
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of aminothiazoles: polymer-supported approaches
R. V. Patil
 ,
J. U. Chavan
and
A. G. Beldar
*
,
J. U. Chavan
and
A. G. Beldar
*
PSGVPM’S Arts, Science & Commerce College, Shahada Dist.- Nandurbar, Maharashtra 425409, India. E-mail: dragbeldar@gmail.com
First published on 3rd May 2017
Abstract
Aminothiazoles and their derivatives are of immense biological importance and have been consistently synthesized via various methods. However, the synthesis of aminothiazole derivatives has some problems such as poor yields, difficult isolation procedures, use of expensive catalysts etc. Recently, polymer or solid-supported synthetic protocols have attracted the attention from the research community because of their easy execution, increased product yields, greater selectivity, simple work-up procedures, and recoverability of the catalysts. In this study, we reported the polymer-supported approaches for the synthesis of differently substituted aminothiazoles.
 R. V. Patil | Rajendra Patil completed his M.Sc. from North Maharashtra University (INDIA) in 2009. He was a CSIR-JRF fellowship awardee. He is currently working as an Assistant Professor in Chemistry at PSGVPM's Arts, Sci. & Comm. College, Shahada (Maharashtra, India) pursuing his PhD under the supervision of Dr. Anil Beldar. His research interests are polymer supported organic synthesis, heterocyclic chemistry and catalysis. |
 J. U. Chavan | Jagdish Chavan completed his M.Sc. from North Maharashtra University (India) in 2009. He was a CSIR-JRF fellowship awardee. He is currently working as an Assistant Professor in Chemistry at PSGVPM's Arts, Sci. & Comm. College, Shahada (Maharashtra, India) pursuing his PhD under the supervision of Dr. Anil Beldar. His research interests are polymer supported organic synthesis, heterocyclic chemistry and catalysis. |
 A. G. Beldar | Anil Beldar completed his M.Sc. from the University of Pune (India) in 2004. He was a JRF fellowship awardee and received his PhD from the D.R.D.E., Ministry of Defense, Government if India, Gwalior (India) in 2008. Then, he worked as a Research Scientist at Jubilient Pharma. Ltd., Noida (India) till 2010. Since 2010, he has been working as an Assistant Professor in Chemistry at PSGVPM's Arts, Sci. & Comm. College, Shahada (Maharashtra, India). His research interests mainly covers polymer supported organic synthesis, heterocyclic chemistry and catalysis. |
Introduction
A number of differently substituted thiazoles exhibit a wide range of biological activities.1 Aminothiazoles belonging to the thiazole family form a potential pharmacophore nucleus. 2-Aminothiazole moieties are useful structural motifs in medicinal chemistry as they have been reported to possess antitumor,2 antiviral,3,4 antibacterial,5–7 anti-prion,8 psychotropic,9 anti-allergic,10 anti-hypertensive,11 anti-inflammatory,12,13 antifungal,14 antitubercular,15 anti-HIV,16 pesticidal,17 antiprotozoal,18 antipyretic,19 antioxidative,20 and analgesic21 activities. Aminothiazoles acts as ligands of estrogen receptors22 and represent a novel class of adenosine receptor antagonists;23 its other analogues are used as fungicides, inhibiting the in vivo growth of Xanthomonas, and as an ingredient of herbicides or as schistosomicidal and anthelmintic drugs.24 The 2-aminothiazole analogue MB06322 has also been used to target the neuropeptide Y5 (NPY5) receptor for the treatment of eating disorders such as hyperphagia.25 This class of compounds is well known as a prodrug for the treatment of type-2 diabetes,26 and aminothiazole-4-carboxylate derivatives are known as active compounds against Mycobacterium tuberculosis H37Rv and α-ketoacyl-ACP synthase mtFabH.27 Aminothiazoles are also useful in the synthesis of polymers28,29 and dyes.30,31 Due to the multi-dimensional importance of aminothiazoles and their derivatives, several methods have been reported in the literature for their synthesis. Aminothiazoles are generally prepared by the condensation of α-halo ketones/α-tosylketones with thioureas32–38 or α-thiocyanato carbonyl compounds with aromatic or aliphatic amine hydrochlorides.39–42 Many of these reported methods suffer from some drawbacks such as harsh reaction conditions, unsatisfactory yields, hectic product isolation procedures, and use of volatile or hazardous organic solvents and expensive catalysts. Many improvisations have been consistently attempted to overcome the routine problems encountered in the synthesis of aminothiazoles. One of them is the use of a polymer or solid-supported synthetic protocol. From the literature, the polymer-supported synthetic approaches have been found to be comparatively effective over traditional processes.In the present review, we attempted to highlight the synthesis of aminothiazoles using polymer-supported strategies. In addition, this review covers the use of polymer-supported catalysts in the synthesis of aminothiazoles and polymer-mediated or co-mediated synthesis. The nature of the polymeric support may be either organic (multi-functionalised resins, polyethylene glycol—PEG etc.) or inorganic (such as silica, alumina etc.). In some of methods, supramolecules, such as β-cyclodextrin, have been used as the catalyst, whereas some syntheses are microwave-assisted. This review will provide useful insights for the researchers from the area of polymer-supported synthesis of bioactive heterocyclic motifs.
Synthesis of aminothiazoles with polymer-supported reagents
Josef Stadlwieser and co-workers43 synthesized structurally complex thiazolylhydantoines using a polymer-supported strategy in 1998. Benzhydrylamine resin modified with 6-aminohexanoic acid 1 was linked with N-butoxycarbonyl (Boc) protected amino acid derivatives 2 [R1 = CH2C6H5, CH2-cyclo-C6H11, CH2C(O)N(CH2CH2)2O] using 1,1,3,3-tetramethyl-O-(2-oxo-1,2-dihydropyridin-1-yl)uranium tetrafluoroborate (TPTU)44 as the coupling reagent to obtain the derivatives 3. After cleavage of the Boc protecting group, the free amines 4 were converted into the thiourea derivatives 6 upon reaction with allyloxycarbonyl isothiocyanate 5,45 which contain the allyloxycarbonyl (Alloc) protecting group. The Alloc group was removed using a Pd(0)-catalyzed reaction to yield thioureas 7, which were further treated with α-bromoketones 8 derived from the N-Boc-protected amino acids to furnish the 2-aminothiazole templates. Further, the synthesis of thiazolylhydantoines was accomplished in a sequence of steps, as shown in Scheme 1. Finally, the removal of the polymer support yielded the products with excellent purity. Although this synthesis involves a multi-step strategy, it was successful towards the synthesis of structurally complex moieties with high purity. | ||
| Scheme 1 | ||
Patrick C. Kearney, Monica Fernandez, and John A. Flygare have demonstrated a new method for the synthesis of 2-aminothiazoles (21)46 from a primary amine and α-bromo ketones (19), and aminothiazoles (21) was obtained in good yield and with a high degree of purity. The backbone of this method is the conversion of a resin-bound amino group to a thiourea 18 using Fmoc-NCS (17). Scheme 2 shows the steps for the synthesis of 2-aminothiazoles.
 | ||
| Scheme 2 | ||
Herein, three different resin types (16) were employed in the same procedure. The use of Rink amide MBHA resin yielded thiazoles that were unsubstituted at the 2-amino position, whereas thiazoles substituted at the 2-amino position were generated from ArgoGel-MB-CHO resin that was reductively aminated in two steps. A compound was generated from glycine linked to Rink amide MBHA resin; in this case, the 2-amino group of thiazole did not serve as the point of attachment to the resin.
In 2000, M. Kidwai, B. Dave, and K. R. Bhushan firstly reported a basic alumina (inorganic polymer)-supported synthesis of 2-aminothiazoles from halo carbonyl compounds and substituted thiourea under solvent-free conditions using microwave radiations (Scheme 3).47
 | ||
| Scheme 3 | ||
The reaction time was drastically reduced with improved yields as compared to that in the conventional method. Basic alumina was added to the solution of phenacyl bromide 22 and N-substituted thiourea 23 dissolved in dichloromethane at room temperature. The reaction mixture was thoroughly mixed, and the adsorbed material was dried in air (beaker) and placed in an alumina bath inside a microwave oven and irradiated.
Roman Baer and Thierry Masquelin in 2001 reported a novel solid-phase synthesis of 2,4-diaminothiazoles from a polymer-bound thiouronium salt 27. The synthetic strategy involved the formation of polymer-bound thioureido–thiourea intermediates 29, which upon treatment with α-bromo-ketones 30 underwent S-alkylation, followed by a base-catalyzed intramolecular ring closure/cleavage to provide 2,4-diaminothiazoles 32 (Scheme 4).48 The attractive feature of this method is the polymer-supported auto-scavenging strategy (PSAS), which provides a clean, high-yielding, and traceless synthesis towards 2,4-diaminothiazoles.
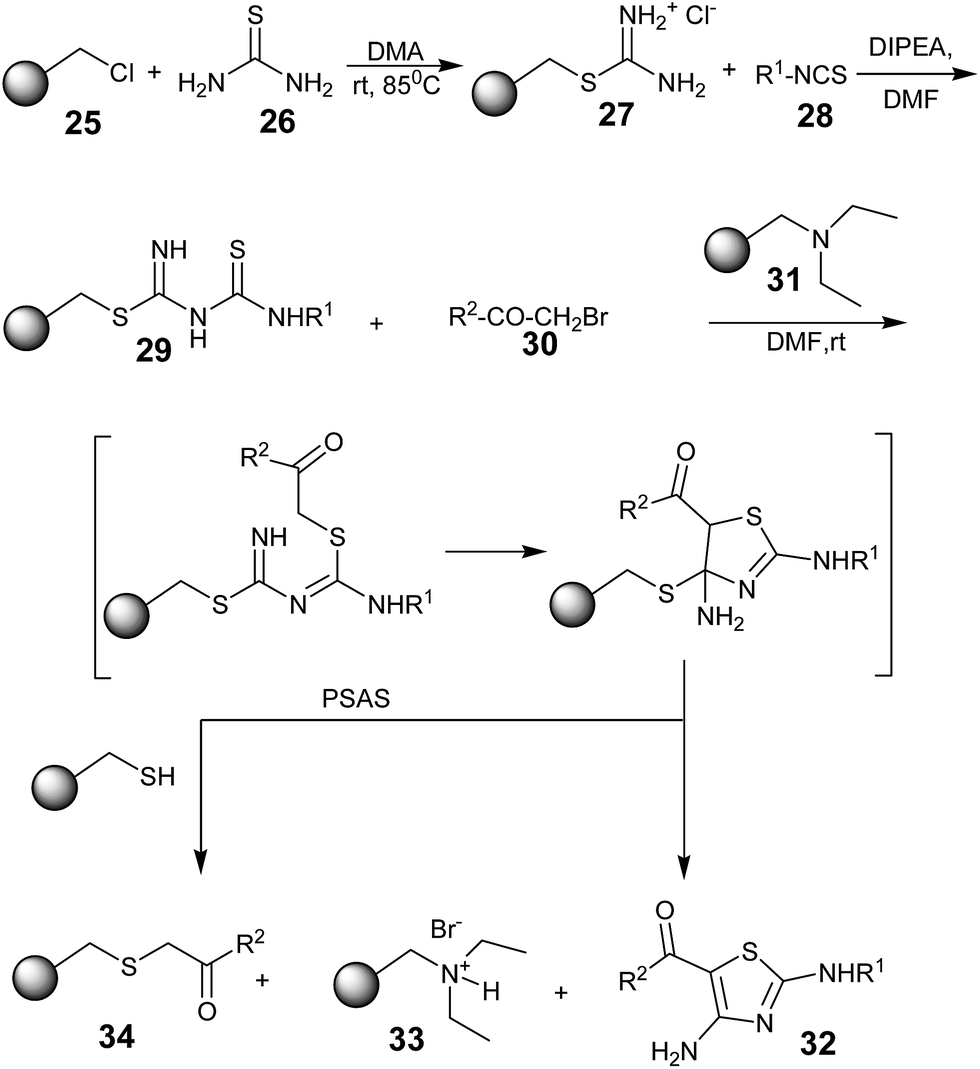 | ||
| Scheme 4 | ||
The diethylaminomethyl polystyrene resin 31 was used to activate the intermediate and isolate the HBr by-product. After the base-catalyzed intramolecular-cyclization/cleavage process, the resulting resin-bound thiol was used to remove the excess of the α-bromo ketones 30, affording the polymer-bound thioether derivatives 34. They effectively illustrated the concept of the polymer-supported auto-scavenging strategy (PSAS), affording highly pure 2,4-diaminothiazoles 32, separated from the polymer-bound derivatives 33 and 34 after simple filtration and evaporation. This was an advantage over other strategies for producing 2,4-diaminothiazoles.
Michael C. Pirrung and Sunil V. Pansare demonstrated the synthesis of 2-aminothiazole-5-carboxylates using trityl chloride resin49 (35) as the starting solid support, which was subsequently converted to the trityl isothiocyanate (TrITC) resin 36. The polymer-supported thiourea 37 prepared from 36 was subjected to condensation with a methyl 2-chloroacetoacetate50 38 and released the 2-aminothiazole-5-carboxylic acid methyl esters as their hydrochloride salts 39, which were converted to their free bases 40 by a solid-supported neutralization with sodium carbonate (Scheme 5).
 | ||
| Scheme 5 | ||
In 2002, Frank Stieber and co-workers reported51 a new method for the polymer-supported synthesis of 2-amino thiazoles, which involved a traceless hydrazide linker,52 as well as the biological evaluation of the synthesized aminothiazoles. The polymer bound amide 43 was synthesized by coupling monomethyl adipicate 42 with amino-functionalized polystyrene 41 and the subsequent saponification of the methyl ester upon treatment with LiOH (Scheme 6). The acid-functionalized resin 43 was then converted into the p-nitrophenylhydrazides 45 upon activation with N,N-diisopropylcarbodiimide (DIC) and N-hydroxybenzotriazole (HOBt) and treatment with differently substituted 4-nitrophenylhydrazines 44. The two hydrazide nitrogen atoms in 45 were acylated with Fmoc-Cl (Fmoc = fluorenylmethoxycarbonyl) before reduction of the nitrobenzene groups to anilines, as shown in Scheme 6. The polymer-bound thiourea 50 was further reacted with α-bromoketone 19 to produce polymer bound aminothiazoles 51. Finally, the targeted aminothiazoles 52 were sequestered from the solid support upon treatment with a catalytic amount of Cu(OAc)2 in n-propylamine and purging with O2 for the reoxidation of Cu+ formed in the oxidation of the linker group. The traceless cleavage of the linker was achieved via oxidation of phenylhydrazide to acyldiazene and subsequent nucleophilic attack on the carbonyl group (Scheme 7). Although this novel synthetic strategy involved a large number of steps to synthesize the aminothiazoles, it has contributed a diverse range of new structural motifs to the library of aminothiazoles, which show potential bioactivity.
 | ||
| Scheme 6 | ||
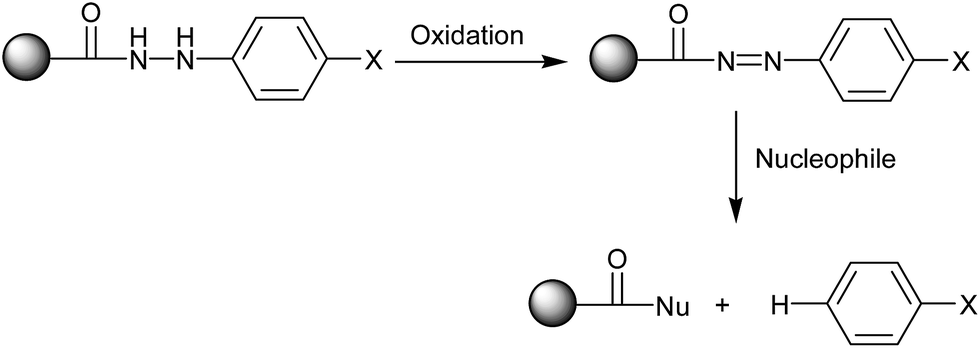 | ||
| Scheme 7 | ||
Mitsuo Kodomari, Tadashi Aoyama, and Yoshitada Suzuki developed a method for the synthesis of 2-aminothiazoles from α-bromo ketones in one-pot using a supported-reagent system, KSCN/SiO2–R1NH3OAc/Al2O3, in which the α-bromo ketone 19 reacted first with KSCN/SiO2 (ref. 53) and the product α-thiocyano ketone reacted with R1NH3OAc/Al2O3 to provide the final product 2-aminothiazole 21 in high yield. This synthesis involved two steps in one-pot (Scheme 8).54 After completion of the reaction, the used solid reagents were removed by filtration. The filtrate was then evaporated to obtain the crude products, which were purified by column chromatography over silica gel. The authors also employed α-chloro ketones as the substrate in the one-pot procedure to produce their corresponding 2-aminothiazoles in high yields as in the case of the α-bromo ketones. Although this solid-supported strategy is simple and efficient, both supported reagents were required in a large excess to obtain high yields.
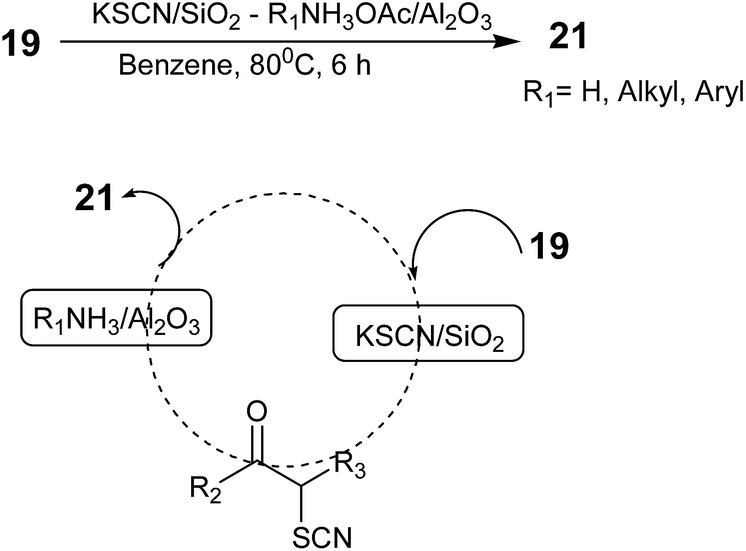 | ||
| Scheme 8 | ||
Said El Kazzouli and co-workers in 2002 demonstrated that the α-bromo ketone derived from the Rink amide resin loaded with 3-iodobenzoic acid after two steps could be subsequently treated with thiourea to obtain a variety of 2-aminothiazoles (Scheme 9).55 The resin 53 was deprotected with 20% piperidine in N,N-dimethylacetamide. 3-Iodobenzoic acid was bound onto the Rink amide resin 54 under standard peptide coupling conditions.56
 | ||
| Scheme 9 | ||
A palladium(0)-mediated coupling reaction was performed on resin 55 in the presence of tributyl(1-ethoxyvinyl)tin, triphenylarsine,56 and tris(dibenzylidene acetone)dipalladium(0) as the catalyst via heating at 50 °C under argon in 1,4-dioxane to obtain the resin 56. The conversion of 56 to α-bromoketone 57 was achieved upon treatment with N-bromosuccinimide in a mixture of THF and water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). 2-Aminothiazoles 58 were obtained by treating the resin 57 with an ethanol solution of thiourea at 50 °C. The resin bound aminothiazoles 58 were derivatized at the –NH2 position upon reaction with an acyl chloride or p-toluenesulfonyl chloride in a mixture of pyridine/dichloromethane (1/1) at room temperature. Resin cleavage using TFA provided 2-aminothiazoles 59. After seven steps, the yields varied from 33 to 78% with excellent purity.
:1). 2-Aminothiazoles 58 were obtained by treating the resin 57 with an ethanol solution of thiourea at 50 °C. The resin bound aminothiazoles 58 were derivatized at the –NH2 position upon reaction with an acyl chloride or p-toluenesulfonyl chloride in a mixture of pyridine/dichloromethane (1/1) at room temperature. Resin cleavage using TFA provided 2-aminothiazoles 59. After seven steps, the yields varied from 33 to 78% with excellent purity.
In 2003, Alan R. Katritzky, Xiaohong Cai, and Boris V. Rogovoy synthesized trisubstituted resin bound thioureas and demonstrated their applications in the synthesis of triazoles and thiazoles.57 Resin-bound amines 63 were obtained in three steps:58 the conversion of Wang resin 60 into the brominated resin 61; its subsequent nucleophilic substitution with 4-hydroxybenzaldehyde in the presence of potassium tert-butoxide; and a standard reductive amination in the presence of an amine and sodium triacetoxyborohydride (Scheme 10). 63 condensed with acyl isothiocyanates to provide N-acylated thiourea resins 64 (R1 = H). Condensation of resins 64 with 2-bromoacetophenones in the presence of TEA affords thiazoles 65. After cleavage of the solid support, the desired substituted aminothiazoles 66 were obtained in 62–80% isolated yields. The steps in Scheme 10 were also performed using Merrifield resin instead of Wang resin and resulted in similar results.
 | ||
| Scheme 10 | ||
Ill Young Lee and co-workers in 2005 reported a solid-phase synthetic strategy for the synthesis of trisubstituted aminothiazoles using resin-bound cyanodithioimidocarbonic acid and α-bromoketone.59 The resin-bound thiazole was treated with acyl chlorides or isocyanates.
After oxidation–activation of a thioether linker to a sulfone linker, traceless cleavage was achieved with nucleophiles to obtain trisubstituted aminothiazoles (Scheme 11). Merrifield resin 25 was treated with dipotassium cyanodithioimidocarbonate, which was prepared from CS2, KOH, and cyanamide in aqueous ethanol.60 The obtained resin 66 requires to be washed with water because the salt form of the resin-bound cyanodithioimidocarbonic acid is difficult to filter with organic solvents. Subsequently, resin 66 was reacted with 2-bromoacetophenone or ethyl bromoacetate and triethylamine to obtain the polymer bound 4-aminothiazoles 67. Sulfonyl resin 68 was obtained by MCPBA oxidation, an application of the solution-phase thiazole synthesis.61,62 The desired thiazoles 69 were liberated from resin 68 using the nucleophilic addition of amines (primary and secondary) and anilines. The reaction of thiazole resin 67 with isocyanate or acid chloride gave the desired resin-bound thiazole urea 70. Then, oxidation and amine addition gave the corresponding thiazoles 71. Some new differently substituted aminothiazoles were synthesized in 28–42% yield after four steps.
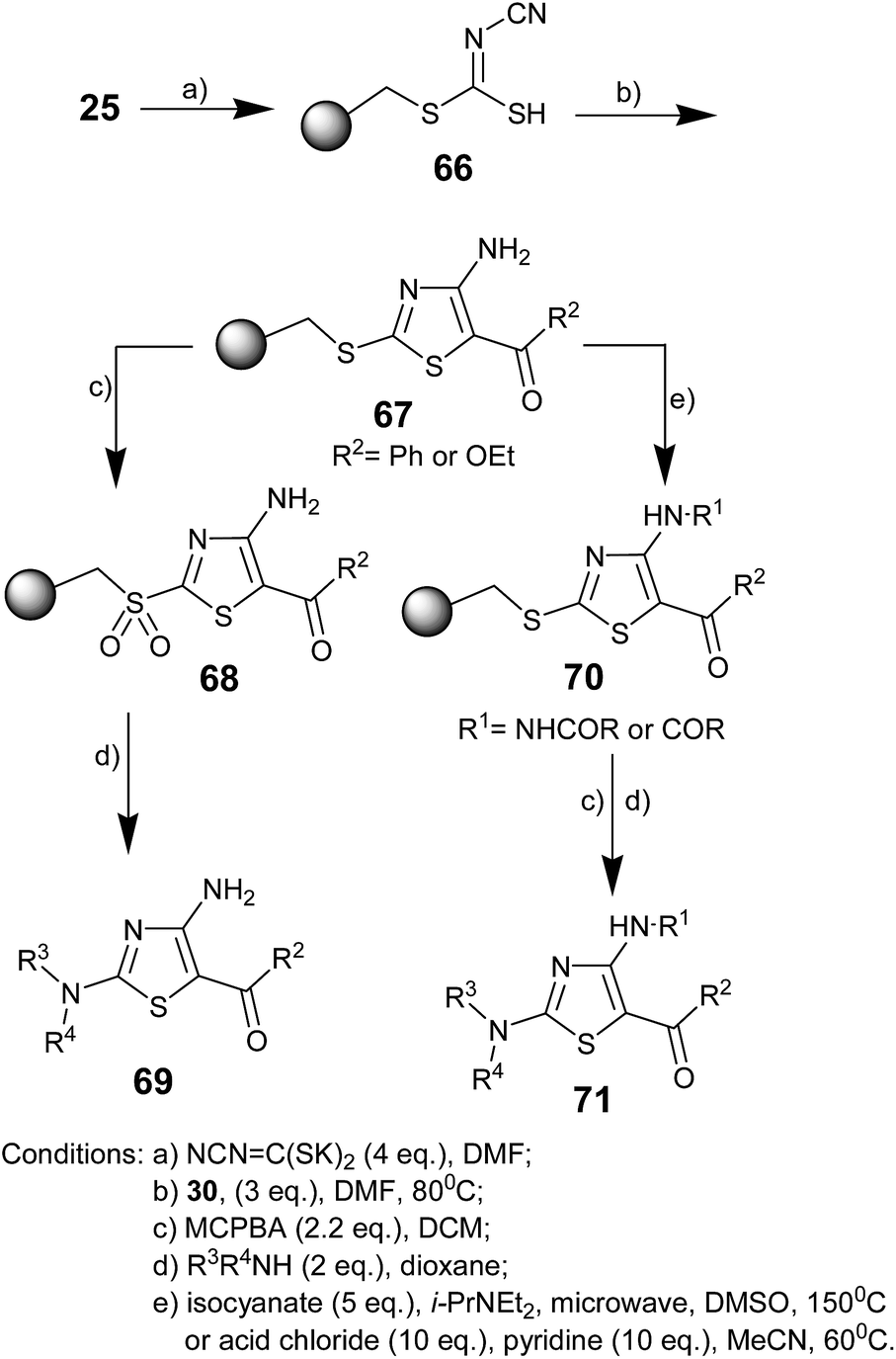 | ||
| Scheme 11 | ||
In 2006, Tadashi Aoyama and co-workers reported a method for the synthesis of 2-aminothiazoles in one pot using a supported reagents system, KSCN/SiO2–NH4OAc/Al2O3, in which an α-halo ketone 72 (X = Cl and Br) reacted first with KSCN/SiO2 and the product α-thiocyanatoketone 73 reacted with NH4OAc/Al2O3 to provide the final product 2-aminothiazoles 74 in good yield (Scheme 12).63
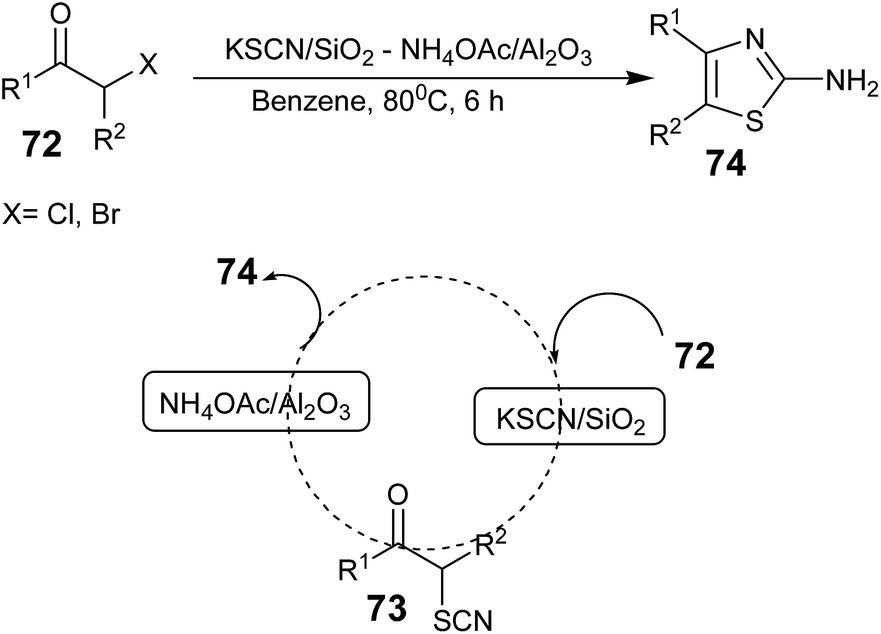 | ||
| Scheme 12 | ||
Kumaran G. Sreejalekshmi and co-workers in 2006, efficiently synthesized 2,4-diamino-5-ketothiazoles 78 using polymer-supported amidinothioureas 77 derived from aminomethylpolystyrene 75. The resin 75 was prepared from Merrifield resin using a standard literature procedure.64 Amidinothioureas 77 were treated with α-haloketones 30 to produce 78, an approach involving traceless cleavage from the support in good yield. It was reported that the second step tracelessly removes the solid support via eliminative aromatization to afford the 2,4-diamino-5-ketothiazoles 78 (Scheme 13).65 Although this strategy involved a solid-support protocol, the targeted products were purified by column chromatography. Reuse of the resin was found to be inefficient and it enormously affected the yield.
 | ||
| Scheme 13 | ||
In 2007, Tadashi Aoyama and co-workers demonstrated a one-pot three-step synthetic strategy for the synthesis of aminothiazoles using an inorganic-supported reagents system.66 The effectiveness of the one-pot three-step reaction was compared with the reaction of 79 with solid-supported reagents in a sequence of steps and also in one pot (Scheme 14); the yields obtained from the one-pot strategy were found to be higher than those obtained from the stepwise method.
 | ||
| Scheme 14 | ||
After optimization of the reagent system, differently substituted diones 83 were subjected to react with all the required reagents in one-pot to form aminothiazoles 84 (Scheme 15). This reported one-pot method provides a highly efficient synthesis of 2-aminothiazoles when compared to the stepwise method, and all three reagents were uneventfully removed from the crude products by simple filtration.
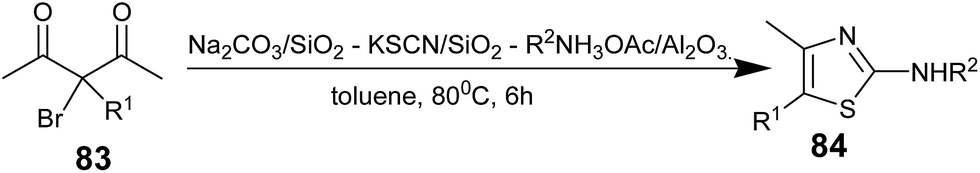 | ||
| Scheme 15 | ||
In 2009, Andrew Cole and co-workers reported the synthesis of 2-amino-5-benzoyl-4-(2-furyl) thiazoles 90, adenosine A2A receptor antagonists.67 The amino-functionalized polystyrene resin 85 was acylated with an acid-cleavable linker, 4-(4′-formyl-3′-methoxy)phenoxybutyric acid, to obtain the aldehyde 86 (Scheme 16).68 A resin-bound secondary amine 87 was generated by reductive alkylation of a series of primary amines (R1NH2) with 86. Subsequently, the reaction of 87 with different acylisothiocyanates gave the N-acyl thioureas 88. The resin-bound 2-aminothiazoles 89 were obtained upon the S-alkylation of 88 with a series of α-bromoketones and their subsequent cyclization. Finally, cleavage of the aminothiazoles from the resin using trifluoroacetic acid provided 2-aminothiazoles 90. The antagonists were further characterized via the A2A binding assay and an A1 selectivity assay.
 | ||
| Scheme 16 | ||
It is observed from the literature that, some research groups utilized polymer-supported brominating reagents, bases, and scavengers to quantitatively produce corresponding aminothiazoles with high degrees of purity. Jorg Habermann and co-workers reported the synthesis of aminothiazoles; the synthesis involved an effective α-bromination of differently substituted acetophenones using polymer-supported pyridinium bromide perbromide 92 (PSPBP).69 The targeted aminothiazoles were prepared in high yield using a sequence of polymer-supported reagents without any chromatographic purification step. The α-bromo acetophenones 93 obtained from 91 using PSPBP 92 were treated with thiourea (26) in the presence of polymer-supported 1,5,7-triazabicyclo[4.4.0]-dec-5-ene 94 (TBD-P)70 as a base in THF and the mixture was refluxed for 30 minutes; then, the cyclodehydration reaction was conducted to obtain the desired aminothiazoles 95 (Scheme 17).
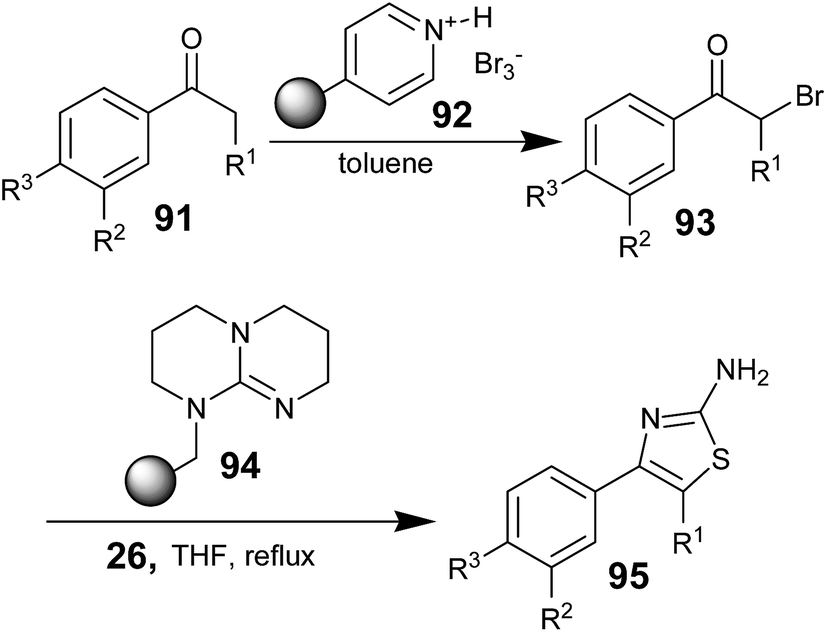 | ||
| Scheme 17 | ||
The same group has also demonstrated the synthesis of aminothiazoles 97 using sulfonamidated α-bromo piperidones 96, which were treated with thiourea 26 in the presence of TBD-P 94 in refluxing THF over 4 hours (Scheme 18). The corresponding 2-amino-1,3-thiazoles 97 were obtained in high yield and high purity.71
 | ||
| Scheme 18 | ||
Young K. Yun and co-workers synthesized 2-aminothiazoles hydrobromide monohydrates (99) by the reaction of thioureas 98 and α-bromoketones 30 in dry acetone using a Quest 205 Organic Synthesizer72 and free-based the compounds (100) in the same reaction vessel using polymer-supported bases (Scheme 19). Herein, two polymeric bases were found to be effective: MP-carbonate (macroporous triethylammonium methyl polystyrene carbonate resin) 101, a resin-bound equivalent of potassium carbonate (K2CO3),73–75 and PS-DIEA (N,N-(diisopropyl)aminomethyl-polystyrene) resin 102.76 The polymer-supported bases successfully neutralized the 2-aminothiazole hydrobromide monohydrates to produce the free-based aminothiazoles.
 | ||
| Scheme 19 | ||
Synthesis of aminothiazoles by polymer-supported catalysis
Some research groups have successfully employed polymer-supported catalysts, oligosaccharides (polysugars) etc. in the synthesis of aminothiazoles. K. Rama Rao and co-workers from India reported a few research findings on the synthesis of aminothiazoles, which involved the use of β-cyclodextrin (β-CD) as the catalyst. The use of aqueous medium for the synthesis is an attractive feature of these synthetic routes. Cyclodextrins are cyclic oligosaccharides, which have attracted interest as enzyme models due to their ability to selectively bind substrates and catalyze chemical reactions via supramolecular catalysis, involving the reversible formation of host–guest complexes with the substrates by non-covalent bonding, as seen in enzyme complexation processes.77 The complexation depends on the size, shape, and hydrophobicity of the guest molecule. Cyclodextrins have been utilized for the biomimetic modeling of the synthesis of thiazoles in water.In 2005, K. Rama Rao and co-workers demonstrated β-cyclodextrin in water as a catalyst for the first time in the synthesis of thiazoles. This method claimed efficient and user-friendly reactions conditions such as the use of a hazard-free solvent, low temperatures, fast reactions, high yields, and no unwanted side products. Phenacyl bromides 103 and thioamide/thiourea 104 react in the presence of β-CD in water, and the β-CD complex of 103 was formed in situ to obtain the corresponding thiazole or aminothiazoles 105 in high yield. It was found that the reaction smoothly proceeded without the formation of by-products or rearranged products (Scheme 20).78 After completion of the reaction, the organic material was extracted with ethyl acetate, the organic phase was separated and washed with brine, dried over Na2SO4, filtered, and the solvent was removed under vacuum. The crude product was purified by silica gel column chromatography.
 | ||
| Scheme 20 | ||
The same group synthesized aminothiazoles in 2007 via the α-halogenation of β-keto esters using N-bromosuccinimide (NBS), followed by cyclization with thiourea in the presence of β-cyclodextrin in water at 50 °C. The β-keto esters 106 forms an in situ complex with β-cyclodextrin after the addition of NBS and thiourea (26); stirring the above mentioned mixture at the same temperature afforded the corresponding thiazoles 107 in excellent yield. The treatment of 106 with NBS may afford α-bromo β-keto esters as intermediates, which then undergo cyclization with thiourea to provide the corresponding thiazole derivatives (Scheme 21).79 The reaction smoothly occurred and succinimide was obtained as the by-product, which could be recycled to obtain NBS.80 After completion of the reaction, the mixture was then extracted with EtOAc and the extract was filtered. The organic layer was dried over anhyd. Na2SO4 and the solvent was removed under reduced pressure. The resulting product was further purified by column chromatography (EtOAc–hexane, 2:8). The aqueous layer was cooled to 5 °C, and β-CD was recovered from it by filtration. To the filtrate that contained succinimide, HBr, NaBrO3, and conc. H2SO4 were added,80 and the mixture was stirred for 30 min. Then, the mixture was extracted with EtOAc and the solvent was removed under vacuum to regenerate NBS in an isolated yield of 75–80%.
 | ||
| Scheme 21 | ||
In the same year, K. Rama Rao and co-workers reported the supramolecular catalytic synthesis of thiazoles/aminothiazoles 109 from β-keto tosylates 108 and thioamide/thiourea 104 in water in the presence of β-cyclodextrin with impressive yields (Scheme 22).81 At room temperature, the planned reaction was performed, and after its completion, the products were isolated and purified by column chromatography. The catalyst β-cyclodextrin was recovered from aqueous solution cooled at 5 °C using filtration.
 | ||
| Scheme 22 | ||
Biswanath Das, V. Saidi Reddy, and R. Ramu reported the synthesis of thiazoles and aminothiazoles via the treatment of phenacyl bromides 103 with thioamides/thiourea 104 in the presence of ammonium-12-molybdophosphate (AMP), [(NH4)3(PMo12O40)]82,83 at room temperature. They observed the completion of the reaction within 20 minutes in excellent yield. Recently, heteropoly acids and their salts have been used in the synthesis because of their interesting catalytic activity and capability of conducting the reaction in a cleaner manner when compared to conventional liquid acid catalysts.84 The heterogeneous catalyst AMP, the ammonium salt of a heteropoly acid, can easily be handled and removed from the reaction mixture using simple filtration.
To a mixture of 103 and 104 (1.2 mmol) in MeOH, AMP was added. The mixture was stirred at room temperature, and after 20 min, the mixture was filtered. The filtrate was concentrated and the residue was subjected to column chromatography over silica gel using hexane–EtOAc (4:1) as the eluent to afford the pure thiazole/aminothiazoles 105 (Scheme 23).85
 | ||
| Scheme 23 | ||
Hitendra Karade, Manisha Sathe, and M.P. Kaushik demonstrated silica chloride as an effective heterogeneous catalyst for the synthesis of aminothiazoles. Many chemical transformations have been reported using silica gel as a support.86,87 The modified silica gel silica chloride (SiO2–Cl) has been reported to be an efficient reagent for the synthesis of many organic compounds.88–91 The silica chloride catalyst was prepared by stirring silica gel in DCM with thionyl chloride (SO2Cl) with the instantaneous liberation of HCl and SO2, and the catalyst prepared retained its activity for 6 months.
Silica chloride was added into ketone 110 and thiourea 26 in acetonitrile at 0 °C. The reaction mixture was brought to room temperature and refluxed for 1 h (Scheme 24).92 After completion of the reaction, the reaction mixture was filtered and the filtrate was washed with sodium bicarbonate solution and brine and extracted with ethyl acetate. The organic layer was separated, dried using anhydrous sodium sulphate, and concentrated under reduced pressure to afford the corresponding 2-aminothiazoles 74 in quantitative yield. It was noticeable that the original activity of the recovered silica chloride from recycle batch 3 could be restored by treatment with thionyl chloride under reflux.92 Studies on the application of this heterogeneous system have shown that silica chloride93,94 is an excellent source for the generation of HCl.
 | ||
| Scheme 24 | ||
Mazaahir Kidwai, Ritika Chauhan, and Divya Bhatnagar employed recyclable solid-supported Nafion-H using a polyethylene glycol (PEG)–water solvent system in the synthesis of 2-aminothiazoles with improved efficiency and reduced waste production.95 The high catalytic activity of Nafion-H, its selectivity, recyclability, inertness, thermal stability, and ease of separation from the reaction mixture makes it an attractive candidate for organic synthesis.96–98 Nafion-H, a perflourinated sulfonic acid resin, is a useful solid acid catalyst for a variety of acid-catalyzed organic transformations.99–101 It consists of a polymeric backbone with highly acidic sulfonic acid groups and possesses hydrophobic (–CF2CF2) as well as hydrophilic regions (–SO3H). Optimization of the reaction medium was performed by comparing the yields obtained with different solvents such as ethanol, acetonitrile, THF, toluene, ethylene glycol, PEG-400, and PEG–H2O. The PEG–H2O (60:40) combination was found to be most impressive as far as the yield was concerned. A mixture of phenacyl bromide 22 (1 mmol), thiourea 26 (1 mmol), and Nafion-H (1 bead) in 5 mL of the PEG:water (60:40) solvent system was stirred at 50 °C for 10–30 min (Scheme 25).
 | ||
| Scheme 25 | ||
After completion of the reaction, the catalyst was physically removed by forceps. Then, to the reaction mixture, 50 mL of ice-cold water was added. The resulting solid 2-aminothiazole products were filtered, then washed with water, and dried. The crude products were purified by column chromatography on silica gel to yield the 2-aminothiazoles 24. The catalyst showed excellent recyclability in this reaction. The catalyst was successively washed with acetone and deionized water and then dried overnight at 105 °C. The obtained catalyst had the same catalytic activity as the fresh catalyst.95
Recently, Javad Safari and Masoud Sadeghi employed starch nanoparticles (NPs) as an efficient catalyst for the synthesis of 2-aminothiazoles using methylcarbonyl compounds 110 and thiourea 26 as the precursors. Starch is a natural, renewable, biocompatible, and biodegradable polymer, which is produced by many plants as a source of stored energy. Starch or amylum is a carbohydrate that consists of glucose monomer units, which have two types of arrangements, amylose and amylopectin. Amylose is a linear polymer of glucose units that are connected to each other through an a-link. Amylopectin is a large and branched polysaccharide whose main structure is similar to that of amylose. In total, starch is formed from 20 to 30% amylose and 70 to 80% amylopectin.102–105
Starch NPs were prepared using the precipitation of amorphous starch.106 A mixture of methylcarbonyl 110, thiourea 26, iodine, DMSO, and 10 mg of starch NPs was stirred at 80 °C (Scheme 26). After completion of the reaction (as monitored by TLC, petroleum:ethyl acetate, 4:1), the reaction was quenched upon the addition of 10 mL distilled water. The aqueous solution was extracted with EtOAc and the combined extract was dried with anhydrous Na2SO4. The solvent was removed by vaccum, and finally, the resulting precipitated aminothiazoles 74 were recrystallized from EtOH.106
 | ||
| Scheme 26 | ||
Polymer-mediated synthesis of aminothiazoles
Polymers can play a distinguished role in organic synthesis in different forms such as the catalyst support, reagent support, co-medium or medium. A lot of work has been reported for the role played by the polymers in their different forms in organic synthesis. Polyethylene glycol (PEG) has been found to be used as a solvent medium support for various transformations.107,108 PEG is a non-toxic, easily available, inexpensive, and non-ionic liquid medium with low volatility. PEG and its monoethyl ethers are inexpensive, thermally stable, reusable, and non-toxic media and are also phase-transfer catalysts.109,110 Many important reactions such as the Heck reaction,111 catalytic hydrogenation,112 asymmetric dihydroxylation reaction,113 Baylis–Hillman reaction,114 Biginelli reaction,115 Suzuki–Miyaura reaction, Stille cross-coupling reaction,116 Wacker reaction,117 and asymmetric aldol reaction118 have been carried out and investigated in PEG. It has also been reported by a few researchers that PEG-mediated synthesis to prepare aminothiazoles is efficient and green method.In 2009, Pei-Ying Lin and co-workers synthesized 2-aminothiazoles from β-keto tosylates and thioureas using PEG-400 as the medium.119 A comparison between acetonitrile and PEG as the medium was performed; the PEG medium results in an enhancement in the product yield. α-Toxyloxyacetophenone 108, thiourea 23, and sodium carbonate were added to PEG-400 (Scheme 27). The resulting mixture was stirred at room temperature for 1 h. Subsequently, the reaction mixture was extracted with Et2O. The remaining PEG suspension was filtered and recycled. The combined ethereal solution was evaporated under reduced pressure. The resulting residue was purified by column chromatography to obtain the aminothiazoles 111.
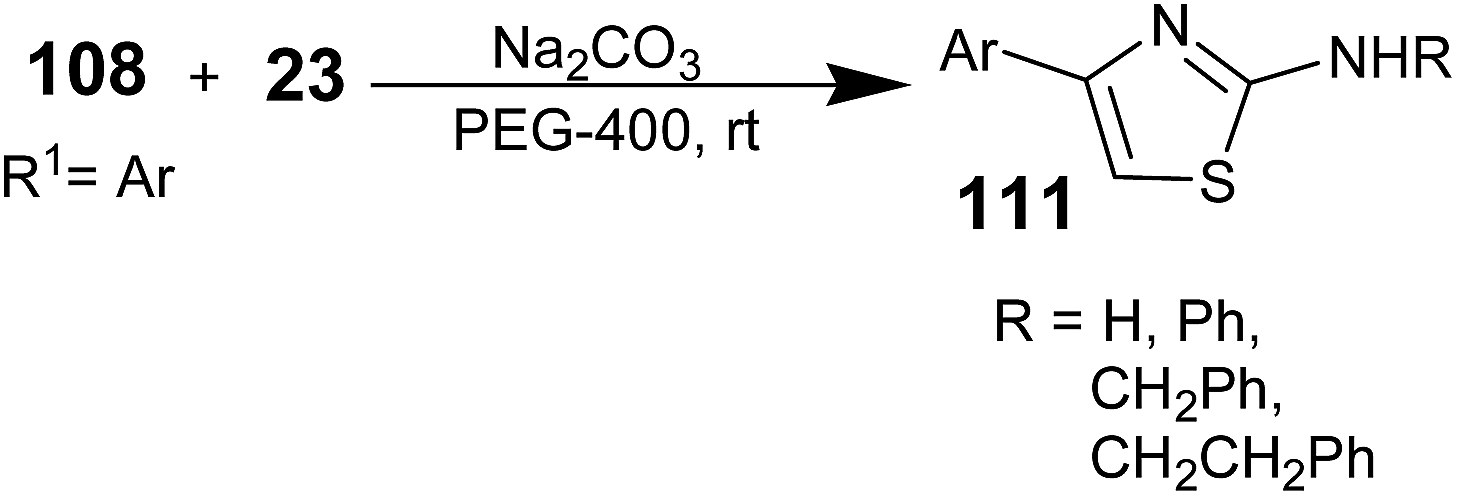 | ||
| Scheme 27 | ||
The PEG-400 can be typically recovered by first extracting the product, and the recovered solvent can be reused with retained activity. The authors reported the synthesis of the anti-inflammatory drug Fanetizole (111, Ar = Ph; R = CH2CH2Ph) with excellent yield using a PEG-mediated approach.
Dhanaji V. Jawale and co-workers reported the reactions of acetophenones 112, N-bromosuccinimide (NBS), and thiourea or aryl thioureas 23 in PEG-400 as a green reaction medium at room temperature (rt) with excellent yields of the corresponding 2-aminothiazole derivatives 113 (Scheme 28).120 In situ α-bromination of 112 was observed with (i) KBr, KBrO3 and a catalytic amount of acetic acid, (ii) bromine, and (iii) NBS. Among these brominating reagents, NBS shows an attractive combination with PEG; no radical initiator was needed and the reaction completed within 6 h. Subsequently, the brominated product underwent cyclocondensation to produce the aminothiazoles 113 in high yield. From these reported synthetic strategies, PEG has been proven as an attractive choice as a mediator.
 | ||
| Scheme 28 | ||
Conclusions
Aminothiazoles, a class of important heterocyclic compounds, often attracts the attention of researchers to upgrade their synthesis in various forms of derivatives. Polymer-supported synthetic protocols result in an improvement of the traditional reaction conditions, with enhancements in the productivity of reactions. From the present review article, it can be concluded that a polymer-supported protocol in which the reagents are loaded on the support will be more efficient than polymer-supported catalysis and polymer-mediated synthesis. The supports can be recycled in an effective manner. Although polymer-supported strategies were employed, the products needed to be purified by chromatographic methods in many cases. A few of the polymer-supported protocols involved numerous steps to achieve the desired aminothiazoles. A multi-step synthetic scheme is a limitation of polymer-supported synthesis when compared to a one pot synthesis. Use of polymers as a support still needs improvements in all aspects such as the supports for reagents need a minimum number of steps, the supports for the catalysts need to improve their recyclability, or newer catalytic supports need to be discovered, and as a medium, to date, only PEG has been reported. This drives researchers to establish other polymeric assistance as a medium. Solid-supported synthesis has established reliable and attractive synthetic approaches although continuous advancements are still needed to make it widely applicable.Perspectives and outlooks
As these polymer-supported synthetic strategies have successfully formed a firm basis for the synthesis of biologically active heterocyclic compounds, there are more opportunities for researchers to apply these polymer-utilized approaches in the nano-level synthesis of many medicinally active motifs. Further, from the available research work, it seems that there is much more scope for polymer-supported nanocatalysis. Wider applications of suitable polymer supports can also be useful to accomplish the total synthesis of essential natural products and also seems to be a challenge for future researchers in this field.Acknowledgements
The authors would like to express their sincere thanks to Principal, Head Department of Chemistry, Poojya Sane Guruji Vidya Prasarak Mandals Arts, Science and Commerce College, Shahada-425409, India for providing research and library facilities.References
- M. Schnürch, B. Waldner, K. Hilber and M. D. Mi-hovilovic, Bioorg. Med. Chem. Lett., 2011, 26, 2149–2154 CrossRef PubMed.
- Y. Komar, R. Green, D. Wise and L. L. Worting, J. Med. Chem., 1988, 23, 501–509 CrossRef.
- C. Liu, A. Phadke, X. Wang and S. Zhang, PCT Int. Appl., WO 2009149436 A1, 2009.
- H. Liebig, H. Pfetzing and A. Grafe, Arzneim.-Forsch., 1974, 24, 887–892 CAS.
- K. K. Thomas and R. Reshmy, Asian J. Chem., 2008, 20, 1457–1463 CAS.
- P. G. More and R. B. Bhalvankar, J. Indian Chem. Soc., 2006, 83, 113–115 CAS.
- Z. Ming, C. Zhen-Feng, H. L. Shao-Ming and S. J. Guan, J. Tianjin Norm. Univ., Nat. Sci. Ed., 2002, 20, 42–48 Search PubMed.
- S. Ghaemmaghami, B. C. H. May, A. R. Renslo and S. B. Prusiner, J. Virol., 2010, 84, 3408–3412 CrossRef CAS PubMed.
- A. I. Zablotskaya, S. Germane, I. Shestakova, I. Domracheva, A. Nesterova, A. Geronikaki and E. Lukevies, Chem. Heterocycl. Compd., 2002, 38, 859–866 CrossRef CAS.
- K. D. Hargrave, F. K. Hess and J. T. Oliver, J. Med. Chem., 1983, 26, 1158–1163 CrossRef CAS PubMed.
- W. C. Patt, H. W. Hamilton, M. D. Taylor, M. J. Ryan, D. J. J. Taylor, C. J. C. Connoly, M. Doherty, S. R. Klutchko, I. Sircar, B. A. Steinbaugh, B. L. Batly, C. A. Painchaud, S. T. Rapundalo, B. M. Michniewicz and S. C. J. Olson, J. Med. Chem., 1992, 35, 2562–2572 CrossRef CAS PubMed.
- F. Haviv, J. D. Ratajczyk, R. W. DeNet, F. A. Kerdesky, R. L. Walters, S. P. Schmidt, J. H. Holms, P. R. Young and G. W. Carter, J. Med. Chem., 1998, 31, 1719–1728 CrossRef.
- B. S. Holla, K. V. Malini, B. S. Rao, B. K. Sarojini and N. S. Kumari, Eur. J. Med. Chem., 2003, 38, 313–318 CrossRef CAS PubMed.
- D. Logu, M. Saddi, M. C. Cardia, R. Borgna, C. Sanna, B. Saddi and E. Maccioni, J. Antimicrob. Chemother., 2005, 55, 692–698 CrossRef PubMed.
- Q. Al-Balas, N. G. Anthony, B. Al-Jaidi, A. Alnimr, G. Abbott, A. K. Brown, R. C. Taylor, G. S. Besra, T. D. McHugh, S. H. Gillespie, B. F. Johnston, S. P. Mackay and G. D. Coxon, PLoS One, 2009, 4, 5617–5621 Search PubMed.
- T. K. Venkatachalam, E. A. Sudbeck, C. Mao and F. M. Uckun, Bioorg. Med. Chem. Lett., 2001, 11, 523–528 CrossRef CAS PubMed.
- M. C. Wilkes, P. B. Lavrik and J. Greenplate, J. Agric. Food Chem., 1991, 39, 1652–1657 CrossRef CAS.
- D. C. Warhurst, I. S. Agadu, D. Nolder and J. F. Rossignol, J. Antimicrob. Chemother., 2002, 49, 1103–1111 Search PubMed.
- D. Geronikaki, C. Chatziopoulos and G. Soloupis, Molecules, 2003, 8, 472–549 CrossRef.
- S. M. Sondhi, N. Singh, A. M. Lahoti, K. Bajaj, A. Kumar, O. Lozach and L. Meijer, Bioorg. Med. Chem., 2005, 13, 4291–4299 CrossRef CAS PubMed.
- H. L. Lu, Z. Li and T. Anthonsen, Molecules, 2000, 5, 1055–1061 CrossRef.
- B. A. Fink, D. S. Mortensen, S. R. Stauffer, Z. D. Aron and J. A. Katzenellenbogen, Chem. Biol., 1999, 6, 205–219 CrossRef CAS PubMed.
- J. E. Van Muijlwijk-Koezen, H. Timmerman, R. C. Vollinga, J. F. Von Drabbe Kunzel, M. De Groote, S. Visser and A. P. Ijzerman, J. Med. Chem., 2001, 44, 749–762 CrossRef CAS PubMed.
- J. V. Metzger, Comprehensive Heterocyclic Chemistry I, Pergamon, New York, NY, 1984, vol. 6, p. 328 Search PubMed.
- M. Nettekoven, W. Guba, W. Neidhart, P. Mattei, P. Pflieger, O. Roche and S. Taylor, Bioorg. Med. Chem. Lett., 2007, 15, 1409–1419 CrossRef PubMed.
- A. Zhang, W. Xiong, J. E. Hilbert, E. K. DeVita, J. M. Bidlack and J. L. Neumeyer, J. Med. Chem., 2004, 47, 1886–1888 CrossRef CAS PubMed.
- M. D. Erionvan, P. D. Poelje, Q. Dang, S. R. Kasibhatla, S. C. Potter, M. R. Reddy, K. R. Reddy, T. Jiang and W. N. Lipscomb, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7970–7975 CrossRef PubMed.
- A. Shockravi, A. Javadi, M. Kamali and S. Hajavi, J. Appl. Polym. Sci., 2012, 125, 1521–1529 CrossRef CAS.
- A. Javadi, A. Shockravi, M. Kamali, A. Rafieimanesh and A. Malek, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3505–3515 CrossRef CAS.
- K. L. Georgiadou and E. G. Tsatsaroni, Dyes Pigm., 2002, 53, 73–78 CrossRef CAS.
- H. R. Maradiya, Chem. Heterocycl. Compd., 2009, 10, 1558–1563 Search PubMed.
- A. R. Hantzsch and J. H. Weber, Ber. Dtsch. Chem. Ges., 1887, 20, 3118–3132 CrossRef.
- E. Garcia-Egido, S. Y. F. Wong and B. H. Warrington, Lab Chip, 2002, 2, 31–33 RSC.
- P. Y. Lin, R. S. Hou, H. M. Wang, I. J. Kang and L. C. Chen, J. Chin. Chem. Soc., 2009, 56, 455–458 CrossRef CAS.
- S. Arutyunyan and A. Nefzi, J. Comb. Chem., 2010, 12, 315–317 CrossRef CAS PubMed.
- D. Kumar, N. M. Kumar, G. Patel, S. Gupta and R. S. Varma, Tetrahedron Lett., 2011, 52, 1983–1986 CrossRef CAS.
- A. Nefzi and S. Arutyunyan, Tetrahedron Lett., 2010, 51, 4797–4800 CrossRef CAS.
- T. M. Potewar, S. A. Ingale and K. V. Srinivasa, Tetrahedron, 2008, 64, 5019–5022 CrossRef CAS.
- N. Baily, A. W. Dean, D. B. Judd, D. Middlemiss, R. Storer and S. P. Watson, Bioorg. Med. Chem. Lett., 1996, 6, 1409–1414 CrossRef.
- P. C. Kearney and M. Fernandez, J. Org. Chem., 1998, 63, 196–200 CrossRef CAS PubMed.
- T. Aoyama, S. Murata, I. Arai, N. Araki, T. Takido, Y. Suzuki and M. Kodomari, Tetrahedron, 2006, 62, 3201–3213 CrossRef CAS.
- J. Abbasi Shiran, A. Yahyazadeh, M. Mamaghani and M. Rassa, J. Mol. Struct., 2013, 1039, 113–118 CrossRef CAS.
- J. Stadlwieser, E. P. Ellmerer-Müller, A. TakoÂ, N. Maslouh and W. Bannwarth, Angew. Chem., Int. Ed., 1998, 37, 1402–1404 CrossRef CAS.
- R. Knorr, A. Trzeciak, W. Bannwarth and D. Gillessen, Tetrahedron Lett., 1989, 30, 1927–1930 CrossRef CAS.
- (a) A. Takamizawa, K. Hirai and K. Matsui, Bull. Chem. Soc. Jpn., 1963, 36, 1214–1220 CrossRef CAS; (b) P. Hebeisen, personal communication.
- P. C. Kearney, M. Fernandez and J. A. Flygare, J. Org. Chem., 1998, 63, 196–200 CrossRef CAS PubMed.
- M. Kidwai, B. Dave and K. R. Bhushan, Chem. Pap., 2000, 54, 231–234 CAS.
- R. Baer and T. Masquelin, J. Comb. Chem., 2001, 3, 16–19 CrossRef CAS PubMed.
- M. C. Pirrung and S. V. Pansare, J. Comb. Chem., 2001, 3, 90–96 CrossRef CAS PubMed.
- (a) A. Barton, S. P. Breukelman, P. T. Kaye, G. D. Meakins and D. J. Morgan, J. Chem. Soc., Perkin Trans. 1, 1982, 159–164 RSC; (b) P. T. Kaye, G. D. Meakins, A. K. Smith and M. D. Tirel, J. Chem. Soc., Perkin Trans. 1, 1983, 1677–1680 RSC.
- F. Stieber, R. Mazitschek, N. Soric, A. Giannis and H. Waldmannwe, Angew. Chem., Int. Ed., 2002, 41, 4757–4761 CrossRef CAS PubMed.
- (a) F. Stieber, U. Grether and H. Waldmann, Angew. Chem., 1999, 111, 1142 (Angew. Chem., Int. Ed., 1999, 38, 1073) CrossRef; (b) H. B. Milne and C. F. Most, Jr, J. Org. Chem., 1968, 33, 169–175 CrossRef CAS PubMed; (c) A. N. Semenov and K. Y. Gordeev, Int. J. Pept. Protein Res., 1995, 45, 303–304 CrossRef CAS PubMed; (d) C. R. Millington, R. Quarrell and G. Lowe, Tetrahedron Lett., 1998, 39, 7201–7204 CrossRef CAS; (e) C. Rosenbaum and H. Waldmann, Tetrahedron Lett., 2001, 42, 5677–5680 CrossRef CAS.
- M. Kodomari, T. Kuzuoka and S. Yoshitomi, Synthesis, 1983, 2, 141–142 CrossRef.
- M. Kodomari, T. Aoyama and Y. Suzuki, Tetrahedron Lett., 2002, 43, 1717–1720 CrossRef CAS.
- S. El Kazzouli, S. Berteina-Raboin, A. Mouaddibb and G. Guillaumeta, Tetrahedron Lett., 2002, 43, 3193–3196 CrossRef CAS.
- V. Farina and B. Krishnan, J. Am. Chem. Soc., 1991, 113, 9585–9895 CrossRef CAS.
- A. R. Katritzky, X. Cai and B. V. Rogovoy, J. Comb. Chem., 2003, 5, 392–399 CrossRef CAS PubMed.
- A. R. Katritzky, B. V. Rogovoy, N. Kirichenko and V. Vvedensky, Bioorg. Med. Chem. Lett., 2002, 12, 1809–1811 CrossRef CAS PubMed.
- I. Y. Lee, J. Y. Lee, H. J. Lee and Y. Gong, Synlett, 2005, 16, 2483–2485 CrossRef.
- R. J. Timmons and L. S. Wittenbrook, J. Org. Chem., 1967, 32, 1566–1572 CrossRef CAS.
- (a) H. Yamanaka, S. Ohba and T. Sakamoto, Heterocycles, 1990, 31, 1115–1127 CrossRef CAS; (b) S. Konno, A. Masaki, S. Mataichi and Y. Hiroshi, Yakugaku Zasshi, 1990, 110, 105–114 CrossRef CAS PubMed.
- G. Vernin, C. Siv and J. Metzger, J. Heterocycl. Chem., 1978, 15, 1361–1366 CrossRef CAS.
- T. Aoyama, S. Murata, I. Arai, N. Araki, T. Takido, Y. Suzuki and M. Kodomari, Tetrahedron, 2006, 62, 3201–3213 CrossRef CAS.
- B. Mathew and V. N. R. Pillai, Eur. Polym. J., 1994, 30, 61–65 CrossRef CAS.
- K. G. Sreejalekshmi, S. K. C. Devi and K. N. Rajasekharan, Tetrahedron Lett., 2006, 47, 6179–6182 CrossRef CAS.
- T. Aoyama, S. Murata, T. Takido and M. Kodomari, Tetrahedron, 2007, 63, 11933–11937 CrossRef CAS.
- A. G. Cole, T. M. Stauffer, L. L. Rokosz, A. Metzger, L. W. Dillard, W. Zeng and I. Henderson, Bioorg. Med. Chem. Lett., 2009, 19, 378–381 CrossRef CAS PubMed.
- A. G. Cole, A. Metzger, G. Ahmed, M.-R. Brescia, R. J. Chan, J. Wen, L. O'Brien, L.-Y. Qin and I. Henderson, Tetrahedron Lett., 2006, 47, 8897–8900 CrossRef CAS.
- J. Habermann, S. V. Ley and J. S. Scott, J. Chem. Soc., Perkin Trans. 1, 1998, 3127–3130 RSC.
- W. Xu, R. Mohan and M. M. Morrissey, Tetrahedron Lett., 1997, 38, 7337–7340 CrossRef CAS.
- J. Habermann, S. V. Ley, J. J. Scicinski, J. S. Scott, R. Smits and A. W. Thomas, J. Chem. Soc., Perkin Trans. 1, 1999, 2425–2427 RSC.
- Y. K. Yun, S. S. W. Leung and J. A. Porco Jr, Biotechnol. Bioeng., 2000, 71, 9–18 CrossRef CAS PubMed.
- G. Cardillo, M. Orena, G. Porzi and S. Sandri, Synthesis, 1981, 10, 793–794 CrossRef.
- A. Bongini, G. Cardillo, M. Orena, G. Porzi and S. Sandri, J. Org. Chem., 1982, 47, 4626–4633 CrossRef CAS.
- C. L. Ruddick, P. Hodge and M. P. Houghton, Synthesis, 1996, 11, 1359–1362 CrossRef.
- C. Hulme, J. Peng, B. Louridas, P. Menard, P. Krolikowski and N. V. Kumar, Tetrahedron Lett., 1998, 39, 8047–8050 CrossRef CAS.
- J. Szejtli and T. Osa, in Comprehensive Supramolecular Chemistry, Pergamon Press, 1996, vol. 3 Search PubMed.
- M. Narender, M. S. Reddy, R. Sridhar, Y. V. D. Nageswar and K. Rama Rao, Tetrahedron Lett., 2005, 46, 5953–5955 CrossRef CAS.
- M. Narender, M. S. Reddy, V. P. Kumar, B. Srinivas, R. Sridhar, Y. V. D. Nageswar and K. Rama Rao, Synthesis, 2007, 22, 3469–3472 Search PubMed.
- S. Fujisaki, S. Hamura, H. Eguchi and A. Nishida, Bull. Chem. Soc. Jpn., 1993, 66, 2426–2428 CrossRef CAS.
- V. P. Kumar, M. Narender, R. Sridhar, Y. V. D. Nageswar and K. Rama Rao, Synth. Commun., 2007, 37, 4331–4336 CrossRef CAS.
- C. Marchal-Roch, N. Laronze, A. Guillou, A. Teze and G. Herve, Appl. Catal., A, 2000, 199, 33–44 CrossRef CAS.
- N. Lingaiah, K. M. Reddy, N. S. Babu, K. N. Rao, I. Suryanarayana and P. S. Prasad, Catal. Commun., 2006, 7, 245–250 CrossRef CAS.
- M. Misino, I. Ono, G. Koyano and A. Aoshima, Pure Appl. Chem., 2000, 72, 1305–1311 Search PubMed.
- B. Das, V. S. Reddy and R. Ramu, J. Mol. Catal. A: Chem., 2006, 252, 235–237 CrossRef CAS.
- D. M. Tal, D. Keinan and Y. Mazur, Tetrahedron, 1981, 37, 4327–4330 CrossRef CAS.
- A. Onofrio and A. Scettri, Synthesis, 1985, 12, 1159–1161 CrossRef.
- Y. Kamitori, M. Hojo, R. Masuda, T. Kimura and T. Yoshida, J. Org. Chem., 1986, 51, 1427–1431 CrossRef CAS.
- H. Firouzabadi, N. Iranpoor, H. Hazarkhani and B. Karimi, J. Org. Chem., 2002, 67, 2572–2576 CrossRef CAS PubMed.
- K. V. N. S. Srinivas, I. Mahender and B. Das, Synthesis, 2003, 16, 2479–2482 Search PubMed.
- M. Sathe, A. K. Gupta and M. P. Kaushik, Tetrahedron Lett., 2006, 47, 3107–3109 CrossRef CAS.
- H. Karade, M. Sathe and M. P. Kaushik, Catal. Commun., 2007, 8, 741–746 CrossRef CAS.
- F. Mohanazadeh, A. R. Momeni and Y. Rangbar, Tetrahedron Lett., 1994, 33, 6127–6128 CrossRef.
- H. Firouzabadi, N. Iranpoor, B. Karimi and H. Hazarkhani, Synlett, 2000, 2, 263–265 CrossRef.
- M. Kidwai, R. Chauhan and D. Bhatnagar, J. Sulfur Chem., 2011, 32, 37–44 CrossRef CAS.
- G. A. Olah, S. I. Pradeep and G. K. S. Prakash, Synthesis, 1986, 7, 513–531 CrossRef.
- D. E. Lopez, J. G. Goodwin Jr and D. A. Bruce, J. Catal., 2007, 245, 381–391 CrossRef CAS.
- R. J. Cozens, P. J. Hogan, M. J. Lalkham, Eur. P.A. 24128, 1981Chem. Abstr., 1981, 95, 150884.
- G. A. Olah, T. Mathew and G. K. S. Prakash, Chem. Commun., 2001, 17, 1696–1697 RSC.
- M. Hachoumy, T. Mathew, E. C. Tongco, Y. D. Vankar, G. K. S. Prakash and G. A. Olah, Synlett, 1999, 3, 363–365 CrossRef.
- T. Yamato, C. Hideshima, G. K. S. Prakash and G. A. Olah, J. Org. Chem., 1991, 56, 3192–3194 CrossRef CAS.
- H. Angellier, L. Choisnard, S. Molina-Boisseau, P. Ozil and A. Dufresne, Biomacromolecules, 2004, 5, 1545–1551 CrossRef CAS PubMed.
- J. N. BeMiller and R. L. Whistler, Starch: Chemistry and technology, 3rd; food science and technology, Academic Press, New York, 2009 Search PubMed.
- A. Buléon, P. Colonna, V. Planchot and S. Ball, Int. J. Biol. Macromol., 1998, 23, 85–112 CrossRef.
- F. Hatamjafari, Helv. Chim. Acta, 2013, 96, 1560–1563 CrossRef CAS.
- J. Safari and M. Sadeghi, Monatsh. Chem., 2017, 148, 745 CrossRef CAS.
- T. J. Dickerson, N. N. Reed and K. D. Janda, Chem. Rev., 2002, 102, 3325–3344 CrossRef CAS PubMed.
- J.-M. Ahn, P. Wentworth Jr and K. D. Janda, Chem. Commun., 2003, 4, 480–481 RSC.
- J. M. Harris, Poly(ethylene glycol) Chemistry, Biotechnological and Biomedical Applications, Plenum Press, New York, 1992, p. 3 Search PubMed.
- J. Harris and S. Zalipsky, Polyethylene Glycol: Chemistry and Biological Application, ACS Books, Washington, DC, 1997 Search PubMed.
- S. Chandrasekhar, C. Narsihmulu, S. S. Sultana and N. R. K. Reddy, Org. Lett., 2002, 4, 4399–4401 CrossRef CAS PubMed.
- S. Chandrasekhar, C. Narsihmulu, S. S. Sultana and N. R. K. Reddy, Chem. Commun., 2003, 14, 1716–1717 RSC.
- S. Chandrasekhar, C. Narsihmulu, G. Chandrasekhar and T. Shyamsunder, Tetrahedron Lett., 2004, 45, 2421–2423 CrossRef CAS.
- S. Chandrasekhar, C. Narsihmulu, B. Saritha and S. S. Sultana, Tetrahedron Lett., 2004, 45, 5865–5867 CrossRef CAS.
- M. Xia and Y.-G. Wang, Tetrahedron Lett., 2002, 43, 7703–7705 CrossRef CAS.
- J.-H. Li, X.-C. Hu, Y. Liang and Y.-X. Xie, Tetrahedron, 2006, 62, 31–38 CrossRef CAS.
- A. Haimov and R. Neumann, Chem. Commun., 2002, 8, 876–877 RSC.
- S. Chandrasekhar, C. Narsihmulu, N. R. K. Reddy and S. S. Sultana, Tetrahedron Lett., 2004, 45, 4581–4582 CrossRef CAS.
- P.-Y. Lin, R.-S. Hou, H.-M. Wang, I.-J. Kang and L.-C. Chen, J. Chin. Chem. Soc., 2009, 56, 455–458 CrossRef CAS.
- D. V. Jawale, D. L. Lingampalle, U. R. Pratap and R. A. Mane, Chin. Chem. Lett., 2010, 21, 412–416 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |