DOI:
10.1039/C6RA28890A
(Paper)
RSC Adv., 2017,
7, 13112-13122
Preparation of dithiocarbamate polymer brush grafted nanocomposites for rapid and enhanced capture of heavy metal ions†
Received
31st December 2016
, Accepted 19th February 2017
First published on 24th February 2017
Abstract
High-density and narrow-distribution dithiocarbamate (DTC) functionalized polymer brush grafted SiO2 nanocomposites (DTC-PGMA@SiO2) were synthesized via surface initiated atom transfer radical polymerization (SI-ATRP) and subsequent DTC functionalization, which could serve as an efficient nanostructured adsorbent material. Systematic characterization was performed to identify the sea anemone like core-brush structure. More importantly, the DTC-PGMA@SiO2 adsorbent exhibited remarkable performance in capturing heavy metal ions from water. The adsorption behaviour, including the effect of pH, adsorption kinetics, adsorption isotherms, adsorption thermodynamics and adsorption mechanism, was investigated in detail. Interestingly, the adsorbent complexes show different color changes depending upon the species of adsorbed ions, indicating that the DTC-PGMA@SiO2 can be potentially used as a sensor for metallic contaminants in water bodies. The regeneration experiments showed that the adsorbent is both cost-effective and sustainable. The high-capacity and rapid adsorption of metallic ions, which are due to the well-defined core-brush structure, large specific surface area and strong binding ability of DTC groups, make this adsorbent material promising in the capture of heavy metal ions from contaminated water.
Introduction
Water is an essential and significant component of the ecosystem, and plays a vital role in Earth's ecological cycle.1 Despite this fact, fresh water systems are directly threatened by heavy metal ions contamination.2–4 Heavy metal ions (such as Pb2+, Cd2+, Cr6+, Hg2+, and Cu2+) can diffuse into surface and ground waters, posing a critical threat to the ecosystem and public health due to their high toxicity, carcinogenicity, non-biodegradability and bioaccumulation in the food chain.5–8 Common mechanisms regarding the toxicity of heavy metal ions have been reviewed by the World Health Organization (WHO). Some of these include modifying the active conformation of enzymes and proteins, displacing essential metals from biomolecules and disturbing the integrity of biomembranes.9,10 Consequently, this has led to the development of highly efficient technologies for the removal of heavy metal ions from water bodies.
For this purpose, various methods have been developed over the past few years including ion exchange,11 chemical precipitation,12 membrane filtration,13 reverse osmosis,14 and adsorption.15 By contrast, adsorption is considered to be the most suitable process for capturing metallic ions due to its ease of operation, low cost, high efficiency, and safety.16,17 In order to improve the adsorbent performance, organic chelating ligands have been developed based on imidazole, thiol, dithiocarbamate (DTC), and thiourea, or carbonyl groups.18,19 It is particularly worth mentioning that the high affinity for metallic ions is a unique characteristic of sulfur containing groups, especially in case of the DTC group.20–22 DTC group is well-known to be an soft Lewis base, which could form stable complexes with heavy metal ions (soft Lewis acids) containing partially and fully filled d orbitals through π-coordinated interactions.
A large number of adsorbent materials have been designed and studied as heavy metal capturing agents. Some of these include biomass materials,23 activated carbon,24 nanoparticles,25–27 polymer resins,28 and hydrogels.29 Compared with other adsorbent materials, nanostructured adsorbent materials exhibit unique properties due to their abundant surface adsorption sites provided by higher specific surface area.30,31 However, the drawbacks of traditional nano-sized adsorbent materials are also obvious. Some of these drawbacks include their natural tendency to agglomerate and the limited monolayer adsorption sites attached to the surface.32 For this reason, the development of a surface modification to achieve improved and optimized adsorption sites is crucial to the advancement of this field.
Surface functionalization using polymer brush can provide a homogeneous and well-defined interface between nanocomposites and has garnered much attention due to its wide range of applications.33–35 Additionally, polymers have been used to effectively reduce the agglomeration of nano-sized materials.36 Being inspired by the structure of sea anemone, which is particularly efficient in capturing plankton, the authors of current study have designed a novel nanostructured adsorbent having a core-brush structure similar to sea anemones. Surface-initiated polymerization (SIP) is a facile means of creating high-density polymer brushes on substrates. More importantly, high-density and well-defined polymer brushes having narrow molecular distribution can be precisely fabricated on the matrix surface by using versatile surface-initiated atom transfer radical polymerization (SI-ATRP).37–40
Poly(glycidyl methacrylate) (PGMA), a very valuable polymer, containing active epoxy group that can undergo ring opening reactions with functional ligands. Therefore, the graft of PGMA brush as reactive anchor is of particular interest. Accordingly, adsorptive groups can be introduced to construct efficient adsorptive polymer brush. Surface modification of the adsorptive polymer brushes can not only offer abundant and spatially-organized adsorption sites (just like the tentacles of sea anemones), but can also improve their performance for capturing heavy metal ions. Some previous studies have reported such a modification in the field of adsorption.41–43 However, few work focused on the fabrication of efficient nanostructured adsorbent materials through SI-ATRP for capturing heavy metal ions.
In this work, a new strategy to design nano-sized heavy metal adsorbent material with core-brush structure is explored by combining SI-ATRP and DTC functionalization. The low-cost bare SiO2 nanoparticles were initially anchored by ATRP initiator, which was further used for grafting PGMA brushes. Subsequently, the DTC groups were introduced to the polymer brushes through ring-opening reaction and DTC functionalization (see Scheme 1). The resultant nanostructured DTC-PGMA@SiO2 adsorbent was characterized by various techniques. Moreover, the reported material was finally applied for efficient removal of Cu2+, Pb2+ and Cd2+ from an aqueous environment. Adsorption behaviours, such as the effect of pH, adsorption kinetics, adsorption isotherms, adsorption thermodynamics and adsorption mechanism were systematically investigated. The regeneration experiments were also conducted to explore the reusability of the studied adsorbent.
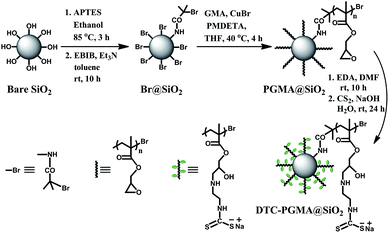 |
| | Scheme 1 The synthetic route of nanostructured DTC-PGMA@SiO2 adsorbent material via SI-ATRP and DTC functionalization. | |
Experimental
Materials and characterization
Chemicals including 3-triethoxysilylpropylamine (APTES), ethylene diamine tetraacetic acid (EDTA), anisole, N,N-dimethylformamide (DMF), triethylamine (Et3N), 2-bromoisobutyryl bromide (BiBB), glycidyl methacrylate (GMA), pentamethyldiethylenetriamine (PMDETA), carbon disulfide (CS2), ethylenediamine (EDA) used are all of analytical grade (J&K Scientific Ltd., Beijing, China). The model sacrificial initiator 2-bromo-2-methyl-N-propylpropanamide (BMPA) was self-prepared (see ESI†). Bare SiO2 nanoparticles and initiator immobilized SiO2 nanoparticles were prepared according to previous reports (see ESI†).44 CuBr was successively washed with acetic acid and methanol before use. All of the aqueous solutions were prepared using ultrapure water (18.25 MΩ cm at 25 °C).
The chemical structures of PGMA brush and synthesized materials were determined by 1H NMR (Bruker AVANCE 300 MHz NMR spectrometer) and FT-IR spectra (Bruker Tensor 27 spectrometer). GPC measurements were performed on a Waters 515 GPC system, which used THF as the eluent (flow rate 1 mL min−1). TGA was measured by a Netzsch STA 449 instrument from 40 °C up to 800 °C. The morphology observations of materials were observed with a Hitachi S4800 scanning electron microscope (SEM) and a JEM 1011 transmission electron microscope (TEM). Water contact angle measurements were performed using a Kruss DSA10 optical contact angle system. Elemental analysis of C, H, N and S was carried out by using an Elementar Vario Cube EL analyser. XPS spectra were acquired by using a Perkin Elmer PHI5300 spectrometer. Zeta potential measurements of adsorbent suspensions (0.2 g DTC-PGMA@SiO2 in 50 mL of 0.1 mol L−1 NaCl solution) were carried out by a Malvern Zetasizer Nano ZS90 system. The concentrations of metal ions in the solution were determined by inductively coupled plasma-atomic emission spectrometer (ICP-AES, Thermo Optima 7000DV).
Growth of PGMA brushes on Br@SiO2 nanoparticles
The surface of prepared Br@SiO2 nanoparticles can provide abundant initiators for triggering the graft of PGMA brushes through SI-ATRP, during which CuBr/PMDETA (in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, respectively) was used as the catalyst. 4 g Br@SiO2, GMA (20 mL; 146.32 mmol), BMPA (101.5 mg; 0.487 mmol) and PMDETA (100 μL; 0.487 mmol) were dissolved in 30 mL anisole. The solution was processed by several freeze–pump–thaw cycles to remove oxygen. CuBr (70 mg; 0.487 mmol) was ultrasonically dispersed in the reaction flask under N2 atmosphere. The system was initiated at 40 °C, and the polymerization was stopped after 5 h by opening the flask. The PGMA brushes grafted SiO2 nanocomposites (PGMA@SiO2) were purified by THF washing, EDTA (0.05 mol L−1 solution) washing, acetone washing and vacuum drying. The free PGMA brushes were precipitated in acetone and were purified through a column of neutral alumina using THF as eluent.
:1 molar ratio, respectively) was used as the catalyst. 4 g Br@SiO2, GMA (20 mL; 146.32 mmol), BMPA (101.5 mg; 0.487 mmol) and PMDETA (100 μL; 0.487 mmol) were dissolved in 30 mL anisole. The solution was processed by several freeze–pump–thaw cycles to remove oxygen. CuBr (70 mg; 0.487 mmol) was ultrasonically dispersed in the reaction flask under N2 atmosphere. The system was initiated at 40 °C, and the polymerization was stopped after 5 h by opening the flask. The PGMA brushes grafted SiO2 nanocomposites (PGMA@SiO2) were purified by THF washing, EDTA (0.05 mol L−1 solution) washing, acetone washing and vacuum drying. The free PGMA brushes were precipitated in acetone and were purified through a column of neutral alumina using THF as eluent.
DTC functionalization of PGMA@SiO2 nanocomposites
4 g PGMA@SiO2 were dispersed and stirred in 50 mL 4 mol L−1 EDA/DMF solution for 10 h at room temperature. The generated amino functionalized PGMA@SiO2 (NH2-PGMA@SiO2) nanocomposites were washed thoroughly with acetone and then, were dried under vacuum. 4 g of prepared NH2-PGMA@SiO2 were dispersed in 40 mL of 2 mol L−1 NaOH aqueous solution. After the addition of 2 mL CS2, the mixture was stirred at room temperature for 24 h. The target DTC-functionalized PGMA@SiO2 (DTC-PGMA@SiO2) adsorbent nanocomposites were thoroughly and successively washed with water and acetone, and then, were dried under vacuum.
Metallic ions adsorption tests
All adsorption experiments were performed in batch mode and using a constant temperature incubator shaker (Julabo SW-22) at 150 rpm. The effect of pH (within the range of 1.0–7.0) was investigated in NaAc-HAc buffer solutions having an initial metal ion concentration of 800 mg L−1. The temperature was kept constant at 25 °C during each 150 minute long adsorption experiment. For kinetic studies, the experiments were carried out at pH = 5.0 with different adsorption time intervals (from 3 to 150 min) at 25 °C, where the initial concentration of the investigated metallic ion was 800 mg L−1. The adsorption isotherms were investigated at pH = 5.0 by varying the initial concentration of heavy metal ions within the range of 200–2000 mg L−1. Furthermore, all isothermal experiments were conducted at three different temperatures (298 K, 308 K and 318 K) to investigate the adsorption thermodynamic parameters. The adsorption capacity (Qe; mmol g−1) and removal efficiency (R; %) were calculated using eqn (1) and (2), respectively.| | |
R = (C0 − Ce)/C0 × 100%
| (2) |
where C0 and Ce represent the initial and equilibrium concentrations of the metallic ions (mmol L−1), respectively; V is the volume of the solution (20 mL), and m is the mass of adsorbent dosage (20 mg).
Regeneration of DTC-PGMA@SiO2 adsorbent
The adsorbent loaded with heavy metal ions was immersed in 0.2 mol L−1 HNO3 solution for 2 h. Next, the adsorbent was successively washed with 0.1 mol L−1 EDTA solution, deionized water and acetone. After the centrifugal separation at 11000 rpm for 5 min, the regenerated DTC-PGMA@SiO2 nanocomposite was dried under vacuum. The adsorption performance of regenerated DTC-PGMA@SiO2 nanocomposite was evaluated in heavy metal ion solution having an initial concentration of 200 mg L−1 (the adsorption was performed as above-mentioned isothermal adsorption experiment at 25 °C, pH = 5.0).
Results and discussion
FTIR analysis
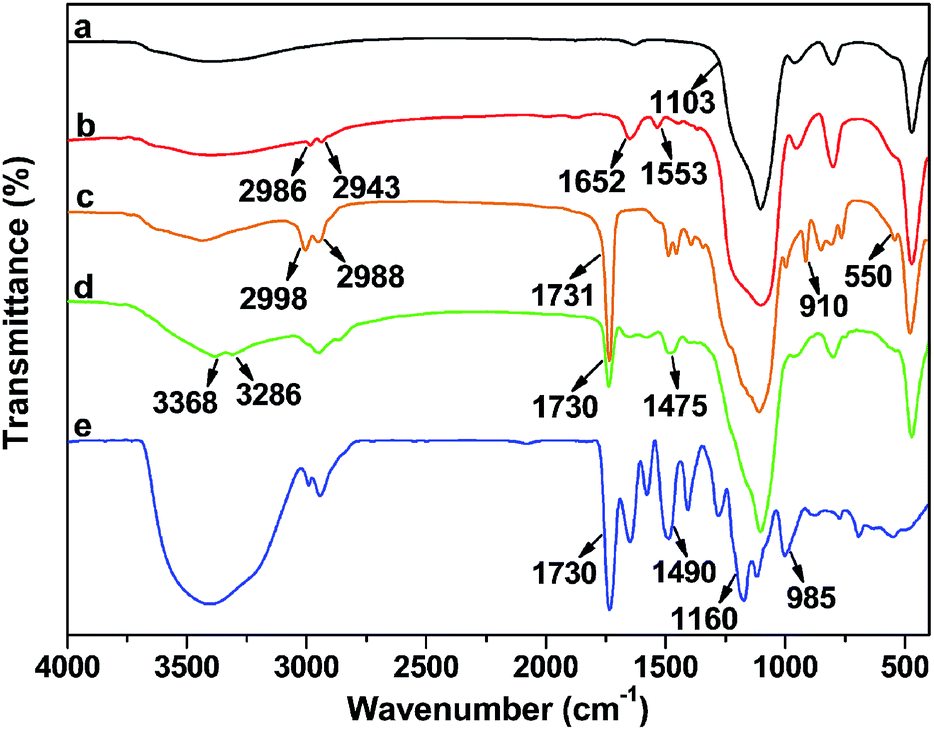
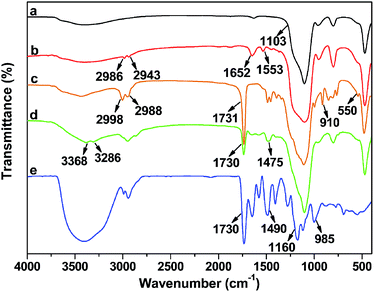
Each step in the preparation of DTC-PGMA@SiO2 adsorbent was monitored by FTIR spectra (see Fig. 1). The strong adsorption band at 1103 cm−1 was assigned to Si–O–Si stretching vibration of SiO2 substrate. For Br@SiO2, the appearance of adsorption bands at 1652 cm−1 and 1553 cm−1 represented the amide condensation reaction between NH2@SiO2 and BiBB. Moreover, the appearance of weak C–Br band at 550 cm−1 also demonstrated the BiBB immobilization on SiO2. Compared with Br@SiO2, the PGMA@SiO2 showed strong adsorption peaks at 2998 and 2988 cm−1, which were attributed to aliphatic C–H stretching vibration of PGMA brushes. In addition, the characteristic signals for GMA at 910 (epoxy group) and 1731 cm−1 (carbonyl stretching vibration of the ester group) were observed.45 For NH2-PGMA@SiO2, both the disappearance of characteristic peak for epoxy group and the appearance of adsorption bands at 3368, 3286 and 1475 cm−1 (N–H stretching vibration of primary amino group and C–N stretching vibration) represented the introduction of amino groups via an exhaustive ring-opening reaction between PGMA and EDA. The spectrum of DTC-PGMA@SiO2 showed remarkable characteristic signals for DTC group at 1490 (N–CS2 vibration), 985 (C–S vibration) and 1160 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S vibration),22 indicating that the DTC groups have been successfully introduced in the polymer brushes.
S vibration),22 indicating that the DTC groups have been successfully introduced in the polymer brushes.
 |
| | Fig. 1 FTIR spectra of bare SiO2 (a), Br@SiO2 (b), PGMA@SiO2 (c), NH2-PGMA@SiO2 (d) and DTC-PGMA@SiO2 (e) samples. | |
XPS determination and elemental analysis of nanocomposites
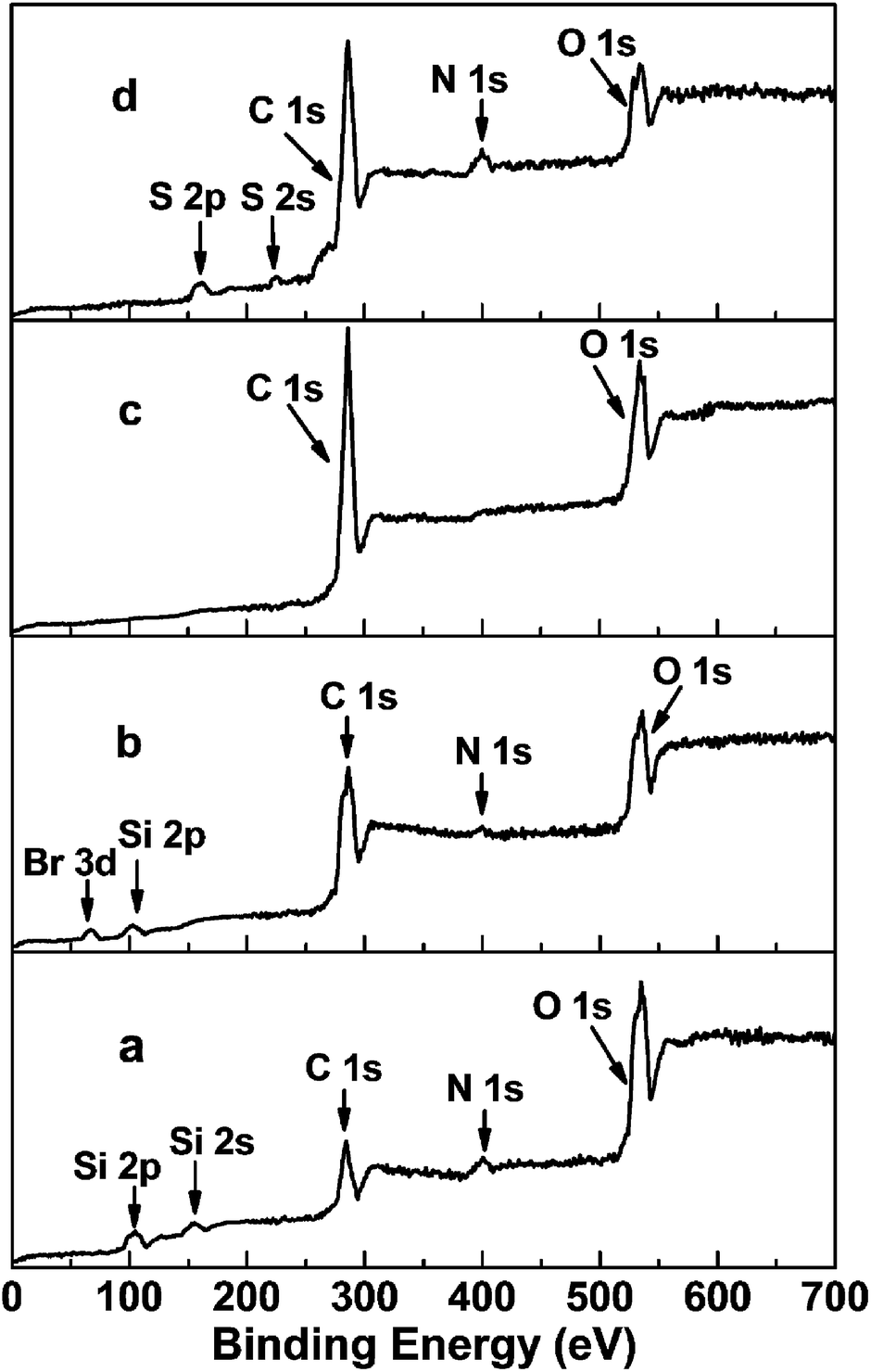
The XPS spectra of NH2@SiO2, Br@SiO2, PGMA@SiO2 and DTC-PGMA@SiO2 have been shown in Fig. 2. The peaks of Si 2p, Si 2s, C 1s, N 1s and O 1s were identified in the NH2@SiO2 spectrum. The survey spectrum of Br@SiO2 confirms the presence of Br (Br 3d peak at about 69 eV).46 For PGMA@SiO2, the remarkable increase in the intensity of C 1s and the disappearance of Si 2p, Si 2s and Br 3d peaks implied that the high-density PGMA brushes were grafted on the surface of SiO2 nanoparticles. The signals of S 2s and N 1s in the XPS spectrum of DTC-PGMA@SiO2 further confirmed the introduction of DTC groups. The elemental analysis results (in Table S1†) showed the content of C increased significantly due to the grafting of PGMA. The N content was strongly enhanced by the ring-opening reaction occurring between GMA and EDA. The drastic increase of S content from zero to 32.38% confirmed the introduction of abundant DTC groups (according to sulphur content, the calculated quantity of DTC group is 6.52 mmol g−1).
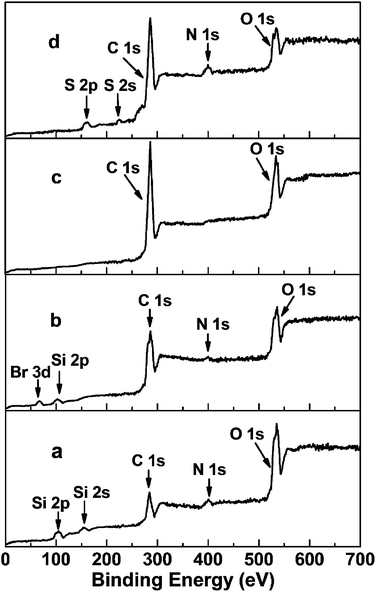 |
| | Fig. 2 XPS survey spectra of NH2@SiO2 (a), Br@SiO2 (b), PGMA@SiO2 (c) and DTC-PGMA@SiO2 (d) samples. | |
Molecular structure characterization of PGMA brushes
The synthesized model sacrificial initiator BMPA has very similar initiating chemical structure with the grafted-initiator (see Fig. S1–S3†). It is widely accepted that the ATRP initiated by the model sacrificial initiator shows almost similar polymerization behaviour as the SI-ATRP performed on a solid support.47–49 Therefore, the obtained free polymer brushes were generally used to evaluate the control of SI-ATRP experiments. The chemical structure of PGMA brushes was confirmed by 1H NMR (see Fig. S4†). The GPC traces of free PGMA brushes with various [GMA]:[BMPA]:[CuBr]:[PMDETA] ratios were detected (see Fig. 3), wherein the molecular weight and polydispersity were found to decrease with the increasing initiator content. As shown in Fig. S5,† the kinetic data of polymerization ([GMA]:[BMPA]:[CuBr]:[PMDETA] is [300]:[1]:[1]:[1]) was found to follow the first-order kinetic model. Fig. S6† displayed the evolution of Mn and PDI with the monomer conversion. It was found that the Mn of PGMA brush increased linearly with the increase of GMA monomer conversion. Additionally, the narrow PDI values illustrated that the SI-ATRP experiments were well-controlled, and therefore well-defined PGMA brushes were expected to be fabricated on the substrate surface. Ultimately, PGMA3 brushes having the highest molecular weight were selected to perform the present research.
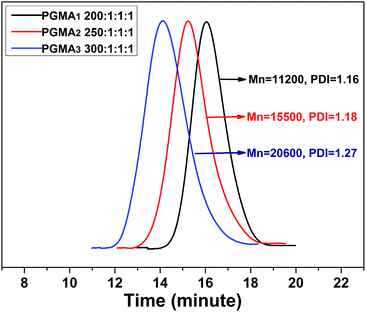 |
| | Fig. 3 GPC traces of free PGMA brushes synthesized at different [GMA]:[BMPA]:[CuBr]:[PMDETA] molar ratio. | |
TGA measurements of nanocomposites
TGA is one of the most appropriate methods to evaluate the thermal stability and graft ratios of the materials.50 As shown in Fig. 4, the weight loss increased stage-by-stage from bare SiO2 substrate to DTC-PGMA@SiO2. A significant weight loss in PGMA@SiO2 (ca. 62.4%) was observed after 240 °C, which demonstrated a high-density grafting of PGMA brushes. Both NH2-PGMA@SiO2 and DTC-PGMA@SiO2 displayed another thermal decomposition at around 320 °C, which implied that a new chemical structure has emerged following the ring-opening reaction. The final weight loss ratios of NH2-PGMA@SiO2 and DTC-PGMA@SiO2 were found to be 76.2% and 84.2%, respectively. These results indicated complete evolution of ring opening reaction and DTC functionalization.
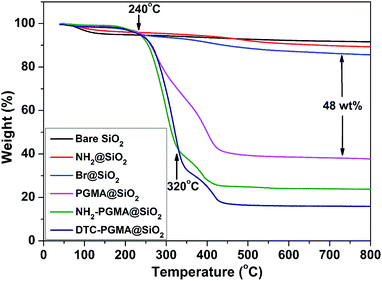 |
| | Fig. 4 TGA curves of synthesized nanocomposites. | |
Morphology observations of nanocomposites
TEM micrographs of bare SiO2, PGMA@SiO2 and DTC-PGMA@SiO2 are displayed in Fig. 5a–c respectively. Compared with the bare SiO2, the PGMA@SiO2 clearly showed the grafted polymer brushes, while the agglomeration of nanocomposites turned obvious. This is due to the reason that water is a poor solvent for PGMA. So, the brushes curl to form links between PGMA@SiO2 nanocomposites in water, which exacerbate the agglomeration. After the DTC modification, all of the nanocomposites in water seemed to be finely dispersed and appeared to be much bigger than before (the size of DTC-PGMA@SiO2 is about 200 nm in water). This is due to the reason that the negatively charged DTC-modified polymer brushes exhibit super hydrophilicity. Besides, these have electrostatic repulsions in between them. Accordingly, these brushes become stretched in water and hence, the dispersibility of nanocomposites is remarkably improved. In this form, the DTC-PGMA@SiO2 adsorbent exhibits unique advantages to capture heavy metal ions from aqueous environments.
 |
| | Fig. 5 TEM images of bare SiO2 nanoparticles (a), PGMA@SiO2 (b) and DTC-PGMA@SiO2 (c) dispersed in water. Water contact angle measurements on bare SiO2 (d), PGMA@SiO2 (e) and DTC-PGMA@SiO2 (f) sample disks are shown. | |
Measurement of water contact angle of nanocomposites
Materials generally show significant difference in wettability following surface modification.51,52 The wettability of sample disks including SiO2, PGMA@SiO2 and DTC-PGMA@SiO2 (Fig. 5d–f, respectively) was assessed. For bare SiO2, the Si–OH groups make the surface hydrophilic (water contact angle of around 28°). However, the majority of Si–OH groups are consumed during the condensation reaction involving Si–OC2H5 groups of APTES. Additionally, the grafting of high-density hydrophobic PGMA brushes leads to a much more hydrophobic surface of PGMA@SiO2 (water contact angle of around 87°). However, the water drops were found to readily wet the surface of DTC-PGMA@SiO2 disk. A transition to a super hydrophilic surface is observed on the DTC-PGMA@SiO2 disk as the contact angle decreases to 9°. Thus, the changes in wettability and TEM observations provide useful complementary information, testifying that the design of nanostructured DTC-PGMA@SiO2 adsorbent occurred as expected.
Effect of pH on adsorption
The surface charge of the adsorbent, ionization of functional groups and speciation of heavy metal ions are strongly affected by the pH of environment. Consequently, the pH effect on the removal of heavy metal ions using DTC-PGMA@SiO2 was investigated. As shown in Fig. 6a, poor uptake of metallic ions was observed in strongly acidic environment (pH < 3.0), while it increased remarkably for a higher pH value. The maximum uptake of Cu2+, Pb2+ and Cd2+ (3.45, 1.97 and 1.56 mmol g−1, respectively) was observed at a pH of 5.0. To elaborate this phenomenon, the pH point of zero charge (pHPZC) of the nanostructured DTC-PGMA@SiO2 adsorbent was measured via zeta potential method (see Fig. 6b). The pHPZC was found to be about 4.3. It is well-known that the adsorbent surface is positively charged at pH < pHPZC, while it is negatively charged at pH > pHPZC.53,54 Under low pH conditions (pH < pHPZC), the functional groups (amine and DTC groups on the polymer brushes) were protonated, indicating that abundant H+ ions would dominantly compete with heavy metal ions for binding these adsorptive sites. Conversely, the removal of metallic ions was facilitated significantly with an increase in the pH value. However, the generation of precipitates of metallic hydroxides obviously disturbed the accurate evaluation of adsorption capacity at higher pH values (pH ≥ 6.0). Therefore, pH = 5.0 was chosen to perform further experiments to study the adsorption behaviour.
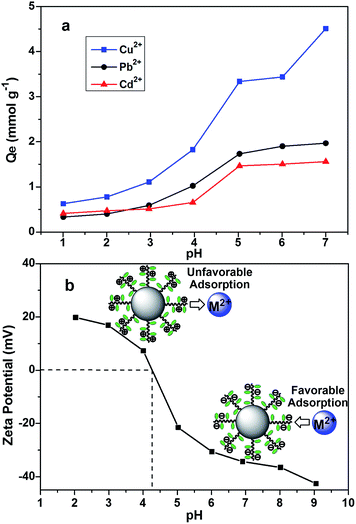 |
| | Fig. 6 (a) Adsorption capacity as a function of the pH for the studied metal ions. (b) Zeta potential of the DTC-PGMA@SiO2 suspension as a function of the pH. | |
Adsorption kinetics
In order to investigate the performance of DTC-PGMA@SiO2 adsorbent, the adsorption kinetics for metallic ions (including Cu2+, Pb2+ and Cd2+) were examined. As shown in Fig. 7a, the time to reach equilibrium was found to be less than 30 min for the investigated ions. Such a rapid adsorption is ascribed to the large specific surface area and the strong metal affinity of DTC groups. Remarkably, for Pb2+, the adsorption equilibrium time is only about 15 min. This is due to the smaller ionic radius of hydrated Pb2+ (4.01 Å) compared to those of Cu2+ (4.19 Å) and Cd2+ (4.26 Å),55,56 resulting in an easier diffusion of Pb2+ to the adsorption sites.22,57 In order to further understand the mechanism of adsorption process, linear pseudo-first-order (PFO) and pseudo-second-order (PSO) models were used to fit the kinetic data. The governing equations for the models are given by eqn (3) and (4), respectively.43,58| | |
ln(Qe − Qt) = lnQe − k1t
| (3) |
| | |
t/Qt = 1/(k2Qe2) + t/Qe
| (4) |
where Qe and Qt (mmol g−1) are the amounts of adsorbed metal ions at equilibrium and at test time t (min), respectively. In eqn (3) and (4), k1 (min−1) and k2 (g mmol−1 min−1) are the relevant rate constants.
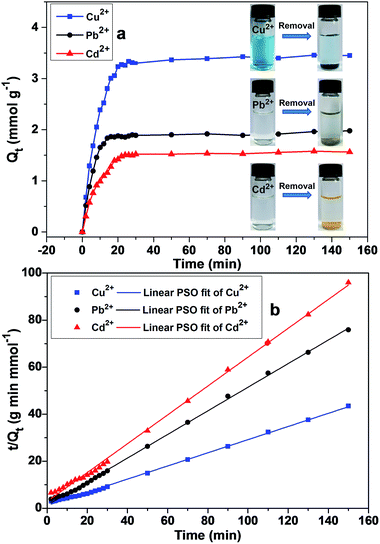 |
| | Fig. 7 (a) Adsorption kinetics of metal ions on DTC-PGMA@SiO2 at 298 K. (b) Linear fitting plots of kinetic data using a pseudo-second-order model. | |
Linear fitting plots were shown in Fig. 7b (fitting plots using PFO model were shown in Fig. S7†) and the corresponding fitting parameters were reported in Table 1. According to R2 values, PSO represented the experimental data better than the PFO model. These results demonstrated that the adsorption process of metallic ions onto the DTC-PGMA@SiO2 adsorbent could well be explained by the pseudo-second-order kinetic model. This also indicated that the rate-limiting step should be the chemisorption of adsorbate on the adsorbent represented by the valence force through an exchange of electrons between DTC ligands and metallic ions.22,56 It is interesting to note that the chelate complexes show different colour changes depending upon the adsorbed ions (see Fig. 7a), which due to the charge transfer between ligand and metal ions.59 Consequently, DTC-PGMA@SiO2 can be used as a potential sensor to identify the contaminating ions in water.
Table 1 Kinetic data obtained by PFO and PSO models for the adsorption of heavy metal ions on the nanostructured DTC-PGMA@SiO2 adsorbent at 298 K
| Model |
Metal ion |
Fitting parameters |
| R2 |
Qe (mmol g−1) |
K1 (min−1) |
| PFO |
Cu2+ |
0.298 |
10.11 |
0.0011 |
| Pb2+ |
0.194 |
11.02 |
0.0037 |
| Cd2+ |
0.314 |
11.51 |
0.0042 |
| Model |
Metal ion |
Fitting parameters |
| R2 |
Qe (mmol g−1) |
K2 (mmol g−1 min−1) |
| PSO |
Cu2+ |
0.998 |
3.57 |
0.167 |
| Pb2+ |
0.999 |
1.99 |
0.203 |
| Cd2+ |
0.998 |
1.60 |
0.131 |
Adsorption isotherms
The study of adsorption isotherms gives a more profound understanding of the adsorbent's performance. Fig. 8 displayed the adsorption isotherms of Cu2+, Pb2+ and Cd2+ on the nanostructured DTC-PGMA@SiO2 at 298 K. It can be seen that the uptake of heavy metal ions showed a steeper curve at low concentrations, which gradually reached a plateau at higher concentrations. The adsorption of Cu2+ (3.60 mmol g−1) was found to be higher than those of Pb2+ (1.96 mmol g−1) and Cd2+ (1.47 mmol g−1). The Langmuir and Freundlich empirical isotherm models were fitted to the experimental data. The Langmuir isotherm describes the adsorption on a homogeneous surface, assuming equal adsorption sites, a monolayer surface coverage of adsorbate and no interaction between the adsorbed ions.59 The Freundlich model depicts an adsorption on a heterogeneous surface, possessing discriminatory adsorption sites and a binding affinity which decreases with the increase in adsorption degree.60 The Langmuir and Freundlich isotherms are represented by eqn (5) and (6), respectively.| | |
Ce/Qe = Ce/Qm + 1/Qm KL
| (5) |
| | |
lnQe = lnKF + 1/nlnCe
| (6) |
where Qe (mmol g−1) is the adsorption capacity, Qm (mmol g−1) is the maximum adsorption capacity, Ce (mmol L−1) is the equilibrium concentration of heavy metal ion in the solution, KF is a constant reflecting the binding affinity and KL is the Langmuir adsorption equilibrium constant.
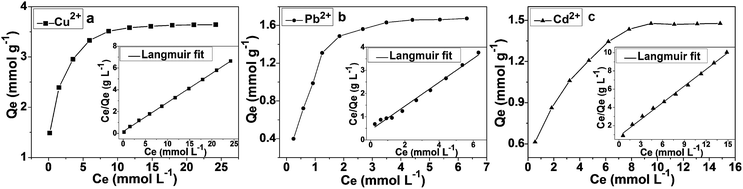 |
| | Fig. 8 Adsorption isotherms and relative Langmuir linear fits for the adsorption of Cu2+ (a), Pb2+ (b) and Cd2+ (c) on DTC-PGMA@SiO2 at 298 K. | |
The corresponding results for the fit of Langmuir and Freundlich models have been shown in Fig. 8 and S8,† respectively. Comparing the R2 values in Table 2, Langmuir model shows better fit for the experimental data, which demonstrates that the adsorption of metal ions on the DTC-PGMA@SiO2 adsorbent is a monolayer, chemisorption onto a homogeneous surface. The Dubinin–Radushkevitch (D–R) eqn (7) was used to further describe the metal ions' adsorption process.
| | |
lnQe = lnQm − kε2
| (7) |
where
k (mol
2 kJ
−2) is a constant related to the average adsorption energy (
E) and
ε is equal to [
RTln(1 + 1/
Ce)]. Furthermore, the
E value can be calculated from the
k value using
eqn (8).
Table 2 Isothermal parameters obtained by using Langmuir and Freundlich models for the adsorption of heavy metal ions on the nanostructured DTC-PGMA@SiO2 adsorbent at 298 K
| Model |
Metal ion |
Fitting parameters |
| R2 |
Qm (mmol g−1) |
KL (L mmol−1) |
| Langmuir |
Cu2+ |
0.999 |
3.75 |
1.526 |
| Pb2+ |
0.990 |
1.94 |
0.991 |
| Cd2+ |
0.996 |
1.64 |
0.727 |
| Model |
Metal ion |
Fitting parameters |
| R2 |
n |
KF (mmol1−1/n L1/n g−1) |
| Freundlich |
Cu2+ |
0.921 |
5.39 |
2.19 |
| Pb2+ |
0.853 |
2.20 |
0.90 |
| Cd2+ |
0.935 |
3.52 |
0.75 |
In the present work, E values for the studied metallic ions were found to lie within a range of 18–23 kJ mol−1 (see Table S2†). This suggests that a strong binding interaction occurs during the adsorption process rather than a simple ion exchange. DTC is well-known to be an anionic dithio ligand, which could form stable complexes with heavy metal ions containing partially and fully filled d orbitals through π-coordinated interactions. From the ligand field stabilization energy theory, d9 type metal ions (such as Cu2+) have stronger stabilization energy with respect to d10 type ions (such as Pb2+ and Cd2+) and therefore, a more significant coordination interaction can be achieved for Cu2+.61 Differences in the adsorption of Pb2+ and Cd2+ mainly rely on their electronegativity, which are 2.33 for Pb and 1.7 for Cd. The higher electronegativity of Pb reflects its higher tendency to share electrons with respect to Cd, thus favouring its adsorption on DTC-PGMA@SiO2 adsorbent.22 Therefore, the capacity of DTC-PGMA@SiO2 adsorbent for capturing the studied metallic ions can be represented as Cu2+ > Pb2+ > Cd2+. The results showed that the reported DTC-PGMA@SiO2 adsorbent shows an outstanding adsorption performance including remarkable adsorption capacity and rapid adsorption rate, which due to the sea anemone like core-brush structure, the strong binding affinity between metal ions and DTC ligand and the high specific area. The comparison between DTC-PGMA@SiO2 nanocomposite and some other nanomaterials regarding to the adsorption performance of Cu2+, Pb2+ and Cd2+ from aqueous environment was listed in Table 3.
Table 3 Comparison of the adsorption performance of DTC-PGMA@SiO2 nanocomposite and some other nanomaterials
| Adsorbent materials |
Adsorption capacity (mmol g−1) |
Equilibrium time (min) |
pH conditions |
Reference |
| Cu2+ |
Pb2+ |
Cd2+ |
| Polyvinyltetrazole grafted resin |
2.65 |
1.52 |
— |
90–120 |
5.0 |
43 |
| Magnesium titanate nanorod |
— |
1.16 |
— |
10 |
5.0 |
53 |
| Mesoporous organosilica embedded with carbon dots |
1.72 |
0.68 |
— |
30 |
7.0 |
56 |
| Graphene oxide membrane |
1.14 |
— |
0.75 |
20 |
5.7 |
62 |
| PVA/graphene oxide nanofiber |
0.51 |
— |
0.40 |
30 |
5.8 |
63 |
| Graphene oxide functionalized with ethylenediamine triacetic acid |
1.71 |
2.19 |
— |
30 |
3.0 for Pb2+; 5.0 for Cu2+ |
64 |
| Hydrogel-supported nanosized hydrous manganese dioxide |
0.857 |
0.972 |
0.835 |
150 |
5.0 for Cu2+; 5.0 for Pb2+; 5.5 for Cd2+ |
65 |
| Soy protein hollow microsphere |
1.02 |
0.62 |
0.74 |
240 |
5.5 |
66 |
| DTC-PGMA@SiO2 |
3.60 |
1.95 |
1.47 |
10–30 |
5.0 |
Present work |
Adsorption thermodynamics
Adsorption thermodynamics were investigated through temperature-dependent isotherms (see Fig. 9a–c). Van't Hoff equation, as given by eqn (9), is used to calculate the adsorption Gibbs free energy (ΔG; kJ mol−1), adsorption enthalpy (ΔH; kJ mol−1) and adsorption entropy (ΔS; J mol−1 K−1)| | |
lnKc = −ΔH/RT + ΔS/R
| (9) |
where Kc is the adsorption equilibrium constant obtained from Langmuir isotherms at various temperatures, R is the ideal gas constant (8.314 J mol−1 K−1), and T is the absolute temperature (K).
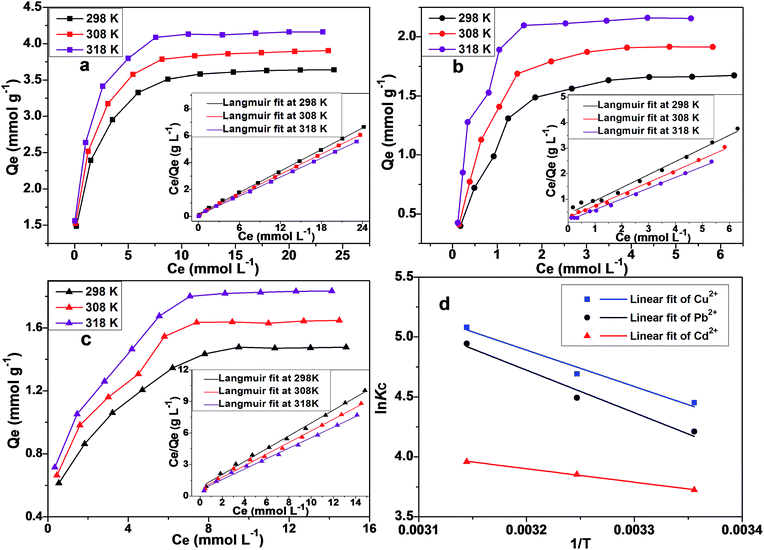 |
| | Fig. 9 Adsorption isotherms and Langmuir linear fits for the adsorption of Cu2+ (a), Pb2+ (b) and Cd2+ (c) at 298 K, 308 K and 318 K. (d) The linear fitting plots for lnKc versus 1/T according to Van't Hoff equation. | |
The values of ΔH and ΔS can be geometrically extracted from the linear fit of lnKc versus 1/T (as shown in Fig. 9d). ΔG can be calculated from ΔH and ΔS values by using eqn (10).
The thermodynamic parameters for the studied metallic ions are tabulated in Table S3.† Since ΔG values are more negative at higher temperatures, therefore the spontaneous adsorption is favoured by high temperatures. Positive ΔH values indicate an endothermic adsorption process.67 The calculated ΔS values also demonstrate that the adsorption process is favoured by the entropy.
Adsorption mechanism
The adsorption mechanism of heavy metal ions using DTC-PGMA@SiO2 adsorbent is relied on synergistic combination of electrostatic interaction (between metallic ions and functional groups such as amino and DTC groups) and some other stronger chemical interactions. On the basis of hard and soft acids and bases theory, the adsorption may be dominated by the coordination interaction between metal ions and DTC groups. In order to confirm this, FTIR and high-resolution XPS analysis of Cu2+/DTC-PGMA@SiO2 complex were performed. The red-shift and the intensity reduction of characteristic FTIR DTC signals (C–S and CS stretching vibrations) following the adsorption of Cu2+, clearly demonstrated that the coordination interactions between Cu2+ and DTC groups actually occurred (see Fig. 10a). In XPS spectrum of Cu 2p (see Fig. 10b), Cu 2p3/2 (932.5 eV) and Cu 2p1/2 (952.2 eV) peaks were identified, indicating the presence of Cu(II).68 The S 2p3/2 (161.9 eV) and S 2p1/2 (163.3 eV) peak doublet contributed to the unique DTC sulphur signal (see Fig. 10c), which is consistent with the reported DTC–metal ion interaction.68–70 Additionally, the highly coloured chelate complex and the discussed adsorption preference could be regarded as other proofs. By comparing the FTIR spectra and S 2p XPS signal of Cu2+/DTC-PGMA@SiO2 complex, Pb2+/DTC-PGMA@SiO2 complex and Cd2+/DTC-PGMA@SiO2 complex, we found no significant differences. Therefore, the adsorption mechanism of the investigated heavy metal ions using DTC-PGMA@SiO2 adsorbent can be summarized as a unified principle. To summarize, these results confirmed that the metallic ions can be adsorbed on DTC-PGMA@SiO2 via electrostatic interaction and coordination interaction with DTC groups.
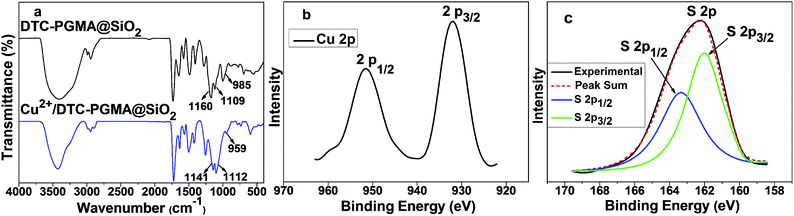 |
| | Fig. 10 (a) FTIR spectra of DTC-PGMA@SiO2 adsorbent and the Cu2+/DTC-PGMA@SiO2 complex. High-resolution XPS data of Cu 2p (b) and S 2p (c) signals of the Cu2+/DTC-PGMA@SiO2 complex. | |
Regeneration of DTC-PGMA@SiO2 adsorbent
The regeneration features of DTC-PGMA@SiO2 adsorbent for repeated use were investigated. The pH factor has obviously been taken into account, due to which, the pH has proven to be a remarkable influencing factor in the binding interactions between DTC groups and metallic ions. Because of the significant chelating ability of EDTA, it was also used to further improve the metallic ions' desorption from the adsorbent. After four reuse cycles, the removal efficiencies for Cu2+, Pb2+ and Cd2+ were 85.7%, 84.3% and 63.5% respectively, which decreased slightly compared with the corresponding values for the first cycle (see Fig. S9†). These results demonstrated that an effective regeneration of DTC-PGMA@SiO2 can be achieved, which makes it sustainable and economically valuable adsorbent for wastewater treatment.
Conclusions
In summary, a novel nanostructured DTC-PGMA@SiO2 adsorbent material containing well-defined core-brush structure was synthesized via SI-ATRP and subsequent DTC group functionalization. The high-density (grafting ratio is 62.4%) and narrow-distribution polymer brushes, which contain abundant DTC groups (6.52 mmol g−1), were identified by combining multiple techniques. The nanostructured DTC-PGMA@SiO2 adsorbent exhibits remarkable adsorption performance for d9 and d10 type heavy metal ions, including considerable adsorption capacities (the adsorption capacities for Cu2+, Pb2+ and Cd2+ were found to be 3.60, 1.96 and 1.47 mmol g−1, respectively) and rapid adsorption equilibriums (within 30 min), which were attributed to the sea anemone-like core-brush structure, large specific surface area and high affinity between DTC groups and metallic ions. The adsorption process can be well-described by the pseudo-second-order and Langmuir models, indicating a spontaneous monolayer chemisorption occurring on the adsorbent surface. The adsorption mechanism between metal ions and DTC-PGMA@SiO2 adsorbent relies on synergistic combination of electrostatic interaction and coordination interaction between heavy metal ions and DTC groups. Additionally, the effective regeneration was achieved. These results highlight the promising potential of DTC-PGMA@SiO2 for treating water bodies contaminated by heavy metal ions.
Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (Grant No. 21374055) and the Major Research of Science and Technology, China (Grant No. 2016ZX05025-003).
Notes and references
- C. J. Vörösmarty, P. B. McIntyre, M. O. Gessner, D. Dudgeon, A. Prusevich, P. Green, S. Glidden, S. E. Bunn, C. A. Sullivan, C. R. Liermann and P. M. Davies, Nature, 2010, 467, 555–561 CrossRef PubMed.
- Q. Peng, J. Guo, Q. Zhang, J. Xiang, B. Liu, A. Zhou, R. Liu and Y. Tian, J. Am. Chem. Soc., 2014, 136, 4113–4116 CrossRef CAS PubMed.
- W.-H. Leung, P.-K. So, W.-T. Wong, W.-H. Lo and P.-H. Chan, RSC Adv., 2016, 6, 106837–106846 RSC.
- A. Eichler, L. Tobler, S. Eyrikh, N. Malygina, T. Papina and M. Schwikowski, Environ. Sci. Technol., 2014, 48, 2635–2642 CrossRef CAS PubMed.
- R. M. Izatt, S. R. Izatt, R. L. Bruening, N. E. Izatt and B. A. Moyer, Chem. Soc. Rev., 2014, 43, 2451–2475 RSC.
- Z. Li, J. Chen, H. Guo, X. Fan, Z. Wen, M. H. Yeh, C. Yu, X. Cao and Z. L. Wang, Adv. Mater., 2016, 28, 2983–2991 CrossRef CAS PubMed.
- J. Tang, B. Mu, M. Zheng and A. Wang, ACS Sustainable Chem. Eng., 2015, 3, 1125–1135 CrossRef CAS.
- J. Yang, K. Yu and C. Liu, J. Hazard. Mater., 2017, 321, 73–80 CrossRef CAS PubMed.
- B. L. Rivas, E. D. Pereira, M. Palencia and J. Sánchez, Prog. Polym. Sci., 2011, 36, 294–322 CrossRef CAS.
- S. A. El-Safty, S. Abdellatef, M. Ismael and A. Shahat, Adv. Healthcare Mater., 2013, 2, 854–862 CrossRef CAS PubMed.
- F. Liu, C. Shan, X. Zhang, Y. Zhang, W. Zhang and B. Pan, J. Hazard. Mater., 2017, 321, 290–298 CrossRef CAS PubMed.
- D. Nagai, M. Yoshida, T. Kishi, H. Morinaga, Y. Hara, M. Mori, S. Kawakami and K. Inoue, Chem. Commun., 2013, 49, 6852–6854 RSC.
- M. M. Nasef and O. Güven, Prog. Polym. Sci., 2012, 37, 1597–1656 CrossRef CAS.
- J. Zhou, V. W.-C. Chang and A. G. Fane, Energy Environ. Sci., 2011, 4, 2267–2278 CAS.
- D. Godinho, D. Dias, M. Bernardo, N. Lapa, I. Fonseca, H. Lopes and F. Pinto, J. Hazard. Mater., 2017, 321, 173–182 CrossRef CAS PubMed.
- M. Liu, B. Zhang, H. Wang, F. Zhao, Y. Chen and Q. Sun, RSC Adv., 2016, 6, 67057–67071 RSC.
- J. Zhao, Z. Li, J. Wang, Q. Li and X. Wang, J. Mater. Chem. A, 2015, 3, 15124–15132 CAS.
- A. Farrukh, A. Akram, A. Ghaffar, S. Hanif, A. Hamid, H. Duran and B. Yameen, ACS Appl. Mater. Interfaces, 2013, 5, 3784–3793 CAS.
- Z. Sekhavat Pour and M. Ghaemy, RSC Adv., 2015, 5, 64106–64118 RSC.
- C. Chen, R. Wang, L. Guo, N. Fu, H. Dong and Y. Yuan, Org. Lett., 2011, 13, 1162–1165 CrossRef CAS PubMed.
- B. Ochiai, T. Ogihara, M. Mashiko and T. Endo, J. Am. Chem. Soc., 2009, 131, 1636–1637 CrossRef CAS PubMed.
- Y. Ge, D. Xiao, Z. Li and X. Cui, J. Mater. Chem. A, 2014, 2, 2136–2145 CAS.
- C. Gai, Y. Guo, N. Peng, T. Liu and Z. Liu, RSC Adv., 2016, 6, 53713–53722 RSC.
- F. Di Natale, A. Erto, A. Lancia and D. Musmarra, J. Hazard. Mater., 2015, 281, 47–55 CrossRef CAS PubMed.
- D. S. Tavares, C. B. Lopes, A. L. Daniel-da-Silva, A. C. Duarte, T. Trindade and E. Pereira, Chem. Eng. J., 2014, 254, 559–570 CrossRef CAS.
- P. Z. Ray and H. J. Shipley, RSC Adv., 2015, 5, 29885–29907 RSC.
- M. M. Khin, A. S. Nair, V. J. Babu, R. Murugan and S. Ramakrishna, Energy Environ. Sci., 2012, 5, 8075 CAS.
- J. Zhang and Y. Chen, RSC Adv., 2016, 6, 69370–69380 RSC.
- H. Gao, Y. Sun, J. Zhou, R. Xu and H. Duan, ACS Appl. Mater. Interfaces, 2013, 5, 425–432 CAS.
- M. Y. Nassar and S. Abdallah, RSC Adv., 2016, 6, 84050–84067 RSC.
- F. Perreault, A. Fonseca de Faria and M. Elimelech, Chem. Soc. Rev., 2015, 44, 5861–5896 RSC.
- J. J. Alcaraz-Espinoza, A. E. Chávez-Guajardo, J. C. Medina-Llamas, C. A. Andrade and C. P. de Melo, ACS Appl. Mater. Interfaces, 2015, 7, 7231–7240 CAS.
- N. Zhang, G. L. Zang, C. Shi, H. Q. Yu and G. P. Sheng, J. Hazard. Mater., 2016, 316, 11–18 CrossRef CAS PubMed.
- D. Yang, X. Pang, Y. He, Y. Wang, G. Chen, W. Wang and Z. Lin, Angew. Chem., Int. Ed., 2015, 54, 12091–12096 CrossRef CAS PubMed.
- D. Zheng, X. Pang, M. Wang, Y. He, C. Lin and Z. Lin, Chem. Mater., 2015, 27, 5271–5278 CrossRef CAS.
- X.-Q. Liu, Y.-L. Li, Y.-W. Lin, S. Yang, X.-F. Guo, Y. Li, J. Yang and E.-Q. Chen, Macromolecules, 2013, 46, 8479–8487 CrossRef CAS.
- D. Kundu, C. Hazra, A. Chatterjee, A. Chaudhari, S. Mishra, A. Kharat and K. Kharat, RSC Adv., 2016, 6, 80438–80454 RSC.
- W. Hu, Y. Liu, T. Chen, Y. Liu and C. M. Li, Adv. Mater., 2015, 27, 181–185 CrossRef CAS PubMed.
- W. Mai, B. Sun, L. Chen, F. Xu, H. Liu, Y. Liang, R. Fu, D. Wu and K. Matyjaszewski, J. Am. Chem. Soc., 2015, 137, 13256–13259 CrossRef CAS PubMed.
- A. F. Hirschbiel, S. Geyer, B. Yameen, A. Welle, P. Nikolov, S. Giselbrecht, S. Scholpp, G. Delaittre and C. Barner-Kowollik, Adv. Mater., 2015, 27, 2621–2626 CrossRef CAS PubMed.
- Y. Yue, R. T. Mayes, J. Kim, P. F. Fulvio, X. G. Sun, C. Tsouris, J. Chen, S. Brown and S. Dai, Angew. Chem., Int. Ed., 2013, 52, 13458–13462 CrossRef CAS PubMed.
- S. Yuan, J. Gu, Y. Zheng, W. Jiang, B. Liang and S. O. Pehkonen, J. Mater. Chem. A, 2015, 3, 4620–4636 CAS.
- Y. Chen, M. He, C. Wang and Y. Wei, J. Mater. Chem. A, 2014, 2, 10444 CAS.
- M. Cvek, M. Mrlik, M. Ilcikova, T. Plachy, M. Sedlacik, J. Mosnacek and V. Pavlinek, J. Mater. Chem. C, 2015, 3, 4646–4656 RSC.
- J. Pan, J. Zeng, Q. Cao, H. Gao, Y. Gen, Y. Peng, X. Dai and Y. Yan, Chem. Eng. J., 2016, 284, 1361–1372 CrossRef CAS.
- B. Ernould, M. Devos, J.-P. Bourgeois, J. Rolland, A. Vlad and J.-F. Gohy, J. Mater. Chem. A, 2015, 3, 8832–8839 CAS.
- J. M. Bak and H.-I. Lee, Polymer, 2012, 53, 4955–4960 CrossRef CAS.
- J. Luo, F. Zhao, X. Fei, X. Liu and J. Liu, Chem. Eng. J., 2016, 293, 171–181 CrossRef CAS.
- S. G. J. Emmerling, L. B. N. Langer, S. A. Pihan, P. Lellig and J. S. Gutmann, Macromolecules, 2010, 43, 5033–5042 CrossRef CAS.
- S. Banerjee, T. K. Paira, A. Kotal and T. K. Mandal, Adv. Funct. Mater., 2012, 22, 4751–4762 CrossRef CAS.
- G. Yin, D. Zhao, L. Zhang, Y. Ren, S. Ji, H. Tang, Z. Zhou and Q. Li, Chem. Eng. J., 2016, 302, 1–11 CrossRef CAS.
- N. R. Ko, G. Sabbatier, A. Cunningham, G. Laroche and J. K. Oh, Macromol. Rapid Commun., 2014, 35, 447–453 CrossRef CAS PubMed.
- X. Wang, J. Cai, Y. Zhang, L. Li, L. Jiang and C. Wang, J. Mater. Chem. A, 2015, 3, 11796–11800 CAS.
- A. S. Krishna Kumar, S.-J. Jiang and W.-L. Tseng, J. Mater. Chem. A, 2015, 3, 7044–7057 CAS.
- E. R. Nightingale, J. Phys. Chem., 1959, 63, 1381–1387 CrossRef CAS.
- L. Wang, C. Cheng, S. Tapas, J. Lei, M. Matsuoka, J. Zhang and F. Zhang, J. Mater. Chem. A, 2015, 3, 13357–13364 CAS.
- X. Jing, F. Liu, X. Yang, P. Ling, L. Li, C. Long and A. Li, J. Hazard. Mater., 2009, 167, 589–596 CrossRef CAS PubMed.
- X. Wang, S. Jing, X. Qiu, S. Zhao, Y. Liu and Y. Tan, Chem. Eng. J., 2016, 304, 493–502 CrossRef CAS.
- M. L. Rahman, S. M. Sarkar, M. M. Yusoff and M. H. Abdullah, RSC Adv., 2016, 6, 745–757 RSC.
- J. Ma, Y. Shen, C. Shen, Y. Wen and W. Liu, Chem. Eng. J., 2014, 248, 98–106 CrossRef CAS.
- W. Liu, D. Wei, J. Mi, Y. Shen, B. Cui and C. Han, Chem. Eng. J., 2015, 277, 312–317 CrossRef CAS.
- P. Tan, J. Sun, Y. Hu, Z. Fang, Q. Bi, Y. Chen and J. Cheng, J. Hazard. Mater., 2015, 297, 251–260 CrossRef CAS PubMed.
- P. Tan, J. Wen, Y. Hu and X. Tan, RSC Adv., 2016, 6, 79641–79650 RSC.
- I. E. M. Carpio, J. D. Mangadlao, H. N. Nguyen, R. C. Advincula and D. F. Rodrigues, Carbon, 2014, 77, 289–301 CrossRef.
- Q. Zhu and Z. Li, Chem. Eng. J., 2015, 281, 69–80 CrossRef CAS.
- D. Liu, Z. Li, W. Li, Z. Zhong, J. Xu, J. Ren and Z. Ma, Ind. Eng. Chem. Res., 2013, 52, 11036–11044 CrossRef CAS.
- H. Chen, X. Wang, J. Li and X. Wang, J. Mater. Chem. A, 2015, 3, 6073–6081 CAS.
- R. Cao Jr, A. Díaz, R. Cao, A. Otero, R. Cea, M. C. Rodríguez-Argüelles and C. Serra, J. Am. Chem. Soc., 2007, 129, 6927–6930 CrossRef PubMed.
- K. Zargoosh, H. Abedini, A. Abdolmaleki and M. R. Molavian, Ind. Eng. Chem. Res., 2013, 52, 14944–14954 CrossRef CAS.
- D. Gao, F. Scholz, H. G. Nothofer, W. E. Ford, U. Scherf, J. M. Wessels, A. Yasuda and F. von Wrochem, J. Am. Chem. Soc., 2011, 133, 5921–5930 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The synthesis method of sacrificial initiator, bare SiO2 and Br@SiO2 nanoparticles, the 1H NMR and FTIR spectra of sacrificial initiator and PGMA (Fig. S1–S4); the kinetic plot of SI-ATRP (Fig. S5); the evolution of Mn and PDI versus monomer conversion (Fig. S6); the fitting plots of kinetic and isothermal data (Fig. S7 and S8); the results of reuse experiments (Fig. S9); the elemental analysis results (Table S1), the fitting parameters obtained by using D–R model (Table S2) and the thermodynamic parameters of adsorption process (Table S3). See DOI: 10.1039/c6ra28890a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab
*ab