
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy of the E–Z stilbene isomerisation in perchlorotriphenyl-methane (PTM) derivatives†
F. Bejarano‡
 a,
I. Alcon‡b,
N. Crivillersa,
M. Mas-Torrent*a,
S. T. Bromleybc,
J. Vecianaa and
C. Rovira*a
a,
I. Alcon‡b,
N. Crivillersa,
M. Mas-Torrent*a,
S. T. Bromleybc,
J. Vecianaa and
C. Rovira*a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Networking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Campus de la UAB, 08193 Bellaterra, Spain. E-mail: cun@icmab.es; mmas@icmab.es
bDepartament de Ciència de Materials I Química Física, Institut de Química Teórica i Computacional, Universitat de Barcelona (IQTCUB), E-08028 Barcelona, Spain
cInstiuició Catalana de Recerca i Estudis Avançats (ICREA), 08010 Barcelona, Spain
First published on 7th March 2017
Abstract
The E–Z isomerisation of two perchlorotriphenylmethane derivatives containing stilbene units has been investigated, both thermally and photochemically. The irreversibility of the E → Z isomerisation in both compounds is experimentally demonstrated and supported by density functional calculations.
The photoinduced E–Z isomerization phenomenon has been a longstanding interesting topic in organic and inorganic chemistry as well as in biology. The change in the functional properties depending on the molecular structure of the different meta-stable isomers has been exploited for many applications in biochemistry, pharmacology and supramolecular chemistry.1 The most representative isomeric switchable systems are azobenzenes2 and stilbenes.3 Both, upon illumination with a specific wavelength, can undergo a reversible photo-isomerisation between the trans and cis conformation. They have been extensively investigated as molecular switches4 and also have led to many different applications such as molecular motors5 or solar-thermal fuels.6 In the latter, solar energy is stored in the chemical bonds by photoconversion and, upon activation, the stored energy is released in the form of heat. Here we study, both photochemically and thermally, the E–Z isomerisation of perchlorotriphenylmethane (PTM) derivatives containing a stilbene unit with one of their phenylene rings substituted with four chlorine atoms and a perchlorodiphenylmethyl group at the para position. This phenomenon is presented herein with the study of two differently substituted stilbene derivatives (Scheme 1). This family of molecules is very appealing since it has been demonstrated that, with a proper design, they can be excellent building blocks to form functional supramolecular nano- and micro-structures driven by Cl⋯Cl and Cl⋯phenyl interactions. Indeed, very recently, it was demonstrated that PTM moieties substituted with p-vinylphenylene groups functionalised with long alkyl chains can be employed to prepare molecular micro-scale objects with unprecedented shapes (i.e., flowers, cones, fibres, etc.) following the so-called “hydrophobic–amphiphilic” approach.7 Moreover, by chemical modification of the phenyl substitution pattern, robust micro-capsules that could encapsulate hydrophilic contents were prepared. Interestingly, such hydrophilic contents were neatly released upon UV irradiation due to photochemical decomposition leading to a phenanthrene derivative.8 Further, chemically bonded self-assembled monolayers (SAMs) on gold were previously prepared using the E isomer of compound 1 (Scheme 1).9 In this case, the SAMs were employed as precursors to form the organic free radical derivative which has been shown to constitute an excellent platform for the fabrication of robust molecular memories due to its electroactive character.10 In view of the interesting properties of the functional materials prepared employing the PTM derivatives as building blocks, we were interested in studying the isomerisation processes that these systems can undergo when they are irradiated or heated.
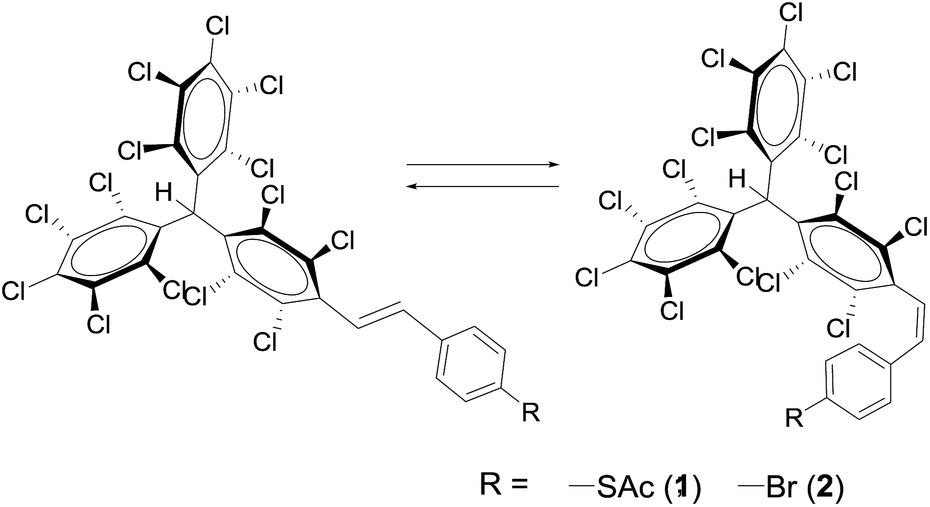 | ||
| Scheme 1 Isomerisation of PTM–SAc (1) and PTM–Br (2); E-1/2 to Z-1/2. | ||
Compounds 1 and 2 were obtained through a Wittig and a Horner–Wadsworth–Emmons reaction, respectively. In the case of 1, the reaction was carried out between the PTM phosphonium bromide salt and the 4-(acetylthio)benzaldehyde (see details on the synthesis in ref. 11). Molecule 2 was prepared through the coupling of PTM phosphonate and 4-bromobenzaldehyde (synthesis adapted from ref. 12). It is known that the 1H-NMR chemical shift of the hydrogen atoms of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups, as well as their coupling constant J, allows for the identification of the two possible isomers that can be obtained from these synthetic routes. For the compounds presented here, the J value of the E isomers is around 17 Hz, while for the Z isomers it is around 12 Hz. It was determined that the E isomer was the major component of the reaction mixtures, with a ratio of ca. 92% and 99% for E-1 and E-2, respectively (determined by 1H-NMR signal integration).
C groups, as well as their coupling constant J, allows for the identification of the two possible isomers that can be obtained from these synthetic routes. For the compounds presented here, the J value of the E isomers is around 17 Hz, while for the Z isomers it is around 12 Hz. It was determined that the E isomer was the major component of the reaction mixtures, with a ratio of ca. 92% and 99% for E-1 and E-2, respectively (determined by 1H-NMR signal integration).
In order to follow and quantify the E/Z isomerisation processes for compounds 1 and 2, High Performance Liquid Chromatography (HPLC) was employed using reversed-phase conditions, at room temperature and under an isocratic regime. Mixtures of acetonitrile/tetrahydrofuran (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and acetonitrile/chloroform (8:2) were found as optimal conditions for an adequate separation of the corresponding isomers for 1 and 2, respectively.
:1) and acetonitrile/chloroform (8:2) were found as optimal conditions for an adequate separation of the corresponding isomers for 1 and 2, respectively.
After reaching a good separation of the two isomers, we proceeded to study the photo-isomerisation conversion from the E to the Z form of 1 and 2. The samples were irradiated with a Hg(Xe) lamp 300 W placed at around 30 cm from the sample. In the case of 1, a 0.07 mM solution in a THF/ACN (1:1) mixture was used for this experiment. To follow the evolution of the isomerisation process, several aliquots were taken during the irradiation time and analysed by HPLC. Fig. 1 shows the corresponding chromatograms and the retention times. The relative peak areas (in %) are depicted for each analysed aliquot in Table 1. See ESI† for the monitoring performed for 2.
 | ||
| Fig. 1 Evolution of the HPLC chromatograms obtained upon irradiation of E-1 in a solvent mixture of THF/ACN (1:1). Experimental conditions: 25 °C, mobile phase THF/ACN (10/90), stationary phase ODS, UV detector with a fixed wavelength of 230 nm and a flow of 1 mL min−1. | ||
| Time (min) | % Area (Z-1) | % Area (E-1) | (Z/E) Peak areas ratio |
|---|---|---|---|
| 0 | 7.5 | 92.5 | 0.08 |
| 10 | 23.0 | 77.0 | 0.30 |
| 46 | 67.7 | 31.3 | 2.16 |
| 102 | 75.5 | 24.5 | 3.08 |
| 115 | 87.9 | 12.1 | 7.26 |
| 145 | 88.8 | 11.2 | 7.93 |
As expected, the Z/E ratio increases in both cases during the irradiation time until the photostationary state is reached. The 1H-NMR (see ESI†) of the sample after irradiation confirmed that the peak at a retention time of 12 minutes corresponds to the expected Z-1 compound. The same analysis was performed for compound 2.
The characterization by ultraviolet-visible (UV-vis) spectroscopy shows that there is an overlap of the absorption spectra of both isomers (see ESI†). In addition, the Z isomer absorbs white light more poorly than the E isomer. For these two reasons the back Z → E phoisomerization could not be carried out.
Since the most thermodynamically favoured isomer for stilbenoid compounds is usually the E isomer, we proceeded with the study of the thermally induced conversion of the Z form back to the E. To do that, the photostationary state mixture obtained by irradiation was refluxed in different solvents: chloroform, tetrahydrofuran, cyclohexane, toluene and o-dichlorobenzene for, at least 3 hours. These solvents do not only allow access to different heating temperatures, but also they provide different polarities of the media, as well as interactions between the solvent and the PTM derivatives through their ability to form π–π and Cl–Cl interactions. Again, the solution evolution was followed by HPLC. In this case, the thermal back conversion was not observed for either compound 1 or 2. This result is in agreement with previous studies which determined an activation barrier for the Z → E isomerisation for stilbenoid compounds of the order of 1.9 eV (43.8 kcal mol−1), which makes the thermal back reaction unlikely.13
We attempted to carry out the PTM Z → E isomerisation process using catalytic amounts of iodine. This is an established methodology to convert isomeric mixtures to the E derivative.14 The obtained photostationary state mixture of 2 was dissolved in toluene (0.022 M) with iodine (0.03 eq.) and the solution was refluxed for 5 hours. The evolution of the mixture was followed by HPLC and no back isomerisation was observed by HPLC monitoring. Then, 0.03 more equivalents of I2 were added and the solution was refluxed 19 additional hours. After such a long period of time at high temperature many products of decomposition appeared (see Fig. S5 in ESI†) but no isomer E formation was observed. Therefore, this suggests that back isomerisation, in the best case, is less favoured than several decomposition pathways.
Density functional theory (DFT) calculations were carried out to better understand the irreversible isomerisation process Z → E observed for both PTM derivatives 1 and 2. It is known that, unlike azobenzene derivatives, in ethylene derivatives photo-activated twisting of the CC bond is the main mechanism for E–Z isomerisation15 as represented with the red arrow on the stilbene structure in Fig. 2a. To obtain the ground state energy profile associated with such a twisting process in compounds 1 and 2, we systematically varied the corresponding CC dihedral angle and performed constrained optimisations of the molecular structure for each conformation. In all cases, the E conformer (φ = 0°, 360°) is slightly more stable than the Z conformer (φ = 180°), with the perpendicular conformation (i.e. φ = 90°, 270°) being the most energetically unstable by 1.9 eV (i.e. the E ↔ Z energetic barrier height, see ESI†). These results are fully in accordance with previous studies.13,14 Time dependent DFT (TD-DFT) calculations were then used to obtain approximate absorption spectra for E and Z conformers for both derivatives 1 and 2. For the E conformers we found the most intense peak at 340 and 345 nm (for 1 and 2, respectively). In the corresponding Z conformers, these peaks slightly shift towards higher energies (315 and 317 nm for 1 and 2, respectively) and decreases in intensity by ∼60%. These calculations coincide with our experimental UV-vis spectroscopy measurements (see ESI Fig. S1†), where the peak at ∼310 nm for the E conformer decreases in intensity upon photo-generating the Z conformer and shifts towards higher energy wavelengths. This UV-vis band variation has been observed for other stilbene derivatives, where such optical absorptions were associated with the photo-activation of the CC moiety.16
 | ||
| Fig. 2 (a) Fully optimized structures of the E-isomers of PTM derivatives 1 and 2, and stilbene. The coloured arrows on the structure of stilbene highlight the important twist angles (red: main CC axis, black: twist angles of the aryl groups bonded to the ethylene unit). (b) and (c) Evolution of the twist angles of the aryl groups bonded to the ethylene unit for each molecule (see legends) during 4 ps of an AIMD run at 450 K for the E and Z conformers respectively. | ||
The higher absorbance of the E conformer might explain the high production of the Z conformer upon light absorption. However, the thermal irreversibility of the photo-isomerisation of Z towards the E isomer (even under I2 catalyzed conditions) suggests that the different UV-vis absorption characteristics of the two conformers may not fully explain the experimental findings.16 In the CC twisting process assessed by constrained DFT optimizations (see ESI†) we found that, for both 1 and 2, the aryl ring groups bonded to the ethylene unit had to be twisted to specifically chosen angles (black arrows on stilbene structure in Fig. 2a) in order to prevent steric interactions and hence allow the rotation around the CC bond axis (red arrow on stilbene structure in Fig. 2a). The key role of such aryl ring twist angles for the E ↔ Z photo-isomerisation has been demonstrated both theoretically17 and experimentally,18 and proven to be more relevant for the Z → E process19 than for the E → Z one.20 Based on this, we performed ab initio molecular dynamics (AIMD) simulations at 450 K (see computational details below) to compare the thermal fluctuations of such aryl ring twist angles in compounds 1 and 2 and stilbene (where the E ↔ Z isomerisation is completely reversible) in each conformation.
Extracting the twist angle of the central ethylene unit (red arrow in Fig. 2a) we could find that, for all cases, the CC bond hardly rotates (see ESI†). This is in full accordance with its associated high twisting energy and corroborates the need for light irradiation to induce the E ↔ Z transition, even at high temperatures (see ESI†). In contrast, by following the variation in the twist angles of the aryl rings with respect to the ethylene unit (black arrows in Fig. 2a), it is possible to observe significant differences between the E and Z conformer profiles (Fig. 2b and c).
For both PTM derivatives and stilbene in the E conformation such aryl ring twist angles fluctuate significantly over time (Fig. 2b) which is consistent with the little steric interaction between the two aryl ring groups bonded to the CC unit. For stilbene in the Z conformation the two aryl rings tend to keep rotating in opposite directions leading to a gradual increase in the magnitude of the associated twist angle (see blue lines in Fig. 2c). Although in this case the two rings are close to each other, by counter rotating in this manner they are able to avoid significant steric hindrance. In the Z conformers of derivatives 1 and 2, however, the aryl ring twist angles quickly converge to fixed values of about 60°. The lack of rotation in these cases comes from two different factors. From one side, the bulky PTM group possesses relatively high inertia to move which impedes the rotation of that bulky group attached to the ethylene unit. Also, the close proximity of the ortho-chlorine atoms within the PTM unit with the opposite aryl ring (see the Z-1 structure in Fig. 2c) gives rise to large steric constraints which prevent the free rotation of the brominated aryl ring. Hence, overall, the rotational freedom of both groups bonded to the ethylene unit in the Z conformation of 1 and 2 is significantly suppressed.
As noted above, when calculating the twisting of derivatives 1 and 2 about the CC bond specific twist angles of the aryl ring groups are needed to avoid significant steric interactions and allow the isomerisation to occur. In the E conformer such specific aryl ring twist angles should be easily accessible due to the freely rotating behaviour in such conformation for 1, 2 and stilbene (see Fig. 2b). However, in the Z conformation, aryl rings in compounds 1 and 2 do not rotate due to steric interactions between the two aryl ring groups and the larger inertia of the PTM unit. Such an effect does not occur for stilbene (blue curve in Fig. 2c), where the E ↔ Z isomerisation is completely reversible.16 These results suggest that the complete and irreversible formation of Z isomers under light irradiation (Fig. 1) for PTM derivatives 1 and 2 could be at least partially explained by the lack of free rotation of their aryl rings in the Z conformation, causing a kinetic blockage. This suggestion may also help explain why even when using a chemical catalyst (I2) the Z → E process does not seem to occur and also the uniqueness of these observations as compared to less hindered stilbene derivatives reported in previous studies.16 Therefore, as represented schematically in Fig. 3a, in stilbene the E and Z conformations stay in an equilibrium under conditions of light irradiation and high temperature, due to the high conformational flexibility of the molecule. Contrary, as sketched in Fig. 3b, in PTM derivatives 1 and 2 the E conformation is kinetically blocked, even when the ethylene unit is electronically excited with light, which impedes the Z → E process and, consequently, shifts the isomerisation equilibrium towards the less stable Z isomer.
 | ||
| Fig. 3 (a) Mechanistic scheme of the isomerisation process taking place in stilbene (a) and the PTM derivative 1 (b), under the effect of light irradiation and temperature. In both cases light opens the central double-bond, which allows the free rotation of the ethylene unit due to thermal fluctuations (RT). Contrary to stilbene, PTM derivatives 1 and 2 get kinetically blocked in the Z conformation, shifting the equilibrium towards such conformation. | ||
In summary, we have studied the photo-induced isomerisation of E-p-vinylphenylene functionalised perchlorotriphenylmethane derivatives to the Z isomer. The isomerisation process has been monitored by HPLC which reveal an almost complete isomerisation. The higher absorption ability of the E isomer as compared to the Z isomer at the same wavelengths prevents the back Z → E isomerisation. No thermal back isomerisation takes place although the E isomer is the most thermodynamically favoured. Theoretical calculations point to the steric hindrance within the Z conformers leading to a possible kinetic blocking of the Z → E transition as the effect contributing to the thermal irreversibility of the E → Z process.
Experimental details
Synthesis of 1 was carried out as previously reported.11Characterization of E-1
UV-vis: λmax (THF)/nm 222 (ε/dm3 mol−1 cm−1 168894), 297 (15709). FT-IR (KBr): νmax./cm−1 3032, 1712 (CO), 1636, 1532, 1493, 1406, 1368, 1351, 1338, 1297, 1240, 1139, 1118, 1115, 1088, 969, 943, 871, 718, 688, 668, 648, 613, 536, 528, 507, 484. 1H-NMR: δH(500 MHz; CDCl3; Me4Si) 7.60 (2H, d, J = 8.5 Hz, ArH), 7.48 (2H, d, J = 8.5 Hz, ArH), 7.13 (1H, d, J = 17 Hz), 7.09 (1H, d, J = 17 Hz, –(C6Cl4)–CHCH–), 7.06 (1H, s, –(C6Cl4)–CHCH–), 2.48 ppm (3H, s, –CH3). 13C-NMR: δC(101 MHz; CDCl3; Me4Si) 137.22, 136.88, 136.82, 136.24, 135.97, 134.82, 134.75, 134.58, 134.53, 133.68, 133.65, 133.37, 133.2, 133.14, 132.21, 132.18, 132.03, 128.19, 127.28, 124.11, 56.34 (α), 30.00 (–CH3). MALDI-ToF (negative mode) (C29Cl14H10SO, M = 902.8) m/z (M − 902.08); (M − 43859.08), (M − 70832.08).
Characterization of Z-1
UV-vis: λmax (THF)/nm 222 (ε/dm3 mol−1 cm−1 104222), 254 (35917), 285 (14087). ATR-IR νmax./cm−1 3125, 1711 (CO), 1534, 1493, 1462, 1358, 1338, 1296, 1262, 1239, 1207, 1188, 1133, 1115, 1090, 1015, 946, 871, 835, 808, 764, 719, 709, 687, 678, 661, 646, 630, 611, 573, 550, 541. 1H-NMR: δH(600 MHz; CD2Cl2; Me4Si) 7.30 (2H, d, J = 8.5 Hz, ArH), 7.02 (2H, m, ArH and 1H αH-PTM), 6.9 (1H, d, J = 12 Hz, –(C6Cl4)–CHCH–), 6.6 (1H, d, J = 12 Hz, –(C6Cl4)–CHCH–), 2.38 (3H, s, CH3) ppm. 13C-NMR: δH(101 MHz; CD2Cl2; Me4Si): δ = 193.87 (CO), 138.32, 137.52, 136.98, 136.96, 135.43, 135.31, 134.84, 134.52, 134.37, 134.30, 134.05, 134.01, 133.93, 133.73, 132.92, 132.68, 129.08, 128.66, 127.88, 125.95, 57.01 (α), 30.48 (–CH3). MS (negative mode) (C29Cl14H10SO, M = 902.8) m/z (M + 4906.4).
Synthesis of 2 (adapted from ref. 12): potassium tert-butoxide (95 mg, 0.87 mmol) was added to a solution of diethyl 4-[bis(2,3,4,5,6-pentachlorophenyl)methyl]-2,3,5,6-tetrachlorobenzyl phosphonate (500 mg, 0.57 mmol) in dry tetrahydrofuran (10 mL) at −90 °C, in a N2/acetone bath. The resulting yellow-orange ylide solution was stirred for 30 minutes. Then, p-bromobenzaldehyde (188 mg, 1.04 mmol) dissolved in 4 mL of dry THF was added drop-wise. The solution was allowed to stir for 24 hours without changing the N2/acetone bath, which reached room temperature along the reaction. The reaction was monitored by thin-layer chromatography (silica-gel). Once the reaction finished, one drop of concentrated HCl was added to the resulting dark purple solution, which turned on a yellow suspension. The reaction mixture was extracted with water (10 mL) and dichloromethane (10 mL). The phases were separated and the aqueous phase was washed with further dichloromethane (2 × 10 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under vacuum. Almost pure E-2 isomer (97%) (473 mg, 92%) was obtained through purification by chromatography (silica gel with hexane as eluent).
Characterization of E-2
UV-vis: λmax (C6H12)/nm 222 (ε/dm3 mol−1 cm−1 107000), 304 (29000). FT-IR (KBr): νmax./cm−1 3015, 1635, 1590, 1535, 1490, 1405, 1365, 1340, 1295, 1240, 1140, 1075, 1010, 965, 940, 870, 805, 790, 715, 645, 532, 495. 1H-NMR: δH(400 MHz; CD2Cl2; Me4Si) 7.59 (2H, m, ArH), 7.48 (2H, m, ArH), 7.12 (1H, d, J = 16.6 Hz, –(C6Cl4)–CHCH–), 7.07 (1H, s, αH-PTM) 7.06 ppm (1H, d, J = 16.6 Hz, –(C6Cl4)–CHCH–). 13C-NMR: δC(101 MHz, CD2Cl2) δ = 137.70, 137.58, 137.05, 137.04, 136.63, 135.55, 135.48, 135.46, 135.26, 134.47, 134.37, 134.00, 133.98, 133.94, 133.91, 133.84, 132.89, 132.72, 132.37, 128.85, 124.22, 123.17, 57.09 (α). MS (C27Cl14H7Br, M = 907.6) (negative mode) (m/z) (M − 1906.4). UV-Vis (C6H12) λ(ε) [nm (M−1 cm−1)] 304 (29000); 222 (107000).
The Z-2 isomer was obtained after irradiating a 3.7 mM solution in THF of the as synthesized 2 for 300 min. Then the solvent was evaporated to characterize the solid.
Characterization of Z-2
UV-vis: λmax(THF)/nm 221 (ε/dm3 mol−1 cm−1 116098), 284 (12337). FT-IR νmax./cm−1 3015, 1725, 1585, 1485, 1370, 1335, 1295, 1240, 1130, 1070, 1010, 830, 805, 760, 710, 680, 655, 640, 550, 530, 460, 425. 1H-RMN: δH(400 MHz; CD2Cl2; Me4Si) 7.39 (2H, m, ArH), 7.00 (1H, s, αH-PTM), 6.91 (2H, d, J = 7.52 Hz, ArH), 6.83 (1H, d, J = 12.01 Hz, –(C6Cl4)–CHCH–), 6.57 ppm (1H, d, J = 12.01 Hz, –(C6Cl4)–CHCH–).13C-RMN: δH(101 MHz; CD2Cl2; Me4Si) 138.25, 136.97, 136.95, 135.56, 135.39, 135.29, 134.52, 134.34, 134.30, 134.07, 134.03, 133.95, 133.71, 133.57, 132.94, 132.65, 132.02, 130.04, 125.48, 122.58, 108.39, 57.01 (α). MS (negative mode) (C27Cl14H7Br, M = 907.6) (m/z) (M − 1906.4).
DFT calculations
For assessing the isomerisation energy profile we twisted the molecular skeleton about the ethylene double-bond axis from 0 to 360 degrees. At each point the molecular structure was optimized while freezing the twist angle of the CC bond. These constrained optimizations were done using the hybrid PBE0 functional21,22 and a 6-311-G++ basis set as implemented in the Gaussian09 code [Gaussian 09, Revision E.01, D. J. Fox, Gaussian, Inc., Wallingford CT, 2009]. Within the same code, the time-dependent DFT method23–29 was used to calculate absorption spectra. Ab initio molecular dynamics (AIMD) simulations at 450 K were run for 5 ps (1 ps of equilibrations plus 4 ps of production) using the Bussi–Donadio–Parrinello thermostat.30 For these calculations the PBE0 functional and a “light” numerical basis set was used as implemented in the FHI-AIMS code.31,32
Acknowledgements
We thank Carlos Franco for his help with the HPLC analysis and Amable Bernabé CTQ2016-80030-R for the MALDI-ToF characterizations. We thank the Networking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN); DGI (Spain) with projects BE-WELL CTQ2013-40480-R and MAT2012-30924, and Generalitat de Catalunya (grant 2009SGR00516, 2014SGR97 and XRQTC). We also thank the EU projects ERC StG 2012-306826 e-GAMES, ITN iSwitch (GA no. 642196), ACMOL (GA no. 618082) and CIG-ELECTROMAGIC (PCIG10-GA-2011-303989). Spanish Ministry of Economy and Competitiveness, through the ‘Severo Ochoa’ Programme for Centres of Excellence in R&D (SEV-2015-0496). N. C. thanks the RyC contract. F. B. acknowledges the FPU fellowship. F. B. is enrolled in the Materials Science PhD program of UAB.References
- B. G. Levine and T. J. Martínez, Annu. Rev. Phys. Chem., 2007, 58, 613–634 CrossRef CAS PubMed.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- D. H. Waldeck, J. Mol. Liq., 1993, 57, 127–148 CrossRef CAS.
- E. Merino and M. Ribagorda, Beilstein J. Org. Chem., 2012, 8, 1071–1090 CrossRef CAS PubMed.
- C. A. Stanier, S. J. Alderman, T. D. W. Claridge and H. L. Anderson, Angew. Chem., Int. Ed., 2002, 41, 1769–1772 CrossRef PubMed.
- E. Durgun and J. C. Grossman, J. Phys. Chem. Lett., 2013, 4, 854–860 CrossRef CAS PubMed.
- F. Vera, M. Mas-Torrent, J. Esquena, C. Rovira, Y. Shen, T. Nakanishi and J. Veciana, Chem. Sci., 2012, 3, 1958–1962 RSC.
- F. Vera, M. Mas-Torrent, C. Avci, J. Arbiol, J. Esquena, C. Rovira and J. Veciana, Chem. Commun., 2013, 49, 7827–7829 RSC.
- N. Crivillers, M. Mas-Torrent, J. Vidal-Gancedo, J. Veciana and C. Rovira, J. Am. Chem. Soc., 2008, 130, 5499–5506 CrossRef CAS PubMed.
- C. Simao, M. Mas-Torrent, N. Crivillers, V. Lloveras, J. Artés, P. Gorostiza, J. Veciana and C. Rovira, Nat. Chem., 2011, 3, 359–364 CrossRef CAS PubMed.
- S. Furukawa, N. Crivillers, A. Minoia, A. Ver Heyen, M. Mas-Torrent, C. Sporer, M. Linares, A. Volodin, C. Van Haesendonck, M. Van Der Auweraer, R. Lazzaroni, S. De Feyter, J. Veciana and C. Rovira, J. Am. Chem. Soc., 2009, 131, 6246–6252 CrossRef PubMed.
- C. Rovira, D. Ruiz-Molina, O. Elsner, J. Vidal-Gancedo, J. Bonvoisin, J. P. Launay and J. Veciana, Chemistry, 2001, 7, 240–250 CrossRef CAS PubMed.
- F. Leyssner, S. Hagen, L. Óvári, J. Dokić, P. Saalfrank, M. V. Peters, S. Hecht, T. Klamroth and P. Tegeder, J. Phys. Chem. C, 2010, 114, 1231–1239 CAS.
- V. Lloveras, J. Vidal-Gancedo, T. M. Figueira-Duarte, J. F. Nierengarten, J. J. Novoa, F. Mota, N. Ventosa, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2011, 133, 5818–5833 CrossRef CAS PubMed.
- H. Meier, Angew. Chem., Int. Ed., 1992, 31, 1399–1420 CrossRef.
- D. Gegiou, K. A. Muszjat and E. Fischer, J. Am. Chem. Soc., 1968, 90, 3907–3918 CrossRef CAS.
- W.-G. Han, T. Lowell, T. Liu and L. Noodleman, ChemPhysChem, 2002, 3, 167–178 CrossRef CAS PubMed.
- D. H. Waldeck, Chem. Rev., 1991, 91, 415–436 CrossRef CAS.
- H. Petek, K. Yoshihara, Y. Fujiwara, Z. Lin, J. H. Penn and J. H. Frederick, J. Phys. Chem., 1990, 94, 7539–7543 CrossRef CAS.
- M. Lee, a. J. Bain, P. J. McCarthy, C. H. Han, J. N. Haseltine, a. B. Smith Iii and R. M. Hochstrasser, J. Chem. Phys., 1986, 85, 4341–4347 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- R. Improta and V. Barone, Chem. Rev., 2004, 104, 1231–1253 CrossRef CAS PubMed.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439 CrossRef CAS.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- C. Van Caillie and R. D. Amos, Chem. Phys. Lett., 1999, 308, 249–255 CrossRef CAS.
- C. Van Caillie and R. D. Amos, Chem. Phys. Lett., 2000, 317, 159–164 CrossRef CAS.
- F. Furche and R. Ahlrichs, J. Chem. Phys., 2002, 117, 7433–7447 CrossRef CAS.
- G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, J. Chem. Phys., 2006, 124, 094107 CrossRef PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS.
- V. Havu, V. Blum, P. Havu and M. Scheffler, J. Comput. Phys., 2009, 228, 8367–8379 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28618f |
| ‡ Equally contributed. |
| This journal is © The Royal Society of Chemistry 2017 |