
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
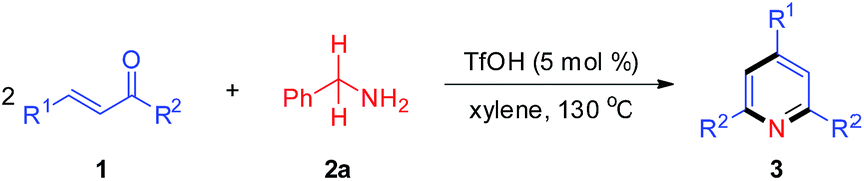
Acid-catalyzed tandem reaction for the synthesis of pyridine derivatives via C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C/C(sp3)–N bond cleavage of enones and primary amines†
C/C(sp3)–N bond cleavage of enones and primary amines†
Zhong-Yuan Mao,
Xiao-Yun Liao,
Heng-Shan Wang ,
Chun-Gu Wang,
Ke-Bin Huang* and
Ying-Ming Pan*
,
Chun-Gu Wang,
Ke-Bin Huang* and
Ying-Ming Pan*
State Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, School of Chemistry and Pharmaceutical Sciences of Guangxi Normal University, Guilin 541004, People's Republic of China. E-mail: hkg2003@163.com; panym2013@hotmail.com
First published on 24th February 2017
Abstract
A one-pot acid-catalyzed tandem reaction has been developed without any metallic reagents or extra oxidants. This reaction involves a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond cleavage of enones via a “masked” reverse Aldol reaction, and C(sp3)–N bond cleavage of primary amines to provide nitrogen sources for the assembly of pyridine derivatives in high yields with excellent functional group tolerance.
C bond cleavage of enones via a “masked” reverse Aldol reaction, and C(sp3)–N bond cleavage of primary amines to provide nitrogen sources for the assembly of pyridine derivatives in high yields with excellent functional group tolerance.
Introduction
Construction of N-heterocycles is one of the most significant areas in synthetic organic chemistry. Among them, the development of efficient methods for building the pyridine motif has attracted considerable attention since pyridine forms the structural core of numerous natural products,1 pharmaceuticals,2 and functionalized materials.3 During the past decades, abundant methodologies have been disclosed for the construction of pyridine rings, in which a variety of nitrogen-containing substrates were used as building blocks.4,5 As we know, the nitrogen source of pyridine rings can be provided by compounds containing C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, CN and C–N bonds.6 Among them, the metal-mediated [2 + 2 + 2] cycloaddition reaction constitutes a powerful tool for the synthesis of pyridines from nitriles and alkynes.7 In addition, the transition-metal-catalyzed intermolecular reactions of compounds bearing a CN group (imines, oximes, oxime esters, etc.) and carbon–carbon unsaturated compounds (e.g., alkynes, alkenes, acrylic acids) have been extensively studied and have become a fascinating method to construct pyridines.8–10 For example, Cheng8 and Ellman9 groups respectively reported the Rh-catalyzed C–H functionalization of α,β-unsaturated ketoximes with alkynes to produce substituted pyridines (Scheme 1a). Another Rh-catalyzed C–H functionalization of unsaturated oxime esters with alkenes or acrylic acids was reported by Rovis' group (Scheme 1b).10 Moreover, compounds tethered with C(sp2)–N and C(sp3)–N bonds have also been widely utilized as nitrogen-containing substrates for building pyridines.11–13 For instance, Wang and colleagues accomplished an impressive work that highly substituted pyridines were efficiently constructed via a Ru-catalyzed dehydrative [4 + 2] cycloaddition of enamides and alkynes (Scheme 1c).11 Cui's group disclosed a base-promoted synthesis of polysubstituted pyridines from 1-arylethylamines and ynones through the β-C(sp3)–H functionalization of enaminones (Scheme 1d).12 Recently, Jiang, Yi and Dhavale groups respectively reported the synthesis of 2,4,6-trisubstituted pyridines from ketones and amines (Scheme 1e).13 Nevertheless, it is rarely reported to construct pyridine rings via C(sp3)–N bond cleavage of diverse primary amines providing the nitrogen source. Therefore, we herein present a route through tandem reverse Aldol reaction/condensation/cyclization/aromatization for the formation of pyridine derivatives via CC/C(sp3)–N bond cleavage of enones/primary amines. This new reaction system avoids the use of costly catalyst and extra oxidant with broad functional group tolerance at the cost of a catalytic amount of Brønsted acid in the absence of any transitional metal (Scheme 1f).
N, CN and C–N bonds.6 Among them, the metal-mediated [2 + 2 + 2] cycloaddition reaction constitutes a powerful tool for the synthesis of pyridines from nitriles and alkynes.7 In addition, the transition-metal-catalyzed intermolecular reactions of compounds bearing a CN group (imines, oximes, oxime esters, etc.) and carbon–carbon unsaturated compounds (e.g., alkynes, alkenes, acrylic acids) have been extensively studied and have become a fascinating method to construct pyridines.8–10 For example, Cheng8 and Ellman9 groups respectively reported the Rh-catalyzed C–H functionalization of α,β-unsaturated ketoximes with alkynes to produce substituted pyridines (Scheme 1a). Another Rh-catalyzed C–H functionalization of unsaturated oxime esters with alkenes or acrylic acids was reported by Rovis' group (Scheme 1b).10 Moreover, compounds tethered with C(sp2)–N and C(sp3)–N bonds have also been widely utilized as nitrogen-containing substrates for building pyridines.11–13 For instance, Wang and colleagues accomplished an impressive work that highly substituted pyridines were efficiently constructed via a Ru-catalyzed dehydrative [4 + 2] cycloaddition of enamides and alkynes (Scheme 1c).11 Cui's group disclosed a base-promoted synthesis of polysubstituted pyridines from 1-arylethylamines and ynones through the β-C(sp3)–H functionalization of enaminones (Scheme 1d).12 Recently, Jiang, Yi and Dhavale groups respectively reported the synthesis of 2,4,6-trisubstituted pyridines from ketones and amines (Scheme 1e).13 Nevertheless, it is rarely reported to construct pyridine rings via C(sp3)–N bond cleavage of diverse primary amines providing the nitrogen source. Therefore, we herein present a route through tandem reverse Aldol reaction/condensation/cyclization/aromatization for the formation of pyridine derivatives via CC/C(sp3)–N bond cleavage of enones/primary amines. This new reaction system avoids the use of costly catalyst and extra oxidant with broad functional group tolerance at the cost of a catalytic amount of Brønsted acid in the absence of any transitional metal (Scheme 1f).
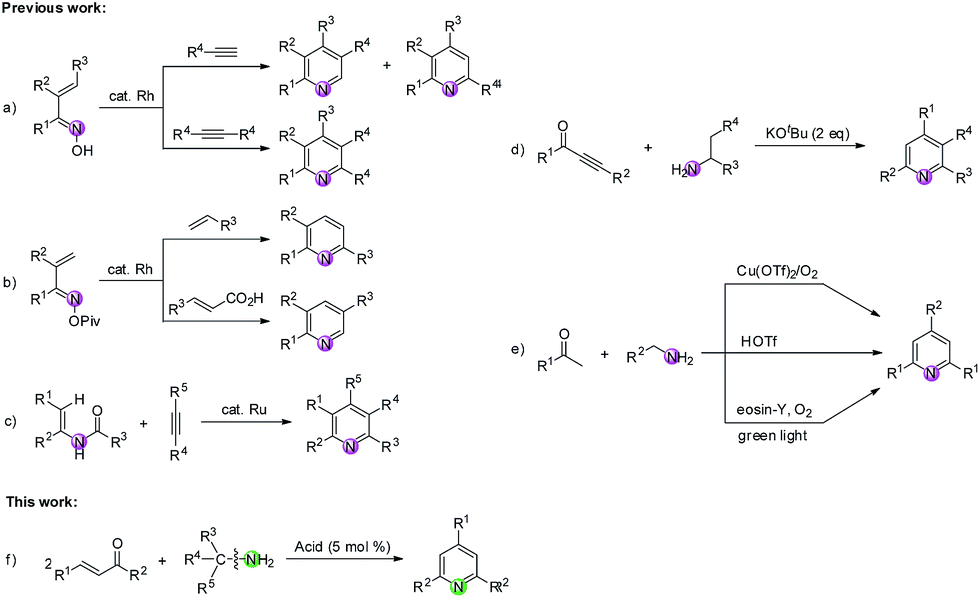 | ||
| Scheme 1 Comparison of previous study and our work. | ||
Results and discussion
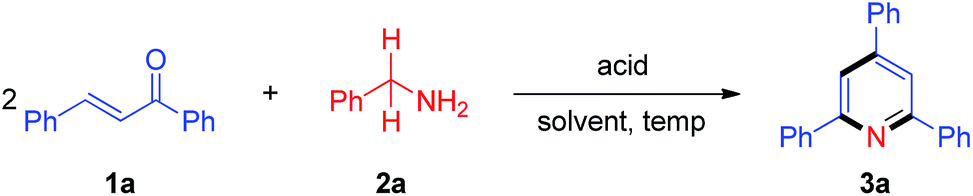
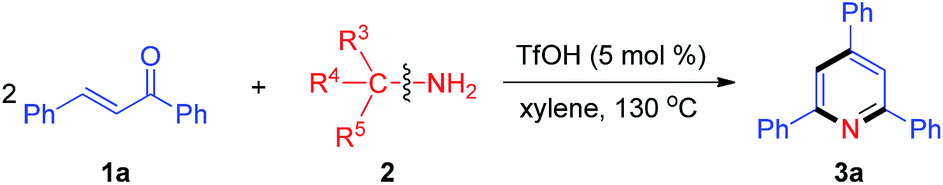
We initiated our research on the model reaction of enone 1a with primary amine 2a for screening optimum reaction conditions (Table 1). The desired product 3a was obtained in 51% isolated yield upon treatment of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1a and 2a with 5 mol% TfOH in xylene at 100 °C for 2 h (Table 1, entry 1). We subsequently varied the reaction temperature from 100 to 130 °C, which showed that 130 °C is optimal for this protocol (Table 1, entries 2–4). Compared with 5 mol% TfOH, the yield of 3a was not distinctly improved as the amount of TfOH was increased to 10 mol% (Table 1, entry 5). When TfOH was replaced by other acids (e.g., PhCO2H, AcOH, PivOH, HCl, TFA, TsOH, MsOH), the yield of 3a remarkably decreased (Table 1, entries 6–12). Without any acids, TLC analysis indicated no desired product formation (Table 1, entry 13). Furthermore, several different solvents were also surveyed (e.g., DMF, DMSO, NMP, MeCN and 1,4-dioxane), and the results revealed that xylene is the optimal solvent for this reaction (Table 1, entry 4 vs. entries 14–18).
:1 mixture of 1a and 2a with 5 mol% TfOH in xylene at 100 °C for 2 h (Table 1, entry 1). We subsequently varied the reaction temperature from 100 to 130 °C, which showed that 130 °C is optimal for this protocol (Table 1, entries 2–4). Compared with 5 mol% TfOH, the yield of 3a was not distinctly improved as the amount of TfOH was increased to 10 mol% (Table 1, entry 5). When TfOH was replaced by other acids (e.g., PhCO2H, AcOH, PivOH, HCl, TFA, TsOH, MsOH), the yield of 3a remarkably decreased (Table 1, entries 6–12). Without any acids, TLC analysis indicated no desired product formation (Table 1, entry 13). Furthermore, several different solvents were also surveyed (e.g., DMF, DMSO, NMP, MeCN and 1,4-dioxane), and the results revealed that xylene is the optimal solvent for this reaction (Table 1, entry 4 vs. entries 14–18).
|
||||
|---|---|---|---|---|
| Entry | Acid | Solvent | Temp. (°C) | Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol), 2a (0.5 mmol), acid (5 mol%) and solvent (1 mL) for 2 h.b Isolated yield.c 10 mol% TfOH.d Not detected. | ||||
| 1 | TfOH | Xylene | 100 | 51 |
| 2 | TfOH | Xylene | 110 | 72 |
| 3 | TfOH | Xylene | 120 | 85 |
| 4 | TfOH | Xylene | 130 | 92 |
| 5c | TfOH | Xylene | 130 | 93 |
| 6 | PhCO2H | Xylene | 130 | 33 |
| 7 | AcOH | Xylene | 130 | 37 |
| 8 | PivOH | Xylene | 130 | 28 |
| 9 | HCl | Xylene | 130 | 68 |
| 10 | TFA | Xylene | 130 | 87 |
| 11 | TsOH | Xylene | 130 | 76 |
| 12 | MsOH | Xylene | 130 | 81 |
| 13 | — | Xylene | 130 | n.d.d |
| 14 | TfOH | DMF | 130 | 46 |
| 15 | TfOH | DMSO | 130 | 33 |
| 16 | TfOH | NMP | 130 | 39 |
| 17 | TfOH | CH3CN | 130 | 70 |
| 18 | TfOH | 1,4-Dioxane | 130 | 82 |
With the optimal reaction conditions in hand, we proceeded to explore the nitrogen source scope for this reaction, as shown in Table 2. The aromatic methylamines (2b–2f) as nitrogen sources gave the desired pyridine 3a in high yields. Moreover, treatment of 1a with various aliphatic amines (2g–2k) produced 3a in 40–71% yields, but tert-butylamine (2l) could not provide the nitrogen atom for the synthesis of pyridine 3a. These results indicate that at least one hydrogen atom at α-position of amino is necessary for this reaction.
|
||||
|---|---|---|---|---|
| [N] source | R3 | R4 | R5 | Yield of 3ab (%) |
| a Reaction conditions: 1a (0.5 mmol), 2 (0.5 mmol), TfOH (5 mol%) and xylene (1 mL) for 2 h.b Isolated yield.c No reaction. | ||||
| 2b | 4-MeOC6H4 | H | H | 95 |
| 2c | 4-F3CC6H4 | H | H | 88 |
| 2d | 2-Furyl | H | H | 81 |
| 2e | 2-Thienyl | H | H | 86 |
| 2f | Bn | H | H | 77 |
| 2g | Et | H | H | 56 |
| 2h | Me | Me | H | 40 |
| 2i | i-Pr | H | H | 63 |
| 2j | i-Bu | H | H | 67 |
| 2k | 3-Heptyl | H | H | 71 |
| 2l | Me | Me | Me | n.r.c |
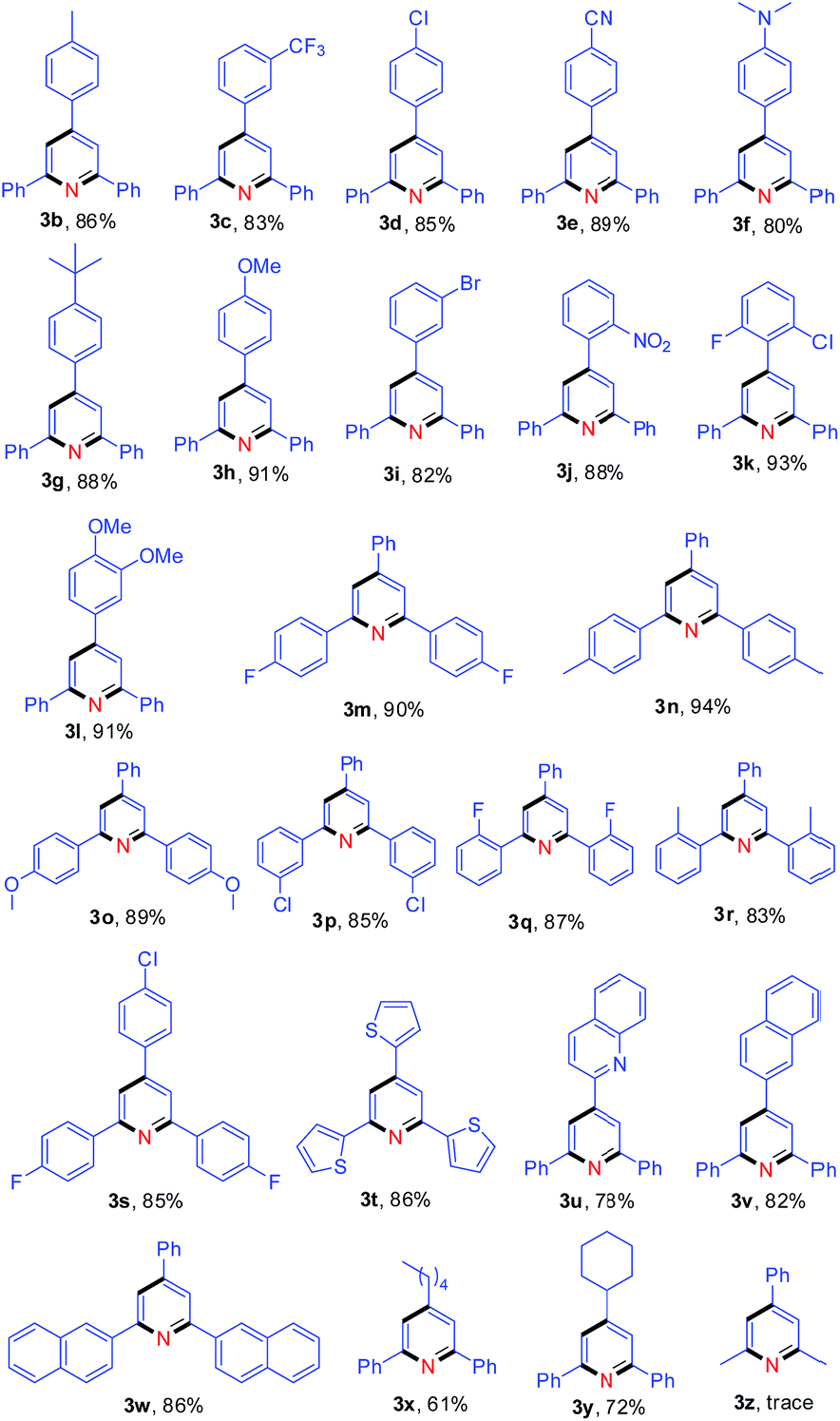
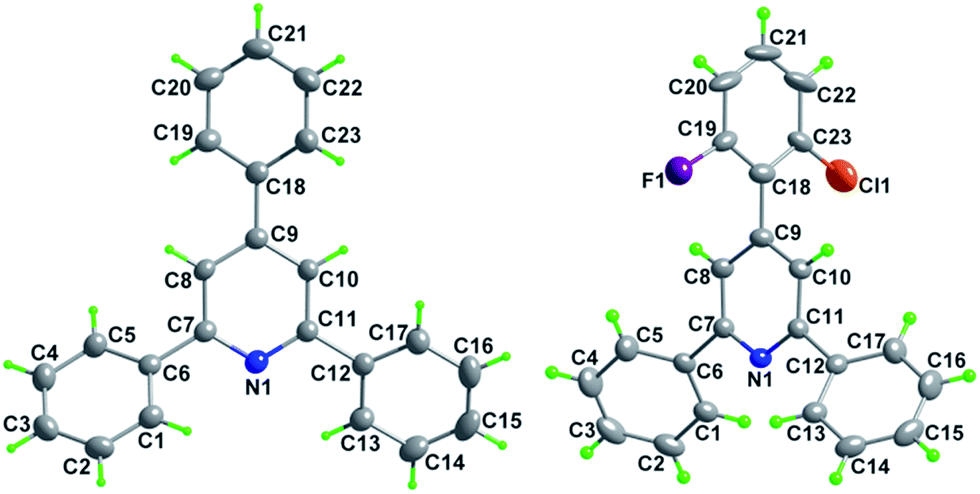
Subsequently, the scope of enones was investigated under the optimal conditions (Table 3). Enones containing one or more electron-withdrawing groups (EWG = F, Cl, Br, CF3, CN, NO2) or electron-donating groups (EDG = Me, tBu, N(Me)2, OMe) on the phenyl ring (R1) are both well-tolerated and afforded excellent yields (80–93%, 3b–3l). Variation of the electronic properties of the substituents on the aromatic ring (R2) has little influence on the reaction efficiency (3m–3r). Notably, 4,4′-dihalogen substituted aryl enone also proceeded smoothly with 85% yield (3s). Gratifyingly, enones bearing heterocycle and fused ring reacted efficiently as well to give the corresponding product 3t–3w in 78–86% yields. With respect to aromatic rings, the alkyl-substituted enones were also tolerated in this transformation, generating the corresponding 2,4,6-trisubstituted pyridines 3x and 3y in 61% and 72% isolated yields, respectively. Unfortunately, benzalacetone failed to afford the desired product 3z. The structures of 3a and 3k were unambiguously confirmed by single-crystal X-ray diffraction analysis (Fig. 1).14
|
|---|
| a Reaction conditions: 1 (0.5 mmol), 2a (0.5 mmol), TfOH (5 mol%) and xylene (1 mL) for 2 h. Isolated yields are shown. |
 |
With the aim of evaluating the practicality of this catalytic process, a gram-scale experiment was performed with 1a (10 mmol) and 2a (15 mmol was added in two portions), yielding the corresponding product 3a in 72% (Scheme 2).
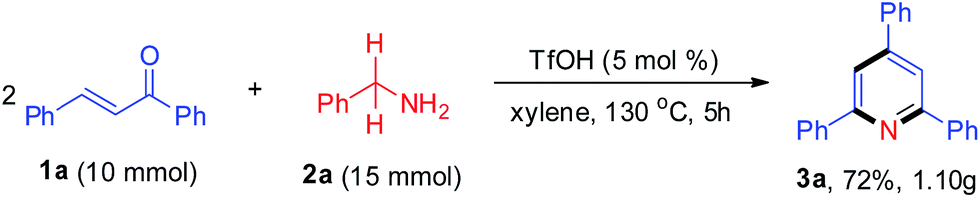 | ||
| Scheme 2 Gram scale synthesis. | ||
To gain insight into the mechanism of this new reaction, we conducted several control experiments (Scheme 3). When the model substrates 1a, and 2a were reacted under argon atmosphere in xylene at 130 °C, we failed to obtain the product 3a. The reaction of 1a with 2a under oxygen atmosphere resulted in the formation of 3a with 91% isolated yield. The results replacing air with Ar or O2 demonstrated that molecular oxygen is essential as an oxidant for this transformation. Under the standard conditions, the reaction of 4 with 1a and 2a afforded 3a in 89% isolated yield, and the reaction of 5 with 1a and 2a afforded 6, 3a and 3n, indicating that acetophenone should be a reactive intermediate in this process.
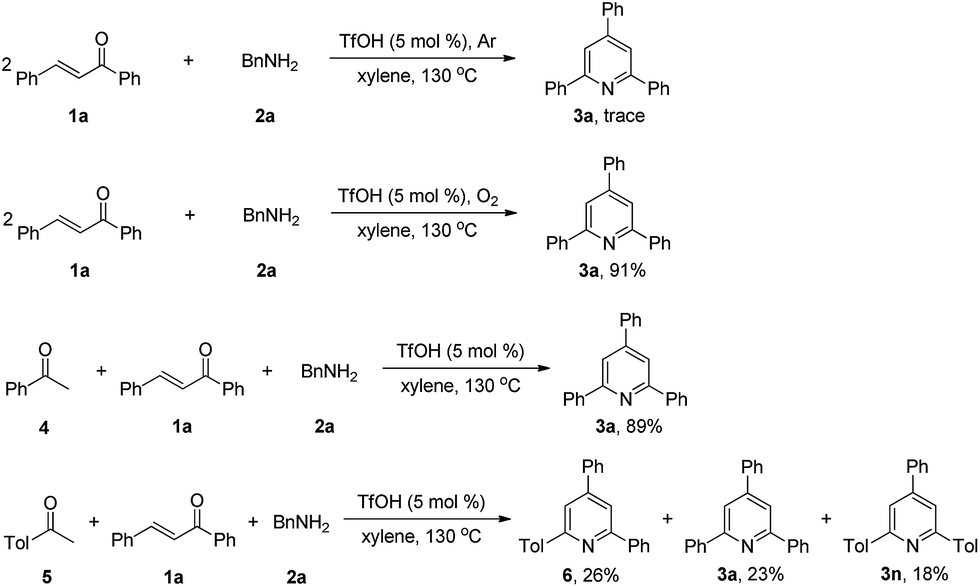 | ||
| Scheme 3 Control experiments. | ||
The above results have led us to propose a reaction mechanism, which is depicted in Scheme 4 using 3a as a representative example. Originally with Brønsted acid catalysis, the reverse Aldol reaction of 1a proceeds to give acetophenone, which reacts with 2a to obtain the dehydrative condensation product imine A. Then, 1,4-addition of imine A to another molecule of 1a generates intermediate B. Subsequent intramolecular nucleophilic addition and dehydration of intermediate B affords 1,4-dihydropyridine C. Finally, aerobic oxidative C(sp3)–N bond cleavage of dihydropyridine C leads to its aromatization, with the latter being the driving force of this oxidation.
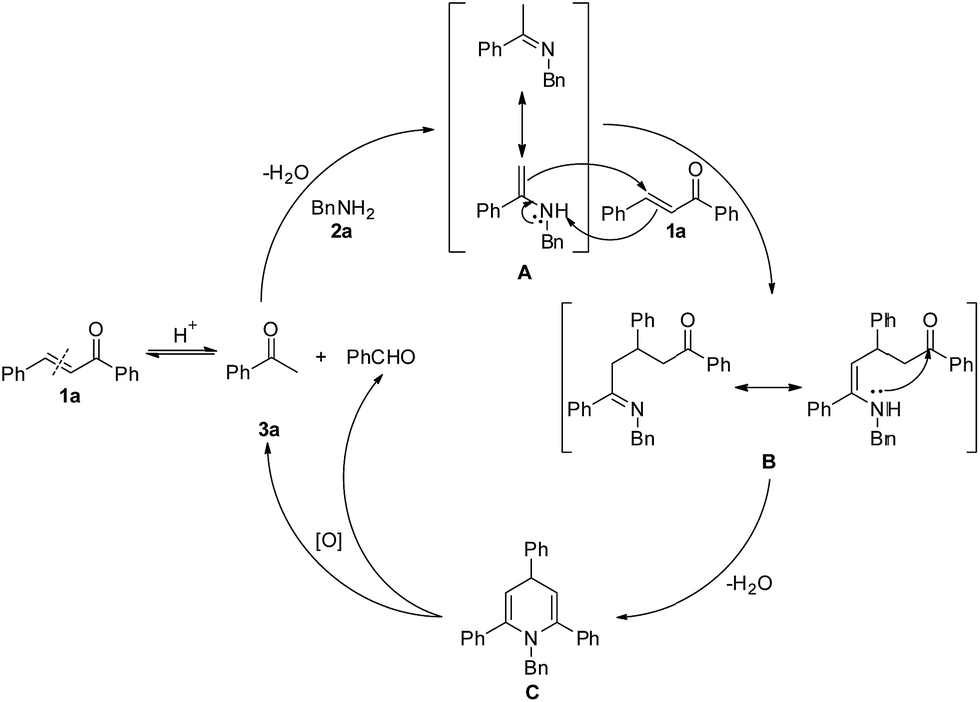 | ||
| Scheme 4 Possible mechanism. | ||
According to the mechanism proposed in Scheme 4, the reaction of (E)-chalcone to acetophenone and benzaldehyde is reversible, the decomposition of C would give the desired pyridine and benzaldehyde. If substituted BnNH2 (such as p-tolylmethanamine) was used, “unsymmetric” chalcone would generated. As a result, the generated “unsymmetric” chalcone would react with acetophenone and benzyl amine to form pyridines with 4-different substituent. In fact, we did not obtain the 4-different substituent pyridines, because it is adverse that ketones with aldehydes perform an Aldol reaction at this reaction temperature (130 °C).
ESI/MS experiments were performed to gain evidence for the possible intermediates in the proposed mechanism. A mixture of 1a (0.5 mmol), 2a (0.5 mmol) and TfOH (5 mol%) in xylene (1 mL) was reacted at 130 °C for 15 min and 50 μL of the mixture was used for the ESI analysis in MeOH. The ESI/MS analyses showed three peaks at m/z 210.1269, m/z 418.2148, and m/z 400.1953 which were respectively identified as imine A, intermediate B, and dihydropyridine C (Fig. 2).
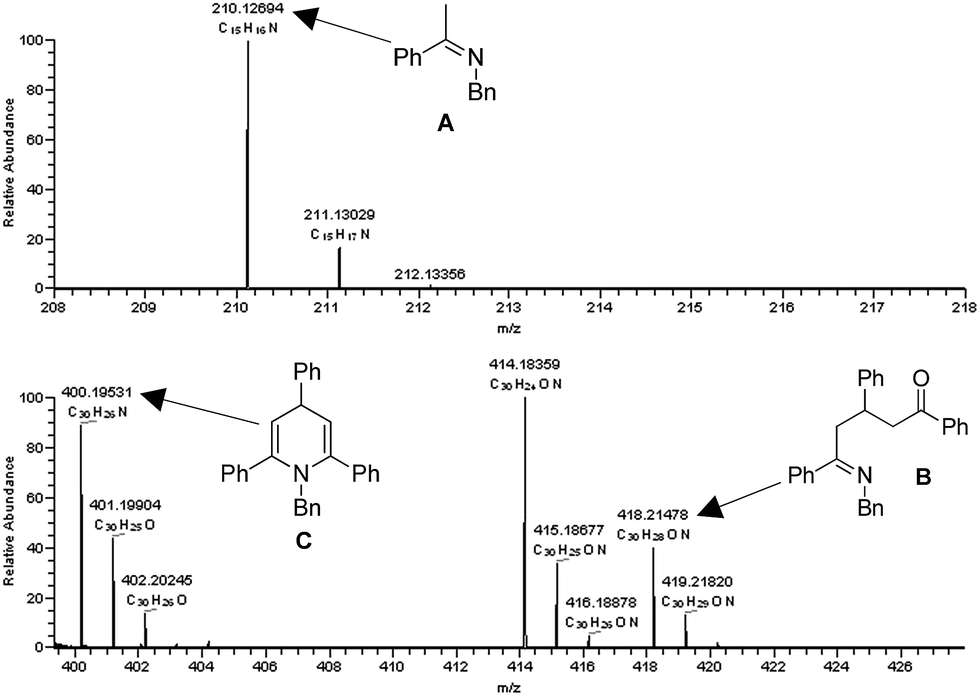 | ||
| Fig. 2 ESI-MS based analysis during the formation of 3a. | ||
Conclusions
In summary, we have demonstrated an expedient approach for the synthesis of 2,4,6-trisubstituted pyridines via sequential reverse Aldol reaction/dehydrative condensation/addition–cyclization/aromatization, catalyzed by Brønsted acid. The use of naturally abundant air as an oxidant, readily available starting materials including the nitrogen source, and experimentally convenient catalytic process are the additional advantages of the present protocol. Further investigations on the synthetic applications of this reaction are ongoing in our laboratory.Experimental
General experimental details
Unless otherwise noted, commercial reagents were used without further purification. All manipulations were performed under an air atmosphere unless otherwise statement. Reaction temperatures were controlled using IKAmag temperature modulator. Column chromatography was performed on silica gel (300–400 mesh). NMR spectra were obtained using a Bruker 400 MHz spectrometer. Chemical shifts were reported in parts per million (ppm), and the residual solvent peak was used as an internal reference: proton (chloroform δ 7.26), carbon (chloroform δ 77.00) or tetramethylsilane (TMS δ 0.00) was used as a reference. High resolution mass spectra (HRMS) were recorded on the Exactive Mass Spectrometer equipped with ESI ionization source.General experimental procedure for the synthesis of pyridines 3
The reaction mixture of enones 1 (0.5 mmol), primary amines 2 (0.5 mmol) and TfOH (5 mol%) in xylene (1 mL) was stirred at 130 °C for 2 h in test tube, and periodically monitored by TLC. Upon completion, the crude product was cooled to room temperature and then directly separated by flash column chromatography on silica gel to give the pure product 3.Acknowledgements
We would like to thank the National Natural Science Foundation of China (21362002 and 81260472), Guangxi Natural Science Foundation of China (2014GXNSFDA118007 and 2016GXNSFEA380001), State Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources (CMEMR2014-A02 and CMEMR2012-A20), the Fund of Guangxi Key Laboratory of Functional Phytochemicals Research and Utilization (FPRU2015-2), and Guangxi's Medicine Talented Persons Small Highland Foundation (1306).Notes and references
- (a) J. P. Michael, Nat. Prod. Rep., 2005, 22, 627 RSC; (b) M. Abass, Heterocycles, 2005, 65, 901 CrossRef CAS.
- (a) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed; (b) T. P. Whitehead, C. Havel, C. Metayer, N. L. Benowitz and P. Jacob, Chem. Res. Toxicol., 2015, 28, 1007 CrossRef CAS PubMed; (c) P. L. Lanza, M. F. Pack, Z. Li, S. A. Krajewski and M. A. Blank, Aliment. Pharmacol. Ther., 2000, 14, 1663 CrossRef PubMed; (d) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC; (e) D. F. Fisher and R. Sarpong, J. Am. Chem. Soc., 2010, 132, 5926 CrossRef PubMed; (f) C. Yuan, C.-T. Chang, A. Axelrod and D. Siegel, J. Am. Chem. Soc., 2010, 132, 5924 CrossRef CAS PubMed; (g) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed.
- (a) H. Sasabe and J. Kido, Chem. Mater., 2011, 23, 621 CrossRef CAS; (b) X.-C. Hang, T. Fleetham, E. Turner, J. Brooks and J. Li, Angew. Chem., Int. Ed., 2013, 52, 6753 CrossRef CAS PubMed; (c) S. J. Su, T. Chiba, T. Takeda and J. Kido, Adv. Mater., 2008, 20, 2125 CrossRef CAS.
- For partial reviews, see: (a) J. A. Varela and C. Saá, Chem. Rev., 2003, 103, 3787 CrossRef CAS PubMed; (b) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127 CrossRef CAS PubMed; (c) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644 CrossRef CAS PubMed; (d) B. Heller and M. Hapke, Chem. Soc. Rev., 2007, 36, 1085 RSC; (e) B. Groenendaal, E. Ruijter and R. V. A. Orru, Chem. Commun., 2008, 5474 RSC; (f) Y. Nakao, Synthesis, 2011, 3209 CrossRef CAS.
- (a) T. J. Donohoe, J. A. Basutto, J. F. Bower and A. Rathi, Org. Lett., 2011, 13, 1036 CrossRef CAS PubMed; (b) D. G. Stark, L. C. Morrill, P.-P. Yeh, A. M. Z. Slawin, T. J. C. ÓRiordan and A. D. Smith, Angew. Chem., Int. Ed., 2013, 52, 11642 CrossRef CAS PubMed; (c) T. Sakai and R. L. Danheiser, J. Am. Chem. Soc., 2010, 132, 13203 CrossRef CAS PubMed; (d) M. Yoshida, T. Mizuguchi and K. Namba, Angew. Chem., Int. Ed., 2014, 53, 14550 CrossRef CAS PubMed; (e) C.-H. Lei, D.-X. Wang, L. Zhao, J. Zhu and M.-X. Wang, J. Am. Chem. Soc., 2013, 135, 4708 CrossRef CAS PubMed; (f) Z. Shi and T.-P. Loh, Angew. Chem., Int. Ed., 2013, 52, 8584 CrossRef CAS PubMed; (g) Y. Jiang, C.-M. Park and T.-P. Loh, Org. Lett., 2014, 16, 3432 CrossRef CAS PubMed; (h) F. Sha and X. Huang, Angew. Chem., Int. Ed., 2009, 48, 3458 CrossRef CAS PubMed; (i) K. Kral and M. Hapke, Angew. Chem., Int. Ed., 2011, 50, 2434 CrossRef CAS PubMed; (j) P. Huang, R. Zhang, Y. Liang and D. Dong, Org. Lett., 2012, 14, 5196 CrossRef CAS PubMed; (k) C. Allais, F. Lieby-Muller, T. Constantieux and J. Rodriguez, Adv. Synth. Catal., 2012, 354, 2537 CrossRef CAS.
- For some selected examples, see: (a) D. A. Colby, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 3645 CrossRef CAS PubMed; (b) S. Duttwyler, C. Lu, A. L. Rheingold, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2012, 134, 4064 CrossRef CAS PubMed; (c) T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 11846 RSC; (d) K. Parthasarathy and C.-H. Cheng, J. Org. Chem., 2009, 74, 9359 CrossRef CAS PubMed; (e) C.-Z. Luo, J. Jayakumar, P. Gandeepan, Y.-C. Wu and C.-H. Cheng, Org. Lett., 2015, 17, 924 CrossRef CAS PubMed; (f) T. Yamakawa and N. Yoshikai, Org. Lett., 2013, 15, 196 CrossRef CAS PubMed; (g) Y. Wei and N. Yoshikai, J. Am. Chem. Soc., 2013, 135, 3756 CrossRef CAS PubMed; (h) G. Cheng, Y. Weng, X. Yang and X. Cui, Org. Lett., 2015, 17, 3790 CrossRef CAS PubMed.
- (a) A. W. Fatland and B. E. Eaton, Org. Lett., 2000, 2, 3131 CrossRef CAS PubMed; (b) T. Takahashi, F. Tsai and M. Kotora, J. Am. Chem. Soc., 2000, 122, 4994 CrossRef CAS; (c) J. Chen, Q. Song, C. Wang and Z. Xi, J. Am. Chem. Soc., 2002, 124, 6238 CrossRef CAS PubMed; (d) B. Heller, B. Sundermann, H. Buschmann, H.-J. Drexler, J. You, U. Holzgrabe, E. Heller and G. Oehme, J. Org. Chem., 2002, 67, 4414 CrossRef CAS PubMed; (e) R. Tanaka, A. Yuza, Y. Watai, D. Suzuki, Y. Takayama, F. Sato and H. Urabe, J. Am. Chem. Soc., 2005, 127, 7774 CrossRef CAS PubMed.
- K. Parthasarathy, M. Jeganmohan and C.-H. Cheng, Org. Lett., 2008, 10, 325 CrossRef CAS PubMed.
- R. M. Martin, R. G. Bergman and J. A. Ellman, J. Org. Chem., 2012, 77, 2501 CrossRef CAS PubMed.
- (a) J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2013, 135, 66 CrossRef CAS PubMed; (b) J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2014, 136, 2735 CrossRef CAS PubMed.
- J. Wu, W. Xu, Z.-X. Yu and J. Wang, J. Am. Chem. Soc., 2015, 137, 9489 CrossRef CAS PubMed.
- J. Shen, D. Cai, C. Kuai, Y. Liu, M. Wei, G. Cheng and X. Cui, J. Org. Chem., 2015, 80, 6584 CrossRef CAS PubMed.
- (a) H. Huang, X. Ji, W. Wu, L. Huang and H. Jiang, J. Org. Chem., 2013, 78, 3774 CrossRef CAS PubMed; (b) X. Zhang, Z. Wang, K. Xu, Y. Feng, W. Zhao, X. Xu, Y. Yan and W. Yi, Green Chem., 2016, 18, 2313 RSC; (c) R. S. Rohokale, B. Koenig and D. D. Dhavale, J. Org. Chem., 2016, 81, 7121 CrossRef CAS PubMed.
- CCDC 1498096-1498097 contain the supplementary crystallographic data for this paper.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, spectral data for all products, and X-ray crystallographic files (CIF) for 3a and 3k. CCDC 1498096 and 1498097. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra00780a |
| This journal is © The Royal Society of Chemistry 2017 |