
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Experimental observation and quantum chemical investigation of thallium(I) (Z)-methanediazotate: synthesis of a long sought and highly reactive species†‡
Neeraj Singha,
Benjamin Fiedler*b,
Joachim Friedrichb and
Klaus Banert *a
*a
aChemnitz University of Technology, Organic Chemistry, Strasse der Nationen 62, 09111 Chemnitz, Germany. E-mail: Klaus.banert@chemie.tu-chemnitz.de
bChemnitz University of Technology, Theoretical Chemistry, Strasse der Nationen 62, 09111 Chemnitz, Germany. E-mail: benjamin.fiedler@chemie.tu-chemnitz.de
First published on 17th March 2017
Abstract
For the first time, successful synthesis and characterisation of the missing (Z)-isomer of thallium(I) methanediazotate has been accomplished, utilising low-temperature NMR monitoring analysis. The title compound was synthesised from N-methyl-N-nitrosourea and thallium(I) propoxide, under sub-ambient temperature conditions, as a highly moisture sensitive entity. Quantum chemical calculations, performed at the CCSD(T) level, depict excellent conformity to experimental results. Indeed, compared to its (E) counterpart, the formation of the title compound is thermodynamically less favoured, but preferred by means of kinetic control owing to a hindered isomerisation.
Alkane diazotates1,2 have long been discussed as highly reactive species, generated in the in vivo metabolism of N-nitrosoureas, and are responsible for the alkylation of DNA, thereby, acting as potent carcinogens.3 Fixation of nitrous oxide (N2O) by frustrated Lewis-pairs (FLP's)4 and N-heterocyclic carbenes5 also leads to the formation of diazotates. The high moisture sensitivity, exhibited by these entities, makes them extremely difficult to isolate and handle at room temperature. In spite of that, certain diazotates, such as 1, 2 and 4–6, have been isolated and characterised by the XRD technique,6 for example, potassium(I) (Z)-methanediazotate (4) (Scheme 1a). It is long known that configurational isomerism exists in the diazotates owing to the restricted rotation about the N–N bond, and that (Z)-isomers are highly reactive when compared to the corresponding (E)-isomers.
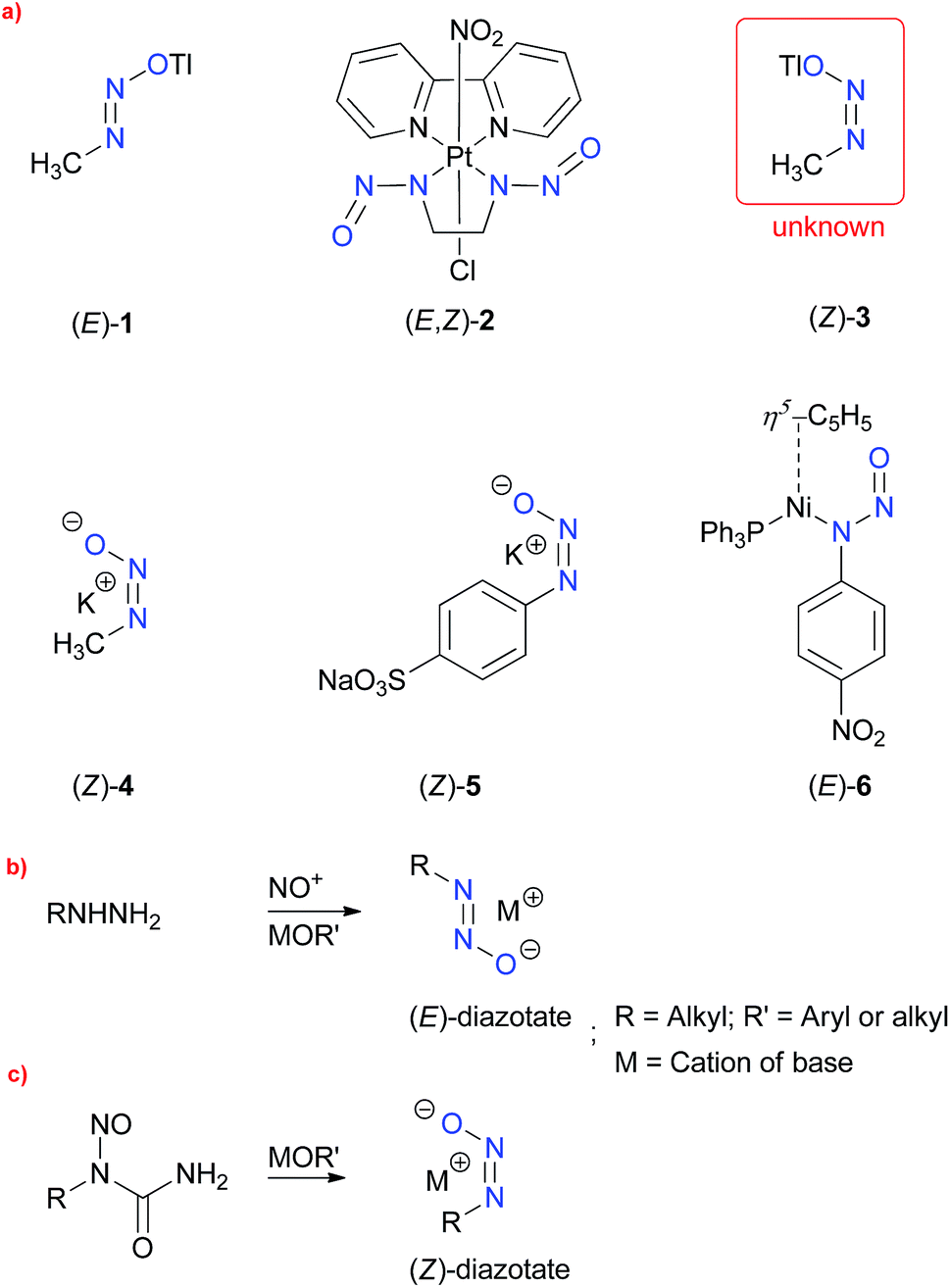 | ||
| Scheme 1 (a) Reported diazotates (1–6) and unknown 4; (b) and (c) established methods for the syntheses of (E)- and (Z)-diazotates, respectively. | ||
Particularly noteworthy are thallium(I) methanediazotates,7 of which heretofore only the (E)-isomer 1 was isolated and characterised by single crystal X-ray analysis, by Keefer et al. Molecule 1 was synthesised by the nitrosation of methylhydrazine in the presence of thallium(I) ethoxide (TlOEt) (Scheme 1b), as a highly crystalline material stable to hydrolysis, a property which is highly contrasting to other similar diazotates and explained by the possible covalent nature of the thallium(I) cation. The configuration was assigned by analogy with the very few (Z)-isomers, obtained by utilising the base induced decomposition of N-alkyl-nitroso-N-acyl derivatives (Scheme 1b). However, the exact mechanism detailing the preference of a particular configurational isomer remains speculative, since contrasting theoretical reports8 exist about the type of the conformation of N-nitrosourea 7 in solvents, and furthermore, no consideration of the solvent was taken into account in determination of the anti/syn existence. Keefer and coworkers,7 however, could not directly characterise thallium(I) (Z)-methanediazotate (3), citing its highly reactive nature, and that still remains a gap in the field of thallium diazotates as well as in the larger case of covalent metal diazotates.
We became interested in the synthesis of (Z)-methanediazotate 3 and also in the theoretical basis for the formation of this diazotate along with the possibility of (Z)–(E) isomerisation under solvation. Thallium(I) alkoxides7,9 are unique bases owing to their covalent character and solubility in a range of organic solvents, even at low-temperatures, which is quintessential for monitoring reactions by low-temperature NMR analysis. Therefore, 15N-labelled N-methyl-N-nitrosourea (7) was synthesised and reacted with TlOEt at −40 °C in CD2Cl2, under constant monitoring by low-temperature NMR spectroscopy.10 In this case, we could only observe the doublet for CH2N15N at δ = 3.34 with 3J(15N,1H) ≈ 1.1 Hz in the 1H NMR spectrum and a signal at δ = 14.17 in the corresponding 15N NMR spectrum. The identity of 15N-labelled diazomethane was further confirmed by 15N,1H gHMBCAD and by comparison with literature known values.11 This result is in complete agreement with the known methods for the synthesis of diazotates and the observed secondary products in the absence of electrophiles (Scheme 2).11
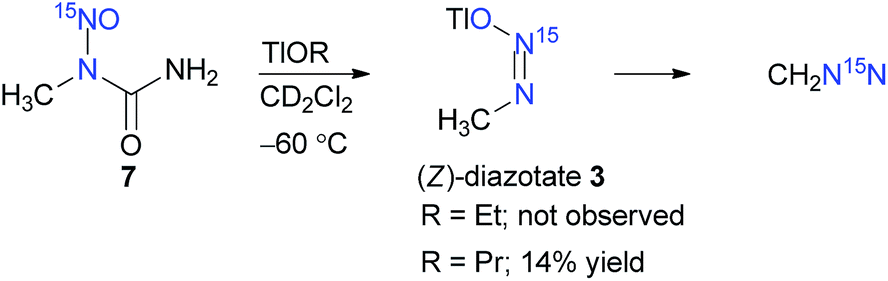 | ||
| Scheme 2 Synthesis of diazotate 3 and subsequent decomposition to CH2N15N. | ||
However, owing to our previous experience with thallium(I) alkoxides, it is known that TlOEt and thallium(I) propoxide (TlOPr)9a react differently, probably because of their varying association nature.9,11 Therefore, to a solution of TlOPr (1.1 eq.) in CD2Cl2, maintained at −60 °C, was added N-nitrosourea 7 (10 mg), and the reaction was analysed by sub-ambient temperature 1H and 15N NMR monitoring technique. To our pleasant surprise, along with the signals of CH2N15N, we also obtained well-defined signals for thallium(I) (Z)-methanediazotate 3 in 14% yield (1H NMR), with a doublet at δ = 3.17 with 3J(15N,1H) = 4.0 Hz in the 1H NMR spectrum (Fig. 1), and a singlet at δ = 104.5 for the 15NOTl unit.10 The structure was further confirmed by 15N,1H gHMBCAD 2D-NMR technique (Fig. 2). Compound 3 was stable for only a few hours at −60 °C, and decomposed completely to CH2N15N (confirmed by NMR as mentioned before), which led to an increase of the total diazomethane yield from 31% to 43%. Since the 1H NMR data and high stability of the corresponding (E)-diazotate7 1 (δ = 3.43), stable at RT, contrasts significantly with the properties of the aforementioned species and because of the additional evidence obtained through 15N NMR as well as 2D-NMR experiments, the structure of the highly elusive (Z)-diazotate 3 is now confirmed.
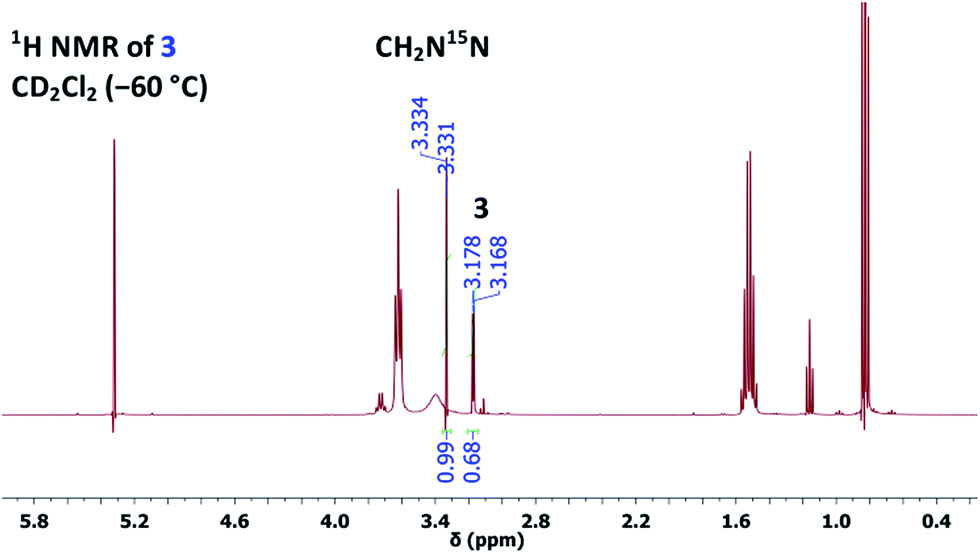 | ||
| Fig. 1 1H NMR spectrum of (Z)-diazotate 3 and CH2N15N. | ||
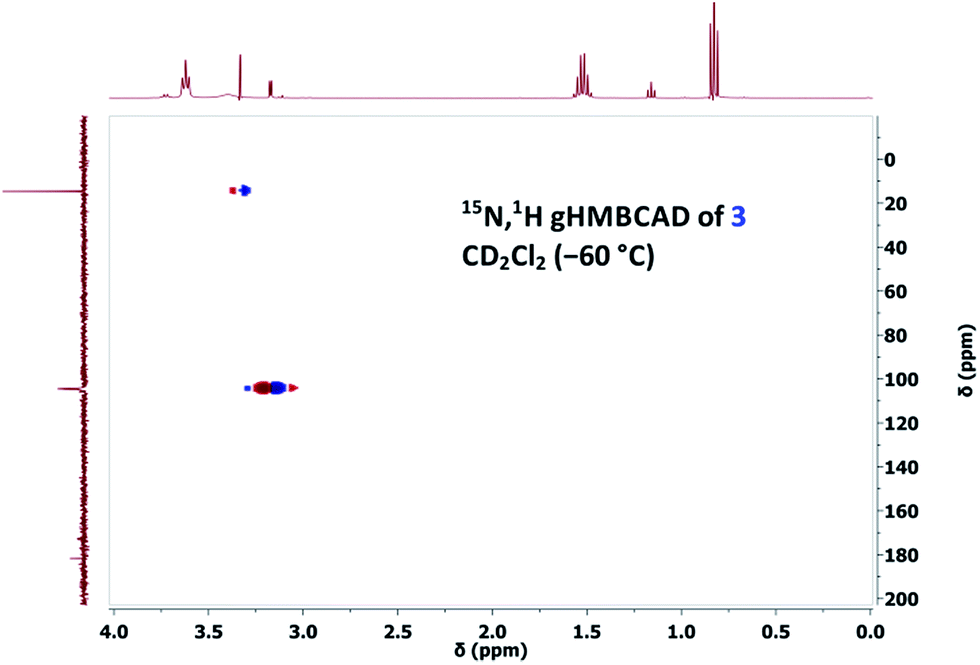 | ||
| Fig. 2 15N,1H gHMBCAD NMR of (Z)-diazotate 3, with nitromethane as external reference. | ||
Moreover, due to the existence of hindered rotation about N–N bond in N-nitrosoureas like 7 in different solvents, thereby, giving rise to syn/anti rotational isomers, it becomes essential to define the preference of a particular rotamer in special solvents. This existence of rotational isomerism plays an important role about the outcome of the stereochemistry of the thallium diazotates 1/3.
Therefore, we performed high-level quantum chemical calculations pertaining to the anti/syn conformational isomerism of N-nitrosourea 7 as well as the (Z)/(E) configurational isomerism of 3/1. Low-level theoretical calculations, without any consideration of solvent contributions, have been performed before, regarding to ionic diazotates, but they cannot be extended to covalent diazotates like 1/3.12 Furthermore, by comparing the isomerisation energetics to the mechanisms for the TlOPr induced formation of both diazotates from the corresponding conformers of 7 (Fig. 3), we explain the occurrence of the thermodynamically less stable (Z)-isomer.
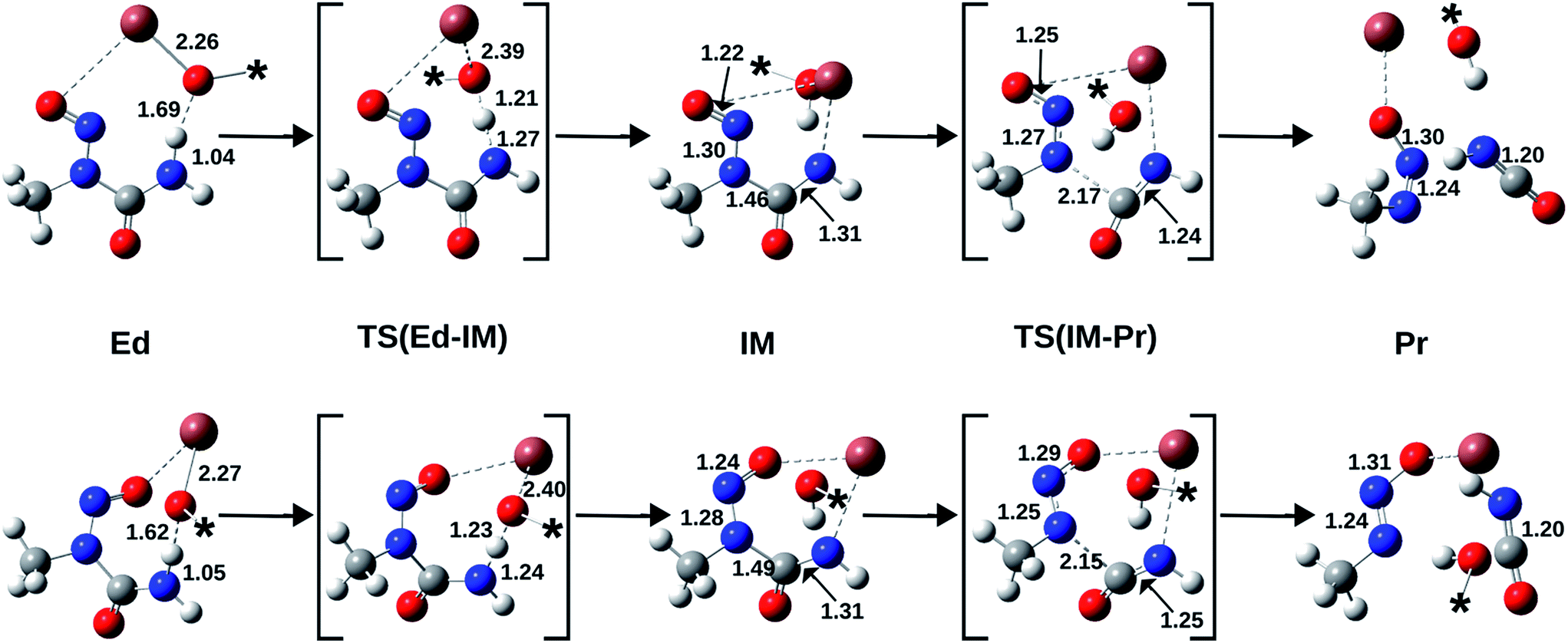 | ||
| Fig. 3 Molecular structures, optimised with PW6B95-D3/TZVPP and COSMO (ε = 8.9), for the reactions anti-7 → 3 (top) and syn-7 → 1 (bottom). Bond lengths (Å) have been depicted and the asterisk (*) represents the n-propyl group, which has been omitted for clear representation. Atoms are H (white), C (grey), N (blue), O (red), Tl (brown). | ||
The molecular geometries were optimised by TURBOMOLE V6.5,13 using RI-DFT14 with the PW6B95 functional,15 the D3 dispersion correction,16 the TZVPP basis set17 (def2-TZVPP with corresponding ECP for Tl18) as well as the COSMO solvation model (ε = 8.9 for CD2Cl2).19 Numerical frequency analyses were performed to characterise the stationary points and to obtain the zero point vibrational energy.20 The freeh module was used to calculate the Gibbs free energy contribution at the reaction temperature (−60 °C). We also performed CCSD(T)/cc-pVXZ21 (X = T, Q) calculations (with pseudopotential for Tl22) and a subsequent CBS(34) extrapolation23 according to eqn (1) and (2) as well as CCSD(T)(F12)/cc-pVTZ-F12 (ref. 24) (aug-cc-pwCVTZ-PP basis for Tl22) calculations to obtain highly accurate electronic energies. Additionally, we applied the focal-point method in eqn (3), using an MP2 increment to approach the CBS limit.
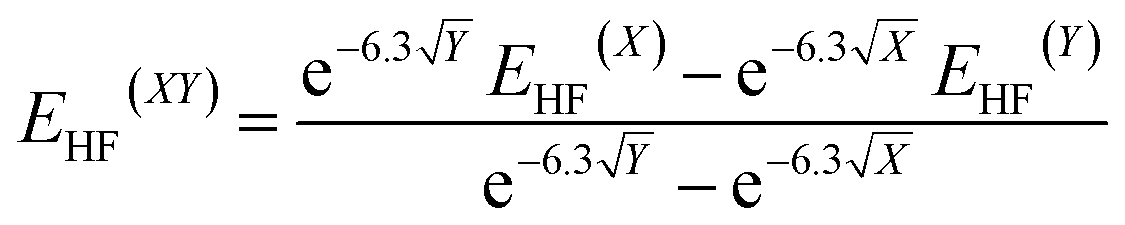 | (1) |
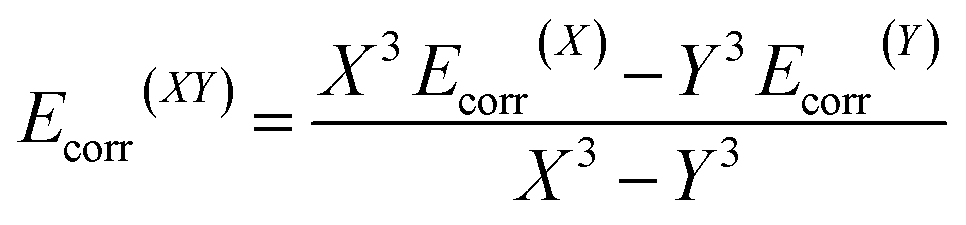 | (2) |
| EfoT/TZ = Ecc-pVTZCCSD(T) + (Ecc-pVTZ-F12MP2-F12 − Ecc-pVTZMP2) | (3) |
With the single-point energy EM, obtained by method M, and a Gibbs free energy contribution G−60°C as well as a COSMO solvation correction ΔEsolv, both at the PW6B95-D3/TZVPP level, the total Gibbs free energy of a structure is obtained as:
| G = EM + G−60°C + ΔEsolv | (4) |
| ΔEsolv = ECOSMO − EnoCOSMO | (5) |
As shown in Table 1, the reaction energy (ΔGreact) for the anti–syn isomerisation of 7 is positive, representing the higher stability of the anti-rotamer. In contrast, the (Z)-diazotate 3, which yields from the anti-rotamer, is less stable than the (E) form. Indeed, the experimental results show the formation of the (Z) isomer, which excludes a thermodynamic reaction control (Fig. 3).
| Method M | DFTa | foT/TZb | CBS(34)c | TZ-F12d | |
|---|---|---|---|---|---|
| a PW6B95-D3/TZVPP.b According to eqn (3), Tl: aug-cc-pwCVTZ-PP basis instead of cc-pVTZ-F12.c CBS(34) extrapolated CCSD(T)/cc-pVXZ according to eqn (1) and (2) with X = 3, Y = 4.d CCSD(T)(F12)/cc-pVTZ-F12, Tl: aug-cc-pwCVTZ-PP basis instead of cc-pVTZ-F12. | |||||
| anti-7 → syn-7 | ΔGact | +103.2 | +89.8 | +91.5 | +90.8 |
| ΔGreact | +20.7 | +18.6 | +18.9 | +18.9 | |
| 3 → 1 | ΔGact | +176.7 | +191.4 | +192.2 | +192.8 |
| ΔGreact | −17.8 | −14.0 | −16.7 | −15.1 | |
Furthermore, the activation barrier (ΔGact) for the formation of the syn-rotamer is around 90 kJ mol−1 and therefore rather large, representing the hindered rotation. However, the direct isomerisation of 3 to 1 needs significantly more activation (>190 kJ mol−1) and can thus be excluded. Also, the covalent nature of the Tl–X (X = O, N) bond is supported by the Tl–O bond length in (Z)-diazotate 3 (2.42 Å; Fig. 4), in accordance with published results.
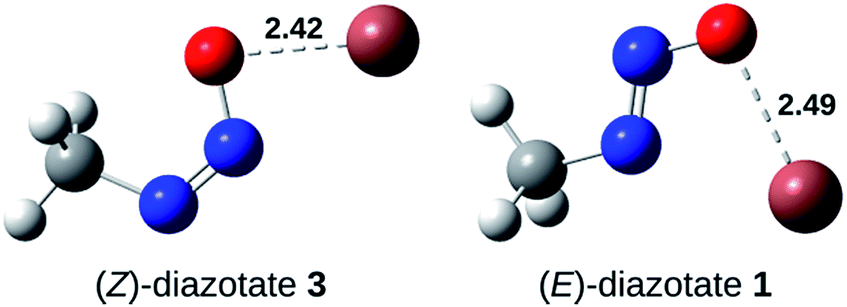 | ||
| Fig. 4 Molecular structures, optimised with PW6B95-D3/TZVPP and COSMO (ε = 8.9), of both thallium(I) diazotate conformers. The lengths (Å) of the Tl–O bonds are shown. Atoms are H (white), C (grey), N (blue), O (red), Tl (brown). | ||
Considering the different highly accurate CCSD(T) methods to approach the complete basis set (CBS) limit, the relative energies of all three (Table 1) are in perfect agreement. Hence, the most efficient of them, the focal-point method in eqn (3), provides a suitable accuracy and expensive CCSD(T)/cc-pVQZ or CCSD(T)-F12/cc-pVTZ-F12 calculations are not necessary for the more extended model system, which is used for the reaction mechanisms.
Therefore, we modelled the mechanisms for the formation of 3 (respectively 1) from anti-7 (respectively syn-7) by using the focal-point method (Fig. 5). Again, the higher stability of the anti-educt and the lower one of the (Z)-product can be seen. Furthermore, the first reaction step, i.e. deprotonation of the urea by propoxide, is exothermic with a very low barrier in both cases. In contrast, the second step, i.e. the N–C bond cleavage, is endothermic for the (Z)-isomer and slightly exothermic for (E). However, this second step is rate-determining as it provides clearly larger activation energies of about 58 kJ mol−1 for (E) and 82 kJ mol−1 for (Z).
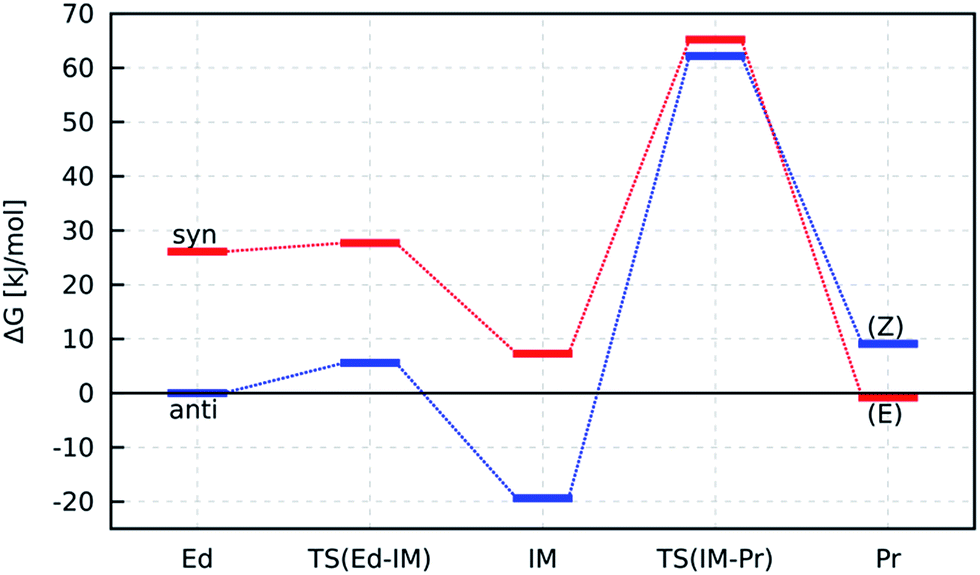 | ||
| Fig. 5 Energy pathways for the reactions anti-7 → 3 (blue) and syn-7 → 1 (red). Gibbs free energies are calculated with the focal-point method (eqn (3)) for EM respectively with PW6B95-D3/TZVPP (G−60°C, ΔEsolv) and shown relative to anti-7. | ||
For completeness, we also calculated the rotation barrier for the intermediate (IM) of the reaction, i.e. the deprotonated N-nitrosourea, and obtained 126 kJ mol−1. Thus, if an isomerisation took place, it would occur at the educt step, as intermediate and product structure provide significantly higher rotation barriers. However, the barrier for the isomerisation of anti-7 (≈90 kJ mol−1) is still larger than the rate-determining step of the formation of 3 from anti-7 (≈82 kJ mol−1). Thus, the formation of the thermodynamically less stable (Z)-diazotate 3 can be explained by kinetic reaction control.
Conclusions
In summary, synthesis and characterisation of the elusive thallium(I) Z-diazotate 3 has been achieved for the first time. It was generated as a highly temperature and moisture sensitive entity by TlOPr induced decomposition of N-nitrosourea 7. The structure of the title compound has been confirmed unambiguously by 1H NMR, 15N NMR and 15N,1H gHMBCAD NMR spectroscopic analytical technique. The decomposition of (Z)-diazotate 3 to CH2N15N, also confirms the general rule about the decomposition of other known (Z)-diazotates to diazomethane, in the presence of a base. Quantum chemical calculations, performed at the CCSD(T) level, reveal a kinetic, rather than a thermodynamic control, for the mechanism of the (Z)-diazotate 3 formation. Indeed, the (Z)-diazotate is thermodynamically less stable than the (E) form, but generated from the more stable anti-conformer of nitrosourea 7. Furthermore, the anti/syn rotational isomerisation is hindered and features even a larger activation energy than the rate-determining step within the formation of title compound 3.N-Methyl-N-nitrosourea is a highly potent carcinogen, therefore, great precautions should be taken while handling it. Thallium(I) propoxide is a toxic compound and should be handled in accordance with safety protocols. Thallium (E)-diazotate can also lead to spontaneous explosions,7 therefore great care must be exercised while handling the highly reactive (Z)-diazotate.
Acknowledgements
We are grateful to Dr M. Hagedorn for his assistance with low-temperature NMR measurements. N. S. thanks DAAD (Deutscher Akademischer Austauschdienst) for a PhD fellowship. B. F. thanks the DFG Research Group FOR 1497 for financial support.Notes and references
- R. A. Moss, Acc. Chem. Res., 1974, 7, 421–427 CrossRef CAS.
- (a) P. Griefs, Justus Liebigs Ann. Chem., 1866, 137, 39–91 CrossRef; (b) A. Wohl, Ber. Dtsch. Chem. Ges., 1892, 25, 3631–3634 CrossRef; (c) A. Engler, Ber. Dtsch. Chem. Ges., 1900, 33, 2188–2190 CrossRef CAS; (d) K. J. P. Orton, J. Chem. Soc., 1903, 83, 796–814 RSC; (e) E. Müller and H. Haiss, Chem. Ber., 1963, 96, 570–583 CrossRef; (f) E. H. White, T. J. Ryan and K. W. Field, J. Am. Chem. Soc., 1972, 94, 1360–1361 CrossRef CAS; (g) E. Müller, H. Haiss and W. Rundel, Chem. Ber., 1960, 93, 1541–1552 CrossRef; (h) E. Müller, W. Hoppe, H. Hagenmaier, H. Haiss, R. Huber, W. Rundel and H. Suhr, Chem. Ber., 1963, 96, 1712–1719 CrossRef.
- (a) W. P. Tong and D. B. Ludlum, Biochem. Pharmacol., 1979, 28, 1175–1179 CrossRef CAS PubMed; (b) W. P. Tong, M. C. Kirk and D. B. Ludlum, Cancer Res., 1982, 42, 3102–3105 CAS; (c) J. A. Montgomery, Cancer Treat. Rep., 1976, 60, 651–664 CAS; (d) B. Singer, Nature, 1976, 264, 333–339 CrossRef CAS PubMed; (e) C. T. Gombar, W. P. Tong and D. B. Ludlum, Biochem. Biophys. Res. Commun., 1979, 90, 878–882 CrossRef CAS PubMed; (f) J. D. Scribner and G. P. Ford, Cancer Lett., 1982, 16, 51–56 CrossRef CAS PubMed; (g) K. K. Park, M. C. Archer and J. S. Wishnok, Chem.-Biol. Interact., 1980, 29, 139–144 CrossRef CAS PubMed; (h) K. Morimoto, A. Tanaka and T. Yamaha, Carcinogenesis, 1983, 4, 1455–1458 CrossRef CAS PubMed.
- E. Otten, R. C. Neu and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 9918–9919 CrossRef CAS PubMed.
- A. G. Tskhovrebov, E. Solari, M. D. Wodrich, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2012, 51, 232–234 CrossRef CAS PubMed.
- (a) W. A. Freeman, J. Am. Chem. Soc., 1983, 105, 2725–2729 CrossRef CAS; (b) R. Huber, R. Langer and W. Hoppe, Acta Crystallogr., 1965, 18, 467–473 CrossRef CAS; (c) L. Malatesta, G. La Monica, M. Manassero and M. Sansoni, Gazz. Chim. Ital., 1980, 110, 113–121 CAS; (d) N. W. Alcock, P. D. Goodman and T. J. Kemp, J. Chem. Soc., Perkin Trans. 2, 1980, 1093–1095 RSC; (e) F. J. Lalor, T. J. Desmond, G. Ferguson and P. Y. Siew, J. Chem. Soc., Dalton Trans., 1982, 1981–1985 RSC.
- L. K. Keefer, S.-m. Wang, T. Anjo, J. C. Fanning and C. S. Day, J. Am. Chem. Soc., 1988, 110, 2800–2806 CrossRef CAS.
- (a) A.-M. Sapse, G. Snyder and L. Osorio, Cancer Res., 1984, 44, 1904–1907 CAS; (b) N. Nakayama and O. Kikuchi, J. Mol. Struct.: THEOCHEM, 2001, 536, 213–218 CrossRef CAS.
- (a) P. J. Burke, R. W. Matthews and D. G. Gillies, J. Chem. Soc., Dalton Trans., 1980, 1439–1442 RSC; (b) C. Janiak, Coord. Chem., 1997, 163, 107–216 CrossRef CAS.
- See ESI for further details.‡.
- K. Banert, N. Singh, B. Fiedler, J. Friedrich, M. Korb and H. Lang, Chem. – Eur. J., 2015, 21, 15092–15099 CrossRef CAS PubMed.
- J. W. Lown, M. S. Chauhan, R. R. Koganty and A.-M. Sapse, J. Am. Chem. Soc., 1984, 106, 6401–6408 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- (a) O. Treutler and R. Ahlrichs, J. Chem. Phys., 1995, 102, 346–354 CrossRef CAS; (b) K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652–660 CrossRef CAS; (c) K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2005, 109, 5656–5667 CrossRef CAS PubMed.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143–152 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- P. Deglmann, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2002, 362, 511–518 CrossRef CAS.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- (a) K. A. Peterson, J. Chem. Phys., 2003, 119, 11099–11112 CrossRef CAS; (b) B. Metz, M. Schweizer, H. Stoll, M. Dolg and W. Liu, Theor. Chem. Acc., 2000, 104, 22–28 CrossRef CAS; (c) K. A. Peterson and K. E. Yousaf, J. Chem. Phys., 2010, 133, 174116 CrossRef PubMed; (d) C. Hättig, G. Schmitz and J. Koßmann, Phys. Chem. Chem. Phys., 2012, 14, 6549–6555 RSC.
- (a) S. J. Zhong, E. C. Barnes and G. A. Petersson, J. Chem. Phys., 2008, 129, 184116 CrossRef PubMed; (b) A. Halkier, T. Helgaker, P. Jørgensen, W. Klopper, H. Koch, J. Olsen and A. K. Wilson, Chem. Phys. Lett., 1998, 286, 243–252 CrossRef CAS.
- (a) D. P. Tew, W. Klopper, C. Neiss and C. Hättig, Phys. Chem. Chem. Phys., 2007, 9, 1921–1930 RSC; (b) H. Fliegl, W. Klopper and C. Hättig, J. Chem. Phys., 2005, 122, 084107 CrossRef PubMed; (c) K. A. Peterson, T. Adler and H.-J. Werner, J. Chem. Phys., 2008, 128, 084102 CrossRef PubMed.
Footnotes |
| † Dedicated to Prof. V. K. Singh, Director, IISER Bhopal, India. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, copies of NMR spectra as well as coordinates and energies from quantum chemical calculations. See DOI: 10.1039/c7ra00872d |
| This journal is © The Royal Society of Chemistry 2017 |