DOI:
10.1039/C7RA00826K
(Paper)
RSC Adv., 2017,
7, 17923-17933
Three novel transition metal complexes of 6-methyl-2-oxo-quinoline-3-carbaldehyde thiosemicarbazone: synthesis, crystal structure, cytotoxicity, and mechanism of action†
Received
19th January 2017
, Accepted 14th March 2017
First published on 24th March 2017
Abstract
Three novel 6-methyl-2-oxo-quinoline-3-carbaldehyde thiosemicarbazone (H-L) transition metal complexes, [Cu(H-L)NO3H2O]·NO3 (1), [Zn(H-L)NO3H2O]·NO3 (2) and [CoL2] (3), were synthesized. In vitro antitumor screening revealed that complex 1 exhibited better inhibitory activities than the commercial anticancer drug cisplatin against SK-OV-3 and MGC80-3 tumor cell lines, with IC50 values of 10.35 ± 1.26 μM and 10.17 ± 0.95 μM, respectively. All three complexes showed low cytotoxicity toward the normal human liver HL-7702 cells compared with cisplatin. Their binding properties to DNA were investigated by various methods. It was found that the complexes interacted with DNA mainly through intercalation, and their binding affinities ranked in the order of 3 > 1 > 2. Complex 1 induced the highest apoptosis rate of MGC80-3 cells, and it caused cell arrest in the S phase according to flow cytometry. Further experiments confirmed that complex 1 triggered MGC80-3 cells apoptosis via a mitochondrial dysfunction pathway.
1. Introduction
Over the past three decades, inspired by the success of cisplatin and related platinum complexes as anticancer agents,1–3 researchers have actively explored transition metal complexes to search for novel compounds with better medicinal activity. Many organic ligands that exhibit good antitumor activity have been designed and combined with transition metals to give novel complexes as anticancer drugs. In our previous work, synthetic heterocyclic compounds that are active ingredients in traditional Chinese medicines were employed as organic ligands to successfully produce new transition metal complexes that possessed stronger anticancer activities.4–8
The anticancer activities of quinoline and its derivatives have been intensively studied in drug discovery and development.9–11 2-Oxo-quinoline is a quinoline alkaloid that is widely found in nature. Some synthetic derivatives of 2-oxo-quinoline were found to exhibit superior anticancer activity.12–14 Besides, thiosemicarbazones and their metal complexes also possess good antitumor activities.15–18 Nevertheless, to the best of our knowledge, the transition metal complexes of thiosemicarbazones derived from 6-methyl-2-oxo-quinoline-3-carbaldehyde have not been examined as potential anticancer agents.
In this work, we choose the biocompatible endogenous metal ions such as Cu(II), Zn(II) and Co(II) which play essential role in biological living system. Zinc has an important role in various biological systems,19 since it is critical for numerous cell processes and is a major regulatory ion in the metabolism of cells.20 In the literature, diverse zinc complexes with biological activity are reported. More specifically, crystal structures of zinc complexes with drugs used for the treatment of Alzheimer disease21 or presenting anticonvulsant,22 antidiabetic,23 anti-inflammatory,24 antimicrobial,25 antiproliferative or antitumor26–28 and bactericidal29 activity are now available. In addition, some Co(II) complexes exhibit interesting biological activity such as antifungal antibacterial and anticancer activity. For instance, Co(II) complexes of acyl-hydrazones are known for wide spectrum of biological and pharmaceutical activities, such as inhibition of tumor growth,30,31 antioxidative effect,32 antimicrobial and antiviral.33 In the previous literature, a number of Cu(II) complexes with biological activities such as antibacterial and anti-cancer properties have been also reported.34,35
In the present work, we synthesized and characterized three transition metal complexes of 6-methyl-2-oxo-quinoline-3-carbaldehyde thiosemicarbazone (H-L), and we measured their in vitro cytotoxicities against five tumor cell lines. The mechanism of their interaction with DNA was also examined.
2. Experimental
2.1. Materials
All chemical reagents, including nitrate salts and solvents, were of analytical grade. The ligand H-L was synthesized according to the literature.36 Ethidium bromide (EB) was purchased from Biotium (ShangHai, China). Calf thymus DNA (ct-DNA) was purchased from Sigma-Aldrich. The pBR322 plasmid DNA was purchased from TaKaRa Biotech. All materials were used as received without further purification unless noted otherwise. The synthetic complexes were dissolved in dimethyl sulfoxide (DMSO) as a stock solution (2.0 mM) for DNA binding studies. The TBS buffer (5 mM Tris, 50 mM NaCl, adjusted to pH = 7.3 with hydrochloric acid) was prepared using double distilled water. A TBS solution of the ct-DNA gave a ratio of UV absorbance at 260–280 nm of ca. 1.85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating that the DNA was sufficiently free of protein. The DNA concentration per nucleotide was determined spectrophotometrically by employing a molar absorption coefficient of 6600 M−1 cm−1 at 260 nm.
:1, indicating that the DNA was sufficiently free of protein. The DNA concentration per nucleotide was determined spectrophotometrically by employing a molar absorption coefficient of 6600 M−1 cm−1 at 260 nm.
2.2. Instrumentation
Elemental analyses (C, H, N) were carried out on a Perkin Elmer Series II CHNS/O 2400 elemental analyzer. NMR spectra were recorded on a Bruker AV-500 NMR spectrometer. Fluorescence measurements were performed on a Shimadzu RF-5301/PC spectrofluorophotometer. The circular dichroism (CD) spectra of the DNA were obtained at 25 °C on a JASCO J-810 spectropolarimeter. The region between 200 and 400 nm was scanned for each sample. The UV-Vis spectra were recorded on a TU-1901 ultraviolet spectrophotometer.
2.3. Synthesis of complex 1
The mixture of ligand H-L (0.26 g, 1 mmol) and Cu(NO3)2·6H2O (0.29 g, 1 mmol) in 20 mL methanol was maintained at reflux (70 °C) for 8 h to afford complex 1 as blue crystals in 60% yield. The blue block crystals of 1 suitable for X-ray diffraction analysis were subsequently harvested. MS m/z: 321.9 [Cu(H-L)]+. Anal. calcd for C12H16N6O9SCu: C, 29.78; H, 3.33; N, 17.37; found: C, 29.80; H, 3.36; N, 17.35.
2.4. Synthesis of complex 2
Through the same procedure, complex 2 was obtained from Zn(NO3)2·6H2O as yellow crystals in 85% yield. The yellow block crystals of 2 suitable for X-ray diffraction analysis were subsequently harvested. 1H NMR (500 MHz, DMSO-d6) δ 11.95 (s, 1H, NH), 11.65 (s, 1H, NH), 8.72 (s, 1H, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 8.33 (s, 1H, NH), 8.28 (s, 1H, CCH), 8.10 (s, 1H, NH), 7.43 (s, 1H, H-Ar), 7.37 (d, J = 8.5 Hz, 1H, H-Ar), 7.23 (d, J = 8.4 Hz, 1H, H-Ar), 2.36 (s, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ 178.50, 161.34, 137.42, 137.38, 135.37, 132.83, 131.82, 128.36, 125.65, 119.64, 115.59, 20.89; MS m/z: 322.9988 [Zn(H-L)]+. Anal. calcd for C12H14N6O8SZn: C, 30.81; H, 3.02; N, 17.97; found: C, 30.79; H, 3.01; N, 17.99.
CH), 8.33 (s, 1H, NH), 8.28 (s, 1H, CCH), 8.10 (s, 1H, NH), 7.43 (s, 1H, H-Ar), 7.37 (d, J = 8.5 Hz, 1H, H-Ar), 7.23 (d, J = 8.4 Hz, 1H, H-Ar), 2.36 (s, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ 178.50, 161.34, 137.42, 137.38, 135.37, 132.83, 131.82, 128.36, 125.65, 119.64, 115.59, 20.89; MS m/z: 322.9988 [Zn(H-L)]+. Anal. calcd for C12H14N6O8SZn: C, 30.81; H, 3.02; N, 17.97; found: C, 30.79; H, 3.01; N, 17.99.
2.5. Synthesis of complex 3
Through the same procedure, complex 3 was obtained from Co(NO3)2·6H2O in 86% yield. The brown color product of 3 was suitable for its structural characterization. 1H NMR (500 MHz, DMSO-d6) δ 13.94 (s, 1H, NH), 8.78 (d, J = 8.6 Hz, 2H, CHN and H-Ar), 7.69 (s, 1H, CHC), 7.58 (d, J = 8.5 Hz, 1H, H-Ar), 7.43 (s, 2H, NH2), 7.39 (d, J = 8.5 Hz, 1H, H-Ar), 2.42 (s, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ 178.41, 158.84, 148.71, 146.60, 136.61, 135.50, 134.65, 128.54, 121.78, 120.73, 116.60, 20.97. MS m/z: 577.1 [Co(L)2]+. Anal. calcd for C24H22CoN8O2S2: C, 49.91; H, 3.84; N, 19.40; found: C, 49.87; H, 3.75; N, 19.29.
2.6. X-ray crystallography
The blue block crystal of complex 1 (0.21 × 0.14 × 0.07 mm) was measured on a Bruker Smart Apex II or a Rigaku Saturn CCD diffractometer with graphite monochromatic Mo-Kα radiation (λ = 0.71074 Å) at 298(2) K, 2.5° < θ < 26.3° for hkl (−16 ≤ h ≤ 15, −14 ≤ k ≤ 16, −16 ≤ l ≤ 14) in the triclinic lattice. The yellow block crystal of complex 2 (0.17 × 0.08 × 0.06 mm) was measured on a Bruker Smart Apex II or a Rigaku Saturn CCD diffractometer with graphite monochromatic Mo-Kα radiation (λ = 0.71074 Å) at 298(2) K, 3.1° < θ < 25.7° for hkl (−10 ≤ h ≤ 8, −11 ≤ k ≤ 11, −14 ≤ l ≤ 14) in the triclinic lattice. The positions and anisotropic thermal parameters of all non-hydrogen atoms were refined on F2 by full-matrix least-squares techniques with the SHELX-97 program package.37 Absorption correction was conducted with semi-empirical methods from equivalents.
2.7. In vitro cytotoxicity
The BEL-7404, Hep-G2, SK-OV-3, MGC80-3 and HeLa cancer cell lines and the normal liver cell HL-7702 were obtained from the Shanghai Cell Bank in the Chinese Academy of Sciences. Cell lines were grown in triplicate in 96-well plates (Gibco, UK) and incubated for 48 h at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air. To investigate the potential activity of complexes 1–3, cisplatin was employed as a reference metallodrug. Cytotoxicity assays were carried out in 96-well, flat bottomed microtitre plates. Specifically, the supplemented culture medium with cell lines was added to the wells. Complexes 1–3 and cisplatin were dissolved in the culture medium at various concentrations (1.25, 2.5, 5.0, 10.0 and 20.0 μM) with 1% DMSO, and the resulting solutions were subsequently added to a set of wells. The control wells contained supplemented medium with 1% DMSO. The microtitre plates were then incubated at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air for 2 days. Cytotoxic screening through 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was conducted. At the end of each incubation period, the MTT solution (10 mL, 5 mg mL−1) was added into each well and the cultures were incubated for a further 48 h (the treatment times were 24, 48 and 72 h for time-dependent cytotoxicity studies) at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air. After removal of the supernatant, DMSO (150 mL) was added to dissolve the formazan crystals. The absorbance at 570 and 630 nm was read from an enzyme labeling instrument. Cytotoxicity was estimated based on the percentage cell survival in a dose-dependent manner relative to the negative control. The final IC50 values were calculated by the Bliss method (n = 5). All tests were repeated in at least three independent experiments.
2.8. Spectroscopic studies on DNA interaction
The stock solution of ct-DNA (2 × 10−3 M) was stored at 4 °C for no more than 5 days before use. For DNA binding studies, stock solutions of the synthesized complexes 1–3 in DMSO (2.0 mM) were diluted with TBS (at ∼11.5:1 v/v) to give the final working solutions that contained no more than 8% DMSO. For the UV-Vis absorption experiments, the stock solutions of 1–3 (2.0 mM in DMSO) were used directly as the working solutions. Increasing amounts of the ct-DNA stock solution were added until saturation was achieved, and the solution was allowed to incubate for 5 min after each addition before the absorption spectra were recorded. The intrinsic binding constant Kb was determined by the equation:38 [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/Kb(εb − εf), where [DNA] is the concentration of the DNA per nucleotide in base pairs. The apparent absorption coefficients εa, εf and εb correspond to Aobsd/[compound], the extinction coefficient for the free compounds, and the extinction coefficient for the compounds fully bound with DNA, respectively. The ratio of the slope to the y intercept of the linear fit of [DNA]/(εa − εf) versus [DNA] gives the intrinsic absorption coefficient Kb of the compounds.
A solution containing 2 × 10−4 M DNA and 2 × 10−5 M EB ([DNA]/[EB] = 10:1) was prepared for EB–DNA competitive binding studies. Fluorescence emission spectra were recorded with a slit width of 5 nm/5 nm for Ex/Em. To compare the quenching efficiency, the quenching constant (Kq) of each compound was obtained by plotting F0/F versus [Q] according to the classic Sterne–Volmer equation:39 F0/F = 1 + Kq × [Q], where F0 and F are the peak emission intensities of the EB–DNA system in the absence and presence of each complex as the quencher, and [Q] is the concentration of the quencher. The CD absorption spectra of the DNA in TBS were measured at 100 nm min−1 scan rate over 200–400 nm, with 1 × 10−4 M DNA in the absence and presence of each compound at various concentrations. The CD signal of TBS was taken as the background and subtracted from the spectra. All spectroscopic experiments were performed at 25 °C.
2.9. Agarose gel electrophoresis assay
For plasmid DNA unwinding experiments, the stock solutions of all complexes (2.0 mM in DMSO) were diluted with TBE buffer (Tris/boric acid/EDTA buffer solution) to 20–100 μM. The complexes at designated concentrations were mixed with 0.5 mg DNA and diluted to 25 mL with TBE buffer so that electrophoresis on each sample could be conducted twice. All samples were incubated at 25 °C in dark for 4 h. Each sample (12 mL) was then mixed with DNA loading buffer (2 mL) and underwent electrophoresis on 0.8% agarose gel immersed in TBE buffer for 90 min at 5 V cm−1, followed by visualization on a BIO-RAD imaging system with a UV-Vis transilluminator.
2.10. Apoptosis assays by flow cytometry
Phosphate buffered saline (PBS) solution (2 mmol KH2PO4, 10 mmol Na2HPO4, 136.7 mmol NaCl, 2.68 mmol KCl, adjusted to pH = 7.4 with hydrochloric acid) was prepared using double distilled water (adjusted to a volume of 1000 mL). The ability of complex 1 to induce apoptosis was evaluated by flow cytometry of a MGC80-3 cell line with annexin V-FITC and PI counterstaining. MGC80-3 cells undergoing exponential growth were inoculated in six-well plates and cultured for 24 h before complex 1 was added at designated final concentrations. After incubation for 24 h, the cells were harvested, washed twice in PBS, and resuspended in 100 mL binding buffer (140 mmol L−1 NaCl, 2.5 mmol L−1 CaCl2, 10 mmol L−1 HEPES/NaOH, pH 7.4) at a concentration of 1 × 106 cells per mL. The cells were then incubated with 5 μL annexin V-FITC (in a buffer containing 10 mmol L−1 NaCl, 1% bovine serum albumin, 0.02% NaN3, and 50 mmol L−1 Tris, pH 7.4) and 10 μL PI (20 μg mL−1) for 15 min at room temperature in dark. Cells were protected from light before analysis by flow cytometry on a Becton Dickinson FACSCalibur analyzer.
2.11. Cell cycle analysis
The MGC80-3 cells were maintained in Dulbecco's modified Eagle's medium with 10% fetal calf serum in 5% CO2 at 37 °C. The cells were harvested by trypsinization and rinsed with PBS and centrifuged at 3000g for 10 min. The pellet (105 to 106 cells) was suspended in PBS (1 mL) and kept on ice for 5 min. The cells suspension was then fixed by the dropwise addition of 9 mL pre-cooled (4 °C) 100% ethanol with vigorous shaking, and the fixed samples were kept at 4 °C until use. For staining, the cells were centrifuged, resuspended in PBS, digested with 150 mL RNase A (250 μg mL−1), treated with 150 mL PI (0.15 mM), and then incubated for 30 min at 4 °C. PI-positive cells were counted with a FACScan fluorescence-activated cell sorter (FACS). The population of cells in each cell cycle was determined in the Cell Modi FIT software (Becton Dickinson).
2.12. Other experimental methods
The ESI† provides the detailed procedures of other experimental methods, including the measurement of mitochondrial membrane potential (by JC-1 staining), ROS generation, intracellular free Ca2+, Western blot, and caspase-3/9 activity. The procedures were similar to those given in the previous work of Chen et al.40
2.13. Statistics
Data were processed in SPSS 13.0. Results were considered statistically significant when P ≤ 0.05 (Student's t-test).
3. Results and discussion
3.1. Chemistry
Complexes 1–3 were synthesized as outlined in Scheme 1. The 6-methyl-2-oxo-quinoline-3-carbaldehyde thiosemicarbazone (H-L) was obtained from 4-methyl-acetanilide in good yield through the Vilsmeier–Haack–Arnold reaction, hydrolysis and condensation.36 Complexes 1–3 were then synthesized by treating ligand H-L with Cu(NO3)2·6H2O, Zn(NO3)2·6H2O, and Co(NO3)2·6H2O in hot methanol, respectively.
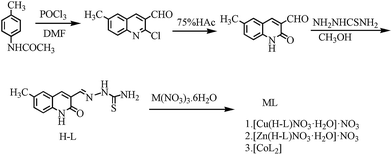 |
| | Scheme 1 Synthesis of the transition metal complexes 1–3. | |
Mass spectrometry (MS), elemental analysis (EA) and crystal diffraction data suggested that the Cu(II) complex was [Cu(H-L)NO3H2O]·NO3 (complex 1). Data from 1H NMR, 13C NMR, MS, EA and crystal diffraction suggested that the Zn(II) complex was [Zn(H-L)NO3H2O]·NO3 (complex 2) and that the Co(II) complex was [CoL2] (complex 3, chemical structure see in Fig. S17†). The 1H NMR of complex 2 showed proton signals corresponding to NH at δ 11.95, 11.65, 8.33 and 8.10, indicating that none of the N atoms of ligand L were involved in coordination. The 1H NMR of complex 3 showed proton signals at δ 13.94 (NH) and 7.43 (NH2), implying that only one N atom of ligand H-L participated in coordination. The results were consistent with the corresponding crystal diffraction and MS data.
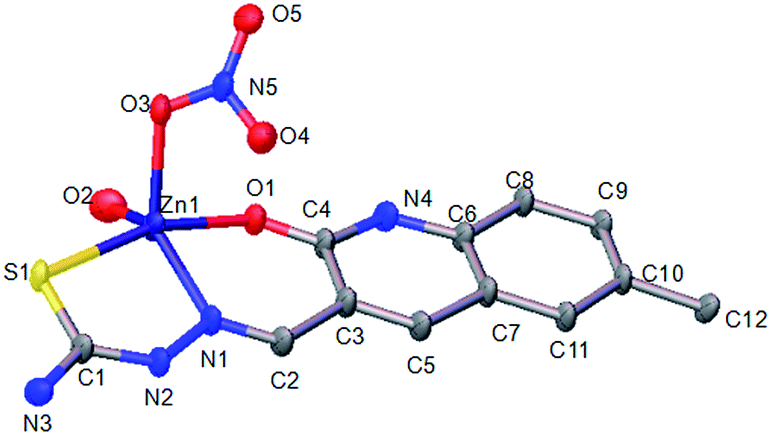
3.2. Crystal structure of complexes 1 and 2
Complexes 1 and 2 both had a mononuclear structure. Fig. 1 shows the structure of 1 and Table S2† lists the selected bond lengths and angles. The coordination of the thiosemicarbazone with Cu(II) resulted in the formation of six-membered (CuNCCCO) and five-membered (CuNNCS) chelating rings. Moreover, one NO3− group took part in coordination. Fig. 2 shows the structure of complex 2 and Table S3† lists the selected bond lengths and angles. The thiosemicarbazone coordinated with Zn(II) to form six-membered (ZnNCCCO) and five-membered (ZnNNCS) chelating rings. One coordinating NO3− group was nearby.
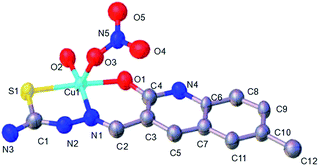 |
| | Fig. 1 ORTEP view of complex 1 showing the atom numbering of scheme and 10% thermal ellipsoids probability for the non-hydrogen atoms. | |
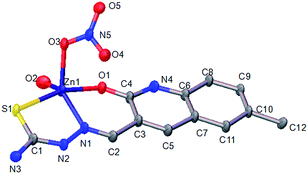 |
| | Fig. 2 ORTEP view of complex 2 showing the atom numbering of scheme and 10% thermal ellipsoids probability for the non-hydrogen atoms. | |
3.3. Stability in solution
The stability of complexes 1–3 in physiological condition (PBS buffer solution containing 1% DMSO, pH 7.35) was tested by UV-Vis spectroscopy (Fig. S1†). Fig. S1† shows the time-dependent (0, 1, 4, 12, 24 and 48 h) UV-Vis spectra of each complex. No obvious changes could be found in the spectral character and the peak absorptions of 1–3 over time, suggesting that they were also stable in their coordinating mode in the aqueous solution. Besides, in the ESI-MS analysis (Fig. S14†), complex 1 consistently gave maximum abundance at m/z = 322.0 for [CuII + (H-L)]+ both in freshly prepared TBS solution and after incubation for 24 h. Therefore, the predominantly coordinated species of 1 was stable, and the coordination state of ligand H-L to Cu(II) could be retained under physiological condition for at least 24 h. Such stability could ensure the adequate cell uptake of 1 during the time course of MTT assay. Taken together, complexes 1–3 could retain their coordinated geometry under physiological condition.
3.4. In vitro cytotoxicity
The in vitro cytotoxic potencies of ligand H-L and complexes 1–3 were evaluated in the MTT assay with SK-OV-3, BEL-7404, HeLa, Hep-G2 and MGC80-3 tumor cell lines and the HL-7702 normal liver cells. The tumor cells were incubated with the test compound at 2 × 10−5 M for 48 h under identical conditions. Cisplatin was used as the positive control. Table S4† lists the inhibitory rates. It was found that 1–3 showed higher inhibitory activity than the free ligand H-L against the SK-OV-3, BEL-7404, HeLa, Hep-G2 and MGC80-3 cell lines. Moreover, both H-L and the complexes 1–3 displayed lower inhibitory rates against normal liver cells (HL-7702) than the commercial anticancer drug cisplatin. The in vitro cytotoxicity of H-L and complexes 1–3 were further quantified by determining their corresponding IC50 values. Table 1 showed that, toward the tested human tumor cells, complexes 1–3 had much stronger cytotoxicity than the free ligand H-L. For instance, complex 1 was more sensitive to MGC80-3 and SK-OV-3 cells and exhibited low IC50 values of 10.17 and 10.35 μM, respectively, thus outperforming H-L, complex 2, complex 3 and cisplatin in the cytotoxicity against MGC80-3 and SK-OV-3 cells. Clearly, a synergistic effect between the free 6-methyl-2-oxo-quinoline-3-carbaldehyde thiosemicarbazone ligand and the transition metal existed and the enhanced cytotoxicity of the complexes 1–3.41
Table 1 IC50a (μM) values of complexes 1–3 against the selected tumor cell lines and the normal liver HL-7702 cells
| Compounds |
SK-OV-3 |
BEL7404 |
HeLa |
Hep-G2 |
MGC80-3 |
HL-7702 |
| IC50 values are presented as the mean ± SD (standard error of the mean) from five separated experiments. Cisplatin was used as a reference metallodrug and DMSO was used as the solvent control. |
| H-L |
>150 |
>150 |
>150 |
>150 |
>150 |
52.36 ± 1.42 |
| 1 |
10.35 ± 1.26 |
>150 |
22.89 ± 1.45 |
24.86 ± 3.45 |
10.17 ± 0.95 |
42.68 ± 1.42 |
| 2 |
>150 |
29.55 ± 2.32 |
>150 |
>150 |
>150 |
56.81 ± 0.95 |
| 3 |
>150 |
43.09 ± 3.66 |
29.36 ± 2.15 |
>150 |
>150 |
66.57 ± 0.75 |
| Cisplatinb |
16.32 ± 1.37 |
10.080 ± 0.52 |
9.45 ± 2.05 |
10.28 ± 2.77 |
12.37 ± 1.53 |
10.83 ± 0.34 |
3.5. Cell cycle analysis
Since complexes 1–3 had IC50 values in the low micromolar range, experiments were then carried out to determine the phase of the cell cycle in which these compounds arrested cell growth. The DNA content of cells was estimated by flow cytometry after the cells were stained with propidium iodide (PI). Fig. 3 shows the flow cytometry data of the MGC80-3 cells treated with complex 1, together with the control data. The proportion of MGC80-3 cells in the S phase was 27.02% in the control, but increased to 38.31% after the cells were treated with 1. Therefore, complex 1 could arrest the MGC80-3 cells in the S phase.
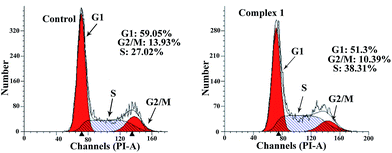 |
| | Fig. 3 Cell cycle analysis by flow cytometry of MGC80-3 cells treated with complex 1 (10 μM). | |
3.6. Apoptosis assays
It is known that apoptosis assays can provide important information on the preliminary mode of action. To determine whether the observed death of the MGC80-3 cells induced by complex 1 resulted from apoptosis or necrosis, common biochemical markers of apoptosis were monitored, including mitochondrial membrane depolarization, chromatin condensation, and phosphatidylserine exposure. Cells were subjected to annexin V-FITC and PI staining, and classified as necrotic (Q1; annexin−/PI+), late apoptotic (Q2; annexin+/PI+), intact (Q3; annexin−/PI−), or early apoptotic (Q4; annexin+/PI−). Fig. 4 shows the assay results. The proportions of late (Q2) and early (Q4) apoptotic cells after treatment with 1 for 24 h amounted to 12.1% and 53.9%, respectively, and the total apoptosis rate (including early and late apoptosis ratios) was 66%. The results from annexin V staining and flow cytometry suggested that complex 1 (10 μM) induced the apoptotic death of MGC80-3 cells. Similarly, the apoptosis rate of MGC80-3 cell after treatment with 2 (10 μM), 3 (10 μM), and cisplatin (10 μM) for 24 h were found to be 2.65%, 4.11% and 14.19%, respectively.
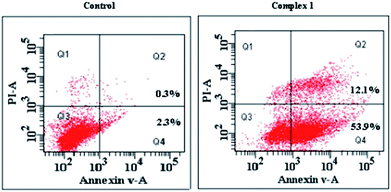 |
| | Fig. 4 Annexin V/propidium iodide assay and flow cytometry assay of MGC80-3 cells treated with complex 1 (10 μM). | |
3.7. DNA binding studies
Although there are other biological targets in tumor cells, including RNA, enzymes and proteins, it is widely believed that DNA is the primary target for many anticancer drugs.42,43 Consequently, the interactions between the small molecules and the DNA play a pivotal role in the drug's antitumor activity. To examine the DNA binding properties of complexes 1–3, UV-Vis, fluorescence and CD spectra were recorded, and gel mobility shift assays were performed.
3.7.1. UV-Vis absorption analysis. UV-Vis absorption spectroscopy is considered to be one of the most useful techniques for studying the binding mode of metal complexes with DNA,44,45 and it was applied to assess the interaction between complexes 1–3 and ct-DNA. Fig. 5 shows the UV-Vis absorption spectra of complexes 1–3 in the absence and presence of ct-DNA. In the presence of ct-DNA, the absorption bands of complex 1 at 242, 270, and 380 nm exhibited hypochromism of about 31.08%, 29.79% and 18.10%, respectively, whereas for complex 2 the bands at 242, 350 and 383 nm exhibited hypochromism of about 29.97%, 30.28% and 29.00%, respectively. Furthermore, the addition of ct-DNA not only caused an evident hypochromicity in the absorption bands of complex 3 at 242 and 299 nm of about 31.86% and 16.56%, but also led to an obvious red-shift for the absorption band at 399 nm. The observed hypochromicity for complexes 1–3 after the addition of ct-DNA firmly suggested their intercalative binding with DNA.38 The intrinsic binding constant Kb of 1, 2, and 3 was calculated according to the classic equation39 to be 4.491 × 104, 1.035 × 104 and 6.496 × 104 L mol−1 respectively.
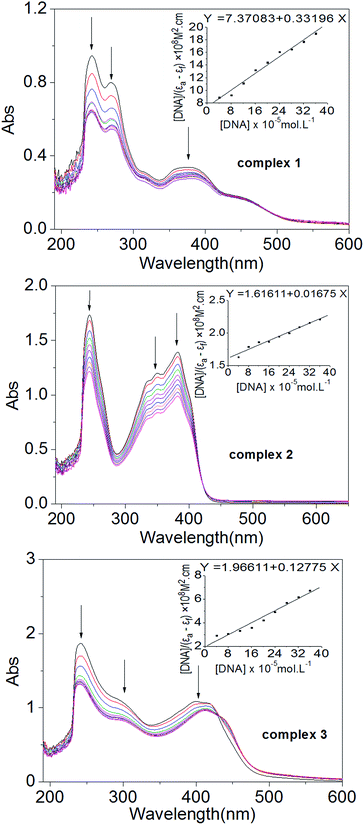 |
| | Fig. 5 UV-Vis absorption spectra of complexes 1–3 in the absence and presence of ct-DNA with the [DNA]/[complex] ratio increasing from 1:1 to 10:1. | |
3.7.2. Fluorescence emission titration. The binding modes of these complexes with ct-DNA were further monitored by a competitive binding assay, in which they acted as an intercalative binding probe to compete with ethidium bromide (EB). EB is well known as a highly sensitive dye that emits intense fluorescence upon its strong, non-specific intercalation between DNA base pairs.46 Competitive binding of other molecules to DNA can replace the bound EB and reduce the fluorescence intensity. Therefore, this fluorescence-based competition technique provides indirect evidence for the DNA binding mode. Fig. 6 shows the emission spectra of the EB–ct-DNA system in the absence and presence of 1–3. When the concentration of complex increased, the characteristic emission of EB at around 580 nm was strongly quenched, and the emission of other bands (around 470 nm) gradually strengthened. Therefore, these three complexes could displace EB from the DNA–EB system, implying that their intercalative binding mode to DNA was similar to that of EB. The total decrease ratios (TDR) of the EB emission intensity for complexes 1, 2 and 3 were found to be 79.49%, 36.96%, and 87.02%, respectively, and the corresponding decrease ratios (DR) at [compound]/[EB] = 1 were found to be 15.70%, 7.39%, and 17.35%, respectively. The quenching constant Kq was derived from the Stern–Volmer equation47 and calculated to be 9.487 × 104, 2.135 × 104 and 1.147 × 105 L mol−1 for 1, 2 and 3, respectively. The Kq values quantified the quenching ability of the complexes and were in agreement with Kb values determined from the UV-Vis absorption spectra.
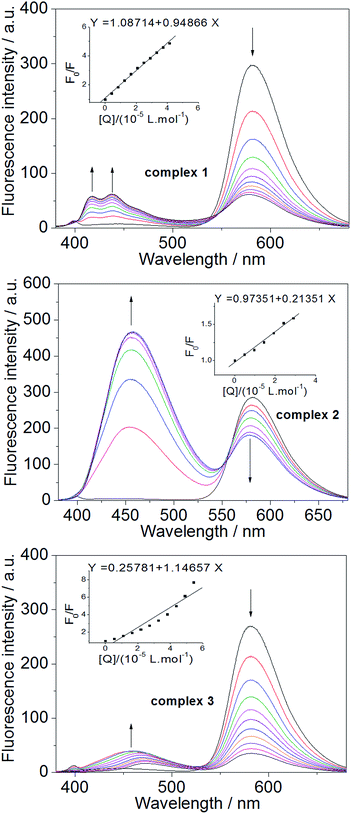 |
| | Fig. 6 Fluorescence emission spectra of EB bound with ct-DNA ([DNA] 2 × 10−4 M, [EB] 2 × 10−5 M) in the absence and presence of complexes 1–3 with the [complex]/[EB] ratio increasing from 1:3 to 10:3. | |
3.7.3. Circular dichroism spectra. Circular dichroism (CD) spectroscopy is a rapid and useful technique to estimate whether nucleic acids have undergone conformational transformation as a result of complex formation or changes in environmental conditions.48 The CD spectroscopy was employed to further assess the interaction between the complexes (1–3) and ct-DNA. As shown in Fig. 7, the CD spectra of ct-DNA (1 × 10−4 M) displayed a positive absorption peak at 277 nm and a negative absorption peak at 243 nm due to π–π base stacking and right-hand helicity, respectively, which were in good agreement with the characteristic B conformation of DNA.49 Upon the addition of complexes 1–3 at 1:15 [complex]/[DNA] ratio, the CD absorption of ct-DNA showed an obvious decrease in the intensity of the positive band. The positive band showed an obvious blue-shift when 1 was added but showed a red-shift when 2 was added, and the red-shift became even more pronounced when 3 was added. The negative band displayed a clear red-shift in all cases, and the intensity of the band decreased markedly after the addition of 1 or 2. Note that upon continued addition of 1–3, both the hypochromism and the red/blue-shift in the CD absorption spectra intensified, and even the shapes of the ct-DNA absorption curves changed markedly (Fig. 7). The results suggested that 1–3 could intercalate between the adjacent base pairs of ct-DNA, since the decrease in the intensity of positive and negative bands in the CD absorption of ct-DNA is usually observed upon the intercalative binding of small molecules to DNA.50 Furthermore, the appreciable red/blue-shifts and the marked changes in the shape of the ct-DNA absorption curve indicated that the secondary structure of the ct-DNA underwent significant transformations.51
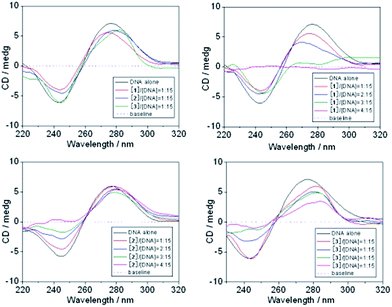 |
| | Fig. 7 CD spectra of ct-DNA in the absence and presence of complexes 1–3. | |
3.7.4. Agarose gel electrophoresis assay. The binding modes of 1–3 to DNA were further investigated by agarose gel electrophoresis assay. The pBR322 plasmid DNA was used to displace ct-DNA, and EB and cisplatin were used as references. When its concentration increased from 20 to 60 μM, EB significantly decreased the electrophoretic mobility of the supercoiled conformation (Form I) of pBR322 DNA (Fig. S16†). Under the same condition, cisplatin did not affect the electrophoretic mobility. However, Fig. 8 shows that as the concentration of 1–3 increased, not only the major form of DNA (supercoiled, Form I) but also the minor forms of DNA (nicked, Form II; linear, Form III) underwent significant changes in migration rate and proportions. It could thus be inferred that 1–3 intercalated to the DNA between neighboring base pairs.52,53 Note that intercalation of complex 1 to the DNA was stronger than that of EB and cisplatin.54,55 Even at a low concentration of 20 μM, complex 1 clearly decreased the migration rate of the supercoiled form of the DNA, indicating that the extent of intercalation between neighboring base pairs was high. In addition, when the concentration of complex 2 was 60 μM, the Form III of pBR322 DNA disappeared completely, and the proportion of Form II DNA decreased. Upon the addition of complex 3, the proportions of all three forms of pBR322 DNA decreased, and the Form II DNA disappeared completely when the concentration of 3 reached 40 μM. These findings were consistent with the results of the UV-Vis absorption analysis, fluorescence spectral analysis, and circular dichroism experiment.
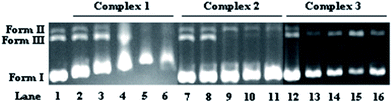 |
| | Fig. 8 Gel electrophoresis mobility shift assay of pBR322 DNA treated with 1–3 (lane 1: DNA alone; lanes 2–6, 7–11, 12–16: each complex with increasing concentrations of 20, 40, 60, 80 and 100 μM, respectively). Form I: supercoiled form, Form II: nicked, Form III: linear. | |
Taken together, it could be inferred that intercalation through the aromatic planar structure of the 6-methyl-2-oxo-quinoline-3-carbaldehyde thiosemicarbazone ligand is the most probable binding mode between the complexes 1–3 and DNA. Since complex 1 gave the highest apoptosis rate in MGC80-3 cells, it was examined in additional cellular tests, including the loss of mitochondrial membrane potential (Δψ), intracellular Ca2+ fluctuation, expressions of apoptotic related proteins, reactive oxygen species (ROS) level, caspase-3/9 activation, and Western blot assay.
3.8. Loss of mitochondrial membrane potential in MGC80-3 cells
The loss of mitochondrial membrane potential (Δψ) or mitochondrial dysfunction is involved in apoptotic cell death due to the cytotoxicity of the antitumor target compound.56,57 Fig. 9 shows the changes in Δψ induced by the copper complex 1. JC-1 staining was used as a fluorescent probe. After the MGC80-3 cells were treated with the copper complex 1 (10 μM) for 24 h, the Δψ decreased significantly. Therefore, the induction of apoptosis by complex 1 was associated with the intrinsic (mitochondrial) pathway.40,56,57
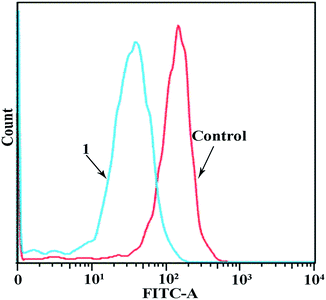 |
| | Fig. 9 Collapse of mitochondrial membrane potential in MGC80-3 cells treated with copper complex 1 (10 μM) for 24 h, as determined by JC-1 staining. | |
3.9. Intracellular Ca2+
The increase in Ca2+ level mediated by various channels can alter the mitochondrial membrane potential Δψ, and thus has been recognized as a factor in cell death, cell apoptosis, and cell injury.40,58 With this in mind, we examined the effects of the copper complex 1 on the mobilization of intracellular Ca2+ in MGC80-3 cells. Fig. 10 shows that the level of intracellular free Ca2+ in the MGC80-3 cells was the lowest in the control group but increased steadily upon addition of complex 1 (10 μM). Therefore, the changes in intracellular Ca2+ level may be involved in the mechanism by which complex 1 induced apoptosis in MGC80-3 cells.
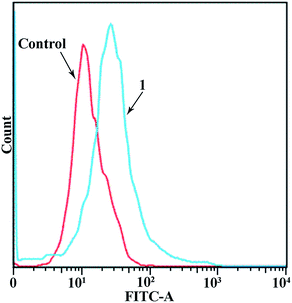 |
| | Fig. 10 Effect of copper complex 1 (10 μM) on intracellular free Ca2+ in MGC80-3 cells. Cells were treated with the copper complex 1 (10 μM) for 24 h. | |
3.10. Expressions of apoptotic related proteins
To further study the mechanism of action of complex 1, the proteins in the mitochondria-related apoptotic pathway, including cytochrome c (Cyt C), bcl-2, bax and apaf-1, were investigated by Western blot.59 Fig. 11 shows that after the MGC80-3 cells were treated with complex 1 (10 μM and 20 μM) for 24 h, the levels of apaf-1, Cyt C, and bax proteins increased significantly and the level of bcl-2 protein decreased significantly. These results further demonstrated that complex 1 may be involved in mitochondria-related apoptosis.59
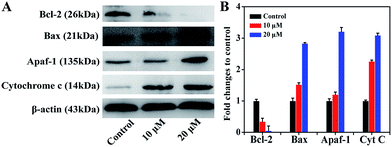 |
| | Fig. 11 (A) Western blot was used to determine the expression levels of bax, cytochrome c, apaf-1 and bcl-2 in MGC80-3 cells treated with copper complex 1 (10 and 20 μM) for 24 h. (B) Densitometric analysis of apoptotic related proteins normalized to β-actin. The relative expression of each protein is represented by the density of the protein band/density of β-actin band. Mean and SD were from three independent measurements. | |
3.11. Reactive oxygen species (ROS) level
It has been reported that the disregulation of ROS generation could dramatically affect cancer cell structure and might result in cell damage, and consequently cell death and apoptosis.60,61 The ROS generation in MGC80-3 cells treated with complex 1 (10 μM) for 24 h was measured by a fluorescent marker to determine whether the apoptosis or death of MGC80-3 cells induced by the copper complex 1 (10 μM) depended on the ROS level. Fig. 12 shows that the MGC80-3 cells treated with the copper complex 1 (10 μM) produced high levels of ROS compared with the control MGC80-3 cells. Therefore, the copper complex 1 (10 μM) stimulated ROS-induced apoptosis in MGC80-3 cells.
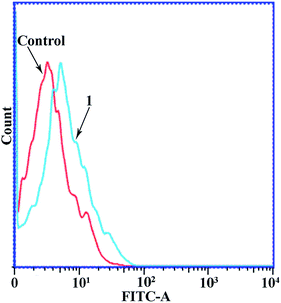 |
| | Fig. 12 The ROS generation assay by flow cytometric analysis of MGC80-3 cells treated with the copper complex 1 (10 μM). Results are expressed as relative fluorescent intensities (from left to right). | |
3.12. Assessment of caspase-3/9 activation
The protein extracts of the MGC80-3 cells treated with the copper complex 1 (10 μM and 20 μM) for 24 h were analyzed by flow cytometry to determine if caspase-3 and caspase-9 were involved in the induced apoptotic cell death. Fig. 13 shows additional peaks of activated caspase-3 (FITC-DEVD-FMK probes) and activated caspase-9 (FITC-LEHD-FMK probes) for the treated cells. Notably, after treatment with 10 μM and 20 μM copper complex 1, the proportion of cells with activated caspase-3 arrived at 34.50% and 45.90%, and the proportion of cells with activated caspase-9 arrived at 16.50% and 26.20%, respectively. Therefore, the copper complex 1 could induce cell apoptosis by triggering the caspase-3/9 activity in MGC80-3 cells.62–66
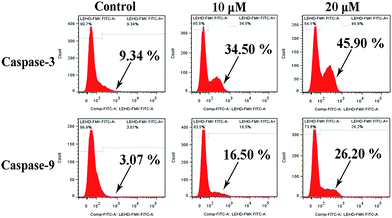 |
| | Fig. 13 The activation of caspase-3 and caspase-9 in MGC80-3 cells treated with the copper complex 1 (10 μM and 20 μM) for 24 h, respectively. | |
Taken together, the copper complex 1 most likely induced apoptosis in MGC80-3 cells by disrupting mitochondrial function, which led to significantly decreased level of the bcl-2 protein and the loss of Δψ, as well as the significant increase in the ROS level, intracellular Ca2+, cytochrome c, apaf-1, activated caspase-3, and activated caspase-9 in MGC80-3 cells.
4. Conclusions
Three transition metal complexes of 6-methyl-2-oxo-quinoline-3-carbaldehyde thiosemicarbazone were synthesized and characterized by NMR, MS, elemental analyses, as well as X-ray single crystal diffraction. Their molecular formulas were found to be [Cu(H-L)NO3H2O]·NO3 (1), [Zn(H-L)NO3H2O]·NO3 (2) and [CoL2] (3), respectively. These complexes exhibited good cytotoxicity against several cancer cell lines including SK-OV-3, BEL-7404, HeLa, Hep-G2, and MGC80-3. In particular, the IC50 values of complex 1 against SKOV-3 and MGC80-3 were even lower than those of cisplatin. Complex 1 could induce apoptosis of MGC80-3 cells and cause cell arrest in the S phase. These complexes most probably interacted with DNA through intercalation. Moreover, it was found that complex 1 induced MGC80-3 cell apoptosis via a mitochondrial dysfunction pathway.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 21501032 and 21431001), Guangxi Natural Science Foundation of China (No. 2016GXNSFAA380300 and 2014GXNSFBA118050), the basic skills improvement project for the young and middle-aged teachers in Guangxi colleges and universities (KY2016YB595), Innovation Project of Guangxi Graduate Education: Synthesis, antitumor activity of heterologous multi-target copper(II) complexes with β-carboline alkaloids and State Key Laboratory Cultivation Base for the Chemistry and Molecular Engineering of Medicinal Resources, Ministry of Science and Technology of China (CMEMR2014-B14).
Notes and references
- C. G. Hartinger, A. A. Nazarov, S. M. Ashraf, P. J. Dyson and B. K. Keppler, Curr. Med. Chem., 2008, 15, 2574–2591 CrossRef CAS PubMed.
- D. R. Richardson, D. S. Kalinowski, V. Richardson, P. C. Sharpe, D. B. Lovejoy, M. Isalam and P. V. Bernhardt, J. Med. Chem., 2009, 52, 1459–1470 CrossRef CAS PubMed.
- Y. Jung and S. J. Lippard, Chem. Rev., 2007, 107, 1387–1407 CrossRef CAS PubMed.
- Z. F. Chen, Y. Q. Gu, X. Y. Song, Y. C. Liu, Y. Peng and H. Liang, Eur. J. Med. Chem., 2013, 59, 194–202 CrossRef CAS PubMed.
- Y. C. Liu, Z. F. Chen, X. Y. Song, Y. Peng, Q. P. Qin and H. Liang, Eur. J. Med. Chem., 2013, 59, 168–175 CrossRef CAS PubMed.
- Z. F. Chen, Y. C. Liu, L. M. Liu, H. S. Wang, S. H. Qin, B. L. Wang, H. D. Bian, B. Yang, H. K. Fun, H. G. Liu, H. Liang and C. Orvig, Dalton Trans., 2009, 262–272 RSC.
- Y. C. Liu, Z. F. Chen, L. M. Liu, Y. Peng, X. Hong, B. Yang, H. G. Liu, H. Liang and C. Orvig, Dalton Trans., 2009, 10813–10823 RSC.
- Z. F. Chen, Y. F. Shi, Y. C. Liu, X. Hong, B. Geng, Y. Peng and H. Liang, Inorg. Chem., 2012, 51, 1998–2009 CrossRef CAS PubMed.
- S. Gemma, C. Camodeca, M. Brindisi, S. Brogi, G. Kukreja, S. Kunjir, E. Gabellieri, L. Lucantoni, A. Habluetzel, D. Taramelli, N. Basilico, R. Gualdani, F. Tadini-Buoninsegni, G. Bartolommei, M. R. Moncelli, R. E. Martin, R. L. Summers, S. Lamponi, L. Savini, I. Fiorini, M. Valoti, E. Novellino, G. Campiani and S. Butini, J. Med. Chem., 2012, 55, 10387–10404 CrossRef CAS PubMed.
- Z. F. Chen, Q. P. Qin, J. L. Qin, J. Zhou, Y. L. Li, N. Li, Y. C. Liu and H. Liang, J. Med. Chem., 2015, 58, 4771–4789 CrossRef CAS PubMed.
- X. Mao, X. Li, R. Sprangers, X. Wang, A. Venugopal, T. Wood, Y. Zhang, D. A. Kuntz, E. Coe, S. Trudel, D. Rose, R. A. Batey, L. E. Kay and A. D. Schimmer, Leukemia, 2009, 23, 585–590 CrossRef CAS PubMed.
- M. Venet, D. End and P. Angibaud, Curr. Top. Med. Chem., 2003, 3, 1095–1102 CrossRef CAS PubMed.
- P. Angibaud, X. Bourdrez and A. Devine, Bioorg. Med. Chem. Lett., 2003, 13, 1543–1547 CrossRef CAS PubMed.
- P. Angibaud, A. K. Saha, X. Bourdrez, D. W. End, E. Freyne, P. Lezouret, G. Mannens, L. Mevellec, C. Meyer, I. Pilatte, V. Poncelet, B. Roux, G. Smets, J. Van Dun, M. Venet and W. Wouters, Bioorg. Med. Chem. Lett., 2003, 24, 4361–43641 CrossRef.
- F. A. French and E. J. J. Blanz, Cancer Res., 1965, 25, 1454–1458 CAS.
- D. S. Kalinowski, P. Quach and D. R. Richardson, Future Med. Chem., 2009, 1, 1143–1151 CrossRef CAS PubMed.
- J. Shao, B. Zhou, A. J. Di Bilio, L. Zhu, T. Wang, C. Qi, J. Shih and Y. Yen, Mol. Cancer Ther., 2006, 5, 586–592 CrossRef CAS PubMed.
- B. M. Paterson and P. S. Donnelly, Chem. Soc. Rev., 2011, 40, 3005–3018 RSC.
- N. Farrell, Coord. Chem. Rev., 2002, 232, 1–4 CrossRef CAS.
- G. K. Walkup, S. C. Burdette, S. J. Lippard and R. Y. Tsien, J. Am. Chem. Soc., 2000, 122, 5644–5645 CrossRef CAS.
- M. D. Vaira, C. Bazzicalupi, P. Orioli, L. Messori, B. Bruni and Z. Paolo, Inorg. Chem., 2004, 43, 3795–3797 CrossRef PubMed.
- J. d'Angelo, G. Morgant, N. E. Ghermani, D. Desmaëlea, B. Fraisse, F. Bonhomme, E. Dichi, M. Sghaier, Y. L. Li, Y. Journauxe and J. R. J. Sorenson, Polyhedron, 2008, 27, 537–546 CrossRef.
- H. Sakurai, Y. Kojima, Y. Yoshikawa, Y. Yoshikawa, K. Kawabea and H. Yasui, Coord. Chem. Rev., 2002, 226, 187–198 CrossRef CAS.
- Q. D. Zhou, T. W. Hambley, B. J. Kennedy, P. A. Lay, P. Turner, B. Warwick, J. R. Biffin and H. L. Regtop, Inorg. Chem., 2000, 39, 3742–3748 CrossRef CAS PubMed.
- N. C. Kasuga, K. Sekino, M. Ishikawa, A. Honda, M. Yokoyama, S. Nakano, N. Shimada, C. Koumo and K. Nomiya, J. Inorg. Biochem., 2003, 96, 298–310 CrossRef CAS PubMed.
- M. B. Ferraria, F. Biscegliea, G. Pelosia, P. Tarasconia, R. Albertinib and S. Pinellib, J. Inorg. Biochem., 2001, 87, 137–147 CrossRef.
- Z. Trávníčeka, V. Kryštofb and M. Šipla, J. Inorg. Biochem., 2006, 100, 214–225 CrossRef PubMed.
- J. S. Casas, E. E. Castellano, M. D. Couce, J. Ellena, A. Sáncheza, J. Sordoa and C. Taboadaa, J. Inorg. Biochem., 2006, 100, 124–132 CrossRef CAS PubMed.
- Z. Q. Li, F. J. Wu, Y. Gong, W. H. Chang, Y. H. Zhang and M. Y. Gan, Chin. J. Chem., 2007, 25, 1809–1814 CrossRef CAS.
- P. V. Bernhardt, J. Mattsson and D. R. Richardson, Inorg. Chem., 2006, 45, 752–760 CrossRef CAS PubMed.
- M. F. Iskander, L. El-Sayed, N. M. H. Salem, W. Haase, H. J. Linder and S. Foro, Polyhedron, 2004, 23, 23–31 CrossRef CAS.
- G. F. Qi, Z. Y. Yang and D. D. Qin, Chem. Pharm. Bull., 2009, 57, 69–73 CrossRef CAS PubMed.
- V. P. Singh and P. Gupta, J. Enzyme Inhib. Med. Chem., 2008, 23, 797–805 CrossRef CAS PubMed.
- S. Tabassum, Spectrochim. Acta, Part A, 2009, 72, 1026–1033 CrossRef PubMed.
- V. Uma, M. Kanthimathi, T. Weyhermuller and B. U. Nair, J. Inorg. Biochem., 2005, 99, 2299–2307 CrossRef CAS PubMed.
- Y. Zhang, Y. L. Fang, H. Liang, H. S. Wang, K. Hu, X. X. Liu, X. H. Yi and Y. Peng, Bioorg. Med. Chem. Lett., 2013, 23, 107–111 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXS-97, Program for Solution of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- R. F. Pasternack, M. Caccam, B. Keogh, T. A. Stephenson, A. P. Williams and E. J. Gibbs, J. Am. Chem. Soc., 1991, 113, 6835–6840 CrossRef CAS.
- Q. P. Qin, Y. C. Liu, H. L. Wang, J. L. Qin, F. J. Cheng, S. F. Tang and H. Liang, Metallomics, 2015, 7, 1124–1136 RSC.
- Q. P. Qin, J. L. Qin, T. Meng, G. A. Yang, Z. Z. Wei, Y. C. Liu, H. Liang and Z. F. Chen, Sci. Rep., 2016, 6, 37644, DOI:10.1038/srep37644.
- V. Rajendiran, M. Murali, E. Suresh, M. Palaniandavar, V. S. Periasamy and M. A. Akbarsha, Dalton Trans., 2008, 2157–2170 RSC.
- C. X. Zhang and S. J. Lippard, Curr. Opin. Chem. Biol., 2003, 7, 481–489 CrossRef CAS PubMed.
- C. H. Wu, D. H. Wu, X. Liu, G. Guoyiqibayi, D. D. Guo, G. Lv, X. M. Wang, H. Yan, H. Jiang and Z. H. Lu, Inorg. Chem., 2009, 48, 2352–2354 CrossRef CAS PubMed.
- A. F. Tanious, D. Y. Ding, D. A. Patrick, C. Bailly, R. R. Tidwell and W. D. Wilson, Biochemistry, 2002, 39, 12091–12101 CrossRef.
- C. Y. Zhong, J. Zhao, Y. B. Wu, C. X. Yin and P. Yang, J. Inorg. Biochem., 2007, 101, 10–18 CrossRef PubMed.
- M. A. Galindo, D. Olea, M. A. Romero, J. Gómez, P. del Castillo, M. J. Hannon, A. Rodger, F. Zamora and J. A. R. Navarro, Chem.–Eur. J., 2007, 13, 5075–5081 CrossRef CAS PubMed.
- R. H. Harsh and J. K. Barton, J. Am. Chem. Soc., 1992, 114, 5919–5925 CrossRef.
- S. Ghosh, A. C. Barve, A. A. Kumbhar, A. S. Kumbhar, V. G. Puranik, P. A. Datar, U. B. Sonawane and R. R. Joshi, J. Inorg. Biochem., 2006, 100, 331–343 CrossRef CAS PubMed.
- S. Schäfe, I. Ott, R. Gust and W. S. Sheldrick, Eur. J. Inorg. Chem., 2007, 3034–3046 CrossRef.
- H. Xu, K. C. Zheng, Y. Chen, Y. Z. Li, L. J. Lin, H. Li, P. X. Zhang and L. N. Ji, Dalton Trans., 2003, 2260–2268 RSC.
- W. Zhong, J. S. Yu, Y. Liang, K. Fan and L. Lai, Spectrochim. Acta, Part A, 2004, 60, 2985–2992 CrossRef PubMed.
- J. G. Liu, Q. L. Zhang, X. F. Shi and L. N. Ji, Inorg. Chem., 2001, 40, 5045–5050 CrossRef CAS PubMed.
- Q. P. Qin, Z. F. Chen, J. L. Qin, X. J. He, Y. L. Li, Y. C. Liu, K. B. Huang and H. Liang, Eur. J. Med. Chem., 2015, 92, 302–313 CrossRef CAS PubMed.
- P. G. Barldi, A. Bovero, F. Fruttarolo, D. Preti, M. A. Tabrizi, M. G. Pavani and R. Romagnoli, Med. Res. Rev., 2004, 24, 475–528 CrossRef PubMed.
- V. G. Vaidyanathan and B. U. Nair, Dalton Trans., 2005, 2842–2848 RSC.
- T. F. Chen and Y. S. Wong, Int. J. Biochem. Cell Biol., 2009, 41, 666–676 CrossRef CAS PubMed.
- Q. P. Qin, J. L. Qin, T. Meng, W. H. Lin, C. H. Zhang, Z. Z. Wei, J. N. Chen, Y. C. Liu, H. Liang and Z. F. Chen, Eur. J. Med. Chem., 2016, 124, 380–392 CrossRef CAS PubMed.
- D. R. Laver, Biophys. J., 2007, 92, 3541–3555 CrossRef CAS PubMed.
- D. Spierings, G. McStay, M. Saleh, C. Bender, J. Chipuk, U. Maurer and D. R. Green, Science, 2005, 310, 66–67 CrossRef CAS PubMed.
- X. Z. Hu, Y. Xu, D. C. Hu, Y. Hui and F. X. Yang, Toxicol. Lett., 2007, 171, 19–28 CrossRef CAS PubMed.
- A. Kawiak, J. Zawacka-Pankau, A. Wasilewska, G. Stasilojc, J. Bigda and E. Lojkowska, J. Nat. Prod., 2012, 75, 9–14 CrossRef CAS PubMed.
- G. S. Salvesen and S. J. Riedl, Adv. Exp. Med. Biol., 2008, 615, 13–23 CrossRef CAS PubMed.
- J. L. Qin, Q. P. Qin, Z. Z. Wei, C. C. Yu, T. Meng, C. X. Wu, Y. L. Liang, H. Liang and Z. F. Chen, Eur. J. Med. Chem., 2016, 124, 417–427 CrossRef CAS PubMed.
- G. H. Zhang, L. J. Cai, Y. F. Wang, Y. H. Zhou, Y. F. An, Y. C. Liu, Y. Peng, Z. F. Chen and H. Liang, Biochem. Pharmacol., 2013, 86, 1721–1730 CrossRef CAS PubMed.
- Z. F. Chen, Q. P. Qin, J. L. Qin, Y. C. Liu, K. B. Huang, Y. L. Li, T. Meng, G. H. Zhang, Y. Peng, X. J. Luo and H. Liang, J. Med. Chem., 2015, 58, 2159–2179 CrossRef CAS PubMed.
- K. B. Huang, Z. F. Chen, Y. C. Liu, X. L. Xie and H. Liang, RSC Adv., 2015, 5, 81313–81323 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Vendor codes for each tested compound, thermal stability experiments, 1H NMR, ESI-MS, IR data, and crystal data. CCDC 1525012 and 1525013 for complexes 1 and 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra00826k |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc,
Meng Wanga,
Yan-Cheng Liua,
Zhen-Feng Chen*a and
Hong Liang*a
*abc,
Meng Wanga,
Yan-Cheng Liua,
Zhen-Feng Chen*a and
Hong Liang*a