
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and antimicrobial evaluation of promising 7-arylamino-5,8-dioxo-5,8-dihydroisoquinoline-4-carboxylates and their halogenated amino compounds for treating Gram-negative bacterial infections†
Juliana S. Novaisa,
Vinicius R. Camposb,
Ana Carolina J. A. Silvaa,
Maria C. B. V. de Souzab,
Vitor F. Ferreirab,
Vitor G. L. Kellerb,
Matheus O. Ferreirab,
Flaviana R. F. Diasb,
Maíra I. Vitorinoa,
Plínio C. Sathlerc,
Marcos V. Santanaa,
Jackson A. L. C. Resendede,
Helena C. Castroa and
Anna C. Cunha *b
*b
aPrograma de Pós-Graduação em Ciências e Biotecnologia, Universidade Federal Fluminense, Niterói, Rio de Janeiro, Brazil
bUniversidade Federal Fluminense, Departamento de Química Orgânica, Programa de Pós-Graduação em Química, Outeiro de São João Batista, 24020-141 Niterói, RJ, Brazil. E-mail: annac@vm.uff.br; Fax: +55 21 26292145; Tel: +55 21 26292148
cUniversidade Federal do Rio de Janeiro, Faculdade de Farmácia, LabTIF, 21941-909, Rio de Janeiro, RJ, Brazil
dUniversidade Federal Fluminense, Laboratório de difração de raios X, Programa de Pós-Graduação em Química, 24020-141, Niterói, RJ, Brazil
eUniversidade Federal do Mato Grosso, Centro Universitário do Araguaia, Instituto de Ciências Exatas e da Terra, 78600-000, Barra do Garças, MT, Brazil
First published on 27th March 2017
Abstract
Pathogenic bacteria may cause serious infections, such as pneumonia, which can be fatal especially to immunosuppressed individuals. Hospitalized patients are particularly susceptible to antibiotic-resistant infections, which are worsened when caused by resistant Gram-negative pathogens due to there being few therapeutic options available. Thus, this work describes the synthesis and in vitro antimicrobial profile of 7-arylamino-5,8-dioxo-5,8-dihydroisoquinoline-4-carboxylates and their halogenated aminoquinones against Gram-positive and Gram-negative bacteria. Interestingly, these bioactive substances have shown promising activity against Gram-negative pathogenic strains. Among these derivatives, two non-halogenated amino compounds exhibited promising MIC and MBC values (MIC = MBC = 1–2 μg mL−1) against Escherichia coli ATCC 25922 and Pseudomonas aeruginosa ATCC 27853, two Gram-negative strains of clinical importance. In addition, mono- and di-brominated aminoquinones were also effective in preventing the growth of E. coli (MIC = MBC = 2–4 μg mL−1). The in vitro hemocompatibility evaluation showed a low toxicity profile for the active aminoquinones in hemolysis assays. These results suggest that these substances have potential for exploring the design of new antimicrobial prototypes against Gram-negative bacteria.
1. Introduction
According to the World Health Organization,1 bacterial infections are among the top ten causes of morbidity and mortality worldwide. The bacterial resistance phenomenon and indiscriminate antibiotic use have contributed to the restriction of the therapeutic options to treat these infections.2 Data from the Center for Disease Control and Prevention (CDC) has revealed that about 2 million patients in the United States (US) develop bacterial infections each year and 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths are directly related to bacterial infections.3
000 deaths are directly related to bacterial infections.3
Based on these facts and due to the high degree of resistance of Gram-negative bacteria with the development of multidrug-resistant (MDR – resistant to three or more classes of antimicrobials) and extensively drug-resistant (XDR – resistant to one or two classes of antimicrobials) strains, and the threatening risk of the development of pan-drug resistant (PDR – resistant to all classes of antimicrobials) strains, researchers have paid particular attention to the infection rate caused by this group.4,5 Usually, Gram-negative bacteria are commonly associated with intra-abdominal infections (IAIs), bacteremia, ventilator-associated pneumonia (VAP) and urinary tract infections (UTIs). The main pathogens involved in these infections are Escherichia coli, Klebsiella pneumoniae and Pseudomonas aeruginosa. Together, these species account for 70% of all Gram-negative bacteria causing health care associated infections in the US.6
These pathogens present resistance mechanisms that involve: (a) the expression of antibiotic-inactivating enzymes, such as carbapenemases, that usually confer resistance to β-lactams,7 and (b) non-enzymatic mechanisms, such as low permeability of the outer membrane and the presence of an efficient efflux system.8 The most severe clinical cases of infections caused by Gram-negative pathogens are often associated with health care acquired infections caused by extended-spectrum β-lactamase (ESBL) producing Enterobacteriaceae (e.g. E. coli), carbapenem-resistant Enterobacteriaceae (CRE) (e.g. K. pneumoniae), MDR Acinetobacter baumannii and MDR P. aeruginosa.9
In nosocomial infections, an important factor for the pathogenicity and persistence potential of Gram-negative bacteria is their ability to form a biofilm.10 Bacteria that produce biofilms become more resistant to antibiotics than those in planktonic form as the biofilm acts as a physical barrier.11 P. aeruginosa and E. coli are the most common Gram-negative bacteria able to form biofilms in medical devices.12,13 P. aeruginosa is also capable of forming biofilms in the lungs of cystic fibrosis patients,14 whereas E. coli is frequently associated with urinary catheter-related infections.15
All of these issues compromise the current strategies for the treatment of infections caused by Gram-negative bacteria, especially in view of the few therapeutic alternatives presently available.16 There are few antibacterial agents on the market, and combining antimicrobials, such as ceftolozane–tazobactam, is the current option for treating infections caused by resistant strains. The ceftolozane–tazobactam combination is effective against resistant Gram-negative bacteria such as P. aeruginosa and Enterobacteriaceae, including extended-spectrum β-lactamase (ESBL) producers.17 Meanwhile, ceftazidime, a broad-spectrum cephalosporin, and avibactam, a β-lactamase inhibitor, are combined to treat resistant infections caused by ESBL and Klebsiella pneumoniae carbapenemase (KPC).18
Based on this scenario and considering the prevalence of infections caused by Gram-negative pathogens resistant to many or all antibacterial agents available, the discovery of new agents with potent activity against these strains is urgently required.19,20
Naturally occurring quinones are well-known as clinically useful drugs for treating cancer.21 Although most of the quinone derivatives show a potential cytotoxic profile against a number of cancer cell lines,22 they are still associated with other pharmacological activities, such as antibacterial,23 antifungal,24 antiviral,25 anti-inflammatory,26 and antimalarial.27
The literature about the rational design of antimicrobials describes several aminoquinone compounds28,29 (1–6) and halo-1,4-naphtoquinone derivatives30 (7–8) with promising inhibitory effects against pathogenic bacteria (Fig. 1).
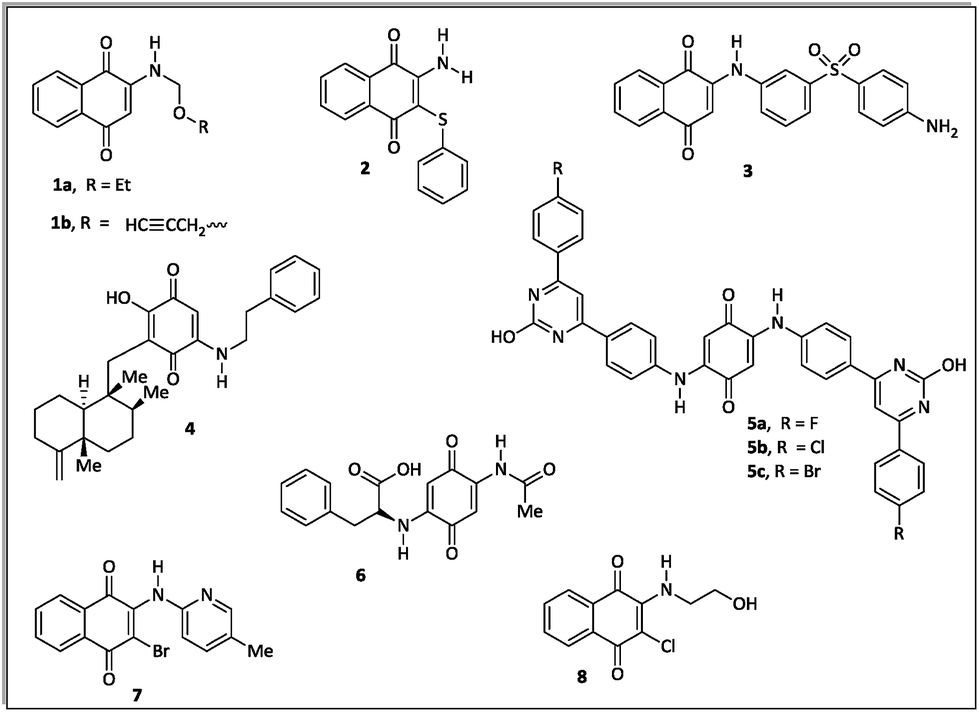 | ||
| Fig. 1 Examples of quinone derivatives as antibacterial agents. | ||
According to the literature, quinones may present different antimicrobial mechanisms.31,32 These substances may act through redox reactions producing reactive oxygen species (ROS), which can lead to cellular damage, oxidative stress and DNA damage.31 Quinone compounds are also able to inhibit DNA topoisomerase, an enzyme that catalyzes changes in DNA topology and is essential for bacterial replication.32 Another mechanism involves inhibition of bacterial cell growth via a competitive electron transport process with interference of endogenous vitamin K or ubiquinone in the electron transfer chain.33
Recently Ryu, Song and Hong34 described 7-arylamino-5,8-dioxo-5,8-dihydroisoquinoline-4-carboxylates as antifungal agents with similar or better profiles than fluconazole, a current therapeutic drug. Interestingly, these authors suggested that the inhibition of the fungal respiratory electron-transfer pathway was a feasible mechanism for active aminoquinone derivatives. Since this pathway is also present in Gram-negative and Gram-positive bacteria,35 in this article we report the synthesis of two homologous series of 7-arylamino-5,8-dioxo-5,8-dihydroisoquinoline-4-carboxylates (9a–d and 10a–b), structurally related to the fungicidal agents 10c–d (Fig. 2),34 and tested them against bacterial strains of clinical importance.
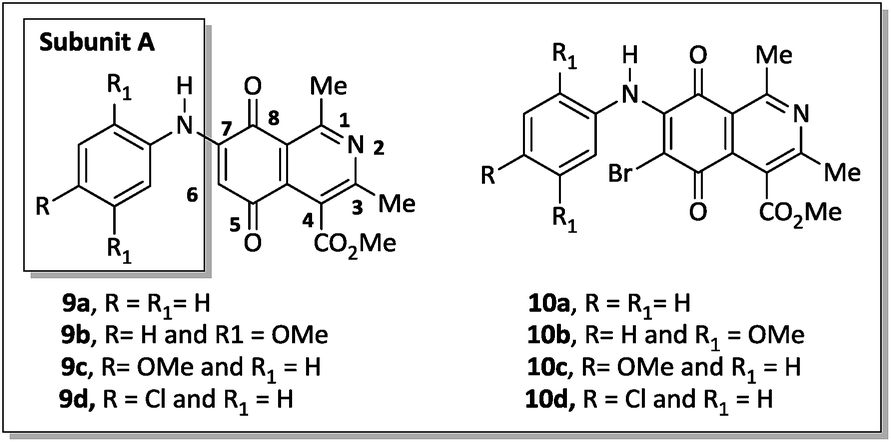 | ||
| Fig. 2 Two homologous series of aminoquinone compounds, 9a–d and 10a–d, which are structurally related to the reported fungicidal agents 10c–d (see ref. 34). | ||
In this work, we also analyzed their mechanism of action (bactericidal and/or bacteriostatic), the hemocompatibility profile and the contribution of different substituents on the phenylamino ring (9a–d), also including the bromo group at the C-6 carbon of the aza-quinone nucleus (10a–d).
2. Results and discussion
2.1 Chemistry
The 7-arylamino-5,8-dioxo-5,8-dihydroisoquinoline-4-carboxylates 9a–d were prepared using the synthetic sequence illustrated in Scheme 1. The reaction between 2,5-dihydroxyacetophenone (11) and methyl 3-aminocrotonate (12) afforded methyl 5,8-dihydroxy-1,3-dimethylisoquinoline-4-carboxylate (13), which was subjected to oxidation with MnO2 to furnish isoquinoline-5,8-dione 14 in 80% yield.36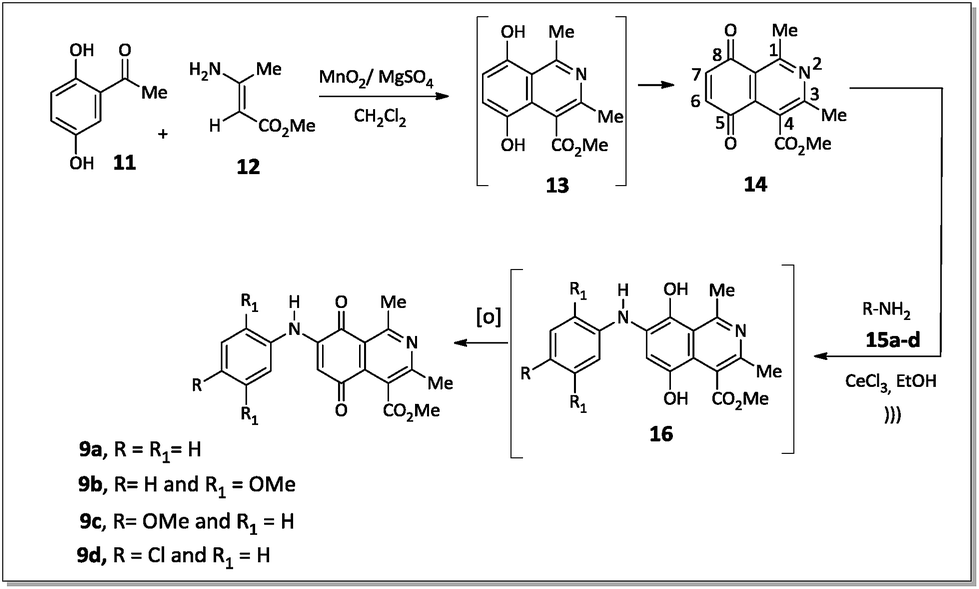 | ||
| Scheme 1 Synthesis of aminoquinones 9a–d. | ||
Aromatic amines 15a–d were transformed into the corresponding amino compounds 9a–d (Scheme 1) by ultrasound-accelerated Michel addition of anilines to electrophilic quinone compound 14, under cerium catalysis, according to the procedure described in the literature34,36 with minor modifications.
The regioselective introduction of aniline substituents at the C-7 position of 14 was confirmed based on the 2D HMBC spectrum of the aminoquinone derivative 9d. For example, the proton from the N–H bond (7.67 ppm) showed long-range correlations (3JCH) with the carbon resonance for C-6 (102.8 ppm), C-2′/C-6′ (124.6 ppm) and C-8 (181.6 ppm). This latter carbon showed correlation with the hydrogen H-6 (6.28 ppm). The observed regioselectivity for products 9a–d is in agreement with that reported by Ryu, Song and Hong.34 The carbonyl carbons C-5 and C-8 were differentiated based on the resonance effect caused by the presence of the amino group bonded at the C-7 position of the quinone-5,8-dione moiety of 9a–d, which stabilizes the C-5 carbonyl carbon making it less electrophilic (Fig. 3).
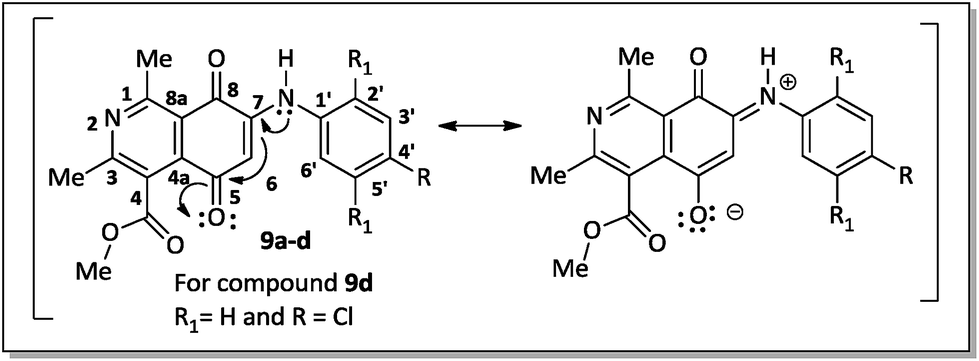 | ||
| Fig. 3 Canonic structures of compounds 9a–d. | ||
The reaction of compounds 9a–d with N-bromosuccinimide (NBS) led to halogenated derivatives 10a–d in poor yields after purification by silica gel column chromatography.
An alternative to the preparation of substances 10a–d consisted of the bromination reaction of 14 with molecular bromine in a buffer solution of sodium acetate in glacial acetic acid, as shown in Scheme 2. This reaction afforded a mixture of di-brominated and tri-brominated compounds (17 and 18, respectively).
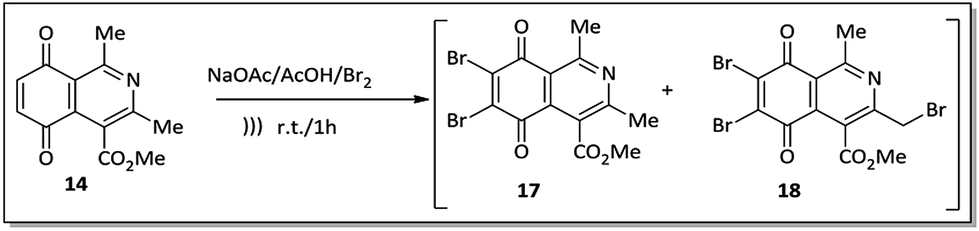 | ||
| Scheme 2 Preparation of brominated compounds 17 and 18. | ||
Compounds 17 and 18 were not isolated due to further degradation during chromatographic processing. For this reason, the crude product, which essentially consists of two major compounds, 17 and 18, was then submitted to nucleophilic substitution reaction with arylamines 15a–d, furnishing a mixture of the corresponding aminoquinone compounds 10a–d and 19a–d (Scheme 3).
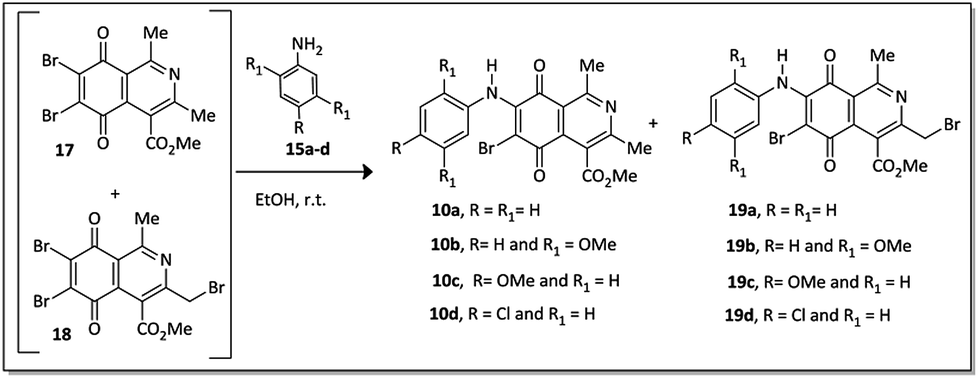 | ||
| Scheme 3 Preparation of quinone derivatives 10a–d and 19a–d. | ||
The X-ray crystallographic analysis of the amino compounds 10d and 19d (Fig. 4) allowed the molecular structures of 17 and 18 to be confirmed (Scheme 2).
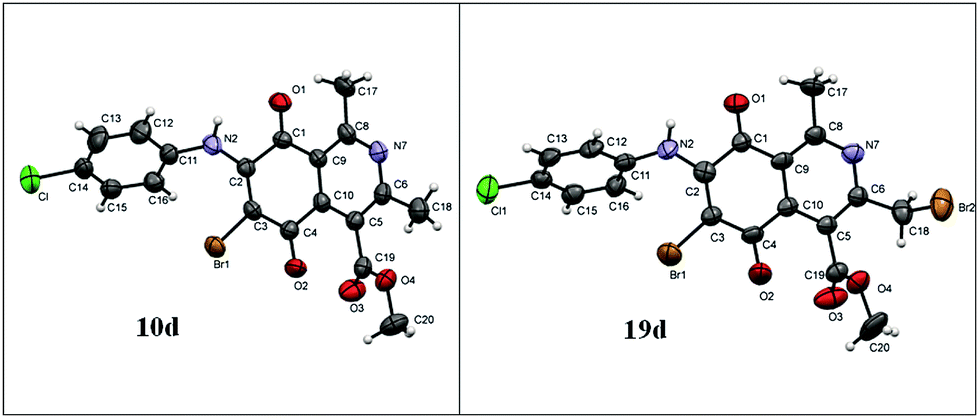 | ||
| Fig. 4 Asymmetric unit representation of quinone derivatives 10d and 19d (ellipsoids at the 50% probability level). | ||
All compounds, except 19a and 19b, were isolated as pure materials by silica gel column chromatography using chloroform/acetone (9.5:0.5) as an eluent mixture. The structures of the novel aminoquinone derivatives 9d, 10a–b and 19c–d were confirmed by spectral data, including one- and two-dimensional 1H and 13C NMR spectra [1H, 13C-APT, COSY-1H × 1H, HSQC and HMBC], IR and mass analysis.
2.2 Biological assay
Initially, the antibacterial profile of the aza-quinone derivatives 9a–d, 10a–d, 14 and 19c–d was evaluated in vitro by measuring the inhibition zone against Gram-positive (Enterococcus faecalis ATCC 29212, Staphylococcus aureus ATCC 25923, Staphylococcus epidermidis ATCC 12228, and Staphylococcus simulans ATCC 27851) and Gram-negative bacteria (Enterobacter cloaceae ATCC 23355, Serratia marcescens ATCC 14756, Escherichia coli ATCC 25922, Proteus mirabilis ATCC 15290, Klebsiella pneumoniae ATCC 4352 and Pseudomonas aeruginosa ATCC 27853).The disk diffusion data showed an inhibition zone (8–18 mm) for these compounds with different patterns. Only compounds 9c and 9d were active against both Gram-positive (S. aureus ATCC 25923, S. epidermidis ATCC 12228 and S. simulans ATCC 27851) and Gram-negative (E. coli ATCC 25922 and P. aeruginosa ATCC 27853) bacteria (Table 1), suggesting that the substitution pattern on the aniline ring is a fundamental requirement for the antimicrobial activity.
| Compound | Inhibition zone (mm) | ||||
|---|---|---|---|---|---|
| Gram-positive | Gram-negative | ||||
| S. aureus ATCC 25923 | S. epidermidis ATCC 12228 | S. simulans ATCC 27851 | E. coli ATCC 25922 | P. aeruginosa ATCC 27853 | |
| a Cip = ciprofloxacin and Van = vancomycin (positive controls); DMSO = dimethyl sulphoxide (negative control). | |||||
| 9a | 0 | 0 | 0 | 0 | 0 |
| 9b | 0 | 0 | 0 | 0 | 0 |
| 9c | 0 | 12 ± 2 | 11 ± 1 | 18 ± 2 | 15 ± 1 |
| 9d | 9 ± 2 | 10 ± 2 | 10 ± 2 | 15 ± 1 | 12 ± 2 |
| 10a | 0 | 0 | 0 | 0 | 9 ± 2 |
| 10b | 0 | 0 | 10 ± 3 | 8 ± 1 | 9 ± 2 |
| 10c | 0 | 0 | 0 | 8 ± 2 | 0 |
| 10d | 12 ± 1 | 0 | 11 ± 2 | 0 | 0 |
| 14 | 15 ± 1 | 14 ± 1 | 16 ± 2 | 25 ± 1 | 23 ± 1 |
| 19c | 0 | 0 | 0 | 0 | 0 |
| 19d | 0 | 0 | 8 ± 2 | 9 ± 2 | 10 ± 2 |
| Cip | — | — | — | 34 ± 2 | 39 ± 3 |
| Van | 14 ± 2 | 15 ± 2 | 12 ± 2 | — | — |
| DMSO | 0 | 0 | 0 | 0 | 0 |
Differently, compound 10d was effective against S. aureus ATCC 25923 and S. simulans ATCC 27851, whereas the related brominated compounds 10a and 10c showed antimicrobial effects against Gram-negative strains (E. coli ATCC 25922 and P. aeruginosa ATCC 27853) (Table 1). The derivative 10b was active against S. simulans ATCC 27851, E. coli ATCC 25922 and P. aeruginosa ATCC 27853.
The selectivity profile of the halogen compounds 10a and 10c was not observed for the non-brominated analogues 9a and 9c, indicating the importance of this type of substituent for the antibacterial activity.
Isoquinoline-5,8-dione (14) exhibited broad spectrum activity (14–25 mm) against Gram-positive and Gram-negative strains. These results showed the importance of the quinonoid ring for antibacterial activity.37
19c, a dibromo analogue of compound 10c, showed no antimicrobial activity, which suggested that the introduction of the second bromine at the benzylic position was responsible for the absence of activity. On the other hand, the dibromo compound 19d, containing a chlorine substituent on the benzene ring of the aniline group, showed antimicrobial activity against S. simulans ATCC 27851, E. coli ATCC 25922 and P. aeruginosa ATCC 27853. This fact is in agreement with previous results that indicate the chlorine atom substituent as a modulating parameter for the antibacterial profile.38
After the disk diffusion analyses, the active compounds were analyzed using an MIC assay to determine the lowest concentration capable of inhibiting visible bacterial growth. The MIC values listed in Table 2 revealed that the non-brominated compounds 9c–d had promising antimicrobial profiles against Gram-negative strains (E. coli ATCC25922 and P. aeruginosa ATCC27853) with MICs (1–2 μg mL−1) close to that observed for ciprofloxacin, an antibacterial currently in use, and within CLSI values (0.05–256 μg mL−1) (Table 2).
| Compound | MIC (μg mL−1) | ||||
|---|---|---|---|---|---|
| S. aureus ATCC 25923 | S. epidermidis ATCC 12228 | S. simulans ATCC 27851 | E. coli ATCC 25922 | P. aeruginosa ATCC 27853 | |
| a Ciprofloxacin: positive control used for Gram-negative strains.b Vancomycin: positive control used for Gram-positive strains.c ND: no detected activity at 256 μg mL−1. | |||||
| 9a | ND | ND | ND | ND | ND |
| 9b | ND | ND | ND | ND | ND |
| 9c | ND | ND | ND | 1 | 2 |
| 9d | ND | ND | ND | 1 | 2 |
| 10a | ND | ND | ND | ND | 32 |
| 10b | ND | ND | 64 | 128 | 128 |
| 10c | ND | ND | ND | 4 | ND |
| 10d | 32 | ND | 16 | ND | ND |
| 14 | ND | 128 | 128 | 4 | 8 |
| 19c | ND | ND | ND | ND | ND |
| 19d | ND | ND | 32 | 2 | 16 |
| Cipa | — | — | — | 0.12 | 0.5 |
| Vanb | 2 | 2 | 2 | — | — |
The analysis of the antibacterial mechanism of these derivatives showed significant minimal bactericidal concentration (MBC) values (Table 3). The MBC is complementary to the MIC, since whereas MIC demonstrates the lowest level of antimicrobial necessary to inhibit growth, MBC represents the lowest level of antimicrobial agent that results in microbial death. According to the ratio of MBC/MIC, it is possible to identify a compound's antibacterial profile (bacteriostatic and/or bactericidal). A ratio of MBC/MIC ≤2 indicates bactericidal activity, whereas a ratio of MBC/MIC ≥4 defines a bacteriostatic effect.39
| Compound | S. aureus ATCC 25923 | S. epidermidis ATCC 12228 | S. simulans ATCC 27851 | E. coli ATCC 25922 | P. aeruginosa ATCC 27853 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| MBC (μg mL−1) | MBC/MIC | MBC (μg mL−1) | MBC/MIC | MBC (μg mL−1) | MBC/MIC | MBC (μg mL−1) | MBC/MIC | MBC (μg mL−1) | MBC/MIC | |
| a ND: no detected activity at 256 μg mL−1. A ratio of MBC/MIC ≤2 indicates bactericidal activity, whereas a ratio of MBC/MIC ≥4 defines a bacteriostatic effect. | ||||||||||
| 9c | ND | — | ND | — | ND | — | 1 | 1 | 2 | 1 |
| 9d | ND | — | ND | — | ND | — | 1 | 1 | 2 | 1 |
| 10a | ND | — | ND | — | ND | — | ND | — | 32 | 1 |
| 10b | ND | — | ND | — | 64 | 1 | 128 | 1 | 128 | 1 |
| 10c | ND | — | ND | — | ND | — | 4 | 1 | ND | — |
| 10d | 32 | 1 | ND | — | 16 | 1 | ND | — | ND | — |
| 14 | ND | — | 128 | 1 | 128 | 1 | 4 | 1 | 8 | 1 |
| 19d | ND | — | ND | — | 32 | 1 | 2 | 1 | 16 | 1 |
Interestingly, the MBC and MIC values of the active derivatives 9c–d, 10c, 14 and 19d were identical against E. coli ATCC 25922 and P. aeruginosa ATCC 27853. This suggests that these compounds can be classified as bactericidal agents against Gram-negative bacteria, which is a more interesting profile.
It is known that Gram-negative bacteria are more resistant to antimicrobial agents than Gram-positive bacteria due to their different morphological features. These bacteria present a more complex biological barrier that avoids the penetration of different antibiotics, whereas their periplasmic space has proteins and enzymes able to break down foreign molecules.40 Therefore, the identification of active molecules/lead compounds against Gram-negative bacteria, such as those identified in this work, is extremely relevant for designing new antimicrobials for medical use. This is even more important in the case of difficult-to-treat infections, like those involving resistant microorganisms. Thus bactericidal compounds that are able to kill microorganisms lead to a faster end of the bacterial infection, also reducing the rates of re-incidence and resistance.41
Most of the quinone analogues showed high MIC values (>256 μg mL−1) against Gram-positive strains, suggesting a selective mechanism for those effective against Gram-negative strains (e.g. 9c–d). Among the mono-brominated compounds tested, the derivative 10b containing methoxy groups at the ortho- and para-positions of the aniline group exhibited the highest MIC (128 μg mL−1), whereas monomethoxylated related compound 10c showed 32 times lower MIC (4 μg mL−1) and MBC (4 μg mL−1) against E. coli ATCC 25922 (Table 2). The comparison between non-brominated compound 9c and brominated compound 10c indicated the negative effect of the bromine atom, which reduced the antibacterial activity against Gram-negative bacteria at least four-fold (Table 2).
A selective antibacterial profile against Gram-negative strains was observed when comparing mono-10d and di-19d brominated compounds. Apparently, the second bromine atom had a great effect on the bacterial activity of 19d (Table 2). This molecule was more potent and selective against E. coli ATCC 25922 and P. aeruginosa ATCC 27853 than derivative 10d.
Compound 14 is a synthetic intermediate that revealed broad-spectrum activity, detected in the disk diffusion test (Table 1). Interestingly, the results of the MIC assay showed better activity against Gram-negative strains (4–8 μg mL−1), indicating that the isoquinoline-5,8-dione is an important medicinal moiety that can be modulated for targeting Gram-negative strains. Additionally, a Michael 1,4-addition type reaction between 14 and p-chloroaniline or p-methoxyaniline led to the formation of the corresponding compounds 9c–d, which exhibited the best antibacterial activity among all of the molecules studied, with MIC values of 1.0 μg mL−1 and 2.0 μg mL−1 against E. coli ATCC 25922 and P. aeruginosa ATCC 27853.
Hemolysis tests on erythrocytes performed with compounds 9c–d, 10a–d, 14 and 19d showed that these quinone derivatives had no hemolytic effect after 3 hours of incubation (0.65–8.6%) (Chart 1). Thus, these compounds can be identified as hemocompatible and non-cytotoxic to erythrocyte membranes, according to Fisher and co-workers (lysis rate <10%).42
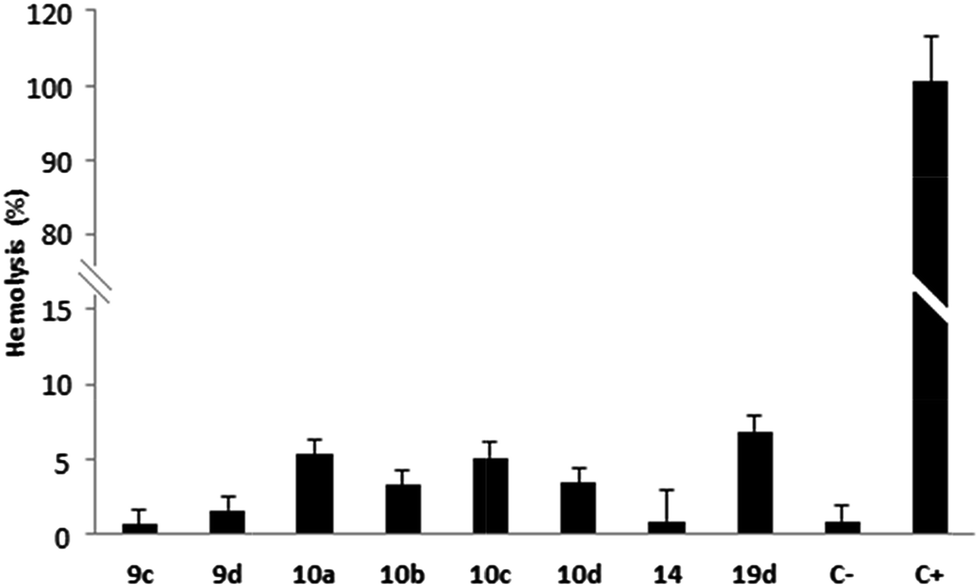 | ||
| Chart 1 Hemolytic profile through hemolysis assay. Values below 10% are considered non-hemolytic. 1% DMSO (C−) and Triton X-100 (C+) are the negative and positive controls, respectively. | ||
It is important to highlight that the aminoquinones 10c–d were recently described due to their antifungal effect against pathogenic fungi strains.34 The results from these studies showed that the substituted phenylamino group at the quinone nucleus plays an important role in the antifungal activity, similar to what we observed in the case of antibacterial activity. The authors34 suggested that 5,8-dioxo-5,8-dihydroisoquinolines related to the compounds 10c–d can act as inhibitors of the cytochrome b complex through the blockade of mitochondrial electron transport in fungi. Considering that this is a pathway also present in bacteria and that quinones and related analogs may act as redox-active molecules, this is also a possible mechanism of action for the active compounds 9c–d, 10a–d, 14 and 19d. The structures of these substances are in agreement with an electron-acceptor profile, which corroborates this hypothesis. The use of orthogonal assays to further explore quinone derivatives as inhibitors of the mitochondrial respiratory chain enzyme cytochrome b will be extremely useful, and will rationally assist the selection of these molecules for further in vivo and in vitro assays.43–45
According to the World Health Organization (WHO), the incidence of fungal and bacterial infections is a serious problem worldwide, especially in immunosuppressed individuals such as those infected with human immunodeficiency virus (HIV).46 This situation has worsened due to the increase of multi-drug resistant pathogen strains (e.g. fungi, bacteria and viruses), which require more and new treatment options.47,48 Therefore, the discovery of molecules with antifungal and antibacterial profiles such as 10c–d, as well as exploring the dual mechanism in new antibacterial derivatives such as 9c–d for treating immunosuppressed individuals such as those with HIV or cancer is interesting.
3. Conclusions
In summary, we have reported the efficient synthesis and in vitro antimicrobial activity of 7-arylamino-5,8-dioxo-5,8-dihydroisoquinoline-4-carboxylates 9a–d and their halogenated amino derivatives 10a–d and 19c–d. Among these aminoquinone compounds, the non-halogenated substances 9c–d showed promising antimicrobial profiles with MIC and MBC values in the range of 1.0 to 2.0 μg mL−1 against Gram-negative strains (E. coli ATCC25922 and P. aeruginosa ATCC27853). In addition, the brominated compounds 10c and 19d and their synthetic precursor 14 showed selective antimicrobial activity against E. coli ATCC25922, with both MIC and MBC values in the range of 2.0 to 4.0 μg mL−1.Our results suggest that the antibacterial effect with a bactericidal profile of these compounds against Gram-negative bacteria is of importance, since the emergence of resistant strains continues and this type of bacteria is of great concern. The derivatives 9c–d, 10c, 14 and 19d had no hemolysis profile when tested on erythrocyte cultures, and constitute promising prototypes for further analysis on antimicrobial therapy.
4. Experimental
4.1 In vitro biological assay
4.2 Synthesis of 7-arylaminoisoquinolinequinone derivatives 9a–d
Into a 50 mL round-bottom flask were added 245 mg (1.0 mmol) of isoquinoline-5,8-dione (14), 2.0 mmol of the required arylamine 15a–d, 18.5 mg (0.05 mmol) of CeCl3·7H2O and 20 mL of anhydrous ethanol. The reaction mixture was stirred at room temperature for 5 hours. After removal of the solvent under reduced pressure, the aminoquinone derivatives 9a–d were purified using a silica gel chromatographic column, using chloroform/acetone (9.5:0.5) as the eluent.
IR νmax (cm−1; KBr): 3300 (N–H), 1715 and 1669 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1074 and 1255 (C–O).
O), 1074 and 1255 (C–O).
1H NMR (500.00 MHz, CDCl3) δ (ppm): 2.61 (s, 3H, CH3), 2.98 (s, 3H, CH3), 4.00 (s, 3H, OCH3), 6.28 (s, 1H, H-6), 7.19 (d, 2H, J = 8.5 Hz, H-2′ and H-6′), 7.39 (d, 2H, J = 8.5 Hz, H-3′ and H-5′), 7.67 (s, 1H, N–H).
13C NMR (125.0 MHz, CDCl3) δ (ppm): 23.1 (CH3), 26.2 (CH3), 53.2 (OCH3), 119.9 (C-8a), 124.2 (C-2′ and C-6′), 125.3 (C-4), 130.1 (C-3′ and C-5′), 131.8 (C-4′), 135.6 (C-1′), 137.7 (C-4a), 102.8 (C-6), 145.3 (C-7), 161.2 (C-3), 161.5 (C-1), 169.1 (C-4b), 181.5 (C-5), 181.6 (C-8).
HRMS [M + H]+: m/z calcd for C19H15N2O4: 371.0799; found: 371.0777.
4.3 Synthesis of 7-arylamino-6-bromo-isoquinolinequinone compounds 10a–d and dibrominated derivatives 19a–d
Into a 50 mL round-bottom flask were added 245 mg (1.0 mmol) of isoquinoline-5,8-dione (14), 660 mg (8.0 mmol) of NaOAc and 8 mL of HOAc. The reaction mixture was stirred at room temperature for 10 min and then 0.1 mL of bromine was added to the reaction medium. The resulting mixture was kept in an ultrasonic water bath for 60 minutes. After this period, the reaction was quenched by addition of 200 mL of water and the crude residue consisting mainly of a mixture of di- and tri-brominated quinones, 17 and 18, respectively, was employed in the next reaction step without prior purification.The products 17 and 18 (200 mg) and the appropriate arylamine (0.15 mmol) were mixed together in EtOH (20 mL). The reaction mixture was stirred at room temperature for 60 min. After removal of the solvent under reduced pressure, the mono- and di-brominated compounds were purified by silica gel chromatography using chloroform:acetone (9.5:0.5) as the eluent.
IR νmax (cm−1; KBr): 3258 (N–H), 1721 and 1671 (CO), 1296 and 1085 (C–O).
1H NMR (500.00 MHz, CDCl3) δ (ppm): 2.97 (s, 3H, CH3), 2.62 (s, 3H, CH3), 4.03 (s, 3H, OCH3), 7.09–7.11 (m, 2H, H-2′ and H-6′), 7.24–7.27 (m, 1H, H-4′), 7.34–7.37 (m, 2H, H-3′ and H-5′), 7.98 (s, 1H, N–H).
13C NMR (125.0 MHz, CDCl3) δ (ppm): 22.7 (CH3), 25.6 (CH3), 53.4 (OCH3), 119.6 (C-8a), 125.2 (C-2′ and C-6′), 126.3 (C-4), 128.7 (C-3′ and C-5′), 126.6 (C-4′), 136.8 (C-1′), 137.0 (C-4a), 104.9 (C-6), 145.0 (C-7), 161.1 (C-3), 161.3 (C-1), 168.2 (C-4b), 175.5 (C-5), 179.5 (C-8).
HRMS [M + H]+: m/z calcd for C19H15BrN2O4: 415.0293; found: 415.0313.
IR νmax (cm−1; KBr): 3278 (N–H), 1727 and 1677 (CO), 1316 and 1157 (C–O).
1H NMR (500.00 MHz, CDCl3) δ (ppm): 3.00 (s, 3H, CH3), 2.70 (s, 3H, CH3), 3.78 (s, 3H, Ph-OCH3), 3.77 (s, 3H, Ph-OCH3), 4.05 (s, 3H, OCH3), 6.64 (d, 1H, J = 2.9 Hz, H-3′), 6.77 (dd, 1H, J = 2.9 and 9.0 Hz, H-4′), 6.82 (d, 1H, J = 9.0 Hz, H-6′), 7.73 (s, 1H, N–H).
13C NMR (125.0 MHz, CDCl3) δ (ppm): 21.8 (CH3), 24.4 (CH3), 53.7 (OCH3), 55.8 (Ph-OCH3), 56.2 (Ph-OCH3), 120.5 (C-8a), 146.7 (C-2′), 111.6 (C-3′), 111.9 (C-6′), 112.3 (C-4′), 153.2 (C-5′), 126.6 (C-4), 126.5 (C-1′), 137.4 (C-4a), 105.0 (C-6), 145.9 (C-7), 160.2 (C-3), 160.3 (C-1), 167.7 (C-4b), 174.7 (C-5), 178.6 (C-8).
HRMS [M + H]+: m/z calcd for C21H19BrN2O6: 475.0505; found: 475.0485.
IR νmax (cm−1; KBr): 3303 (N–H), 1732 and 1675 (CO), 1239 and 1165 (C–O).
1H NMR (300.00 MHz, CDCl3) δ (ppm): 2.96 (s, 3H, CH3), 2.61 (s, 3H, CH3), 3.83 (s, 3H, Ph-OCH3), 4.02 (s, 3H, OCH3), 6.88 (d, 2H, J = 8.9 Hz, H-2′ and H-6′), 7.05 (d, 2H, J = 8.5 Hz, H-3′ and H-5′), 7.94 (s, 1H, N–H).
IR νmax (cm−1; KBr): 3306 (N–H), 1732 and 1677 (CO), 1244 and 1083 (C–O).
1H NMR (500.00 MHz, CDCl3) δ (ppm): 2.61 (s, 3H, CH3), 2.96 (s, 3H, CH3), 4.03 (s, 3H, OCH3), 7.03 (d, 2H, J = 8.4 Hz, H-2′ and H-6′), 7.33 (d, 2H, J = 8.4 Hz, H-3′ and H-5′), 7.89 (s, 1H, N–H).
IR νmax (cm−1; KBr): 3286 (N–H), 1728 and 1677 (CO), 1297 and 1079 (C–O).
1H NMR (500.00 MHz, CDCl3) δ (ppm): 2.98 (s, 3H, CH3), 3.83 (s, 3H, Ph-OCH3), 4.04 (s, 3H, OCH3), 4.55 (s, 2H, C3–CH2–Br), 6.88 (d, 2H, J = 8.8 Hz, H-2′ and H-6′), 7.05 (d, 2H, J = 8.8 Hz, H-3′ and H-5′), 7.92 (s, 1H, N–H).
13C NMR (125.0 MHz, CDCl3) δ (ppm): 25.9 (CH3), 29.2 (CH2), 53.3 (OCH3), 55.5 (Ph-OCH3), 120.9 (C-8a), 113.8 (C-2′ and C-6′), 125.9 (C-4), 126.9 (C-3′ and C-5′), 129.3 (C-1′), 158.3 (C-4′), 138.2 (C-4a), 103.5 (C-6), 144.9 (C-7), 158.5 (C-3), 161.8 (C-1), 167.3 (C-4b), 174.8 (C-5), 179.4 (C-8).
HRMS [M + H]+: m/z calcd for C20H16Br2N2O5: 522.9504; found: 522.9487.
IR νmax (cm−1; KBr): 3295 (N–H), 1736 and 1679 (CO), 1300 and 1092 (C–O).
1H NMR (500.00 MHz, CDCl3) δ (ppm): 2.89 (s, 3H, CH3), 3.95 (s, 3H, OCH3), 4.47 (s, 2H, C3–CH2–Br), 6.95 (d, 2H, J = 8.5 Hz, H-2′ and H-6′), 7.24 (d, 2H, J = 8.7 Hz, H-3′ and H-5′), 7.85 (s, 1H, N–H).
13C NMR (125.0 MHz, CDCl3) δ (ppm): 26.2 (CH3), 29.5 (CH2), 53.5 (OCH3), 120.8 (C-8a), 126.3 (C-2′ and C-6′), 125.8 (C-4), 128.8 (C-3′ and C-5′), 132.2 (C-4′), 135.4 (C-1′), 137.9 (C-4a), 105.4 (C-6), 144.8 (C-7), 158.6 (C-3), 162.2 (C-1), 167.4 (C-4b), 175.2 (C-5), 179.4 (C-8).
HRMS (M+): m/z calcd for C19H13Br2N2O4: 526.9009; found: 526.9003.
Acknowledgements
This work was supported by the Brazilian agency FAPERJ. Fellowships granted to UFF, by CAPES, CNPq, CNPq-PIBIC and FAPERJ are gratefully acknowledged.References
- World Health Organization, Resistance Global Report on Surveillance, Geneva, 2014, http://www.who.int/drugresistance/documents/surveillancereport/en/, accessed April 2016 Search PubMed.
- I. Roca, M. Akova, F. Baquero, J. Carlet, M. Cavaleri, S. Coenen, J. Cohen, D. Findlay, I. Gyssens, O. E. Heure, G. Kahlmeter, H. Kruse, R. Laxminarayan, E. Liébana, L. López-Cerero, A. MacGowan, M. Martins, J. Rodríguez-Baño, J. M. Rolain, C. Segovia, B. Sigauque, E. Taconelli, E. Wellington and J. Vila, New Microbes New Infect., 2015, 6, 22–29 CrossRef CAS PubMed.
- Centers for Disease Control and Prevention (CDC), Antibiotic/Antimicrobial Resistance, https://www.cdc.gov/drugresistance/, accessed, August 2016 Search PubMed.
- A. P. Magiorakos, A. Srinivasan, R. B. Carey, Y. Carmeli, M. E. Falagas, C. G. Giske, S. Harbarth, J. F. Hindler, G. Kahlmeter, B. Olsson-Liljequist, D. L. Paterson, L. B. Rice, J. Stelling, M. J. Struelens, A. Vatopoulos, J. T. Weber and D. L. Monnet, Clin. Microbiol. Infect., 2012, 18, 268–281 CrossRef CAS PubMed.
- L. J. V. Piddock, Lancet Infect. Dis., 2012, 12, 249–253 CrossRef PubMed.
- E. Ruppé, P. L. Woerther and F. Barbier, Ann. Intensive Care, 2015, 5, 21 CrossRef PubMed.
- C. S. Lee and Y. Doi, Infect. Chemother., 2014, 46, 149–164 CrossRef PubMed.
- X. Li, P. Plésiat and H. Nikaido, Clin. Microbiol. Rev., 2015, 28, 337–418 CrossRef CAS PubMed.
- B. Mehrad, N. M. Clark, G. G. Zhanel and J. P. Lynch, Chest, 2015, 147, 1413–1421 CrossRef PubMed.
- N. Rabin, Y. Zheng, C. Opoku-Temeng, Y. Du, E. Bonsu and H. O. Sintim, Future Med. Chem., 2015, 5, 647–671 CrossRef PubMed.
- A. Penesyan, M. Gillings and I. T. Paulsen, Molecules, 2015, 20, 5286–5298 CrossRef CAS PubMed.
- S. J. Cole, A. R. Records, M. W. Orr, S. B. Linden and V. T. Lee, Infect. Immun., 2014, 82, 2048–2058 CrossRef PubMed.
- C. Beloin, A. Roux and J. Ghigo, Curr. Top. Microbiol. Immunol., 2008, 322, 249–289 CAS.
- J. G. Malone, Infect. Drug Resist., 2015, 8, 237–247 CrossRef PubMed.
- G. Laverty, S. P. Gorman and B. F. Gilmore, Pathogens, 2014, 3, 596–632 CrossRef CAS PubMed.
- M. Bassetti and E. Righi, Curr. Opin. Crit. Care, 2015, 5, 402–411 CrossRef PubMed.
- K. S. Kaye and J. M. Pogue, Pharmacotherapy, 2015, 35, 949–962 CrossRef CAS PubMed.
- J. M. Pagès, S. Peslier, T. A. Keating, J. P. Lavigne and W. W. Nichols, Antimicrob. Agents Chemother., 2015, 60, 1349–1359 CrossRef PubMed.
- K. J. Swapnaja, S. Yennam, M. Chavali, Y. Poornachandra, C. G. Kumar, K. Muthusamy, V. B. Jayaraman, P. Arumugam, S. Balasubramanian and K. K. Sriram, Eur. J. Med. Chem., 2016, 117, 85–98 CrossRef CAS PubMed.
- L. Macedo, T. Fernandes, L. Silveira, A. Mesquita, A. A. Franchitti and E. A. Ximenes, Phytomedicine, 2013, 21, 25–29 CrossRef CAS PubMed.
- (a) Y. Kumagai, Y. Shinkai, T. Miura and A. K. Cho, Annu. Rev. Pharmacol. Toxicol., 2012, 52, 221–247 CrossRef CAS PubMed; (b) E. Sailaja, S. Bhavani, D. Rambabu, M. V. B. Rao and M. Pal, Curr. Catal., 2014, 3, 310–315 CrossRef CAS; (c) M. N. da Silva, V. F. Ferreira and M. C. B. V. de Souza, Quim. Nova, 2003, 26, 407–416 CrossRef CAS; (d) E. Pérez-Sacau, R. G. Díaz-Penate, A. Estévez-Braun, A. G. Ravelo, J. M. Garcia-Castellano, L. Pardo and M. Campillo, J. Med. Chem., 2007, 50, 696–706 CrossRef PubMed.
- (a) J. Kang, P. Zhang, Z. Gao, J. Zhang, Z. Yan, H. Wang and R. Chen, Phytochemistry, 2016, 130, 144–151 CrossRef CAS PubMed; (b) V. R. Campos, W. A. Silva, V. F. Ferreira, C. S. Souza, P. D. Fernandes, V. N. Moreira, D. R. Rocha, F. R. F. Dias, R. C. Montenegro, M. C. B. V. Souza, F. C. S. Boechat, C. F. J. Franco, J. A. L. C. Resende and A. C. Cunha, RSC Adv., 2015, 5, 96222–96229 RSC; (c) V. R. Campos, E. A. Santos, V. F. Ferreira, R. C. Montenegro, M. C. B. V. Souza, L. V. Costa-Lotufo, M. O. Moraes, A. K. P. Regufe, A. K. Jordão, A. C. Pinto, J. A. L. C. Resende and A. C. Cunha, RSC Adv., 2012, 2, 11438–11448 RSC.
- P. Ravichandiran, D. Premnath and S. Vasanthkumar, J. Biol. Chem., 2014, 7, 93–101 CrossRef CAS PubMed.
- M. A. Castro, A. M. Gamito, V. Tangarife-Castaño, B. Zapata, J. M. Miguel del Corral, A. C. Mesa-Arango, L. Betancur-Galvis and A. San Feliciano, Eur. J. Med. Chem., 2013, 67, 19–27 CrossRef PubMed.
- I. T. Crosby, D. G. Bourke, E. D. Jones, P. J. de Bruyn, D. Rhodes, N. Vandegraaff, S. Cox, J. A. V. Coates and A. D. Robertson, Bioorg. Med. Chem., 2010, 18, 6442–6450 CrossRef CAS PubMed.
- E. M. Malik and C. E. Müller, Med. Res. Rev., 2016, 36, 705–748 CrossRef CAS PubMed.
- P. F. Carneiro, M. C. Pinto, R. K. Marra, F. C da Silva, J. A. Resende, E. S. L. F. Rocha, H. G. Alves, G. S. Barbosa, M. C. de Vasconcellos, E. S. Lima, A. M. Pohlit and V. F. Ferreira, Eur. J. Med. Chem., 2016, 108, 134–140 CrossRef CAS PubMed.
- (a) X. Zhang, H. Y. Xu, A. M. Huang, L. Wang, Q. Wang, P. Y. Cao and P. M. Yang, Chem. Pharm. Bull., 2016, 64, 1036–1042 CrossRef CAS PubMed; (b) D. Schulz, P. Beese, B. Ohlendorf, A. Erhard, H. Zinecker, C. Dorador and J. F. Imhoff, J. Antibiot., 2011, 64, 763–768 CrossRef CAS PubMed; (c) A. K. Jordão, J. Novais, B. Leal, A. C. Escobar, H. M. dos Santos Jr, H. C. Castro and V. F. Ferreira, Eur. J. Med. Chem., 2013, 63, 196–201 CrossRef PubMed.
- (a) P. Ravichandiran, A. Jegan, D. Premnath, V. S. Periasamy, S. Muthusubramanian and S. Vasanthkumar, Bioorg. Chem., 2014, 53, 24–36 CrossRef CAS PubMed; (b) J. A. Mohammed, J. H. Ahmed and H. A. Al-Hazam, Res. J. Pharm., Biol. Chem. Sci., 2014, 5, 1328–1339 Search PubMed; (c) S. A. A. Osman, A. A. Abdalla and M. O. Alaib, J. Pharm. Sci., 1983, 72, 68–71 CrossRef CAS PubMed.
- (a) V. K. Tandon, H. K. Maurya, N. N. Mishra and P. K. Shukla, Eur. J. Med. Chem., 2009, 44, 3130–3137 CrossRef CAS PubMed; (b) C. Ryu and D. Kim, Arch. Pharmacal Res., 1992, 15, 263–268 CrossRef CAS.
- R. S. Medina, M. P. Barros, R. S. Galhardo, L. E. S. Netto, P. Colepicolo and C. F. M. Menck, Res. Microbiol., 2006, 157, 275–281 CrossRef PubMed.
- S. Karkare, T. T. H. Chung, F. Collin, L. A. Mitchenall, A. R. McKay, S. J. Greive, J. J. M. Meyer, N. Lall and A. Maxwell, J. Biol. Chem., 2013, 288, 5149–5156 CrossRef CAS PubMed.
- M. Jeya, H. J. Moon, J. L. Lee, I. W. Kim and J. K. Lee, Appl. Microbiol. Biotechnol., 2010, 85, 1653–1663 CrossRef CAS PubMed.
- C. Ryu, A. L. Song and J. A. Hong, Bioorg. Med. Chem. Lett., 2013, 23, 2065–2068 CrossRef CAS PubMed.
- J. A. Barnett, Yeast, 2003, 12, 1015–1044 CrossRef PubMed.
- J. A. Valderrama, J. A. Ibacache, V. Arancibia, J. Rodriguez and C. Theoduloz, Bioorg. Med. Chem., 2009, 17, 2894–2901 CrossRef CAS PubMed.
- A. Marella, O. P. Tanwar, R. Saha, M. R. Ali, S. Srivastava, M. Akhter, M. Shaquiquzzaman and M. M. Alam, Saudi Pharm. J., 2013, 21, 1–12 CrossRef PubMed.
- X. J. Fang, P. Jeyakkumar, S. R. Avula, Q. Zhou and C. H. Zhou, Bioorg. Med. Chem. Lett., 2016, 26, 2584–2588 CrossRef CAS PubMed.
- K. Konaté, J. F. Mavoungou, A. N. Lepengué, R. R. R. A. Samseny, A. Hilou, A. Souza, M. H. Dicko and B. M'Batchi, Ann. Clin. Microbiol. Antimicrob., 2012, 11, 18 CrossRef PubMed.
- L. Martínez-Martínez and J. Calvo, Enfermedades Infecciosas y Microbiologia Clinica, 2010, 28, 25–31 CrossRef.
- F. Silva, O. Lourenço, J. A. Queiroz and F. C. Domingues, J. Antibiot., 2011, 64, 321–325 CrossRef CAS PubMed.
- D. Fisher, Y. Li, B. Ahlemeyer, J. Krieglstein and T. Kissel, Biomaterials, 2003, 24, 1121–1131 CrossRef.
- J. B. Baell, Future Med. Chem., 2010, 10, 1529–1546 CrossRef PubMed.
- R. F. Bruns and I. A. Watson, J. Med. Chem., 2012, 55, 9763–9772 CrossRef CAS PubMed.
- J. Baell and M. A. Walters, Nature, 2014, 513, 481–483 CrossRef CAS PubMed.
- World Health Organization, Antimicrobial resistance, http://www.who.int/mediacentre/factsheets/fs194/en/, accessed, February 2016 Search PubMed.
- C. Ibis, A. F. Tuyun, Z. Ozsoy-Gunes, H. Bahar, M. V. Stasevych, R. Y. Musyanovych, O. Komarovska-Porokhnyavets and V. Novikov, Eur. J. Med. Chem., 2011, 46, 5861–5867 CrossRef CAS PubMed.
- G. Alebachew, B. Teka, M. Endris, Y. Shiferaw and B. Tessema, BioMed Res. Int., 2016, 1–8 CrossRef PubMed.
- CLSI, Clinical and Laboratory Standard Institute.M100-S25 Performance Standards for Antimicrobial Susceptibility Testing, Twenty-fifth Informational Supplement 35 (3): 1–240, 2015 Search PubMed.
- E. L. Chuah, Z. A. Zakaria, Z. Suhaili, S. Abu Bakar and M. N. Desa, J. Microbiol. Res., 2014, 4, 6–13 Search PubMed.
- K. Konaté, P. Zerbo, M. Ouédraogo, C. I. Dibala, H. Adama, O. Sytar, M. Brestic, N. Barro, G. A. Pankey and L. D. Sabath, Ann. Clin. Microbiol. Antimicrob., 2013, 12–14 Search PubMed.
- P. C. Sathler, A. L. Lourenço, C. R. Rodrigues, L. C. da Silva, L. M. Cabral, A. K. Jordão, A. C. Cunha, M. C. Vieira, V. F. Ferreira, C. E. Carvalho-Pinto, H. C. Kang and H. C. Castro, Thromb. Res., 2014, 134, 376–383 CrossRef CAS PubMed.
- M. J. Parnham and H. Wetzig, Chem. Phys. Lipids, 1993, 64, 263–274 CrossRef CAS PubMed.
- M. Bauer, C. Lautenschlaeger, K. Kempe, L. Tauhardt, U. S. Schubert and D. Fischer, Macromol. Biosci., 2012, 7, 986–998 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1528385 and 1528399. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra00825b |
| This journal is © The Royal Society of Chemistry 2017 |