates that a high temperature favors deoxygenation to generate a large amount of benzene for anisole HDO.
| T (K) | Con. (%) | TOFb (s−1) | Selectivity (%) | |||
|---|---|---|---|---|---|---|
| Cyclohexane | Benzene | Methoxyl cyclohexane | Phenol | |||
| a Reaction condition: 1.5 MPa, liquid hour space velocity (LHSV) 1.85 h−1, particle diameter 0.18 mm, H2/anisole mole ratio 45.b The turnover frequency (TOF) was calculated based on linear-type CO chemisorption. | ||||||
| 523 | 21.7 | 0.04 | 63.6 | 5.4 | 31.0 | 0 |
| 573 | 97.0 | 0.18 | 92.6 | 5.8 | 1.6 | 0 |
| 623 | 97.5 | 0.19 | 88.0 | 12.0 | 0 | 0 |
| 673 | 100 | 0.19 | 39.4 | 60.6 | 0 | 0 |
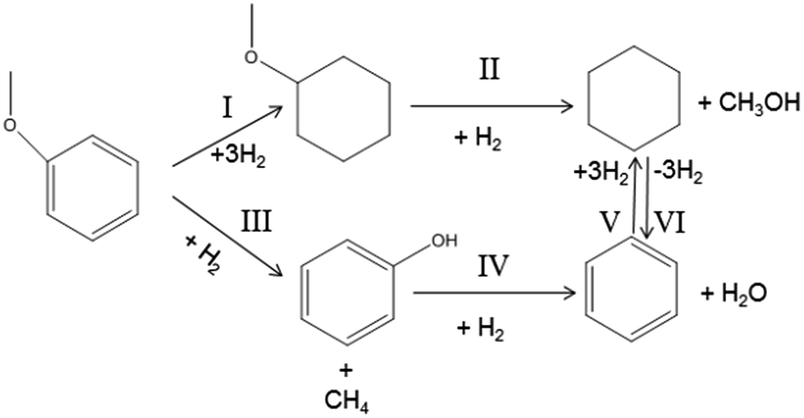 | ||
| Fig. 3 Reaction pathway for anisole HDO. | ||
To distinctly understand the HDO reaction intrinsic process, we changed the space velocities and reduced the particle sizes to determine the influence of internal and external diffusion on the macroscopic rate of anisole HDO. When the liquid hourly space velocity exceeds 0.93 h−1 and the particle diameter is less than 0.25 mm at 673 K (Fig. S2†), the conversion of anisole remains constant. In other words, the catalytic results in Table 1 are obtained under the circumstance of negligible internal and external diffusion. Furthermore, the intrinsic activation energy of anisole HDO is presented in Fig. 4, which is similar to the research of H. Y. Zhao.18 It is lower than expected for carbon–oxygen bond rupture (>240 kJ mol−1), which reflects the high activity of the transition metal phosphide for anisole HDO.
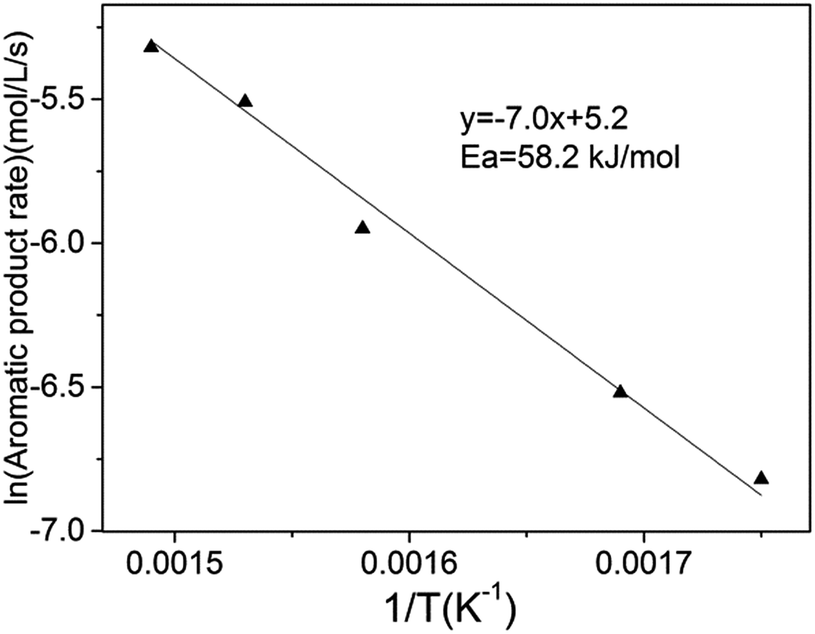 | ||
| Fig. 4 Intrinsic activation energy for anisole HDO over the Ni2P/SiO2 catalyst. Reaction conditions: 573–673 K, 1.5 MPa, LHSV 1.85 h−1, and particle diameter 0.18 mm. | ||
The effects of H2 pressure on the product distribution in anisole HDO were also studied, and the results presented in Table 2. As demonstrated in Table 2, anisole is partially converted into cyclohexane and phenol under atmospheric pressure. With an increment in H2 pressure, anisole is consumed completely and the main products become benzene and cyclohexane. The higher the H2 pressure, the more cyclohexane is generated together with the less benzene. This result confirms the existence of reaction process V in Fig. 2. In addition, at 673 K and 0.5 MPa, cyclohexane as a reactant has 20.1% conversion to benzene. This is reduced to 10.0% at the same temperature and 1.5 MPa.
| Pressure (MPa) | Conversion (%) | Selectivity (%) | |||
|---|---|---|---|---|---|
| Cyclohexane | Benzene | Methoxyl cyclohexane | Phenol | ||
| a Reaction conditions: 673 K, LHSV 1.85 h−1, particle diameter 0.18 mm, and H2/anisole mole ratio 45. | |||||
| 0.1 | 70.8 | 0 | 54.6 | 0 | 45.4 |
| 0.5 | 100 | 3.6 | 96.4 | 0 | 0 |
| 1.5 | 100 | 39.4 | 60.6 | 0 | 0 |
| 2.0 | 100 | 53.6 | 46.4 | 0 | 0 |
Based on these results, we speculate the reaction pathways for anisole HDO at different temperatures in Fig. 3. At about 573 K and 1.5 MPa the anisole HDO reaction occurs following the pathways I and II, whereas the HDO reaction mainly proceeds according to pathways III, IV, V and VI at 673 K and 0.5 MPa (Table 2).
A mixture of anisole and phenol (mass ratio of anisole and phenol is equal to 1.0), and guaiacol were used as reactants to further investigate the HDO properties of the model chemicals from lignin pyrolysis oil, and the results are presented in Table 3. From Table 3, it can be seen that phenol has similar reaction features to anisole under the same reaction conditions. Specifically, at 573 K, the benzene ring is easily hydrogenated, whereas at 673 K, Caromatic–O preferentially cleaves to benzene over the Ni2P catalyst. In addition, the evaluation results from the HDO of guaiacol with methoxyl and phenolic hydroxyl groups (Table 3) show that the consumption of guaiacol follows a similar HDO pathway to anisole and phenol under the same reaction conditions despite the relatively lower conversion. Guaiacol HDO is inhibited in comparison with anisole and phenol, which may result from the steric hindrance effect of its functional groups39 in the reaction process. In addition, elemental analysis presents that 4.6% carbon is generated over Ni2P/SiO2 after the guaiacol HDO reaction (Table S2†).
| Reactant | Temperature (K) | Pressure (MPa) | Con.b (%) | Selectivity (%) | ||
|---|---|---|---|---|---|---|
| Cyclohexane | Benzene | Phenolc | ||||
| a Reaction conditions: LHSV 1.85 h−1, particle diameter 0.18 mm, and H2/anisole (phenol or guaiacol) mole ratio 45.b Conversions of anisole and phenol are equal to 100% for the HDO reaction of the mixture containing anisole and phenol.c Phenol selectivity is for guaiacol HDO. | ||||||
| Anisole + phenol | 573 | 1.5 | 100 | 96.7 | 3.3 | 0.0 |
| 673 | 0.5 | 100 | 4.3 | 95.7 | 0.0 | |
| Guaiacol | 573 | 1.5 | 91.0 | 96.5 | 3.5 | 0 |
| 673 | 0.5 | 91.7 | 5.0 | 74.0 | 21.0 | |
We also further checked the effect of LHSV on product distribution in anisole HDO (Table 4). In the range of LHSV from 0.93 to 7.4 h−1, the conversion of anisole initially remains almost constant and then decreases gradually, whereas the selectivity of products varies slightly. With the increase in LHSV from 0.9 to 1.8 h−1, the excellent catalytic activity of Ni2P results in the high conversion of anisole. However, some of the anisole is left in the reaction system as LHSV further increases. A high LHSV means a short reaction time, which does not contribute to all of the anisole contacting with the catalyst surface. Thus, anisole conversion initially remains constant and then drops slowly.
| LHSV (h−1) | Conversion (%) | Selectivity (%) | |||
|---|---|---|---|---|---|
| Cyclohexane | Benzene | Methoxyl cyclohexane | Phenol | ||
| a Reaction conditions: 673 K, 0.5 MPa, particle diameter 0.18 mm, and H2/anisole mole ratio 45. | |||||
| 0.9 | 98.8 | 4.3 | 95.7 | 0 | 0 |
| 1.8 | 100 | 3.6 | 96.4 | 0 | 0 |
| 3.7 | 98.0 | 3.8 | 96.2 | 0 | 0 |
| 7.4 | 95.6 | 2.6 | 97.4 | 0 | 0 |
Since Ni2P/SiO2 has high benzene selectivity for anisole HDO, the stability of the catalyst also becomes a significant issue in the presence of a large amount of benzene. Herein, anisole HDO was run for 36 h (Fig. 5) and then an element analysis was performed to determine the carbon content on the catalyst surface before and after the reaction (Table S2†). As shown Fig. 5 and Table S2,† the conversion and selectivity within 36 h have no obvious decline and the formation of carbon cannot be observed after the reaction. These results show that the Ni2P/SiO2 catalyst has relatively good stability for anisole HDO to benzene.
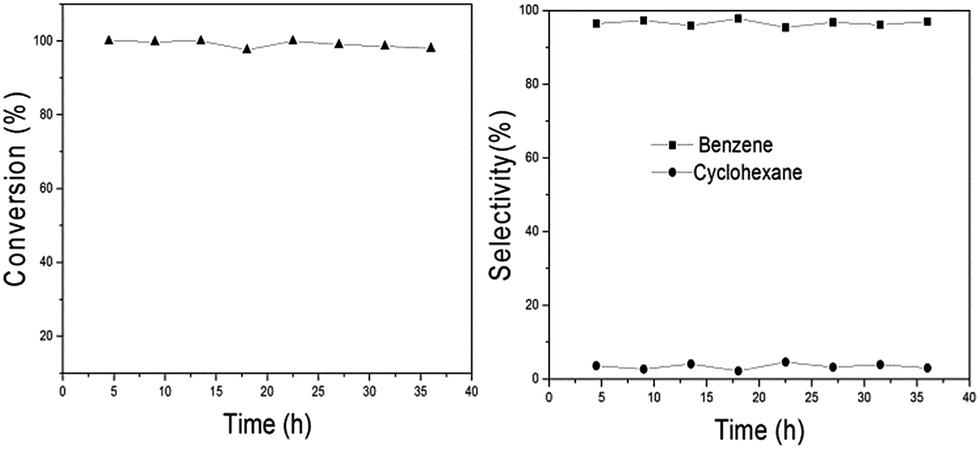 | ||
| Fig. 5 Stability for anisole HDO over Ni2P/SiO2. Reaction conditions: 673 K, LHSV 1.85 h−1, particle diameter 0.18 mm, and H2/anisole mole ratio 45. | ||
Conclusions
In this study, the HDO of anisole, guaiacol and phenol was investigated to obtain high benzene yield over Ni2P/SiO2. The results show that with an increase in the reaction temperature from 573 K to 673 K, the selectivity of benzene prominently increases from 5.8% to 60.6%. In addition, the benzene yield reaches 96.0% from anisole or phenol HDO with a decrease in reaction pressure. In other words, a higher reaction temperature and lower H2 pressure favor the conversion of anisole to benzene. Conversely, hydrogenation of the benzene ring occurs. 58.2 kJ mol−1 of intrinsic activation energy at 573–673 K further accounts for the high catalytic activity of nickel phosphide. The long-run evaluation of anisole HDO to benzene shows that the catalytic performances change negligibly for 36 h. Although guaiacol HDO shows a low conversion due to the space steric effect of its substituent, it has similar reaction characteristics to anisole and phenol conversion to benzene under the same conditions. This work demonstrates that the Ni2P supported catalyst is an efficient and promising catalyst for HDO in industrial application. At the same time, it also provides important information for the development of other catalysts for the conversion of lignin model chemicals to benzene.Acknowledgements
This study was supported by National Natural Science Foundation of China (Grant no. 21476188, 21106118).Notes and references
- L. E. Manzer, Top. Catal., 2010, 53, 1193–1196 CrossRef CAS.
- X. Chen, B. Zhang, Y. Wang and N. Yan, Chimia, 2015, 69, 120–124 CrossRef CAS PubMed.
- S. De, B. Saha and R. Luque, Bioresour. Technol., 2015, 178, 108–118 CrossRef CAS PubMed.
- H. Liu, T. Chen, X. Zhang, J. Li, D. Chang and L. Song, Chin. J. Catal., 2010, 31, 409–414 CrossRef CAS.
- P. S. Rezaei, H. Shafaghat and W. M. A. W. Daud, Appl. Catal., A, 2014, 469, 490–511 CrossRef CAS.
- G. Liang, A. Wang, X. Zhao, N. Lei and T. Zhang, Green Chem., 2016, 18, 3430–3438 RSC.
- D. Nguyen Thanh, O. Kikhtyanin, R. Ramos, M. Kothari, P. Ulbrich, T. Munshi and D. Kubička, Catal. Today, 2016, 277, 97–107 CrossRef CAS.
- K. C. Kwon, H. Mayfield, T. Marolla, B. Nichols and M. Mashburn, Renewable Energy, 2011, 36, 907–915 CrossRef CAS.
- M. Badawi, J. F. Paul, S. Cristol, E. Payen, Y. Romero, F. Richard, S. Brunet, D. Lambert, X. Portier, A. Popov, E. Kondratieva, J. M. Goupil, J. El Fallah, J. P. Gilson, L. Mariey, A. Travert and F. Maugé, J. Catal., 2011, 282, 155–164 CrossRef CAS.
- V. N. Bui, D. Laurenti, P. Afanasiev and C. Geantet, Appl. Catal., B, 2011, 101, 239–245 CrossRef CAS.
- M. C. Samolada, W. Baldauf and I. A. Vasalos, Fuel, 1998, 77, 1667–1675 CrossRef CAS.
- O. İ. Şenol, E. M. Ryymin, T. R. Viljava and A. O. I. Krause, J. Mol. Catal. A: Chem., 2007, 277, 107–112 CrossRef.
- O. İ. Şenol, T. R. Viljava and A. O. I. Krause, Appl. Catal., A, 2007, 326, 236–244 CrossRef.
- Y. Huang, L. Wei, X. Zhao, J. Julson, C. Qiu, S. Dharmarajan, J. Kiratu, D. Raynie, A. Dubey and Q. Qiao, Int. J. Energy Res., 2016, 40, 1724–1730 CrossRef CAS.
- M. S. Zanuttini, B. O. Dalla Costa, C. A. Querini and M. A. Peralta, Appl. Catal., A, 2014, 482, 352–361 CrossRef CAS.
- J. K. Hedlund, D. C. Cronauer, G. Jacobs, A. J. Kropf, J. A. Libera, J. W. Elam, C. L. Marshall, V. R. R. Pendyala and B. H. Davis, Catal. Lett., 2016, 146, 525–539 CrossRef CAS.
- C. A. Teles, R. C. Rabelo-Neto, J. R. de Lima, L. V. Mattos, D. E. Resasco and F. B. Noronha, Catal. Lett., 2016, 146, 1848–1857 CrossRef CAS.
- H. Y. Zhao, D. Li, P. Bui and S. T. Oyama, Appl. Catal., A, 2011, 391, 305–310 CrossRef CAS.
- K. Li, R. Wang and J. Chen, Energy Fuels, 2011, 25, 854–863 CrossRef CAS.
- Y. Shu, Y. Lee and S. Oyama, J. Catal., 2005, 236, 112–121 CrossRef CAS.
- D. J. Rensel, S. Rouvimov, M. E. Gin and J. C. Hicks, J. Catal., 2013, 305, 256–263 CrossRef CAS.
- W. Fu, L. Zhang, D. Wu, Q. Yu, T. Tang and T. Tang, Ind. Eng. Chem. Res., 2016, 55, 7085–7095 CrossRef CAS.
- B. M. Wyvratt, J. R. Gaudet, D. B. Pardue, A. Marton, S. Rudić, E. A. Mader, T. R. Cundari, J. M. Mayer and L. T. Thompson, ACS Catal., 2016, 6(9), 5797–5806 CrossRef CAS.
- S. Verma, R. B. N. Baig, M. N. Nadagouda and R. S. Varma, Green Chem., 2016, 18, 1327–1331 RSC.
- A. L. Jongerius, R. W. Gosselink, J. Dijkstra, J. H. Bitter, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemCatChem, 2013, 5, 2964–2972 CrossRef CAS.
- W.-S. Lee, Z. Wang, R. J. Wu and A. Bhan, J. Catal., 2014, 319, 44–53 CrossRef CAS.
- S. Jin, Z. Xiao, C. Li, X. Chen, L. Wang, J. Xing, W. Li and C. Liang, Catal. Today, 2014, 234, 125–132 CrossRef CAS.
- S. Qi, W. Feng and X. Jie, Chem. Commun., 2012, 48, 7019–7021 RSC.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 CAS.
- S. T. Oyama, T. Onkawa, A. Takagaki, R. Kikuchi, S. Hosokai, Y. Suzuki and K. K. Bando, Top. Catal., 2015, 58, 201–210 CrossRef.
- P. Bui, J. Catal., 2012, 294, 184–198 CrossRef CAS.
- K. Wu, W. Wang, S. Tan, G. Zhu, L. Tan and Y. Yang, RSC Adv., 2016, 6, 80641–80648 RSC.
- S. K. Wu, P. C. Lai, Y. C. Lin, H. P. Wan, H. T. Lee and Y. H. Chang, ACS Sustainable Chem. Eng., 2013, 1, 349–358 CrossRef CAS.
- S. K. Wu, P. C. Lai and Y. C. Lin, Catal. Lett., 2014, 144, 878–889 CrossRef CAS.
- Z. Luo, Z. Zheng, Y. Wang, G. Sun, H. Jiang and C. Zhao, Green Chem., 2016, 18, 5845–5858 RSC.
- Y. Li, X. Yang, L. Zhu, H. Zhang and B. Chen, RSC Adv., 2015, 5, 80388–80396 RSC.
- K. Yan, Y. Li, X. Zhang, X. Yang, N. Zhang, J. Zheng, B. Chen and K. J. Smith, Int. J. Hydrogen Energy, 2015, 40, 16137–16146 CrossRef CAS.
- S. Wang, K. Wang and X. Wang, Appl. Surf. Sci., 2016, 386, 442–450 CrossRef CAS.
- S. F. Guo, A. K. Rogers, M. M. Yung and C. Sievers, ACS Catal., 2016, 1292–1307 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00989e |
| This journal is © The Royal Society of Chemistry 2017 |